

Abstract
To date, most antibiotics have primarily been developed to target bacteria in the planktonic state. However, biofilm formation allows bacteria to develop tolerance to antibiotics and provides a mechanism to evade innate immune systems. Therefore, there is a significant need to identify small molecules to prevent biofilm formation and, more importantly, to disperse or eradicate preattached biofilms, which are a major source of bacterial persistence in nosocomial infections. We now present a modular high-throughput 384-well image-based screening platform to identify Pseudomonas aeruginosa biofilm inhibitors and dispersal agents. Biofilm coverage measurements were accomplished using non-z-stack epifluorescence microscopy to image a constitutively expressing green fluorescent protein (GFP)-tagged strain of P. aeruginosa and quantified using an automated image analysis script. Using the redox-sensitive dye XTT, bacterial cellular metabolic activity was measured in conjunction with biofilm coverage to differentiate between classical antibiotics and nonantibiotic biofilm inhibitors/dispersers. By measuring biofilm coverage and cellular activity, this screen identifies compounds that eradicate biofilms through mechanisms that are disparate from traditional antibiotic-mediated biofilm clearance. Screening of 312 natural-product prefractions identified the cyclic depsipeptide natural products skyllamycins B and C as nonantibiotic biofilm inhibitors with 50% effective concentrations (EC50s) of 30 and 60 μM, respectively. Codosing experiments of skyllamycin B and azithromycin, an antibiotic unable to clear preattached biofilms, demonstrated that, in combination, these compounds were able to eliminate surface-associated biofilms and depress cellular metabolic activity. The skyllamycins represent the first known class of cyclic depsipeptide biofilm inhibitors/dispersers.
INTRODUCTION
Bacterial biofilms are structurally complex biological systems, exhibiting altered phenotypes from their planktonic state (1). Biofilms are made up of an extracellular polymeric substance (EPS) matrix that is composed primarily of polysaccharides but also contains both proteins and nucleic acids (2). Biofilms provide bacteria with a protection mechanism against extreme temperatures, rapid variations in pH, and exposure to UV light, metal ions, and antibiotics (3). Consequently, biofilm-mediated infections tend to be difficult to clear in patients with infections of native tissues (4) or on indwelling medical devices (5–7). To put the impact of biofilms on patients and medical costs into perspective, biofilm-mediated catheter infections annually account for as many as 10,000 deaths and more than $11 billion in increased hospital costs (8).
Pseudomonas aeruginosa is a biofilm-forming Gram-negative proteobacterium. Its biofilm components include a mannose-rich polysaccharide, Psl, a glucose-rich polysaccharide, Pel, and a mannose-derived biopolymer, alginate (9). P. aeruginosa is one of the most common nosocomial pathogens (10) and is of particular concern for cystic fibrosis (CF) patients. CF is an autosomal recessive inherited disease that reduces life expectancy; the reduction in life expectancy is primarily due to respiratory failure, often driven by P. aeruginosa biofilm-mediated chronic pulmonary infection (11, 12).
To discover new therapeutic options for P. aeruginosa biofilm-associated infections, we have developed an image-based screen to directly measure surface-associated biofilm coverage in a high-throughput manner. While there are a number of published screens for biofilm inhibition, there are no reports of a high-throughput screen that directly quantifies biofilm coverage and that is able to model both inhibition and dispersion under similar conditions (13–18). To create such a system, we employed an automated epifluorescence microscope using an approach analogous to that of our previously reported image-based screen for Vibrio cholerae biofilm inhibition (19). To model inhibition of initial biofilm attachment, compounds were pinned into test plates immediately after dispensing a bacterial culture. To model induction of biofilm dispersion, cultures were allowed to initiate biofilm formation by incubation for 2 h prior to compound addition using the 384-well pin transfer robot. Using this delayed pinning approach, we were able to recapitulate the antibiotic tolerance phenotype of bacterial biofilms reported from both laboratory and clinical observations and to profile our natural-product library for the presence of compounds capable of clearing preattached biofilm infections.
In addition to screening for biofilm inhibition/dispersal, it is important to be able to accurately differentiate between compounds with selective biofilm-modulating activities and traditional antibiotics. This is not possible from imaging data alone, as both classes of compounds give low biofilm coverage readings at the end of the incubation period. In order to distinguish between these two biological behaviors, we incorporated a colorimetric measurement of bacterial cellular activity (XTT) and demonstrated that this approach is both compatible with image-based screening and capable of easily differentiating between biofilm modulators and classical antibiotics. Holistically, this modular screen therefore provides a model for the identification both of antibiotics that are active in biofilm states and of new compounds that are capable of inhibiting initial biofilm formation and/or dispersal of preattached biofilms.
Using this screening method, 312 marine-derived microbial prefractions were screened for both biofilm inhibition and dispersal, leading to the identification of five hits in the biofilm inhibition model (BIM) assay and five hits in the biofilm dispersion model (BDM) assay. Only two of the prefractions were active in both the BIM and BDM assays. Examination of prefraction 1675D (Fig. 1) revealed the active constituents of this extract to be the cyclic depsipeptides skyllamycins A to C (20, 21). Further biological evaluation of skyllamycin B, the only analog to show BDM activity, demonstrated promising codose activity with azithromycin leading to direct removal of biofilm coverage and clearance of planktonic cell populations, a result that was not possible using either compound independently.
FIG 1.
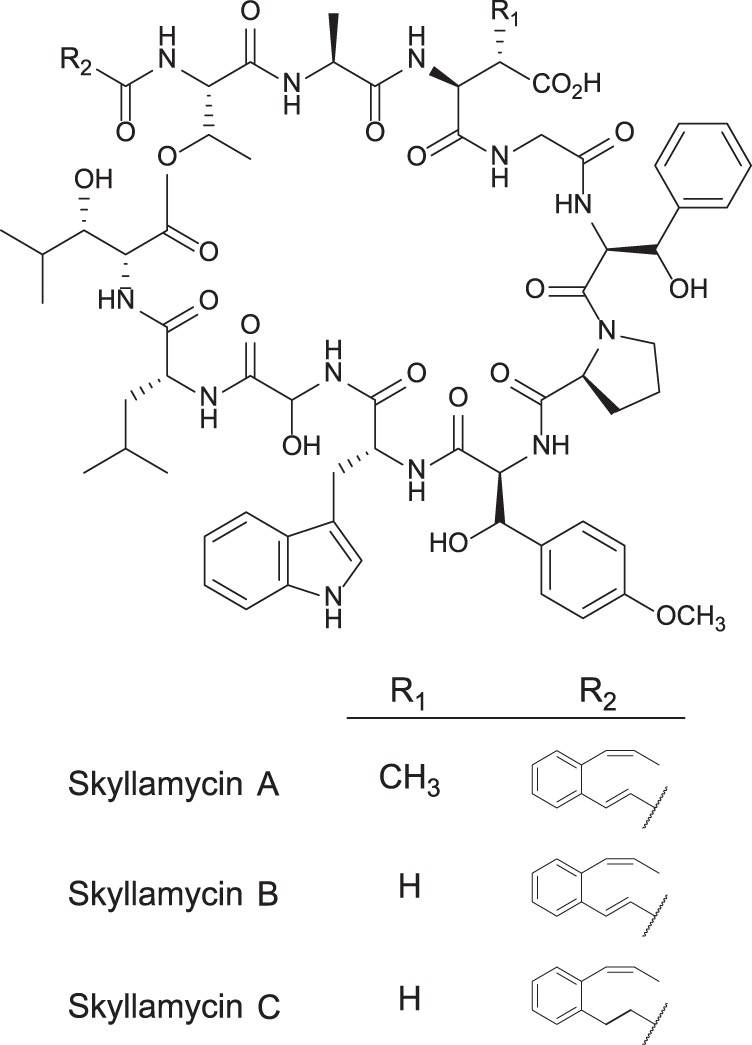
Cyclic depsipeptides skyllamycins A to C. Skyllamycins A and B differ by the presence or absence of β-methylation on the aspartate residue. Skyllamycins B and C differ by the oxidation state of the α-β carbons on the polyketide-derived portion.
MATERIALS AND METHODS
Strains and medium conditions.
Streptomyces sp. strain 1675 was isolated from a marine sediment sample collected at Westport Jetty, WA, under permit number 10-395 from the Washington Department of Fish and Wildlife. The sediment sample was stamped onto AIS media (18.0 g/liter actinomycete isolation agar [Difco], 5.0 g/liter glycerol [Difco], 50 mg/liter nalidixic acid [Sigma], 50 mg/liter cycloheximide [Acros]), and a single colony of Streptomyces sp. strain 1675 was isolated and plated on MB media (31.2 g/liter Instant Ocean mix [Instant Ocean], 20 g/liter agar [Fisher], 5 g/liter peptone [Difco], 1 g/liter yeast extract [Difco]) (22–24). For PCR amplification of the 16S rRNA gene, primers 8F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) were used. Sequencing was performed by Sequetech Corporation using the PCR primers described above plus an additional middle primer, 341F (5′-CCTACGGGAGGCAGCAG-3′) (25). The strain was identified as Streptomyces sp. using 16S rRNA gene analysis of a 1,360-bp sequence (GenBank accession number KF734086) with 100% similarity to the closest homolog and is stored at the University of California Santa Cruz (UCSC) under strain code RL10-429-AIS-A. Skyllamycins A to C were extracted and purified from Streptomyces sp. strain 1675 as described in the supplemental material. P. aeruginosa strains PAO1 ΔwspF and ΔwspF ΔpelA ΔpslBCD were generously provided by the laboratory of Matthew Parsek.
GFP tagging of P. aeruginosa strains.
Insertion of the GFP gene into the P. aeruginosa genome was delivered by electroporation of mini-Tn7. A plasmid encoding a transposase gene (pUX-BF13) and the other mini-Tn7 vector carrying the GFP cassette constitutively expressed from a rRNA promoter (pMCM11) were introduced into ΔwspF and ΔwspF ΔpelA ΔpslBCD mutants by electroporation. Conjugants were selected on LB agar plates containing gentamicin at 30 μg/ml. Insertion of the mini-Tn7 cassette into the genome was biased toward several base pairs between the PA5549 and PA5548 loci. Confirmation of the GFP insertion was performed by colony PCR using primers Tn7-R (5′-CACAGCATAACTGGACTGATTTC-3′) and glmS-F (5′-GCACATCGGCGACGTGCTCTC-3′).
Biofilm inhibition/dispersal assay.
After overnight LB liquid culture growth, cultures were diluted 1:25 into 50 ml of 5% LB (0.2 g/liter tryptone [Fisher], 0.1 g/liter yeast extract [Difco], 10 g/liter NaCl [Fisher]) and allowed to acclimate to nutrient-poor media for approximately 30 min before they were dispensed into 384-well plates (Corning; catalog no. 3712). A 40-μl volume of the liquid culture was dispensed into each well, and the wells were covered with an air-permeable membrane (E&K Scientific T896100-S) and incubated for 6 h at 30°C. Due to the heterogeneous properties of biofilm-state bacteria, it is critical to maintain a light swirling motion to keep bacteria evenly distributed in liquid media while dispensing. When BIM assays were performed, compounds were pinned immediately after dispensing of the culture. When BDM assays were performed, compounds were pinned after 2 h of incubation at 30°C. After a total of 6 h of incubation, 30 μl of 0.5 mg/ml of XTT–phosphate-buffered saline (PBS) buffer with 200 μM menadione (MEN) (CAS; catalog no. 58-27-5) was added. Measurements of the background absorption at 490 nm were taken immediately after dispensing XTT/MEN and again after 2 h of incubation at 30°C.
Biofilm quantification.
For biofilm measurements, each well was quantified by the acquisition of 8 images at ×20 magnification. The combined ×20 images account for ∼20% of the total well surface area, which is critical for reproducible and accurate quantification of biofilm coverage. The images were analyzed using MetaXpress image software (Molecular Devices) to quantify biofilm coverage. Biofilms were identified using a modified version of the existing “count nuclei” script. This modified script compares the intensities of all pixels in the image to identify pixel clusters with intensities above a user-defined threshold (see Fig. S1 in the supplemental material). These bright pixel clusters are defined as representing biofilm coverage. From past experience, we knew that the total biofilm coverage for the negative control must be greater than 25% for a reproducible and accurate 50% effective concentration (EC50) for the polymyxin B dilution series control.
Nucleotide sequence accession number.
The Streptomyces sp. strain sequence identified in this work has been submitted to GenBank under accession number KF734086.
RESULTS
Screen design and implementation.
Having recently developed a 384-well plate high-content high-throughput biofilm screen for Vibrio cholerae, we elected to use a similar approach to study P. aeruginosa biofilms (19). To extend the utility of the screening platform for examining both biofilm inhibition and disruption of bacterial cell viability, we elected to develop a pair of complementary assays that could be run in the same screening plate to measure simultaneously both bacterial biofilm coverage and cellular activity. The advantage of this approach is that it provides direct evaluation of biofilm clearance due to inhibition of biofilm formation versus bacteriostatic or bactericidal activities and can therefore clearly differentiate between classical antibiotics and compounds that decrease biofilm coverage without impacting cell viability. We hypothesized that this method could identify compounds with modes of action orthogonal to those of traditional antibiotics, delivering new molecular scaffolds to treat biofilm-mediated infections.
Measurement of biofilm coverage was accomplished by imaging a P. aeruginosa GFP-tagged strain with enhanced ability to form biofilms in the 384-well format using non-z-stack epifluorescence microscopy. Imaged biofilms were quantified using an automated script to segment and measure bright regions of biofilm coverage. Bacterial cellular activity was measured with the use of an optimized XTT assay (26). With respect to statistical screening quality control, the addition of dimethyl sulfoxide (DMSO) (vehicle) to 32 control wells served as a negative control for the BIM and BDM assays (Fig. 2A). For positive controls, we chose both chemical and biological methods. The biological-screening control was a ΔpelA ΔpslBCD strain of P. aeruginosa incapable of producing biofilms (27, 28). For the chemical control, we included a dilution series of polymyxin B, which was used to quantify plate-to-plate variability of the assay. Biofilm quantification of the negative-control images resulted in an average of 35% biofilm coverage of the total imaged surface area and Z′-factor scores of greater than 0.7 per assay. For the XTT cellular activity assay, these conditions resulted in Z′-factor scores of greater than 0.6 per assay.
FIG 2.

Biofilm images directly from wells of a 384-well assay plate. White bars represent 100 μm. (A) Non-z-stack epifluorescence biofilm images at ×20 magnification for quality control statistical metrics. Negative control: healthy biofilm formation with addition of DMSO vehicle. Genetic positive control: the ΔpelA ΔpslBCD strain shows no biofilm formation due to knockout of biofilm genes. Chemical positive control: no biofilm formation due to treatment of polymyxin B at final concentration of 2 μM. Biofilm inhibitor: biofilm inhibition with treatment of skyllamycin B at final concentration of 102 μM. (B) Confocal images of biofilms at ×10 magnification. Healthy biofilm formation under assay conditions with DMSO and without DMSO is shown.
To directly compare the imaging techniques in our assay wells to traditional confocal imaging techniques, we imaged the biofilms directly in the 384-well plate with a confocal microscope. Confocal images of the negative-control P. aeruginosa biofilms show that the biofilm integrity was similar to that seen with traditional culture conditions (Fig. 2B). In addition, biofilm morphologies in the absence of DMSO developed identically to those seen with wells with DMSO (negative control), suggesting that the addition of up to 1% DMSO had no effect on biofilm growth.
Simultaneous acquisition of both biofilm coverage and cellular activity measurements provides a fast and efficient method to differentiate between antibiotic and biofilm inhibitor/disperser activities. Compound activity plots were divided into three subgroups: (i) compounds that did not remove biofilm coverage but did prevent cellular activity, representing bacteriostatic antibiotics; (ii) compounds that removed biofilm coverage and decreased cellular activity, representing a classical phenotype associated with bactericidal antibiotics; and (iii) compounds that removed biofilm coverage but did not affect cellular activity. The compounds in the third subgroup were of the highest value to this project, as these include both biofilm inhibitors and dispersers. Compounds that either inhibit biofilm formation or disrupt preattached biofilms are of particular interest as they remove biofilm coverage through a mechanism that does not modulate cellular metabolic activity (as measured by XTT reactivity), thus potentially decreasing the selective pressure to develop resistance.
Evaluation of antibiotic tolerance.
Clinically, one of the most significant challenges in the treatment of biofilm-mediated infections is that of tolerance to antibiotics imparted by biofilm formation (29). To be of relevance to this issue, the BDM assay should recapitulate the antibiotic tolerance observed under clinical settings. To examine whether the BDM assay was a suitable model for evaluating this behavior, we screened an array of antibiotics in both the BIM and BDM assays and calculated EC50 shifts in biofilm coverage between the two assays as a measure of antibiotic tolerance. We elected to screen members of three commonly used classes of antibiotics (aminoglycosides, macrolides, and tetracyclines), as well as our positive control, polymyxin B, in both assays (see Table S2 in the supplemental material) (30). Figure 3 presents the dose-response curves for the most active members of each of the tested antibacterial classes: tobramycin (aminoglycoside), azithromycin (macrolide), demeclocycline (tetracycline), and polymyxin B.
FIG 3.
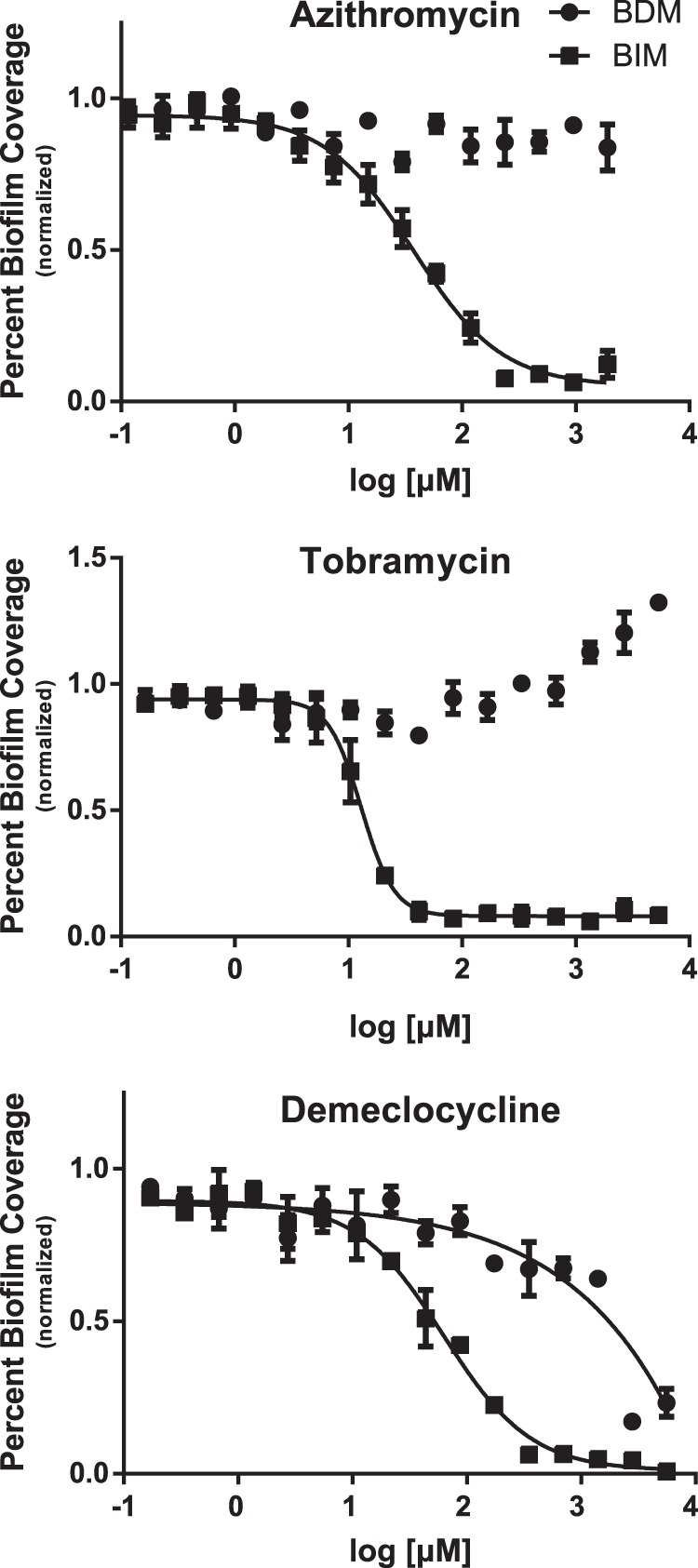
Antibiotic biofilm coverage measurements for BIM and BDM assays, demonstrating a general increase in antibiotic tolerance for the BDM assay. The BIM EC50s for azithromycin, tobramycin, and demeclocycline were 30, 9.0, and 75 μM, respectively. Demeclocycline showed an EC50 of 2,500 μM in the BDM assay, while azithromycin and tobramycin were inactive up to the highest tested concentrations (3,800 and 5,350 μM, respectively). Antibiotic tolerance increased by values of greater than 120- and 590-fold for azithromycin and tobramycin, respectively, while demeclocycline displayed a 33-fold increase in antibiotic tolerance. Assays were performed with 3 technical replicates and 3 biological replicates. For the presented replicate, the Z′ scores were 0.70 for the BIM assay and 0.63 for the BDM assay.
Encouragingly, all three antibiotic classes displayed significant EC50 shifts between the BIM and BDM assays, indicating that these compounds are much less effective at clearing preattached biofilms than inhibiting initial attachment under these test conditions. For azithromycin and tobramycin, the BIM EC50s were 30 and 9.0 μM, respectively, whereas the highest attainable soluble concentrations in DMSO (3,800 and 5,350 μM) did not affect biofilm coverage in the BDM assay. This represents developments in antibiotic tolerances in the BDM assay of greater than 120- and 590-fold for azithromycin and tobramycin, respectively. Demeclocycline had a BIM EC50 = 75 μM and a BDM EC50 = 2,500 μM, representing an antibiotic tolerance of 33-fold. These data show that antibiotic tolerance, a characteristic of biofilm-state bacteria (29), was present in the BDM assay, ranging anywhere from 33-fold to greater than 590-fold with respect to that of the BIM assay. As previously reported, biofilm coverage increases with the amount of tobramycin added as a defense mechanism against aminoglycoside antibiotics (31). Notably, the same phenomenon was observed under our 384-well plate culture conditions, suggesting that the results from this primary screening platform could have direct relevance to clinical issues surrounding antibiotic resistance.
Natural-product screening.
After establishing both assays and validating their utility for quantifying biofilm-mediated antibiotic tolerance, we initiated a screening campaign to identify novel small-molecule-based biofilm inhibitors with activity against P. aeruginosa. To accomplish this, we screened a subset of our marine microbial natural-product library using both the BIM and BDM assays. Hits from this initial screen were categorized into four bins, as illustrated by the four segments in Fig. 4: in bin 1, inactive compounds, with normalized biofilm coverage ≥ 0.55 and normalized cellular activity ≥ 0.8; in bin 2, nonantibiotic biofilm inhibitors, with low normalized biofilm coverage (<0.55) but no effect on cellular activity; in bin 3, bactericidal antibiotics with both low biofilm coverage and decreased cellular metabolic activity; and in bin 4, bacteriostatic antibiotics with biofilm coverage values similar to those of the negative controls but with low cellular metabolic activities.
FIG 4.

BIM and BDM assay plots of results from natural-product screening. The y axis values represent percentages of biofilm coverage normalized to DMSO controls. The x axis values represent absorption at 490 nm normalized to DMSO controls. Wells with less than 55% biofilm coverage were defined as extracts that modulate biofilm formation. Wells with normalized cellular metabolic activity greater than 0.8 were considered to be nonantibiotic.
Generally, we observed fewer prefractions capable of reducing biofilm coverage in the BDM assay than in the BIM assay. This is seen by a decrease in the number of extracts in bin 3 and a concomitant increase in the number of extracts in bin 4 for the BDM versus the BIM assays. This is perhaps to be expected, given that bacterial cells are known to enter a quiescent state inside biofilm microcolonies, making them less susceptible to drug treatment (32). Therefore, the antibiotic leads from the BDM assay are of particular interest, as they possess the ability to affect biofilm clearance even in the presence of preattached biofilms. However, the objective of this initial study was to identify new lead compounds as inhibitors/dispersers of biofilm formation for use against P. aeruginosa; thus, leads from bin 2 were prioritized and profiled using our standard liquid chromatography-mass spectrometry (LC-MS) analysis methods (see Fig. S2 in the supplemental material). From these analyses, one prefraction containing a unique metabolite profile (1675D) was selected for further characterization.
Isolation and structure elucidation of skyllamycins A to C.
LC-MS analysis of the selected prefraction (1675D) identified three major components with similar UV absorbance profiles (see Fig. S2 in the supplemental material). High-resolution ionization–time of flight mass spectroscopy (HRESITOFMS) analysis revealed that two of these metabolites possessed mass spectrometric data consistent with the known cyclic depsipeptides skyllamycins A and B, while data for the third metabolite suggested a skyllamycin B derivative with a mass gain of 2 atomic mass units. Using standard isolation procedures (see the supplemental material), approximately 2 mg of each of the major components was isolated from 6 liters of the producing strain for spectroscopic analysis and biological follow-up.
1H nuclear magnetic resonance (NMR) comparison to published structures (21) revealed close agreement with the original assignments for skyllamycins A and B (see Fig. S3 in the supplemental material). Furthermore, correlation spectroscopy (COSY) and total correlation spectroscopy (TOCSY) data identified all of the spin systems associated with the predicted proteinogenic and nonproteinogenic amino acids, which were assembled through heteronuclear multiple-bond correlations (HMBC) to verify that each compound contained the skyllamycin core. To further verify the identities of these metabolites, skyllamycin A was compared to an authentic sample (20) by LC-MS coinjection, which demonstrated that our sample and the authentic material shared identical retention times (see Fig. S4 in the supplemental material). In order to prove that the isolated structures were not enantiomeric with respect to previously reported structures, Marfey's analysis (33) was conducted on skyllamycins A and B. The absolute configurations of all the proteinogenic amino acids corroborated those originally assigned by biosynthetic studies (see Fig. S5 in the supplemental material). Taken together with optical rotation measurements, the absolute configuration of these isolated skyllamycin analogs can therefore be confirmed as representing the previously published structures (20, 21).
Substantiating the HRESITOFMS data showing a gain of two protons, the one-dimensional (1D) and 2D NMR skyllamycin C data revealed a skyllamycin B analog with a reduced olefin on the polyketide synthase (PKS)-derived portion of the molecule (see Fig. S3 in the supplemental material). This interpretation was supported by the loss of two vinyl protons associated with the PKS-derived portion and the gain of four new aliphatic protons. Utilizing the heteronuclear single-quantum correlation (HSQC) and HMBC data, these new aliphatic signals were identified as part of the reduction product of the trans-olefin. Stereochemistry of the whole molecule was inferred to be identical to that of skyllamycin A and B due to Marfey's analysis (see Fig. S5), optical rotation, and biosynthetic stereospecificity (20). Therefore, skyllamycin B differs from skyllamycin A by the absence of β-methylation on the aspartic acid residue (R1 = H versus R1 = CH3; Fig. 1), while skyllamycin C differs from skyllamycin A by the inclusion of one fewer double bond in the polyketide portion of the sidechain (R2; Fig. 1).
Biological evaluation of skyllamycins A to C.
Biological activity evaluation of skyllamycins A to C in the BIM assay revealed that skyllamycin B had an EC50 = 30 μM, skyllamycin C had an EC50 = 60 μM, and skyllamycin A possessed only marginal activity (>250 μM) (Fig. 5). From these data, we can postulate that the aspartate residue plays an important role in a molecular contact, while the cinnamic acid is not as critical. In the cellular activity assay, none of the analogs displayed any reduced cellular activity up to the highest tested concentrations. In the BDM assay, only skyllamycin B showed biofilm clearance, with an EC50 = 60 μM, with no effect on cellular activity up to the highest tested concentrations.
FIG 5.
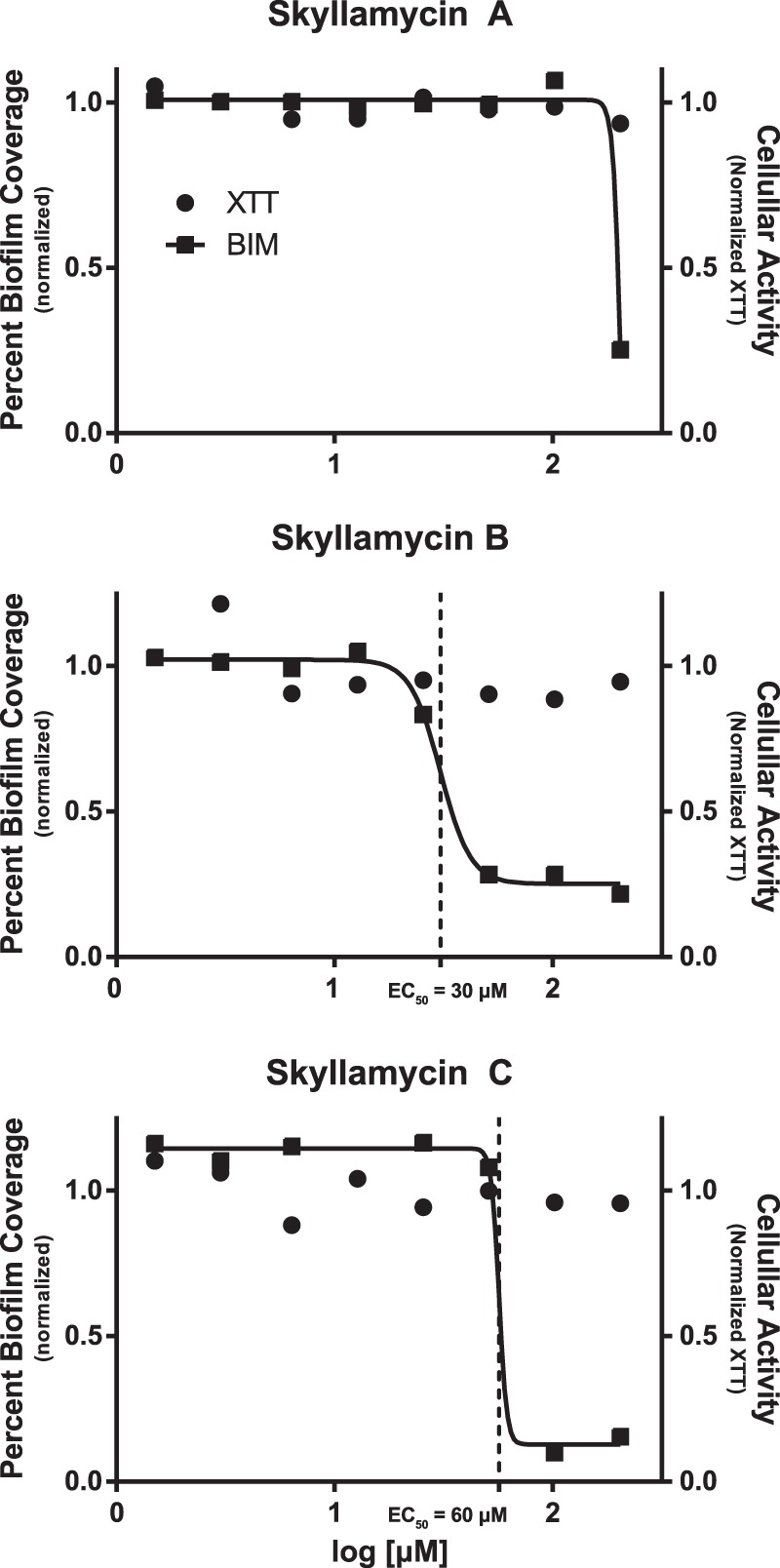
Dose response curves for skyllamycins A to C against percent biofilm coverage (■) and cellular activity (•).
Structurally, the skyllamycins are a unique family of P. aeruginosa biofilm inhibitors. While a number of P. aeruginosa biofilm inhibitors have been published to date (34–38), the majority of inhibitors with nonmicrobicidal effects have been identified as quorum-sensing mimics (36, 37) or as related to quorum modulation (38). As skyllamycin B is structurally unrelated to quorum mimics, its activity is most likely not directly quorum mediated. The skyllamycins are the first reported cyclic depsipeptides with P. aeruginosa biofilm inhibition and dispersal activity, making the elucidation of the mechanism of action for biofilm inhibition a very attractive subject for future work.
Azithromycin and skyllamycin B codose experiments.
The ability to codose antibiotics with biofilm dispersers to clear biofilm-mediated infections is potentially of significant therapeutic value. Codosing has been proposed previously as an attractive alternative to antibiotic monotherapy treatments for biofilm-mediated infections, since it has been suggested that codose experiments could prevent or eliminate bacterial resistance (39). To assess the utility of skyllamycin B for eliminating established biofilm infections by coadministration with current antibiotics, we conducted a codose experiment in the BDM assay. Using protocol conditions identical to those used for the BDM assay, we pinned a concentration array of skyllamycin B, ranging from EC100 to EC10, immediately followed by pinning of three different concentrations of azithromycin, all at concentrations above the EC50 in the BIM assay (see Table S3 in the supplemental material).
Confocal images of BDM assay codose experiments (Fig. 6) highlight the potential of combination therapies for biofilm-mediated infections. Skyllamycin B, while able to detach biofilms independently, could not suppress bacterial cellular metabolic activity. Similarly, azithromycin alone had no effect on biofilm coverage at the tested concentrations. However, skyllamycin B with azithromycin resulted in both the detachment of biofilms and the concomitant reduction of cellular metabolic activity. Since current treatment of biofilm-mediated infections depends on high dosages of antibiotics, procuring antibiotic resistance to the etiological pathogen, the codose results provide encouraging alternative conditions for biofilm detachment and bacterial cellular activity depression, a method that could ameliorate or circumvent acquisition of antibiotic resistance.
FIG 6.

Confocal images and cellular activities of BDM assay codose experiments with skyllamycin B and azithromycin taken at ×10 magnification. White bars represent 100 μm. (A) Azithromycin at 99 μM. (B) Skyllamycin B at 100 μM. (C) Azithromycin at 99 μM and skyllamycin B at 100 μM (codose).
DISCUSSION
Given the significant impact of biofilm-mediated infections on human health, and the biomedical costs associated with P. aeruginosa biofilm-mediated infections, new treatment options are vital to address biofilm-mediated infections. In order to identify biofilm inhibitors and dispersal agents, we have developed a modular assay that models both biofilm inhibition and dispersal and that can recapitulate clinically observed biofilm-mediated antibiotic tolerance. Performing the BIM and BDM assays in parallel promotes the identification of lead compounds that can be directly evaluated for their potential either as prophylactic treatments for inhibiting initial biofilm formation (e.g., as coatings for medical implant devices) or as treatments for eliminating established biofilm infections.
Inclusion of two independent measurements to quantify both cellular activity and biofilm coverage increased the ability of this screen to differentiate between different biological behaviors. Notably, if the marine microbial natural-product library had been screened for antibiotic activity using XTT alone, it would not have been possible to differentiate between the bacteriostatic and bactericidal antibiotics from the extracts found in bins 3 and 4. However, by directly measuring bacterial colonies attached to well surfaces, we can distinguish between bacteriostatic (bin 4) and bactericidal (bin 3) activities. Likewise, compounds in bins 1 and 2 would have been overlooked as inactive compounds because they show little to no effect on cellular metabolic activity. However, by measuring surface-associated bacterial cells, it is possible to directly differentiate between biofilm inhibitors/dispersers and inactive compounds. This combination of image-based biofilm screening and XTT-based cellular metabolic activity screening therefore provides a more detailed and complete view of the roles of lead compounds as antibiotics or antibiofilm agents.
One advantage of this whole-cell screening strategy is that the screen is configured to discover biofilm inhibitors in a target-independent manner. This is in contrast to target-based screening approaches, which report directly on the ability of test compounds to inactivate predefined target proteins of interest. This screen employs a ΔwspF mutant of P. aeruginosa, which overproduces cyclic-di-GMP and therefore forms strong, competent biofilms. Consequently, hits discovered from this screen are inherently robust leads, given that biofilm formation occurs at elevated levels in the ΔwspF mutant compared to the WT strain. However, because the platform is target independent, it is not possible to extract mechanistic or mode-of-action information directly from the primary screen. Therefore, although we would expect to identify inhibitors from a variety of targets, including quorum sensing, cyclic-di-GMP production, and matrix protein production, additional experiments are required to further delineate mechanistic information for lead compounds.
Screening a subset of our marine natural-product library has identified multiple inhibitors/dispersers that eliminate biofilms while having little or no impact on cellular survival. These leads are promising as molecular probes or as potential therapeutics, as they do not directly kill the bacterial cells but rather prevent/disperse the more persistent biofilm state. From these initial leads, we have identified the skyllamycin cyclic depsipeptide family as a unique and enticing new class of biofilm inhibitor/dispersal agents and identified one new natural product, skyllamycin C. Given the nonantibiotic nature of skyllamycin B, the mechanism of action by which it clears biofilm coverage is likely to be different from that of antibiotic-mediated biofilm clearance. Skyllamycin B, a large cyclic depsipeptide, is structurally distinct from known P. aeruginosa biofilm inhibitors/dispersers, which have been almost exclusively quorum-sensing-molecule mimics. Skyllamycin B also showed promising codose activity with azithromycin, demonstrating that combination treatment using the two drugs is able to remove biofilm coverage and suppress cellular activity, a result not possible using either drug independently.
Overall, this screen successfully identified compounds that clear biofilms, highlighted by the discovery of skyllamycin B, raising the possibility of developing new therapeutic options to treat P. aeruginosa biofilm-mediated infections.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH-R21AI098836-01 (R.G.L. and F.H.Y.) and an NSF Graduate Research Fellowship (G.N.).
We thank M. Parsek for providing P. aeruginosa strains, R. Süssmuth for providing an authentic sample of skyllamycin A, and R. S. Lokey for providing access to the UCSC Chemical Screening Center.
Footnotes
Published ahead of print 2 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01781-13.
REFERENCES
- 1.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. 10.1126/science.284.5418.1318 [DOI] [PubMed] [Google Scholar]
- 2.Wingender J, Neu T, Flemming H. 1999. What are bacterial extracellular polymeric substances?, p 1–19 In Wingender J, Neu T, Flemming H. (ed), Microbial extracellular polymeric substance characterization, structure and function, 1st ed. Springer, Berlin, Germany [Google Scholar]
- 3.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2:95–108. 10.1038/nrmicro821 [DOI] [PubMed] [Google Scholar]
- 4.Lynch AS, Robertson GT. 2008. Bacterial and fungal biofilm infections. Annu. Rev. Med. 59:415–428. 10.1146/annurev.med.59.110106.132000 [DOI] [PubMed] [Google Scholar]
- 5.Fux CA, Costerton JW, Stewart PS, Stoodley P. 2005. Survival strategies of infectious biofilms. Trends Microbiol. 13:34–40. 10.1016/j.tim.2004.11.010 [DOI] [PubMed] [Google Scholar]
- 6.Donlan RM. 2001. Biofilms and device-associated infections. Emerg. Infect. Dis. 7:277–281. 10.3201/eid0702.010226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khoury AE, Lam K, Ellis B, Costerton JW. 1992. Prevention and control of bacterial infections associated with medical devices. ASAIO J. 38:M174–M178. 10.1097/00002480-199207000-00013 [DOI] [PubMed] [Google Scholar]
- 8.Scott RD. 2009. The direct medical costs of healthcare-associated infections in US hospitals and the benefits of prevention. National Center for Preparedness, Detection, and Control of Infectious Diseases (U.S.), Division of Healthcare Quality Promotion, Atlanta, GA [Google Scholar]
- 9.Franklin MJ, Nivens DE, Weadge JT, Howell PL. 2011. Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2:167. 10.3389/fmicb.2011.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obritsch MD, Fish DN, MacLaren R, Jung R. 2005. Nosocomial infections due to multidrug-resistant Pseudomonas aeruginosa: epidemiology and treatment options. Pharmacotherapy 25:1353–1364. 10.1592/phco.2005.25.10.1353 [DOI] [PubMed] [Google Scholar]
- 11.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. 1996. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85:229–236. 10.1016/S0092-8674(00)81099-5 [DOI] [PubMed] [Google Scholar]
- 12.Donlan RM, Costerton JW. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15:167–193. 10.1128/CMR.15.2.167-193.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müsken M, DiFiore S, Römling U, Häussler S. 2010. A 96-well-plate-based optical method for the quantitative and qualitative evaluation of Pseudomonas aeruginosa biofilm formation and its application to susceptibility testing. Nat. Protoc. 5:1460–1469. 10.1038/nprot.2010.110 [DOI] [PubMed] [Google Scholar]
- 14.O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461. 10.1046/j.1365-2958.1998.00797.x [DOI] [PubMed] [Google Scholar]
- 15.Müsken M, DiFiore S, Dötsch A, Fischer R, Häussler S. 2010. Genetic determinants of Pseudomonas aeruginosa biofilm establishment. Microbiology 156:431–441. 10.1099/mic.0.033290-0 [DOI] [PubMed] [Google Scholar]
- 16.Peng F, Hoek EMV, Damoiseaux R. 2010. High-content screening for biofilm assays. J. Biomol. Screen. 15:748–754. 10.1177/1087057110374992 [DOI] [PubMed] [Google Scholar]
- 17.Junker LM, Clardy J. 2007. High-throughput screens for small-molecule inhibitors of Pseudomonas aeruginosa biofilm development. Antimicrob. Agents Chemother. 51:3582–3590. 10.1128/AAC.00506-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandberg M, Määttänen A, Peltonen J, Vuorela PM, Fallarero A. 2008. Automating a 96-well microtitre plate model for Staphylococcus aureus biofilms: an approach to screening of natural antimicrobial compounds. Int. J. Antimicrob. Agents 32:233–240. 10.1016/j.ijantimicag.2008.04.022 [DOI] [PubMed] [Google Scholar]
- 19.Peach KC, Bray WM, Shikuma NJ, Gassner NC, Lokey RS, Yildiz FH, Linington RG. 2011. An image-based 384-well high-throughput screening method for the discovery of biofilm inhibitors in Vibrio cholerae. Mol. Biosyst. 7:1176–1184. 10.1039/c0mb00276c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pohle S, Appelt C, Roux M, Fiedler H-P, Süssmuth RD. 2011. Biosynthetic gene cluster of the non-ribosomally synthesized cyclodepsipeptide skyllamycin: deciphering unprecedented ways of unusual hydroxylation reactions. J. Am. Chem. Soc. 133:6194–6205. 10.1021/ja108971p [DOI] [PubMed] [Google Scholar]
- 21.Toki S, Agatsuma T, Ochiai K, Saitoh Y, Ando K, Nakanishi S, Lokker NA, Giese NA, Matsuda Y. 2001. RP-1776, a novel cyclic peptide produced by Streptomyces sp., inhibits the binding of PDGF to the extracellular domain of its receptor. J. Antibiot. (Tokyo) 54:405–414. 10.7164/antibiotics.54.405 [DOI] [PubMed] [Google Scholar]
- 22.Peach KC, Cheng AT, Oliver AG, Yildiz FH, Linington RG. 17 September 2013. Discovery and biological characterization of the auromomycin chromophore as an inhibitor of biofilm formation in Vibrio cholerae. Chembiochem 16:2209–2215. 10.1002/cbic.201300131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong WR, Oliver AG, Linington RG. 2012. Development of antibiotic activity profile screening for the classification and discovery of natural product antibiotics. Chem. Biol. 19:1483–1495. 10.1016/j.chembiol.2012.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulze CJ, Bray WM, Woerhmann MH, Stuart J, Lokey RS, Linington RG. 2013. “Function-first” lead discovery: mode of action profiling of natural product libraries using image-based screening. Chem. Biol. 20:285–295. 10.1016/j.chembiol.2012.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe K, Kodama Y, Harayama S. 2001. Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J. Microbiol. Methods 44:253–262. 10.1016/S0167-7012(01)00220-2 [DOI] [PubMed] [Google Scholar]
- 26.Tunney MM, Ramage G, Field TR, Moriarty TF, Storey DG. 2004. Rapid colorimetric assay for antimicrobial susceptibility testing of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48:1879–1881. 10.1128/AAC.48.5.1879-1881.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirisits MJ, Prost L, Starkey M, Parsek MR. 2005. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 71:4809–4821. 10.1128/AEM.71.8.4809-4821.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Argenio DA, Calfee MW, Rainey PB, Pesci EC. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J. Bacteriol. 184:6481–6489. 10.1128/JB.184.23.6481-6489.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donlan RM. 2002. Biofilms: microbial life on surfaces. Emerg. Infect. Dis. 8:881–890. 10.3201/eid0809.020063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feeley TW, Du Moulin GC, Hedley-Whyte J, Bushnell LS, Gilbert JP, Feingold DS. 1975. Aerosol polymyxin and pneumonia in seriously ill patients. N. Engl. J. Med. 293:471–475. 10.1056/NEJM197509042931003 [DOI] [PubMed] [Google Scholar]
- 31.Hoffman LR, D'Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. 2005. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 436:1171–1175. 10.1038/nature03912 [DOI] [PubMed] [Google Scholar]
- 32.Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5:48–56. 10.1038/nrmicro1557 [DOI] [PubMed] [Google Scholar]
- 33.Bhushan R, Brückner H. 2004. Marfey's reagent for chiral amino acid analysis: a review. Amino Acids 27:231–247. 10.1007/s00726-004-0118-0 [DOI] [PubMed] [Google Scholar]
- 34.Huigens RW, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R, Melander C. 2007. Inhibition of Pseudomonas aeruginosa biofilm formation with bromoageliferin analogues. J. Am. Chem. Soc. 129:6966–6967. 10.1021/ja069017t [DOI] [PubMed] [Google Scholar]
- 35.Reyes S, Huigens RW, Su Z, Simon ML, Melander C. 2011. Synthesis and biological activity of 2-aminoimidazole triazoles accessed by Suzuki-Miyaura cross-coupling. Org. Biomol. Chem. 9:3041–3049. 10.1039/c0ob00925c [DOI] [PubMed] [Google Scholar]
- 36.Geske GD, O'Neill JC, Miller DM, Mattmann ME, Blackwell HE. 2007. Modulation of bacterial quorum sensing with synthetic ligands: systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanisms of action. J. Am. Chem. Soc. 129:13613–13625. 10.1021/ja074135h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. 2005. Small molecule inhibitors of bacterial quorum sensing and biofilm formation. J. Am. Chem. Soc. 127:12762–12763. 10.1021/ja0530321 [DOI] [PubMed] [Google Scholar]
- 38.Stowe SD, Richards JJ, Tucker AT, Thompson R, Melander C, Cavanagh J. 2011. Anti-biofilm compounds derived from marine sponges. Mar. Drugs 9:2010–2035. 10.3390/md9102010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Worthington RJ, Richards JJ, Melander C. 2012. Small molecule control of bacterial biofilms. Org. Biomol. Chem. 10:7457–7474. 10.1039/c2ob25835h [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.