

Abstract
Since the discovery and use of penicillin, the increase of antibiotic resistance among bacterial pathogens has become a major health concern. The most prevalent resistance mechanism in Gram-negative bacteria is due to β-lactamase expression. Class D β-lactamases are of particular importance due to their presence in multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. The class D enzymes were initially characterized by their ability to efficiently hydrolyze isoxazolyl-type β-lactams like oxacillin. Due to this substrate preference, these enzymes are traditionally referred to as oxacillinases or OXAs. However, this class is comprised of subfamilies characterized by diverse activities that include oxacillinase, carbapenemase, or cephalosporinase substrate specificity. OXA-1 represents one subtype of class D enzyme that efficiently hydrolyzes oxacillin, and OXA-24/40 represents another with weak oxacillinase, but increased carbapenemase, activity. To examine the structural basis for the substrate selectivity differences between OXA-1 and OXA-24/40, the X-ray crystal structures of deacylation-deficient mutants of these enzymes (Lys70Asp for OXA-1; Lys84Asp for OXA-24) in complexes with oxacillin were determined to 1.4 Å and 2.4 Å, respectively. In the OXA-24/40/oxacillin structure, the hydrophobic R1 side chain of oxacillin disrupts the bridge between Tyr112 and Met223 present in the apo OXA-24/40 structure, causing the main chain of the Met223-containing loop to adopt a completely different conformation. In contrast, in the OXA-1/oxacillin structure, a hydrophobic pocket consisting of Trp102, Met99, Phe217, Leu161, and Leu255 nicely complements oxacillin's nonpolar R1 side chain. Comparison of the OXA-1/oxacillin and OXA-24/40/oxacillin complexes provides novel insight on how substrate selectivity is achieved among subtypes of class D β-lactamases. By elucidating important active site interactions, these findings can also inform the design of novel antibiotics and inhibitors.
INTRODUCTION
β-Lactams, like penicillin and the cephalosporins, are the most widely prescribed class of antibiotics. However, their clinical use is threatened by the overuse and misuse of these drugs (1). Many bacteria are now able to evade and/or destroy β-lactams, thus giving rise to resistant strains of bacteria (2). Expression of β-lactamase enzymes is the most widespread mechanism of resistance to β-lactams (3). These enzymes efficiently hydrolyze the amide bond present in the defining four-membered β-lactam ring for which these molecules are named. β-Lactamases are categorized into four classes (A, B, C, and D) (4, 5). Classes A, C, and D utilize a nucleophilic serine-based mechanism of catalysis involving a two-step acylation and deacylation reaction, whereas the class B enzymes use a zinc-based mechanism for hydrolysis.
Class D β-lactamases are responsible for much of the β-lactam resistance in several clinically relevant pathogens, such as Acinetobacter spp. and Pseudomonas aeruginosa (6). OXA-1 was identified in the early 1970s (7). Due to their substrate preference for isoxazolyl-type penicillins, like oxacillin, the class D enzymes were referred to as oxacillinases, or OXAs (8). However, as more class D β-lactamases were discovered and characterized, the diversity in their substrate selectivity profiles became apparent. To date, >250 class D β-lactamases have been identified and are classified into three subfamilies according to these profiles (9). Narrow-spectrum enzymes (like OXA-1) exhibit a preference for penicillins, such as ampicillin and oxacillin. Extended-spectrum β-lactamases (ESBLs) are able to hydrolyze advanced-generation cephalosporins, in addition to penicillins (like OXA-14). Of particular concern is the increasing number of carbapenem-hydrolyzing class D β-lactamases (CHDLs), members of which include OXA-24/40, OXA-48, and OXA-23.
OXA-1 and OXA-24/40 are important class D enzymes that highlight the functional differences between the narrow-spectrum and CHDL subfamilies. Oxacillin is the preferred substrate for many class D β-lactamases, and excellent oxacillinase activity is observed for OXA-1 (kcat = 577 s−1), which also binds oxacillin with high affinity (Km = 31 μM), for a catalytic efficiency of 18.6 μM−1 s−1 (10). The reasons behind this substrate preference for the bulky oxacillin have remained elusive. Conversely for OXA-24/40, oxacillin has been reported to be a relatively poor substrate (kcat = 0.18 s−1) that binds with low affinity (Km of >1,000 μM), which is reflected in a 1.3 × 106-fold decrease in catalytic efficiency compared to OXA-1 (OXA-24 kcat/Km = 1.42 × 10−4 μM−1 s−1) (11).
The structural bases for oxacillinase activity and its distinction from carbapenemase activity are not well understood and remain areas of intense investigation. In OXA-24/40, a hydrophobic bridge formed by Tyr112 and Met223 is suggested to play a role in carbapenemase activity for this enzyme based on site-directed mutagenesis of residue Tyr112 (11). However, the subsequent structure determination of the CHDL OXA-48 revealed that a hydrophobic bridge was not present (12) and instead suggested that the β5-β6 loop may determine carbapenemase activity. Recent studies have further confirmed the importance of the β5-β6 loop in carbapenemases (13). Furthermore, the poor oxacillinase activity of OXA-24/40 is similarly attributed to the presence of the Tyr112-Met223 bridge. Modeling of oxacillin in the OXA-24/40 active site revealed that oxacillin would sterically clash with the hydrophobic barrier formed by the bridge residues (11).
To elucidate the structural bases behind the functional differences observed between the two enzymes and to further explore the role of the OXA-24/40 bridge, we determined the X-ray crystal structures of the oxacillinase OXA-1 and the CHDL OXA-24/40 in complexes with oxacillin to 1.4 Å and 2.4 Å, respectively. The overarching goal of these studies is to provide insight into substrate specificity in order to better inform β-lactam and β-lactamase inhibitor design against this diverse class of enzymes.
MATERIALS AND METHODS
Expression, purification, and crystallization of K70D OXA-1 and K84D OXA-24/40.
In order to capture the acyl-enzyme complexes with oxacillin, deacylation-deficient mutant enzymes, where the general base residues (Lys70 for OXA-1; Lys84 for OXA-24/40) were mutated to aspartate, were used in crystallographic experiments. K70D OXA-1 and K84D OXA-24/40 were expressed and purified as previously described (14, 15). Protein was concentrated to ∼10 mg/ml in 50 mM sodium phosphate, pH 7.0, and stored at 4°C. Several commercially available crystallization screens were used to identify crystallization conditions for K70D OXA-1. An initial hit from Molecular Dimensions Pact Premier Screen (MDSR-29-P1-19) yielded diffracting protein crystals with no microseeding required. Large single crystals grew at room temperature by hanging drop vapor diffusion, in which a 1:1 dilution was made with well buffer (20% polyethylene glycol 6000 [PEG 6000], 200 mM NaCl, 100 mM morpholineethanesulfonic acid [MES], pH 7.0), to a final enzyme concentration of 3.5 mg/ml. Resulting crystals were soaked in oxacillin at 1.2 mM and replaced over the equilibration buffer for 5 days. Crystals were harvested from the drop, briefly soaked in a cryoprotectant solution of well buffer supplemented with 25% (vol/vol) 2-methyl-2,4-pentanediol (MPD), and flash-cooled in liquid nitrogen. Crystals of K84D OXA-24/40 were grown by hanging drop vapor diffusion in a 6-μl drop containing 2.5 mg/ml of K84D mixed 1:1 with well buffer (100 mM HEPES, pH 7.5, 2% [vol/vol] PEG 400, 2.0 M ammonium sulfate). Crystals appeared within 2 days at 25°C. Crystals were harvested and soaked in a solution containing 0.5 mM oxacillin and 5% sucrose for approximately 3 min before flash-cooling in liquid nitrogen.
Structure determination.
Diffraction data were measured on the LS-CAT 21-ID-D beamline at the Advanced Photon Source (Argonne, IL) at 100K using a MarCCD detector. Reflections were indexed, integrated, and scaled using HKL2000 (16). The space group for K70D OXA-1 was P1 with four OXA-1 monomers in the asymmetric unit (Table 1). The space group for K84D OXA-24/40 was P41212 with one OXA-24/40 monomer in the asymmetric unit. The structures of both complexes were determined with Phaser (17). The wild-type apo structures of OXA-1 (PDB entry 1M6K) and OXA-24/40 (PDB entry 2JC7) were used as the initial phasing models (all water and ligand molecules removed). Refinement and electron density map calculations were done with REFMAC5 (18) in the CCP4 program suite (19). Manual rebuilding of the model was done with Coot (20, 21). The four monomers in the final model of the K70D/oxacillin complex contained a total of 1,002 amino acid residues, 945 water molecules, seven phosphate ions, and three 2-methyl-2,4,-pentanediol (MPD) molecules. The K84D/oxacillin complex contained 245 residues in the monomer, 110 water molecules, and 4 sulfate molecules. Oxacillin was modeled into each of the active sites based on initial Fo-Fc difference electron density maps (contoured at 3 σ). The quality of the final models was analyzed with MolProbity (22). For OXA-1, 97.3% of the residues were in the favored region, with no outliers of the Ramachandran plot, and for OXA-24/40, 97.9% of residues were in the favored region with no outliers.
TABLE 1.
Crystallographic data collection and refinement parameters
| Data type or parameter | OXA-1 K70D/oxacillin | OXA-24/40 K84D/oxacillin |
|---|---|---|
| Cell constants (a, b, c = Å; α, β, γ = °) | 50.92, 72.61, 73.45; 80.90, 69.87, 71.44 | 102.33, 102.33, 87.44; 90, 90, 90 |
| Space group | P1 | P41212 |
| Resolution (Å) | 1.37 (1.42–1.37)a | 2.4 (2.49–2.40) |
| No. of reflections | ||
| Unique | 187,539 | 18,309 |
| Total | 643,495 | 131,630 |
| Rmerge (%) | 6.5 (42.1) | 15.1 (61.5) |
| Completeness (%)b | 94.9 (91.6) | 97.5 (100) |
| <I/σ(I)> | 9.5 (3.2) | 9.4 (3.7) |
| Resolution range for refinement (Å) | 50–1.37 | 30–2.4 |
| No. of protein residues | 1,002 | 245 |
| No. of water molecules | 945 | 110 |
| RMSD | ||
| For bond lengths (Å) | 0.012 | 0.019 |
| For bond angles (°) | 1.66 | 2.16 |
| R-factor (%) | 16.3 | 17.8 |
| Rfree (%)c | 19.6 | 21.4 |
| Avg B-factor (Å2) | ||
| Protein atoms | 20.4 | 31.2 |
| Water molecules | 32.1 | 31.4 |
| Oxacillin atoms | 25.2 | 41.9 |
Values in parentheses are for the highest-resolution shell.
Fraction of theoretically possible reflections observed.
Rfree was calculated with 5% of reflections set aside randomly.
Protein structure accession numbers.
Final models and structure factors have been deposited with the Protein Data Bank (4MLL for K70D OXA-1/oxacillin; 4F94 for K84D OXA-24/40/oxacillin).
RESULTS
Structure of K70D OXA-1/oxacillin complex.
Since wild-type OXA-1 efficiently hydrolyzes oxacillin, a deacylation-deficient mutant (K70D OXA-1) (14) was used in crystallographic experiments to capture the acyl-enzyme complex with oxacillin. Initial electron density in the active sites of all four monomers was unambiguous for oxacillin bound to the catalytic serine residue, Ser67; specifically, continuous electron density was observed between the Ser67O-γ and the carbonyl carbon of the former lactam ring, indicating the presence of a covalent bond between these atoms and formation of an acyl-enzyme complex (Fig. 1A). Oxacillin was bound in a similar conformation in three of the four active sites. In the fourth (B monomer), the major differences were located at the C-4′ carboxylate and the nitrogen of the former lactam ring (N-13). In the B monomer, the carboxylate carbon atom (C-10) is shifted by ∼2.4 Å and N-10 is shifted by ∼1.7 Å. However, the orientation of the R1 side chain in the B monomer matches that of the other R1 side chains (Fig. 2).
FIG 1.

(A) Chemical structure and atom numbering scheme of the β-lactam oxacillin. (B) Fo-Fc omit maps contoured at 3 σ around oxacillin, which is covalently bound as an acyl-enzyme intermediate in the K70D OXA-1/oxacillin complex. (C) K84D OXA-24/40/oxacillin complex. Nitrogen atoms are colored blue, oxygens are colored red, and sulfurs are colored yellow. The carbon atoms of the OXA-1 complex are colored violet, and for the OXA-24/40 complex, carbons are green. This and all subsequent figures were made with PyMOL (38).
FIG 2.
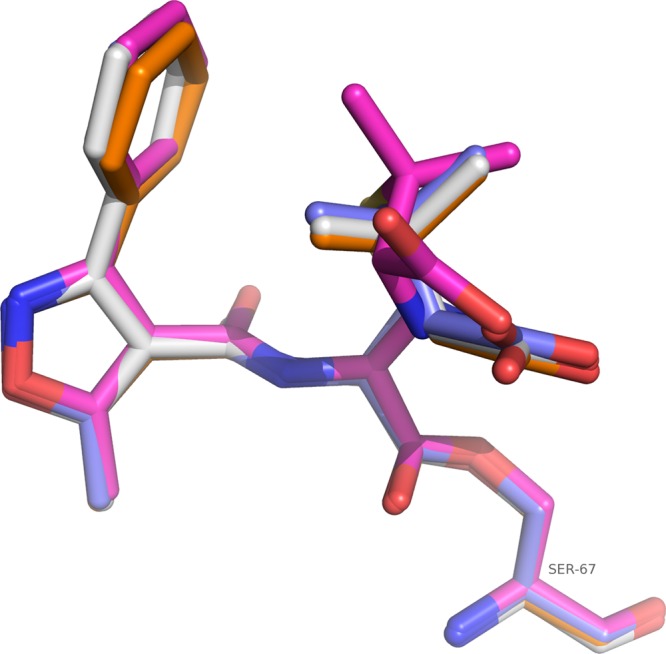
Superposition of acyl oxacillins from each of the four OXA-1 monomers. Atoms are colored as described for Fig. 1, except for carbon atoms, which are colored white for the A monomer, magenta for the B monomer, purple for the C monomer, and orange for the D monomer.
Comparisons between the four monomers of K70D/oxacillin complex indicate that monomers A and D more closely resemble each other (root mean square deviation [RMSD] for all C-α atoms is 0.24 Å for A matched to D versus RMSD of ∼0.5 Å for A matched to B or C) whereas monomers B and C are more similar (RMSD, 0.20 Å). RMSD calculations were performed using the secondary structure matching algorithm in Coot (20). These differences are the result of different packing environments in the crystal. Regions of the B and C monomers (residues 236 to 238) are observed near the phenylisoxazolyl portion of the R1 side chain of oxacillin in the active sites of monomers A and D, respectively, whereas the B and C monomers pack next to the same region of symmetrically related A and D molecules. The average B factors for each of the protein chains were calculated (A chain = 19.7 Å2, B = 21.5 Å2, C = 19.4 Å2, and D = 21.1 Å2). For clarity, the C monomer was chosen to be the representative molecule due to having the lowest protein B factor. Additionally, the conformation of oxacillin in the C monomer is similar to that in two of the other three monomers (Fig. 2).
In the acyl-enzyme complex, oxacillin makes several interactions with OXA-1 that are observed in other class D complexes with β-lactams (Fig. 3A) (23, 24). The carbonyl oxygen of oxacillin (O-2) is bound in the presumed oxyanion hole, making hydrogen bonding interactions with the main chain nitrogen atoms of Ser67 (2.8 Å) and Ala215 (2.9 Å). In the R1 amide group of oxacillin, the amide nitrogen forms a hydrogen bond with the main chain carbonyl oxygen of Ala215 (3.2 Å). In other β-lactamase β-lactam complexes, the amide oxygen usually forms a hydrogen bond with a polar residue (Asn132 in TEM-1 and Asn152 in AmpC) (25–27). This is not possible in OXA-1, where this residue is replaced with the small, hydrophobic Val117. Instead, the amide oxygen of the R1 amide group hydrogen bonds with a water molecule (W555; Fig. 3A). The C-4′ carboxylate of oxacillin hydrogen bonds with Ser258O-γ (2.6 Å) and Thr213O-γ1 (2.6 Å) and two waters; in the A and D monomers, this carboxylate also hydrogen bonds with Ser115O-γ (2.9 to 3.0 Å).
FIG 3.

Hydrogen bonding interactions made between oxacillin and the enzyme. (A) K70D OXA-1/oxacillin. Note that the side chain of Ser115 is shown in dual conformations. (B) K84D OXA-24/40/oxacillin. For clarity, the side chain of Trp221 is not shown. Hydrogen bonds are displayed as dashed yellow lines and represent distances between 2.5 and 3.2 Å. Water molecules are drawn as red spheres.
Beyond the amide group, the rest of the largely hydrophobic R1 side chain of oxacillin forms favorable van der Waals interactions with the enzyme. Met99, Trp102, Leu161, Phe217, and Leu255 form a hydrophobic pocket that recognizes this group (Fig. 4A) (10). This conformation of oxacillin is also stabilized by favorable intramolecular van der Waals interactions between the phenyl ring of the oxacillin R1 side chain and the thiazine ring.
FIG 4.

Binding site for oxacillin in the K70D OXA-1/oxacillin complex (A) and the K84D OXA-24/40 complex (B). In panel A, the hydrophobic residues that line the binding pocket for the R1 side chain of oxacillin are drawn in gold and labeled. In panel B, hydrophobic residues are drawn in bright green and labeled. Oxacillin is shown as a stick representation surrounded by a transparent space-filling model.
Whereas the crystal packing of OXA-1 does not appear to have much of an impact on the conformation of oxacillin, differences in two residues are worth mentioning. Ser115O-γ is observed to hydrogen bond to Lys212N-ζ in all monomers; however, an alternate conformation is also present in the B and C monomers. In the alternate conformation, Ser115O-γ swings away from Lys212 and forms a hydrogen bond to the nitrogen of the lactam ring (N-13; 3.0 Å) in the C monomer. This interaction is not observed in the B monomer due to the unique oxacillin conformation. Alternate conformations of Ser115 and, in some cases, even more significant movement of this residue have been noted in structures of other OXA enzymes (24, 28–31). Ser115 is implicated in playing a role in proton transfer in the hydrolytic mechanism (28, 32, 33). In acylation, Ser115 is likely involved in a relay system that transfers a proton from the general base (i.e., the carboxy-Lys70) to the leaving group nitrogen of the lactam ring, and in deacylation Ser115 could transfer this proton to Ser67 (33). Ser115 must rotate to accomplish this. The dual conformations of Ser115 in our structure provide support for this role.
In monomers B, C, and D, Met99 adopts two conformations that resemble the two that are also found in the wild-type OXA-1 apo structure (1M6K) (34). In one of these two conformations, Met99 clashes with the phenyl ring of oxacillin's R1 side chain. This Met99 clash is not present in the other conformation.
Structure of K84D OXA-24/oxacillin complex.
Since wild-type OXA-24/40 hydrolyzes oxacillin, and for a closer comparison with the K70D OXA-1/oxacillin complex, a deacylation-deficient mutant (K84D OXA-24/40) (15) was used in crystallographic experiments to capture the acyl-enzyme complex with oxacillin. Initial electron density in the active sites was unambiguous for oxacillin bound to the catalytic serine residue, Ser81. Continuous electron density was observed between the Ser81O-γ and the carbonyl carbon of the former lactam ring, suggesting the presence of a covalent bond between these atoms and formation of the acyl-enzyme complex (Fig. 1B).
Several of the canonical β-lactam–β-lactamase interactions are also observed in the complex between OXA-24/40 and oxacillin (Fig. 3B). The carbonyl oxygen of the former lactam ring hydrogen bonds with the main chain nitrogens of Ser81 (2.7 Å) and Trp221 (2.8 Å), the residues comprising the oxyanion hole in OXA-24/40. The nitrogen of the former lactam ring forms a hydrogen bond with Ser128 (3.0 Å). The R1 side chain amide nitrogen of oxacillin makes a hydrogen bond with the main chain oxygen of Trp221 (3.1 Å). The amide oxygen is not involved in any interaction, either with enzyme or with water molecules in the site. Just as in OXA-1, there are no polar residues in proximity to hydrogen bond with this group, and Val130 is the residue equivalent to Val117 in OXA-1. The C-4′ carboxylate group is shown to interact with the carboxylate recognition residue Arg261 (3.2 Å), as well as with Ser219 (2.8 Å) and a water molecule (W488; 2.9 Å).
In contrast to its location in OXA-1, the conformation of the isoxazolyl R1 side chain in OXA-24/40 is more extended out to bulk solvent (Fig. 4B). However, the side chain is somewhat accommodated there by hydrophobic interactions with several residues near the lip of the active site, placing the terminal phenyl ring within van der Waals distance of Leu168 and Val130 (3.9 to 4.4 Å).
Notably, the hydrophobic bridge between Tyr112 and Met223 is not present in the OXA-24/40/oxacillin complex. Met223 is displaced out of the active site so that it can no longer interact with Tyr112. The displacement of Met223 involves an entire reorganization of the β5-β6 loop on which Met223 resides (residues 222 to 228). The RMSD is 1.8 Å for C-α atoms of loop residues 222 to 228, with a maximum RMSD of 2.5 Å and a minimum of 0.6 Å.
DISCUSSION
Oxacillin is the preferred substrate for many class D β-lactamases, and this activity provided the basis behind the naming of the entire class D enzymes as oxacillinases (OXAs). We determined the structures of the oxacillinase OXA-1 and the carbapenemase OXA-24/40 in complexes with oxacillin, and a comparison of the two provides a structural basis for the functional substrate selectivity differences observed. Superposition of the complexes indicates that most of the oxacillin ligand binds similarly between the two structures. The largest difference between the two is observed in the distal portion of the R1 side chain. The conformation of the phenylisoxazolyl group is noticeably different between OXA-1 and OXA-24/40, with an ∼130° rotation around the C-15–C-17 bond that accounts for the more extended conformation of oxacillin in OXA-24/40 (Fig. 5).
FIG 5.
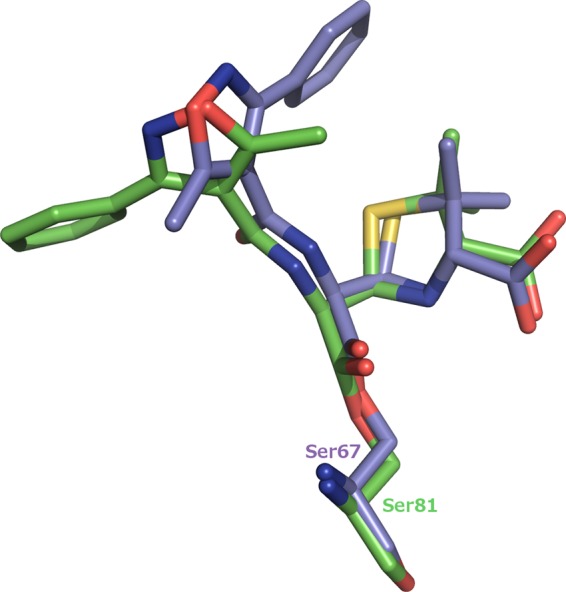
Differences in the conformation of the oxacillin R1 side chain between OXA-1 and OXA-24/40. Carbon atoms of the OXA-1 complex are colored violet, and carbons for the OXA-24/40 complex are green.
In general, class D β-lactamases possess more hydrophobic active sites than other β-lactamase classes. This feature facilitates the formation of the carbamate general base through carboxylation of an active site lysine residue (30, 31). Based on our comparison, the hydrophobic environment also correlates with oxacillinase activity. In our OXA-1 complex, residues Met99, Trp102, Leu161, Phe217, and Leu255 form a hydrophobic pocket that complements the predominantly hydrophobic R1 side chain of its preferred substrate, oxacillin, thereby explaining the low Km of OXA-1 for oxacillin. Specifically, Phe217 and Leu255 in OXA-1 overlay analogous hydrophobic regions in several other oxacillinases: Trp208 and Gly210 in OXA-2 (PDB 1K38); Phe208, Gly210, and Val211 in OXA-10 (31) and OXA-13 (28); and Trp213 and Gly215 in OXA-46 (29). The K70D OXA-1/oxacillin structure was superposed with each of these β-lactamases (RMSDs of 1.6 to 1.7 Å). For OXA-10, OXA-13, and OXA-46, the oxacillin conformation observed in K70D OXA-1 could presumably form similar interactions with the enzyme without steric collisions. This is also the case with OXA-2, although a slight rotation of the Trp208 side chain would be required to accommodate oxacillin in the OXA-1 observed conformation. The conservation of this hydrophobic patch may provide the structural basis for the efficient oxacillinase activity in these other class D enzymes.
In contrast to the exposed active site of OXA-1, OXA-24/40 is characterized by a hydrophobic bridge that occludes the active site, presumably restricting access of substrates and inhibitors alike. Residues Tyr112 and Met223 form the bridge that was suggested to facilitate carbapenemase activity in OXA-24/40 (11). The conformation of oxacillin in the OXA-1 complex is not possible in OXA-24/40, due to steric collisions between the R1 phenylisoxazolyl group and Met223. The 130° rotation of the R1 side chain of oxacillin in the OXA-24/40 complex is necessary to help relieve steric clashes with the bridge. In doing this, the more extended oxacillin loses the intramolecular stabilization observed in the OXA-1 conformation, but the hydrophobic phenylisoxazolyl group picks up van der Waals interactions with hydrophobic residues, Leu168 and Val130, near the edge of the active site.
The OXA-24/40 oxacillin conformation is not observed in OXA-1 because oxacillin is stabilized by favorable interactions made within its hydrophobic pocket, in addition to the intramolecular interactions observed. Although there are a few hydrophobic residues at the edge of the OXA-1 active site that might interact with the distal phenyl ring, the negatively charged Glu158 is also present. The presence of this polar residue probably disfavors an extended OXA-24/40 conformation in OXA-1.
However, simple rotational changes in the oxacillin R1 side chain are not sufficient to prevent collision with the OXA-24/40 bridge, and a significant change in the enzyme structure itself is required. Earlier analysis implicated Tyr112 as the bridge residue that would collide with oxacillin (11). Oxacillinase activity for the single bridge mutant Tyr112Ala was about the same as that for wild-type OXA-24/40, but the double bridge mutant (Tyr112Ala/Met223Ala) became a more efficient oxacillinase.
In our K84D OXA-24/40/oxacillin complex, the OXA-24/40 bridge is disrupted, but Tyr112 overlays almost exactly its position in apo OXA-24/40 (Fig. 6). Met223 is the residue displaced out of the active site to relieve steric collision and accommodate oxacillin binding. The displacement is quite significant and involves an entire reorganization of the β5-β6 loop on which Met223 resides (residues 222 to 228; Fig. 6). The RMSD was calculated to be 1.8 Å for C-α atoms of loop residues 222 to 228, with a maximum RMSD of 2.5 Å and a minimum of 0.6 Å. It is likely that this extensive backbone rearrangement is the major factor that weakens oxacillin binding in OXA-24/40. The bridge methionine is found in many other class D carbapenemases, and in one case, that residue and the β5-β6 loop have been shown to adopt a structure highly similar to that observed in OXA-24/40 (35). It is therefore likely that this structural element may be a general determinant of oxacillin and carbapenem binding across the entire class. Previous investigations suggested that the extent to which this loop projects in toward the active site is a determinant for the acquisition of carbapenemase activity (12, 13). This OXA-24/40/oxacillin complex suggests that the loop conformation that is required for carbapenemase activity is not possible when oxacillin is bound. From this, we conclude that in order to maintain the proper loop conformation for carbapenemase activity, OXA-24/40 sacrifices oxacillinase activity. For OXA-24/40 specifically, carbapenemase and oxacillinase activities are mutually exclusive. Additionally, the bridge positions are occupied by two bulky hydrophobic residues throughout most of the carbapenemase subfamilies (with the exception of the OXA-48 subfamily): Phe/Met in OXA-23, Tyr/Met in OXA-24, Phe/Trp in OXA-51, and Phe/Met in OXA-58 (36). Therefore, it seems likely that the clash between the bridge residues and the R1 side chain is broadly applicable to most carbapenemases.
FIG 6.

Bridge disruption and β5-β6 loop movement in the OXA-24/40/oxacillin complex. (A) Overall fold of the protein. (B) Closeup view of the active site. The OXA-24/40 structure (PDB 3PAE) is superposed on the complex for comparison. Carbon atoms for OXA-24/40 (3PAE) are colored salmon.
In addition to the effects observed on binding of oxacillin (Km), the kinetic parameters of OXA-1 and OXA-24/40 suggest that there are also differences between the two enzymes in their ability to hydrolyze oxacillin (i.e., kcat). The structures determined are complexes with deacylation-deficient mutants, which allow us to focus on side chain interactions but not deacylation. Without the presence of the carboxy-lysine, it is difficult to know how the structure and the deacylation process might be impacted in the wild-type enzyme. An accurate explanation of how deacylation is affected would depend on observation of the deacylating water molecule in the active sites, and they are not observed in the structures presented here or in any class D structure to date. However, it is tempting to speculate that minor structural changes might impact the position of the deacylating water into a mode that may affect kcat.
Our structures of class D β-lactamases that represent the oxacillinase and CHDL subfamilies in complexes with the β-lactam oxacillin provide the structural basis to support the role of the β5-β6 loop in determining substrate selectivity. The hydrophobic patch presented by the β5-β6 loop is conserved among proficient oxacillinases because it provides a complementary region for the hydrophobic R1 side chain of oxacillin and explains the low Km observed for OXA-1. OXA-24/40, on the other hand, is a poor oxacillinase due to the complete reorganization of the entire β5-β6 loop that must occur to disrupt the Tyr112-Met223 bridge and allow oxacillin to bind. Given the high Km of OXA-24/40 for oxacillin, the acquisition of carbapenemase activity in this subfamily comes at a cost of oxacillinase activity. We close with the observation that these structures with oxacillin also reveal important insights that may bring us closer to designing novel broad-spectrum serine β-lactamase inhibitors. Penicillinase-resistant penicillins like oxacillin were previously recognized as having the ability to inhibit class A and class C enzymes. In the studies presented here, the interaction with important hydrophobic features of the active site pocket of OXA-1 and OXA-24/40 stimulates interest in the possibility of using selective hydrophobic side chains on the β-lactam scaffold to create compounds with enhanced activity against class A, class C, and possibly even class D β-lactamases (5, 37). Further investigation will be required to test this hypothesis.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health grant R15AI094489 (R.A.P.). This work was also supported in part by the Veterans Affairs Merit Review Program (R.A.B.), the National Institutes of Health (grant AI072219-05 and AI063517-07 to R.A.B.), and the Geriatric Research Education and Clinical Center VISN 10 (R.A.B.). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817).
Footnotes
Published ahead of print 28 October 2013
REFERENCES
- 1.Neu HC. 1992. The crisis in antibiotic resistance. Science 257:1064–1073. 10.1126/science.257.5073.1064 [DOI] [PubMed] [Google Scholar]
- 2.Davies J. 1994. Inactivation of antibiotics and the dissemination of resistance genes. Science 264:375–382. 10.1126/science.8153624 [DOI] [PubMed] [Google Scholar]
- 3.Fisher JF, Meroueh SO, Mobashery S. 2005. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105:395–424. 10.1021/cr030102i [DOI] [PubMed] [Google Scholar]
- 4.Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54:969–976. 10.1128/AAC.01009-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush K, Jacoby GA, Medeiros AA. 1995. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39:1211–1233. 10.1128/AAC.39.6.1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 51:3471–3484. 10.1128/AAC.01464-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hedges RW, Datta N, Kontomichalou P, Smith JT. 1974. Molecular specificities of R factor-determined β-lactamases: correlation with plasmid compatibility. J. Bacteriol. 117:56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ledent P, Raquet X, Joris B, Van Beeumen J, Frere JM. 1993. A comparative study of class D β-lactamases. Biochem. J. 292:555–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bush K. 2013. Proliferation and significance of clinically relevant β-lactamases. Ann. N. Y. Acad. Sci. 1277:84–90. 10.1111/nyas.12023 [DOI] [PubMed] [Google Scholar]
- 10.Drawz SM, Bethel CR, Doppalapudi VR, Sheri A, Pagadala SR, Hujer AM, Skalweit MJ, Anderson VE, Chen SG, Buynak JD, Bonomo RA. 2010. Penicillin sulfone inhibitors of class D β-lactamases. Antimicrob. Agents Chemother. 54:1414–1424. 10.1128/AAC.00743-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santillana E, Beceiro A, Bou G, Romero A. 2007. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 104:5354–5359. 10.1073/pnas.0607557104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Docquier JD, Calderone V, De Luca F, Benvenuti M, Giuliani F, Bellucci L, Tafi A, Nordmann P, Botta M, Rossolini GM, Mangani S. 2009. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem. Biol. 16:540–547. 10.1016/j.chembiol.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 13.De Luca F, Benvenuti M, Carboni F, Pozzi C, Rossolini GM, Mangani S, Docquier JD. 2011. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc. Natl. Acad. Sci. U. S. A. 108:18424–18429. 10.1073/pnas.1110530108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider KD, Bethel CR, Distler AM, Hujer AM, Bonomo RA, Leonard DA. 2009. Mutation of the active site carboxy-lysine (K70) of OXA-1 β-lactamase results in a deacylation-deficient enzyme. Biochemistry 48:6136–6145. 10.1021/bi900448u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. 2011. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J. Mol. Biol. 406:583–594. 10.1016/j.jmb.2010.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 17.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project N 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763. 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21. 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baurin S, Vercheval L, Bouillenne F, Falzone C, Brans A, Jacquamet L, Ferrer JL, Sauvage E, Dehareng D, Frere JM, Charlier P, Galleni M, Kerff F. 2009. Critical role of tryptophan 154 for the activity and stability of class D β-lactamases. Biochemistry 48:11252–11263. 10.1021/bi901548c [DOI] [PubMed] [Google Scholar]
- 24.Vercheval L, Bauvois C, di Paolo A, Borel F, Ferrer JL, Sauvage E, Matagne A, Frere JM, Charlier P, Galleni M, Kerff F. 2010. Three factors that modulate the activity of class D β-lactamases and interfere with the post-translational carboxylation of Lys70. Biochem. J. 432:495–504. 10.1042/BJ20101122 [DOI] [PubMed] [Google Scholar]
- 25.Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. 1992. Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 Å resolution. Nature 359:700–705. 10.1038/359700a0 [DOI] [PubMed] [Google Scholar]
- 26.Beadle BM, Trehan I, Focia PJ, Shoichet BK. 2002. Structural milestones in the reaction pathway of an amide hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC β-lactamase. Structure 10:413–442. 10.1016/S0969-2126(02)00725-6 [DOI] [PubMed] [Google Scholar]
- 27.Patera A, Blaszczak LC, Shoichet BK. 2000. Crystal structures of substrate and inhibitor complexes with AmpC β-lactamase: possible implications for substrate-assisted catalysis. J. Am. Chem. Soc. 122:10504–10512. 10.1021/ja001676x [DOI] [Google Scholar]
- 28.Pernot L, Frenois F, Rybkine T, L'Hermite G, Petrella S, Delettre J, Jarlier V, Collatz E, Sougakoff W. 2001. Crystal structures of the class D β-lactamase OXA-13 in the native form and in complex with meropenem. J. Mol. Biol. 310:859–874. 10.1006/jmbi.2001.4805 [DOI] [PubMed] [Google Scholar]
- 29.Docquier JD, Benvenuti M, Calderone V, Giuliani F, Kapetis D, De Luca F, Rossolini GM, Mangani S. 2010. Crystal structure of the narrow-spectrum OXA-46 class D β-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob. Agents Chemother. 54:2167–2174. 10.1128/AAC.01517-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. U. S. A. 98:14280–14285. 10.1073/pnas.241442898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maveyraud L, Golemi D, Kotra LP, Tranier S, Vakulenko S, Mobashery S, Samama JP. 2000. Insights into class D β-lactamases are revealed by the crystal structure of the OXA-10 enzyme from Pseudomonas aeruginosa. Structure 8:1289–1298. 10.1016/S0969-2126(00)00534-7 [DOI] [PubMed] [Google Scholar]
- 32.Bethel CR, Distler AM, Ruszczycky MW, Carey MP, Carey PR, Hujer AM, Taracila M, Helfand MS, Thomson JM, Kalp M, Anderson VE, Leonard DA, Hujer KM, Abe T, Venkatesan AM, Mansour TS, Bonomo RA. 2008. Inhibition of OXA-1 β-lactamase by penems. Antimicrob. Agents Chemother. 52:3135–3143. 10.1128/AAC.01677-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard DA, Bonomo RA, Powers RA. 31 July 2013. Class D β-lactamases: a reappraisal after five decades. Acc. Chem. Res. 10.1021/ar300327a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun T, Nukaga M, Mayama K, Braswell EH, Knox JR. 2003. Comparison of β-lactamases of classes A and D: 1.5 Å crystallographic structure of the class D OXA-1 oxacillinase. Protein Sci. 12:82–91. 10.1110/ps.0224303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaitany KC, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA. 2013. Structures of the class D carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins and aztreonam. Antimicrob. Agents Chemother. 57:4848–4855. 10.1128/AAC.00762-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poirel L, Naas T, Nordmann P. 2010. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob. Agents Chemother. 54:24–38. 10.1128/AAC.01512-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saves I, Burlet-Schiltz O, Maveyraud L, Samama JP, Prome JC, Masson JM. 1995. Mass spectral kinetic study of acylation and deacylation during the hydrolysis of penicillins and cefotaxime by β-lactamase TEM-1 and the G238S mutant. Biochemistry 34:11660–11667. 10.1021/bi00037a003 [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. 2002. The PyMOL molecular graphics system. Schrodinger, LLC, Cambridge, MA [Google Scholar]