

Abstract
Quinine and other cinchona-derived alkaloids, although recently supplanted by the artemisinins (ARTs), continue to be important for treatment of severe malaria. Quinine and quinidine have narrow therapeutic indices, and a safer quinine analog is desirable, particularly with the continued threat of antimalarial drug resistance. Hydroxyethylapoquinine (HEAQ), used at 8 g a day for dosing in humans in the 1930s and halving mortality from bacterial pneumonias, was shown to cure bird malaria in the 1940s and was also reported as treatment for human malaria cases. Here we describe synthesis of HEAQ and its novel stereoisomer hydroxyethylapoquinidine (HEAQD) along with two intermediates, hydroxyethylquinine (HEQ) and hydroxyethylquinidine (HEQD), and demonstrate comparable but elevated antimalarial 50% inhibitory concentrations (IC50) of 100 to 200 nM against Plasmodium falciparum quinine-sensitive strain 3D7 (IC50, 56 nM). Only HEAQD demonstrated activity against quinine-tolerant P. falciparum strains Dd2 and INDO with IC50s of 300 to 700 nM. HEQD had activity only against Dd2 with an IC50 of 313 nM. In the lethal mouse malaria model Plasmodium berghei ANKA, only HEQD had activity at 20 mg/kg of body weight comparable to that of the parent quinine or quinidine drugs measured by parasite inhibition and 30-day survival. In addition, HEQ, HEQD, and HEAQ (IC50 ≥ 90 μM) have little to no human ether-à-go-go-related gene (hERG) channel inhibition expressed in CHO cells compared to HEAQD, quinine, and quinidine (hERG IC50s of 27, 42, and 4 μM, respectively). HEQD more closely resembled quinine in vitro and in vivo for Plasmodium inhibition and demonstrated little hERG channel inhibition, suggesting that further optimization and preclinical studies are warranted for this molecule.
INTRODUCTION
Malaria, caused by the Plasmodium parasite, is a devastating disease that has plagued mankind for centuries and continues to wreak havoc across continents, with almost half of the global population being at risk for the disease every year (1). Malaria is the leading cause of death of children under 5 years of age in sub-Saharan Africa and was responsible for over 1 million deaths in Africa alone in 2010 (1, 2). Due to the large reservoir of asymptomatic cases and the spread of antimalarial drug resistance, new strategies of intervention and effective treatment are rapidly becoming more urgent to achieve disease elimination. In particular, new, economically feasible drugs that can rapidly kill the parasite are especially crucial in light of the fact that definitive drug resistance or delayed parasite clearance has been reported for all classes of antimalarials available, including artemisinin-based combined therapy (ACT) (3–6).
Quinine was discovered 4 centuries ago as the first antimalarial drug and was recently supplanted by the artemisinins as the gold standard in the treatment of severe malaria. Presently, quinine or quinidine is still widely used for treating severe malaria because of the lack of availability of intravenous artesunate (7–9). Periodic reports of drug resistance to quinine do exist, mainly in Southeast Asia and South America, but the development of resistance is slow and not sustained, classified as “low grade,” with no “high-grade” resistance being noted for severe malaria cases (7). In many cases, quinine treatment failure is due to noncompliance to a treatment regimen rather than an actual increase in the 50% inhibitory concentration (IC50) (8, 10).
Although quinine and quinidine have proven to be effective antimalarial drugs, both compounds have narrow therapeutic indices; that is, the effective dose is very close to doses associated with toxicity. In particular, higher doses of quinine and quinidine may result in cardiotoxicity associated with delayed ventricular depolarization and, in the case of quinidine, repolarization, leading to prolonged QRS and QT intervals, respectively (11). Blindness is another severe adverse event associated with quinine overdose, while other side effects include tinnitus, hearing loss, headache, and loss of taste sensation (8). In light of these quinine-associated toxicities, a novel compound, similar to quinine but less toxic, would be ideal as either a partner drug with artemisinin-related compounds or a replacement for other antimalarial drugs that have become ineffective due to resistance.
Hydroxyethylapoquinine (HEAQ), also known as hydroxyethylapocupreine, was discovered in the 1930s and cited as a compound “as good as, or better than quinine” with noted antipneumococcal activity but was not pursued due to the advent of penicillin and chloroquine (CQ) as successful antibacterial and antimalarial drugs. Specifically, HEAQ was used at doses of 8 grams per day as an effective antibacterial compound from 1936 to 1939 in the treatment of over 500 pneumonia cases in Pittsburgh, PA, resulting in a 50% reduction in mortality, with no visual disturbances or severe adverse effects, including atrial fibrillation, noted for the drug (12–14). Additional studies confirmed the lower toxicity of HEAQ than quinine and other alkaloids such as ethylapocupreine, showing no visual toxicity in dogs (15). Hegner et al. reported the efficacy of HEAQ in the treatment of three strains of bird malaria, demonstrating that HEAQ was as effective as but less potent than quinine against Plasmodium lophurae, P. relictum, and P. cathemerium (16–18). Most intriguing is a report by W. W. G. Machlachlan in 1963, in which he states, “We were aware of the fact that in malaria hydroxyethylapocupreine [HEAQ] was as effective as quinine as observed in some clinical cases in veterans from Korea and also in Venezuela, in addition to experimental studies recorded by Hegner et al.” (19). Because no formal human trials have been conducted for the use of HEAQ against Plasmodium falciparum, we sought to examine activity in vitro with human P. falciparum and in vivo with the mouse malaria model as well as the important chemical property of quinolines to inhibit human ether-à-go-go-related gene (hERG) channels.
Here we resynthesized and tested HEAQ, in addition to three novel compounds, hydroxyethylquinine (HEQ) and the diastereomers hydroxyethylquinidine (HEQD) and hydroxyethylapoquinidine (HEAQD), against P. falciparum in vitro as well as in a murine malaria model to determine the efficacy of these drugs compared to those of the parent compounds quinine and quinidine. Further characterization in regard to heme crystal inhibition and chemical properties of these derivatives was also conducted along with cytotoxicity and hERG channel studies using a human fibroblast cell line and Chinese hamster ovary (CHO) cells, respectively.
MATERIALS AND METHODS
General synthesis information.
All reagents and solvents were used as supplied by commercial sources, without further purification. All dry solvents were purchased from Aldrich as Sure Seal bottles. Reactions involving air- and/or moisture-sensitive reagents were carried out under an argon atmosphere using glassware that was dried under a vacuum with a heat gun. The evacuated flask was then filled with argon. The reactions were monitored by thin-layer chromatography using Analtech chromatography plates (silica gel hard-layer inorganic binder plus calcium sulfate with fluorescent indicator UV254 [GHLF], 250 μm). Visualization was performed by UV light (254 and 365 nm) and/or by staining with potassium permanganate. Flash chromatography was performed by using a Grace Reveleris flash purification system and Grace silica cartridges (average particle size, 40 μm). 1H (500-MHz) and 13C (125-MHz) nuclear magnetic resonance (NMR) spectra of compounds were obtained by using a Varian Mercury spectrometer. 1H NMR spectra recorded in deuterated methanol (CD3OD) were referenced to 3.310 ppm. 13C NMR spectra recorded in CD3OD were referenced to 39.15 ppm. Accurate mass determinations were recorded by the Mass Spectrometry Facility located at the University of California, Riverside.
Preparation of demethyl quinine.
Preparation of demethyl quinine (cupreine) was a modified procedure adapted from procedures reported by Xu et al. (36) and Furuya et al. (37). A 500-ml three-necked flask equipped with an argon inlet, a reflux condenser, and a large magnetic stir bar (38 by 16 mm) was charged with N,N-dimethylformamide (DMF) (100 ml) and 95% sodium hydride (5.92 g, 247 mmol, 8 equivalents). Ethanethiol (Stench!) (18.3 ml, 247 mmol, 8 equivalents) was added dropwise with cooling with an ice water bath. Note that after the addition of approximately 10 ml of ethanethiol, stirring became difficult. The reaction mixture was allowed to warm to room temperature, and quinine (10.0 g, 30.8 mmol, 1 equivalent) was added in one portion. Once the reaction mixture could be efficiently stirred, the remainder of the ethanethiol was added dropwise at room temperature. Once the addition of ethanethiol was complete, the reaction mixture was stirred at 100°C for 24 h. The reaction mixture was cooled to ambient temperature and quenched with saturated aqueous NH4Cl and water, and the aqueous layer was extracted with ethyl acetate (3 by 200 ml). The organic phase was dried with anhydrous MgSO4, and approximately 300 ml of volatiles was removed by simple distillation in a well-ventilated hood (Stench!). During cooling to ambient temperature, an off-white crystalline solid formed in the still pot and was filtered via vacuum filtration to obtain 6.85 g (72% yield). The product was consistent with previously reported characterization data. 1H NMR (500 MHz, CD3OD) δ 8.60 (d, J = 4.56 Hz, 1H), 7.91 (d, J = 9.12 Hz, 1H), 7.63 (d, J = 4.56 Hz, 1H), 7.34 (dd, J = 2.52, 9.12 Hz, 1H), 7.30 (d, J = 2.52 Hz, 1H), 5.76 (ddd, J = 7.47, 10.14, 17.29 Hz, 1H), 5.54 (d, J = 3.14 Hz, 1H), 4.99 (td, J = 1.49, 17.13 Hz, 1H), 4.92 (td, J = 1.34, 10.38 Hz, 1H), 3.68 to 3.80 (m, 1H), 3.08 to 3.20 (m, 2H), 2.68 to 2.82 (m, 2H), 2.40 (br. s., 1H), 1.85 to 1.97 (m, 2H), 1.79 to 1.85 (m, 1H), 1.56 to 1.68 (m, 1H), 1.46 (tdd, J = 3.30, 10.06, 13.20 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 158.1, 149.6, 147.6, 144.1, 142.4, 131.6, 128.5, 123.5, 120.0, 115.3, 105.3, 72.0, 61.1, 57.5, 44.5, 40.8, 29.2, 28.0, 21.7.
Preparation of demethyl quinidine.
The same procedure for the conversion of quinine to demethyl quinine was used for the conversion of quinidine (5.00 g; 15.4 mmol) to demethyl quinidine, but the product did not crystallize. Oil was purified via flash chromatography (gradient of 99% CHCl3 plus 1% Et3N to 10% methanol [MeOH]–89% CHCl3 plus 1% Et3N) to obtain 3.47 g of a yellow glass. This material contained approximately 10% quinidine and was carried on to the next step without further purification.
Preparation of hydroxyethylapoquinine (HEAQ) and hydroxyethylapoquinidine (HEAQD) was carried out as described previously by Carlson and Cretcher (21), as follows.
Preparation of HEQ.
Ethylene carbonate (5.25 g, 59.6 mmol, 20.0 equivalents), potassium carbonate (824 mg, 5.96 mmol, 2.00 equivalents), demethyl quinine 2 (926 mg, 2.98 mmol, 1.00 equivalent), and 5 ml anhydrous tert-butanol were added to a single-necked 50-ml round-bottom flask equipped with a reflux condenser. The reaction mixture was placed into a preheated oil bath (95°C) for 1 h. The reaction mixture was then poured while still hot onto ice and approximately 10 to 20 ml of 5 M aqueous NaOH. The reaction mixture was extracted with dichloromethane, and volatiles were removed to obtain a brown oil. This material was purified via flash chromatography (gradient of 99% CHCl3 plus 1% Et3N to 10% MeOH–89% CHCl3 plus 1% Et3N) to obtain 854 mg of a brown glass. The brown glass was dissolved in ca. 5 ml of hot 95% ethyl alcohol (EtOH), allowed to cool to ambient temperature, and then placed into a −20°C freezer. Pink-tan crystals formed (726 mg) and were collected by vacuum filtration. 1H NMR (500 MHz, CD3OD) δ 8.66 (d, J = 4.56 Hz, 1H), 7.96 (d, J = 9.12 Hz, 1H), 7.68 (d, J = 4.56 Hz, 1H), 7.42 to 7.51 (m, 2H), 5.76 (ddd, J = 7.62, 10.14, 17.29 Hz, 1H), 5.58 (d, J = 3.14 Hz, 1H), 4.97 (td, J = 1.49, 17.13 Hz, 1H), 4.90 (td, J = 1.34, 10.37 Hz, 1H), 4.20 to 4.29 (m, 2H), 3.93 to 4.01 (m, 2H), 3.69 (dddd, J = 2.36, 5.03, 10.65, 13.24 Hz, 1H), 3.06 to 3.17 (m, 2H), 2.64 to 2.78 (m, 2H), 2.36 (br. s., 1H), 1.83 to 1.96 (m, 2H), 1.76 to 1.83 (m, 1H), 1.54 to 1.65 (m, 1H), 1.46 (tdd, J = 3.22, 9.96, 13.15 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 159.1, 150.7, 148.3, 144.9, 142.8, 131.6, 128.2, 123.8, 120.2, 115.1, 103.4, 72.3, 71.4, 61.7, 61.2, 57.7, 44.3, 41.0, 29.3, 28.3, 21.8. high-resolution mass spectrometry (HRMS) (m/z): [MH+] calculated for C21H27N2O3 355.2016, found 355.2012.
Preparation of HEQD.
Ethylene carbonate (19.7 g, 224 mmol, 20.0 equivalents), potassium carbonate (3.09 g, 22.4 mmol, 2.00 equivalents), crude demethyl quinidine (3.47 g, 11.2 mmol, 1.00 equivalent), and 18.6 ml anhydrous tert-butanol were added to a single-necked 100 ml RBF equipped with a reflux condenser. The reaction mixture was placed into a preheated oil bath (95°C) for 3 h. The reaction mixture was then poured while still hot onto ice and 10 to 20 ml of 5 M aqueous NaOH. The reaction mixture was extracted with dichloromethane to obtain a brown oil, which was purified via flash chromatography (gradient of 99% CHCl3 plus 1% Et3N to 10% MeOH–89% CHCl3 plus 1% Et3N) to obtain a reddish-orange glass. This material was crystallized from hot ethyl acetate to obtain 1.79 g of an off-white crystalline solid. 1H NMR (500 MHz, CD3OD) δ 8.66 (d, J = 4.56 Hz, 1H), 7.95 (d, J = 9.12 Hz, 1H), 7.69 (d, J = 4.56 Hz, 1H), 7.41 to 7.48 (m, 2H), 6.14 to 6.21 (m, 1H), 5.63 (d, J = 3.14 Hz, 1H), 5.05 to 5.14 (m, 2H), 4.20 to 4.27 (m, 2H), 3.96 (t, J = 4.72 Hz, 2H), 3.56 (ddd, J = 2.12, 7.78, 13.52 Hz, 1H), 3.05 (dt, J = 2.59, 9.08 Hz, 1H), 2.88 to 2.95 (m, 2H), 2.77 to 2.86 (m, 1H), 2.20 to 2.35 (m, 2H), 1.73 (br. s., 1H), 1.53 to 1.64 (m, 2H), 1.08 (ddd, J = 4.09, 9.43, 13.52 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 159.0, 150.9, 148.3, 144.9, 142.0, 131.5, 128.2, 123.7, 120.1, 115.2, 103.3, 72.6, 71.4, 61.7, 60.9, 51.0, 50.5, 41.6, 29.9, 27.4, 21.3. HRMS (m/z): [MH+] calculated for C21H27N2O3 355.2016, found 355.2022.
The general procedure for the preparation of HEAQ or HEAQD was carried out as described by Portlock et al. (analytical-scale double-bond isomerization) (38).
HEAQ.
To a solution of HEQ (250.0 mg, 0.705 mmol, 1.00 equivalent) dissolved in 12 ml of 50% aqueous ethanol and concentrated (37%, 12 N) HCl (0.59 ml, 7.05 mmol, 10.0 equivalents) was added 12.5 mg of 5 wt% rhodium catalyst on activated carbon. The mixture was heated to reflux for 24 h. After allowing the reaction mixture to cool to ambient temperature, the reaction mixture was vacuum filtered through Celite, and the volatiles of the filtrate were removed in vacuo. The residue was taken up in water, and the pH was made basic with concentrated NH4OH. The resulting white precipitate was vacuum filtered and dried under high vacuum to obtain 192 mg (76%) of a white amorphous solid. 1H NMR (500 MHz, CD3OD) δ 8.66 (d, J = 4.56 Hz, 1H), 7.95 (d, J = 8.80 Hz, 1H), 7.68 (d, J = 4.56 Hz, 1H), 7.36 to 7.58 (m, 2H), 5.60 to 5.72 (m, 1H), 5.08 to 5.28 (m, 1H), 4.15 to 4.39 (m, 2H), 3.89 to 4.06 (m, 2H), 3.69 to 3.87 (m, 1H), 3.38 to 3.59 (m, 2H), 3.02 to 3.15 (m, 1H), 2.68 to 2.87 (m, 1H), 2.35 (br. s., 1H), 2.07 to 2.19 (m, 1H), 1.83 to 1.98 (m, 1H), 1.56 to 1.71 (m, 1H), 1.53 (d, J = 6.76 Hz, 1H), 1.47 (d, J = 6.76 Hz, 2H), 1.16 to 1.38 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 159.1, 150.8, 148.3, 144.9, 141.5, 140.4, 131.5, 128.1, 123.7, 120.1, 115.9, 115.7, 103.4, 103.4, 72.3, 71.4, 71.4, 62.0, 61.7, 61.7, 59.9, 57.6, 45.0, 45.0, 34.7, 28.6, 28.4, 27.5, 27.3, 27.2, 12.9, 12.6. HRMS (m/z): [MH+] calculated for C21H27N2O3 355.2016, found 355.2017.
HEAQD.
The same procedure was used for HEQD to obtain 264 mg (88%) of a white amorphous solid. 1H NMR (500 MHz, CD3OD) δ 8.65 (d, J = 4.56 Hz, 1H), 7.95 (d, J = 9.59 Hz, 1H), 7.59 to 7.73 (m, 1H), 7.39 to 7.53 (m, 2H), 5.52 to 5.72 (m, 1H), 5.09 to 5.32 (m, 1H), 4.17 to 4.40 (m, 3H), 3.36 (d, J = 17.13 Hz, 1H), 3.14 to 3.28 (m, 1H), 2.88 to 3.04 (m, 1H), 2.70 to 2.88 (m, 1H), 2.35 (br. s., 1H), 1.98 to 2.13 (m, 1H), 1.58 to 1.71 (m, 3H), 1.55 (d, J = 6.76 Hz, 3H), 1.32 to 1.50 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 159.0, 159.0, 150.7, 150.7, 148.3, 144.9, 144.9, 142.5, 141.4, 131.5, 128.2, 128.2, 123.8, 123.7, 120.2, 120.1, 114.3, 114.0, 103.4, 103.3, 72.6, 72.5, 71.4, 71.4, 61.7, 60.7, 60.7, 53.3, 52.2, 51.8, 51.0, 34.9, 28.2, 28.0, 27.4, 27.4, 27.0, 12.9, 12.6. HRMS (m/z): [MH+] calculated for C21H27N2O3 355.2016, found 355.2029.
Heme crystallization inhibition assay.
The heme crystallization assay was designed to mimic hemozoin crystal formation in the parasite digestive vacuole, using acidic pH and lipids to initiate crystallization of monomeric heme (22, 23). Drug dilutions were made from a 10 mM stock with 100 mM sodium acetate (pH 4.8) and aliquoted in a 96-well plate (catalog number 3595; Costar) in triplicate, with five serial, 2-fold dilutions per drug. A heme stock (10 mM) was made in dimethyl sulfoxide (DMSO) and was diluted to 50 μM with 100 mM sodium acetate (pH 4.8). A 10 mM 1-monooleoyl-rac-glycerol (MOG) stock was made in ethanol and sonicated before addition to a 50 μM heme stock to make 25 μM MOG–50 μM heme in 100 mM sodium acetate (pH 4.8). The 25 μM MOG–50 μM heme solution was sonicated and added to the assay plate at 100 μl/well. The plates were incubated at 37°C for 2 h to allow crystallization, followed by addition of 100 μl 100 mM sodium bicarbonate (pH 9.1) to solubilize any remaining monomeric heme. After incubation for 30 min at room temperature, the amount of solubilized monomeric heme was determined by measuring the absorbance at 405 nm and calculating the nanomoles of heme based on a previously determined standard curve. Finally, 20 μl of 1 M sodium hydroxide was added to the plates to dissolve any crystals that formed, and the absorbance was read at 405 nm to determine the total amount of heme present in each well. Data were exported to Microsoft Excel, and inhibition of heme crystallization was determined as a function of the nanomoles of monomeric heme not crystallized divided by the total nanomoles of heme present in the assay mixture.
Fluorescence determination.
To verify that the quinoline derivatives retained fluorescent chemical properties similar to those of quinine and quinidine, 1 M drug stocks of quinine, quinidine, HEAQ, and HEAQD were made in 1 M sulfuric acid. The solutions were diluted 1:100 in distilled water, and 5 dilutions of these stocks were made by using 0.05 M sulfuric acid. The fluorescence of these compounds was determined at 350 and 450 nm.
Seventy-two-hour SYBR green I parasite inhibition assay.
A 72-h SYBR green assay (20, 24) was used to determine the sensitivity of three strains of P. falciparum (3D7, INDO, and Dd2, obtained from the Malaria Research Reference Reagent Resource) to quinine, quinidine, ART, CQ, and the derivatives HEQ, HEAQ, HEQD, and HEAQD. Drug stocks were prepared at 10 mM concentrations in DMSO or water, filter sterilized, and stored at −20°C. Dilutions were made in RPMI 1640 medium to the appropriate starting concentration, followed by serial 2-fold dilutions to generate 5 to 10 concentrations for each drug. Drugs (10 μl) were aliquoted in triplicate into 96-well plates (catalog number 3595; Costar) at 10 times the final concentration. P. falciparum cultures were synchronized and diluted to 2% ring stage and adjusted to 1% hematocrit with uninfected red blood cells (RBCs). The cultures (90 μl) were then added to the assay plates, and the plates were incubated in a gassed chamber at 37°C for 72 h until the no-drug control parasitemia reached between 10 and 15% (trophozoite or schizont stage). Assay plates were frozen at −80°C for at least 1 h or overnight, followed by thawing at 37°C for 1 to 2 h. Immediately after the freeze-thaw, 100 μl of 2× SYBR green I in lysis buffer (20 mM Tris [pH 7.5], 5 mM EDTA, 0.008% [wt/vol] saponin, 0.08% [vol/vol] Triton X-100) was added to each well for a total volume of 200 μl/well and mixed by pipetting up and down. The plates were allowed to incubate, protected from light, for at least 1 h. Fluorescence was measured at 485 and 535 nm by using an HTS 7000 Plus BioAssay reader, adjusting the gain between 80 and 90 for optimal reads. Data were exported to Microsoft Excel, where background fluorescence from the positive controls (1 mM chloroquine) was subtracted from each sample, and percent inhibitions were calculated by dividing the sample fluorescence by the that of no-drug controls and multiplying by 100. IC50s were calculated as the concentration of drug required to inhibit 50% of the parasite growth in the no-drug control. At least three replicates were completed for each strain of P. falciparum and each drug unless otherwise noted.
Murine malaria model.
We obtained 70 C57BL/6 mice, 5 weeks old and weighing between 15 to 23 g, from the Jackson Laboratory for our experiment (n = 5 mice per group; 14 groups [4 mice were used in the artesunate-alone group]). Mice were kept in the Johns Hopkins Bloomberg School of Public Health mouse facility according to ACUC animal protocol number MO09H401. Mice were infected intraperitoneally with Plasmodium berghei ANKA (1 × 107 infected erythrocytes), and drugs were administered orally twice a day by using sterile plastic feeding tubes (catalog number FTP 2038; Instech Solomon Scientific) for 5 days beginning at 24 h postinfection (25, 26). All quinoline salts were dissolved in distilled water (8 mg/ml or 2 mg/ml), and artesunate was dissolved in 100% ethanol (100 mg/ml) and diluted 1:100 in distilled water. The effects of compounds on parasite levels and mouse survival were determined by measuring weekly parasitemia levels by Giemsa-stained blood smears and checking mouse survival daily for up to 30 days.
hERG channel inhibition Ionworks patch clamp assay.
hERG channels were stably expressed in Chinese hamster ovary (CHO) cells which were held at −70 mV, and hERG currents were evoked by two voltage pulses (27). During the first pulse, cells were depolarized to +40 mV for 2 s and hyperpolarized to −30 mV for 2 s. This was repeated after a 3-s hold at −70 mV. The difference of tail currents at the second pulse between pre-compound and post-compound addition was used to measure compound activity, with dofetilide and buffer as positive and negative controls, respectively. Compounds were dissolved in DMSO and diluted 1:3 in an 8-point gradient with the maximum concentration at 100 μM. In order to be classified as a hERG inhibitor, the compounds caused >3 standard deviations of reduction in hERG currents.
Forty-eight-hour alamarBlue assay with human foreskin fibroblasts.
Human foreskin fibroblasts (ATCC CRL-1635) were grown and harvested at log phase (day 3 after passage). Cells were plated at 10,000 cells per well in 200 μl of minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% l-glutamine in a 96-well plate (catalog number 3595; Costar) and allowed to incubate for 1 day at 37°C in 5% CO2 until cells again reached log phase. After 24 h, 100 μl MEM was removed and replaced with fresh medium. After another 24-h incubation, 150 μl medium was removed from wells, and 150-μl drug dilutions made in MEM from 10 mM stocks were added to the assay plate in triplicate, with four 2-fold serial dilutions of drug. Plates were incubated for 2 days at 37°C in 5% CO2. After 48 h, 20 μl of alamarBlue was added to each well, including a no-drug control as well as a blank well with medium only. Sample fluorescence was read on an HTS 700 Plus Bio Assay reader at 550 and 595 nm, and data were exported to Microsoft Excel. Cytotoxicity of drugs was calculated as the percentage of the no-drug growth control after 48 h.
RESULTS
Synthesis.
We modified the original synthetic approach of Butler and Cretcher in 1937 and 1938 (39, 40) to prepare HEAQ from quinine and HEAQD from quinidine (Fig. 1). According to Xu et al. (36) and Furuya et al. (37), quinine underwent demethylation in the presence of sodium ethanethiolate to form cupreine. Formation of the hydroxyethyl ether was accomplished by reacting cupreine with ethylene carbonate and potassium carbonate, yielding HEQ according to methods described by Carlson and Cretcher (21). Finally, rhodium-catalyzed positional isomerization of the terminal alkene of HEQ resulted in the final product, HEAQ(E,Z), as a mixture of E and Z geometric isomers. A similar approach was used to produce HEQD and HEAQD(E,Z), using quinidine as the parent compound. The crystalline alkaloids of HEAQ, HEQD, and HEAQD were initially obtained from solution as free bases and later converted to hydrochloride salts for use in animal studies.
FIG 1.
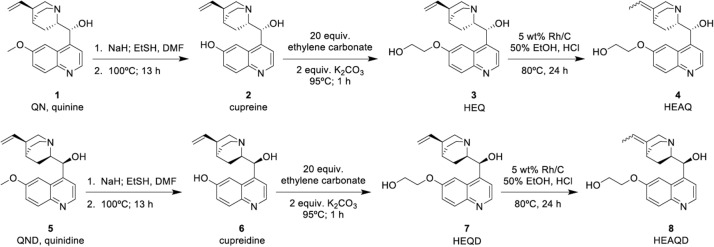
Scheme for synthesis of derivatives.
Heme crystal inhibition.
Like other cinchona alkaloids with antimalarial activity, quinolines accumulate in the parasite digestive vacuole and are thought to inhibit Plasmodium growth by forming an intramolecular hydrogen bond with heme, disrupting formation of the hemozoin crystals in the digestive vacuole, resulting in more free heme, which is toxic to the parasite (28, 29) (Fig. 2). Only HEQD (IC50, 24 μM) demonstrated activities comparable to those of quinine and quinidine (both at an IC50 of 16 μM) in inhibiting heme crystallization in vitro, while HEQ, HEAQ, and HEAQD were effective only at IC50s above 50 μM (Fig. 2). In addition, the derivatives HEQ, HEAQ, HEQD, and HEAQD were found to fluoresce at 465 nm when excited at 360 nm, similarly to quinine and quinidine. Fluorescence at 350/450 nm does not interfere with SYBR green I fluorescence at 485/535 nm.
FIG 2.
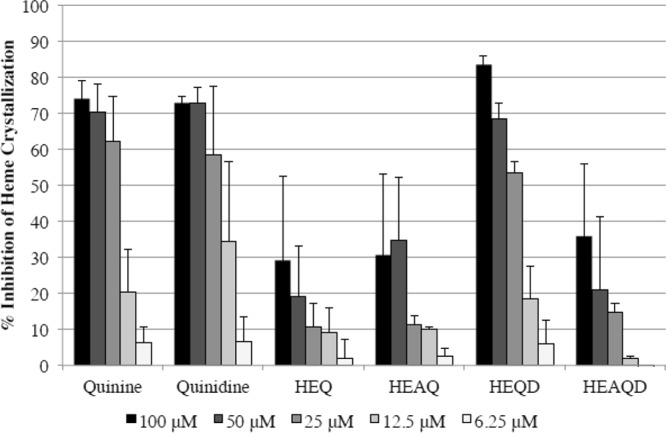
Heme crystallization inhibition by quinine, quinidine, and their derivatives. Only the compound HEQD appreciably inhibited heme crystallization similarly to quinine and quinidine after 16 h. Two to five independent experiments were completed for each compound with each concentration in triplicate per experiment, reported with the corresponding standard errors of the means. Mean IC50s calculated for all compounds in the order listed in graph were 16.3, 16.0, 208, 59.3, 24.0, and 155 μM, respectively.
P. falciparum activity.
Quinoline derivatives were evaluated for antimalarial efficacy against three strains of P. falciparum in a standard 72-h SYBR green assay, and results are shown in Table 1. All derivatives were effective at inhibiting a quinine-sensitive strain, 3D7, at <300 nM, with an IC50 approximately three to five times higher than that of the parent compound quinine or quinidine. Of note, the quinidine derivative HEQD (IC50, 111 nM) had the most activity against 3D7, comparable with quinine at an IC50 of 56 nM. Against quinine-tolerant strains Dd2 and INDO, HEAQ and HEQ had μM inactivity, while HEAQD had IC50s of 333 nM and 725 nM for Dd2 and INDO, respectively, with HEQD having only submicromolar activity for Dd2 at an IC50 of 313 nM. Artemisinin functioned as a control, consistently inhibiting all three strains at low-nanomolar concentrations.
TABLE 1.
Average IC50s of quinine and quinidine derivatives against three strains of P. falciparum determined by using a 72-h SYBR green assaya
| Compound | Avg IC50 (nM) (SEM) |
||
|---|---|---|---|
| 3D7 | INDO | Dd2 | |
| Quinine | 56 (6) | 263 (25) | 92.0 (10) |
| HEQ | 258 (33) | 7,100 (276) | 2,800 (363) |
| HEAQ | 255 (29) | 3,470 (429) | 1,133 (133) |
| HEAQ-HCl salt | 240 (77) | — | 1,250 (189) |
| Quinidine | 24 (2) | 153 (279) | 42.5 (1) |
| HEQD | 111 (22) | 1,875 (427) | 313 (13)b |
| HEAQD | 168 (25) | 725 (170) | 333 (44) |
| HEAQD-HCl salt | 148 (55) | — | 233 (17) |
| Artemisinin | 8.3 (1) | 5.1 (1) | 5.5 (1) |
Three or more biological replicates were completed for all compounds unless otherwise noted, with each compound in triplicate per experiment, reported with corresponding standards error of the means. —, no data for these compounds.
Two independent experiments were completed for this compound.
In vivo murine malaria model.
Due to previous data on HEAQ suggesting that this compound was much less toxic than quinine, we next evaluated the in vivo antimalarial parasite inhibition and 30-day survival of HEAQ, HEQD, and HEAQD in a murine malaria model at doses similar to those of the parent drugs, at 20 mg/kg of body weight, and 4 times higher, at 80 mg/kg. Seventy C57BL/6 mice were infected intraperitoneally with P. berghei ANKA (1 × 107 infected erythrocytes), and compounds were administered orally twice a day for 5 days beginning at 24 h postinfection. Parasitemia was reduced by all derivatives except 20 mg/kg HEAQ at 3 and 6 days (Fig. 3). HEQD at 20 mg/kg was comparable in parasitemia inhibition to quinine at 20 mg/kg. Only 80 mg/kg HEAQD and 20 mg/kg HEQD, both in combination with artesunate, retained parasite suppression at day 13 similar to that of quinidine and artesunate in combination. As single compounds, the derivatives all delayed death to approximately days 20 to 25 compared to the early day 9 deaths for nontreated controls (Fig. 4). In combination with artesunate at 10 mg/kg, 20 mg/kg HEQD enabled 3 of 5 mice to live parasite free for 30 days, compared to 4 of 5 mice in the group treated with the combination of 80 mg/kg HEAQD and artesunate. Quinidine at 20 mg/kg plus artesunate cured 1 of 5 mice by day 30. Only HEQD at 20 mg/kg was similar to quinine and quinidine in this mouse malaria model.
FIG 3.

Dose-dependent clearance of P. berghei ANKA by the compounds quinine (QN), HEAQ, quinidine (QND), HEQD, and HEAQD, alone at either 20 mg/kg or 80 mg/kg and in combination with artesunate (AS) (10 mg/kg), in C57BL/6 mice. Percent parasitemia was calculated as the percentage of infected RBCs out of 500 RBCs for ≥1% parasitemia and out of 10,000 for <1% parasitemia. All compounds are graphed with standard errors of the means. Compounds are listed first, with dosages following (e.g., QN 20 mg/kg). (A) Day 3 parasitemia, after 2 days of dosing. (B) Day 6 parasitemia, 1 day after the final dose was administered. (C) Day 13 parasitemia, 1 week after the final dose was administered.
FIG 4.
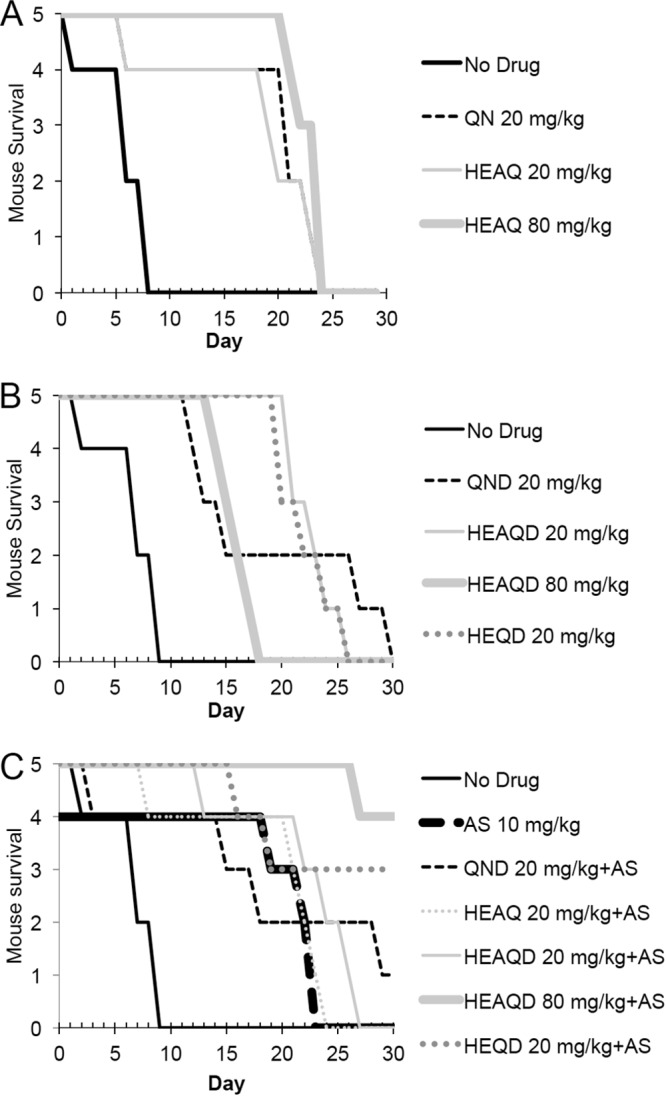
Dose-dependent survival of C57BL/6 mice infected with P. berghei ANKA after treatment with quinine, HEAQ, quinidine, HEQD, and HEAQD, alone or in combination with AS (10 mg/kg). (A) HEAQ at either 20 mg/kg or 80 mg/kg was comparable to quinine in survival, with all mice dying between days 20 and 25. (B) HEAQD and HEQD were comparable to quinidine, with mice surviving to days 20 to 25 as with quinine and HEAQ. Despite the finding that mice treated with 80 mg/kg of HEAQD had a lower parasitemia on day 16, these mice had a surge in parasitemia and died quickly compared to mice treated with the lower dose of 20 mg/kg HEAQD. (C) Drugs in combination with AS demonstrated that 3 of 5 mice given 20 mg/kg of HEQD survived, comparable to the group given 80 mg/kg of HEAQD, with 4 of 5 mice surviving without parasites until day 30. Only 1 mouse of 5 treated with quinidine at 20 mg/kg survived until day 30 but did not clear parasites, unlike treatment with HEAQD and HEQD.
hERG channel inhibition.
The most compelling evidence reported for HEAQ, besides its antimalarial efficacy, is the decreased toxicity associated with its historic human use compared to that of other quinine derivatives. An important clinical adverse drug reaction of the quinoline class is prolongation of the QRS heart interval associated with inhibition of the hERG channel (11). hERG inhibition studies were conducted by using Chinese hamster ovary (CHO) cells expressing hERG channels and an Ionworks automatic patch clamp to determine hERG IC50s for all derivatives, HEQ, HEAQ, HEQD, and HEAQD, compared to the parent compounds (Table 2 and Fig. 5). Strikingly, the compounds HEQ, HEAQ, and HEAQD demonstrated IC50s of approximately 100 μM, compared to 42.2 μM for quinine, while the compound HEAQD was seven times less inhibitory than quinidine, inhibiting 50% of the CHO hERG channels at 27 μM, compared to 4 μM quinidine. Both quinine and quinidine had a less steep slope, with the IC25 and IC75 being more apart than for the derivatives with a steeper slope.
TABLE 2.
Inhibition by quinine, quinidine, and derivatives of hERG channels expressed in CHO cells using the Ionworks patch clamp assaya
| Compound | IC50 (μM) |
Hill coefficient (SD) | % at max concn | hERG inhibitor | |
|---|---|---|---|---|---|
| Reportedb | Experimental (SD) | ||||
| Dofetilide | 0.1 | 0.05 (0.01) | 2.27 (0.79) | −100 | Yes |
| Quinine | 57 | 42.2 (7.41) | 1.3 (0.3) | −83.4 | Yes |
| HEQ | >100 | −35.2 | No | ||
| HEAQ | 109 (20.9) | 1.8 (0.83) | −49.4 | Yes | |
| Quinidine | 4.6 | 3.95 (0.75) | 0.96 (0.18) | −95.8 | Yes |
| HEQD | 94.3 (8.12) | 4 (0) | −70.1 | Yes | |
| HEAQD | 27.3 (7.57) | 3 (0) | −87.0 | Yes | |
Compounds were tested in quadruplicate. SD, standard deviation.
Reported values of hERG inhibition from reference 11.
FIG 5.

IC50s were determined for parent compounds and derivatives against hERG channels expressed in CHO cells, as measured by the Ionworks patch clamp assay. Representative graphs of quinine (A), HEAQ (B), quinidine (C), HEAQD (D), and HEQD (E) are pictured. The positive control, dofetilide, has an IC50 of 0.053 μM. Compounds were tested in quadruplicate in an 8-point gradient with a maximum concentration of 100 μM with serial 1:3 dilutions.
Human fibroblast cytotoxicity.
In vitro cell toxicity studies were also completed by observing the effects of different μM concentrations of quinine, quinidine, and the derivatives on human foreskin fibroblasts using a 48-h alamarBlue assay to determine cell viability. While all compounds showed no toxicity at 100 μM, quinine had a 50% lethal dose (LD50) of 200 μM. Quinidine and HEAQ were slightly less toxic but comparable to quinine, while HEQD and HEAQD showed no cytotoxicity (100% growth compared to the control) at 200 μM.
DISCUSSION
Antimalarial drug resistance continues to threaten global public health measures to treat, contain, and eventually eliminate malaria. Building on research that began in the 1930s but was not pursued due to the advent of chloroquine and other effective antimalarials, we sought to determine whether HEAQ and three derivatives would be effective against P. falciparum in an era of increasing antimalarial drug resistance and also to test hERG channel inhibition. We were successful in synthesizing HEAQ as well as the novel diastereomer HEAQD, and we also produced intermediates without the isomerized vinyl group, HEQ and HEQD.
Our in vitro and in vivo antimalarial results correspond with data previously reported by Hegner et al. for the activity of HEAQ against three strains of bird malaria, with the quinine and quinidine derivatives displaying decreased activity per mg/kg but which at 4-fold-higher doses had action comparable to that of the parent compounds (18). In the present work, HEQ and HEAQ were less potent than quinine in vitro against quinine-sensitive strain 3D7, but HEAQ was equally effective at controlling parasitemia in the in vivo mouse malaria model at 4-fold-higher doses. Similar results were noted for HEQD and HEAQD compared to quinidine. Regarding drug resistance, HEQD and HEAQD demonstrated promising submicromolar activity against quinine-tolerant isolates, with IC50s of <400 nM for Dd2 and below 700 nM for INDO for HEAQD, which are below or within reported cutoffs for quinine tolerance that range from either above 500 or 800 nM (30–32). Overall, HEQD showed the greatest efficacy in the in vivo model when combined with artesunate, with no adverse side effects being observable in the mice, making it a promising potential alternative to quinine or quinidine as an antimalarial drug.
The reported hERG channel inhibition data suggest that all four derivatives have less hERG channel activity than the parent compound quinine or quinidine, with HEAQ, HEQ, and HEQD inhibiting 50% of hERG channels at approximately 100 μM, twice the IC50 of quinine (42 μM). Interestingly, both the intermediates HEQ and HEQD had higher IC50s for hERG channels than did HEAQ and HEAQD. These results suggest that isomerization of the vinyl group did not decrease this cardiac potassium channel inhibition, whereas the hydroxyethyl substituent may slightly decrease parasite activity while also importantly decreasing the hERG potassium channel inhibition. Cretcher and Renfrew previously commented on the effect of the resulting modifications on antipneumococcal activity, stating that “greater antipneumococcic action appears to be associated with the ethylidene group,” and they observed an increased dosage of drug required to obtain the same toxic effects of quinine (33). While we did not observe increased potency with the ethylidene or isomerized vinyl group, we did observe a decrease in hERG channel inhibition associated with the hydroxyethyl substitution, suggesting that this modification and not isomerization of the vinyl group can be credited for the great reduction in overall cardiac disturbances for these compounds seen in historic manuscripts in the 1930s and 1940s. Our fibroblast cell line cytotoxicity data also agree with data from previous studies on cell lines conducted by Kominos and Machlachlan, with HEAQ and HEAQD showing less toxicity than quinine and quinidine and no toxicity at 100 μM after 48 h (19).
Our data suggest that modifications of the methoxy side chain (R) (Fig. 6) with hydroxylated alkyl groups may be effective at decreasing cardiotoxic events associated with quinine and quinidine in addition to the decrease in quinine-associated toxicities such as eye damage in dogs, which decreased with hydroxyalkation of R, as mentioned in other studies. Previous studies have shown that similar alkylations of the side chain without a hydroxyl group such as optochin (R=O-CH2-CH3) are more potent but also more toxic than quinine. Interestingly, increased lengths of the alkylated side chain also increased malaria activity up to 4 to 5 carbons, which then decreased as the chain length continued (34). In recent studies on P. falciparum, substitutions at the R′ group with aromatic groups resulted in increased sensitivity against quinine-resistant strains in some cases, while most resulted in decreased potency compared to that of parent compounds, including quinine, cinchonine, quinidine, and cinchonidine (35). When the R′ group was reduced to the dihydro group (optochin and others), a slight decrease in potency was noted. Compound SN-8707 has the dihydro R′ group as well as a hydroxyethyl R group, with a reported quinine ratio of 0.6 in P. lophurae, which is more potent than our reported derivatives (∼0.2 to 0.25). Toxicity studies of SN-8707 and the quinidine derivative may provide more insight into the role of these modifications in decreasing cardiotoxicity as well as other quinine-associated toxicities.
FIG 6.
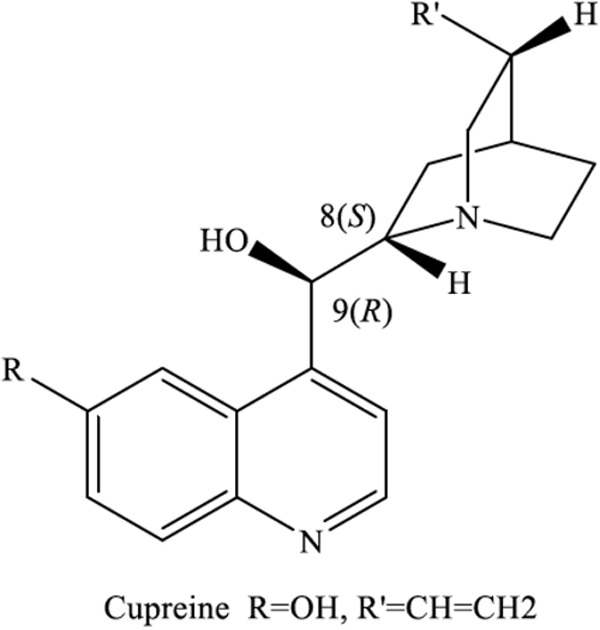
Basic structure of levorotatory alkaloids (8S and 9R).
While we focused initially on the compound HEAQ and its stereoisomer HEAQD due to previous demonstrations of the use in humans of HEAQ at high doses with little noted human clinical toxicity, our data suggest that fluorescent hydroxyethylquinidine or HEQD inhibits heme crystallization, does not appreciably inhibit hERG channels, and is comparable to quinine in vitro against human P. falciparum and in vivo against mouse P. berghei ANKA, making it a potential candidate for further derivations to increase potency against quinine-tolerant strains as well for further in vivo characterizations.
ACKNOWLEDGMENTS
D.J.M. acknowledges NIH Roadmap for Medical Research grant number UL1 RR 025005 and the Flight Attendants Medical Research Fund. We thank the Johns Hopkins Malaria Research Institute and The Bloomberg Family Foundation (D.J.S.).
Footnotes
Published ahead of print 18 November 2013
REFERENCES
- 1.WHO 17 December 2012, posting date World malaria report. WHO, Geneva, Switzerland: http://www.who.int/malaria/publications/world_malaria_report_2012/en/ [Google Scholar]
- 2.Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. 2012. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379:413–431. 10.1016/S0140-6736(12)60034-8 [DOI] [PubMed] [Google Scholar]
- 3.Frosch AE, Venkatesan M, Laufer MK. 2011. Patterns of chloroquine use and resistance in sub-Saharan Africa: a systematic review of household survey and molecular data. Malar. J. 10:116. 10.1186/1475-2875-10-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Brien C, Henrich PP, Passi N, Fidock DA. 2012. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr. Opin. Infect. Dis. 24:570–577. 10.1097/QCO.0b013e32834cd3ed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parija SC, Praharaj I. 2011. Drug resistance in malaria. Indian J. Med. Microbiol. 29:243–248. 10.4103/0255-0857.83906 [DOI] [PubMed] [Google Scholar]
- 6.Sridaran S, McClintock SK, Syphard LM, Herman KM, Barnwell JW. 2010. Anti-folate drug resistance in Africa: meta-analysis of reported dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) mutant genotype frequencies in African Plasmodium falciparum parasite populations. Malar. J. 9:247. 10.1186/1475-2875-9-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achan J, Talisuna AO, Erhart A, Yeka A, Tibenderana JK, Baliraine FN, Rosenthal PJ, D'Alessandro U. 2011. Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar. J. 10:144. 10.1186/1475-2875-10-144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan DJ. 2012. Cinchona alkaloids: quinine and quinidine, p 45–68 In Staines HM, Krishna S. (ed), Treatment and prevention of malaria. Springer Basel AG, Basel, Switzerland [Google Scholar]
- 9.WHO 9 March 2010, posting date Guidelines for the treatment of malaria, 2nd ed. WHO, Geneva, Switzerland: http://www.who.int/malaria/publications/atoz/9789241547925/en/index.html [Google Scholar]
- 10.Griffith KS, Lewis LS, Mali S, Parise ME. 2007. Treatment of malaria in the United States: a systematic review. JAMA 297:2264–2277. 10.1001/jama.297.20.2264 [DOI] [PubMed] [Google Scholar]
- 11.White NJ. 2007. Cardiotoxicity of antimalarial drugs. Lancet Infect. Dis. 7:549–558. 10.1016/S1473-3099(07)70187-1 [DOI] [PubMed] [Google Scholar]
- 12.Maclachlan WWG, Johnston JM, Bracken MM, Crum GE. 1939. The treatment of pneumococcic pneumonia by hydroxyethylapocupreine. Am. J. Med. Sci. 197:31–38. 10.1097/00000441-193901000-00004 [DOI] [Google Scholar]
- 13.Maclachlan WW, Bracken MM, Bailey WR., Jr 1949. Prognosis of pneumonia. Am. J. Med. Sci. 217:438–444. 10.1097/00000441-194904000-00012 [DOI] [PubMed] [Google Scholar]
- 14.Maclachlan WWG, Johnston JM, Bracken MM, Pierce LS. 1941. A comparison of the mortality in pneumococcic pneumonia treated by hydroxyethylapocupreine and by sulfapyradine. Am. J. Med. Sci. 201:367–374. 10.1097/00000441-194103000-00007 [DOI] [Google Scholar]
- 15.Dawson WT, Permar HH, Johnston JM, Maclachlan WWG. 1937. An experimental study of the variations in the production of visual disturbance of certain new cinchona derivatives. Am. J. Med. Sci. 193:543–547. 10.1097/00000441-193704000-00014 [DOI] [Google Scholar]
- 16.Hegner R, Shaw EH, Manwell RD. 1928. Methods and results of experiments on the effects of drugs on bird malaria. Am. J. Hyg. 8:564–582 [Google Scholar]
- 17.Hegner R, West E, Dobler M. 1939. A new drug effective against bird malaria. Am. J. Epidemiol. 33:101–111 [Google Scholar]
- 18.Hegner R, West E, Dobler M. 1941. Further studies of hydroxyethylapocupreine against bird malaria. Am. J. Hyg. 34:132–139 [Google Scholar]
- 19.Kominos S, Maclachlan WW. 1963. The cytotoxic effect of quinine, quinidine and hydroxyethylapocupreine upon mammalian cell cultures. Am. J. Med. Sci. 245:569–572 [PubMed] [Google Scholar]
- 20.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. 2004. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob. Agents Chemother. 48:1807–1810. 10.1128/AAC.48.5.1807-1810.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson WW, Cretcher LH. 1947. Hydroxylalkylation with cyclic alkylene esters. I. Synthesis of hydroxyethylapocupreine. J. Am. Chem. Soc. 69:1952–1956 [DOI] [PubMed] [Google Scholar]
- 22.Pisciotta JM, Coppens I, Tripathi AK, Scholl PF, Shuman J, Bajad S, Shulaev V, Sullivan DJ., Jr 2007. The role of neutral lipid nanospheres in Plasmodium falciparum haem crystallization. Biochem. J. 402:197–204. 10.1042/BJ20060986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. 10.1038/nature09099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE, Waters NC. 2007. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob. Agents Chemother. 51:1926–1933. 10.1128/AAC.01607-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moon DK, Tripathi A, Sullivan D, Siegler MA, Parkin S, Posner GH. 2011. A single, low, oral dose of a 5-carbon-linked trioxane dimer orthoester plus mefloquine cures malaria-infected mice. Bioorg. Med. Chem. Lett. 21:2773–2775. 10.1016/j.bmcl.2010.09.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mott BT, Tripathi A, Siegler MA, Moore CD, Sullivan DJ, Posner GH. 2013. Synthesis and antimalarial efficacy of two-carbon-linked, artemisinin-derived trioxane dimers in combination with known antimalarial drugs. J. Med. Chem. 56:2630–2641. 10.1021/jm400058j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou B, Yu H, Babcock JJ, Chanda P, Bader JS, McManus OB, Li M. 2010. Profiling diverse compounds by flux- and electrophysiology-based primary screens for inhibition of human Ether-a-go-go related gene potassium channels. Assay Drug Dev. Technol. 8:743–754. 10.1089/adt.2010.0339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warhurst DC, Craig JC, Adagu IS, Meyer DJ, Lee SY. 2003. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malar. J. 2:26. 10.1186/1475-2875-2-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Villiers KA, Gildenhuys J, le Roex T. 2012. Iron(III) protoporphyrin IX complexes of the antimalarial cinchona alkaloids quinine and quinidine. ACS Chem. Biol. 7:666–671. 10.1021/cb200528z [DOI] [PubMed] [Google Scholar]
- 30.Pradines B, Hovette P, Fusai T, Atanda HL, Baret E, Cheval P, Mosnier J, Callec A, Cren J, Amalvict R, Gardair JP, Rogier C. 2006. Prevalence of in vitro resistance to eleven standard or new antimalarial drugs among Plasmodium falciparum isolates from Pointe-Noire, Republic of the Congo. J. Clin. Microbiol. 44:2404–2408. 10.1128/JCM.00623-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pradines B, Pistone T, Ezzedine K, Briolant S, Bertaux L, Receveur MC, Parzy D, Millet P, Rogier C, Malvy D. 2010. Quinine-resistant malaria in traveler returning from Senegal, 2007. Emerg. Infect. Dis. 16:546–548. 10.3201/eid1603.091669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pradines B, Mabika Mamfoumbi M, Parzy D, Owono Medang M, Lebeau C, Mourou Mbina JR, Doury JC, Kombila M. 1999. In vitro susceptibility of African isolates of Plasmodium falciparum from Gabon to pyronaridine. Am. J. Trop. Med. Hyg. 60:105–108 [DOI] [PubMed] [Google Scholar]
- 33.Renfew AG, Cretcher LH. 1941. Structure and antipneumococcic activity in the cinchona series. Chem. Rev. 30:49–68 [Google Scholar]
- 34.Buttle GAH, Henry TA, Solomon W, Trevan JW, Gibbs EM. 1938. The action of the cinchona and certain other alkaloids in bird malaria. Biochem. J. 32:47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinio T, Gorka AP, McGinniss A, Roepe PD, Morgan JB. 2012. Investigating the activity of quinine analogues versus chloroquine resistant Plasmodium falciparum. Bioorg. Med. Chem. 20:3292–3297. 10.1016/j.bmc.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu F, Corley E, Zacuto M, Conlon DA, Pipik B, Humphrey G, Murry J, Tschaen D. 2010. Asymmetric synthesis of a potent, aminopiperidine-fused imidazopyridine dipeptidyl peptidase IV inhibitor. J. Org. Chem. 75:1343–1353 [DOI] [PubMed] [Google Scholar]
- 37.Furuya T, Strom AE, Ritter T. 2009. Silver-mediated fluorination of functionalized aryl stannanes. J. Am. Chem. Soc. 131:1662–1663 [DOI] [PubMed] [Google Scholar]
- 38.Portlock DE, Naskar D, West L, Seibel WL, Gu T, Krauss HJ, Peng XS, Dybas PM, Soyke EG, Ashton SB, Burton J. 2003. Positional isomerization of quinine and quinidine via rhodium on alumina catalysis: practical one-step synthesis of D3,10-isoquinine and D3,10-isoquinidine. Tet. Lett. 44:5365–5368 [Google Scholar]
- 39.Butler CL, Hostler M, Cretcher L. 1937. Cinchona alkaloids in pneumonia. V. Alkyl ethers of apocupreine. J. Am. Chem. Soc. 445:5–6 [Google Scholar]
- 40.Butler CL, Renfew AG. 1938. Cinchona alkaloids in pneumonia. VI. A new method for the hydroxylation of phenolic cinchona alkaloids. J. Am. Chem. Soc. 60:1472–1475 [Google Scholar]