

Abstract
Host defense peptides (HDPs) are short antimicrobial peptides of the innate immune system. Deficiencies in HDPs contribute to enhanced susceptibility to infections, e.g., in cystic fibrosis (CF). Exogenous HDPs can compensate for these deficiencies, but their development as antimicrobials is limited by cytotoxicity. Three HDP prodrugs were designed so their net positive charge is masked by a promoiety containing a substrate for the enzyme neutrophil elastase (NE). This approach can confine activation to sites with high NE levels. Enzyme-labile peptides were synthesized, and their activation was investigated using purified NE. Susceptibilities of Pseudomonas aeruginosa to parent and prodrug peptides in the presence and absence of NE-rich CF human bronchoalveolar lavage (BAL) fluid and different NaCl concentrations were compared. The effect of the HDP promoiety on cytotoxicity was determined with cystic fibrosis bronchial epithelial (CFBE41o-) cells. NE in CF BAL fluids activated the HDP prodrugs, restoring bactericidal activity against reference and clinical isolates of P. aeruginosa. However, activation also required the addition of 300 mM NaCl. Under these conditions, the bactericidal activity levels of the HDP prodrugs differed, with pro-P18 demonstrating the greatest activity (90% to 100% of that of the parent, P18, at 6.25 μg/ml). Cytotoxic effects on CFBE41o- cells were reduced by the addition of the promoiety to HDPs. We demonstrate here for the first time the selective activation of novel HDP prodrugs by a host disease-associated enzyme at in vivo concentrations of the CF lung. This approach may lead to the development of novel therapeutic agents with low toxicity that are active under the challenging conditions of the CF lung.
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive disorder characterized by chronic lung disease. Bacterial infection begins soon after birth and is followed by an intense neutrophilic response in the peribronchial and endobronchial spaces, releasing large amounts of neutrophil elastase (NE) (1, 2). By far the most significant pathogen in adult CF infection is Pseudomonas aeruginosa, often causing chronic, endobronchial, drug-resistant infection (3–6). Chronic infection leads to pulmonary exacerbations which, in addition to an exaggerated inflammatory host response to infections, lead to long-term reductions in lung function and premature death (1, 2, 6).
Despite the substantial CF inflammatory response to infection, many components of immunity are compromised in the disease. The high protease levels found in the CF lung, especially of NE (μM concentrations), can degrade lactoferrin, elafin, hemoglobin, and host defense peptides (HDPs) such as the defensins and human cathelicidin (LL-37) (7–10), with members of the latter group being the subject of extensive research as novel alternative antimicrobials. HDPs are short, cationic, amphipathic peptides that play a crucial role in the innate immune system (11). Many of these highly conserved peptides possess substantial antimicrobial activity (12). The antibacterial activity of HDPs is initiated through electrostatic interactions with the anionic phospholipid head groups of the cell envelope. This can lead to either membrane perturbations or translocation across the membrane and interaction with various intracellular targets (13–15). This is also the basis of their selectivity, with characteristics of bacterial membranes such as high levels of anionic lipids and high transmembrane potential allowing endogenous HDPs to target them over eukaryotic membranes (16). Because these peptides have multiple modes of action, including depolarization of the bacterial membrane, pore formation, induction of degradative enzymes, and disruption of intracellular targets, bacteria may have a propensity for the development of resistance to HDPs that is lower than that seen with conventional antibiotics. This would potentially increase the lifetime of any exogenous HDP in clinical use (17) and provide another option for antipseudomonal therapy, should the issue of protease degradation be resolved. As a result of this, HDPs have been the subject of increasing interest.
While a number of HDPs have been the subject of early-phase clinical trials, they have yet to progress further, and the majority of these trials focus on topical applications (17, 18). Furthermore, rapid metabolism and a lack of affinity for their target mean that HDPs often have very narrow therapeutic indices (17, 19). Increased activity due to increased hydrophobicity is often accomplished at the expense of selectivity between eukaryotic and prokaryotic membranes, resulting in host toxicity. Even endogenous HDPs such as LL-37 can be cytotoxic at high concentrations (20). This represents a major obstacle in the use of HDPs in CF; the primary aim of chronic P. aeruginosa infection treatment is the preservation of lung function (5, 21). Research has focused on overcoming these issues by modifying the active HDPs themselves. Approaches that may improve specificity or reduce toxicity include the conjugation of HDPs to conventional antibiotics. For example, magainin 2 has been conjugated to vancomycin, resulting in an increase in permeabilization efficiency via the lipid II-targeting properties of the antibiotic (22). Other strategies include conjugating a HDP to a bacterium-specific pheromone, or a pathogen-specific antibody, resulting in a conjugate active against only the bacterium of interest (23, 24).
Alternatively, a prodrug approach has been used previously by our group to reduce toxicity and increase specificity through selective activation. This was achieved by a HDP modification involving the addition of an oligoglutamic acid promoiety to the HDP P18, linked by a NE-labile trialanine linker, generating a prodrug with reduced antimicrobial activity and net charge compared to the active peptide, P18 (+3 versus +8). Antimicrobial activity was restored by the addition of NE. The pro-HDP also had lower hemolytic activity than P18. However, the peptide resulting from NE activation, AA-P18, had antibacterial activity inferior to that of P18 itself (25).
We describe here the development and evaluation of a further series of NE-targeted prodrugs using an alternative linker moiety, AAAG. Glycine was added between the heterochiral sequences (l-amino acid promoiety and d-amino acid active HDP), as it is achiral, the goal being a cleavage product with comparable rather than inferior bactericidal activity. Three peptides were modified to contain the linker motif and the anionic oligoglutamic acid promoiety.
The pro-HDPs have been developed for local delivery to the CF lung similarly to the method described for tobramycin therapy (3). This would potentially limit cleavage of the pro-HDPs to the bronchioles, where mucus, infection, and the resulting neutrophilic response with NE are localized (1, 5). To simulate the conditions of the airway surface liquid (ASL) with which the pro-HDPs interact, CF bronchoalveolar lavage (BAL) fluid was added to bactericidal assays. Incubation of the pro-HDPs with BAL fluid provides endogenous NE for pro-HDP cleavage and, in addition, is a robust test of the feasibility of local lung delivery. Components of BAL fluid have previously been shown to inhibit the activity of some HDPs (26–31). In this study, we show that this inhibition may be overcome and, for the first time, that an environment similar to that found in the CF lung may be effectively used in the targeted application of pro-HDPs. We also demonstrate that the reversible reduction of the peptide's net charge may reduce the epithelial cell cytotoxicity of HDPs. This report demonstrates a HDP modification with potential for delivery to the CF lung in a manner that reduces cytotoxicity and increases specificity.
MATERIALS AND METHODS
Clinical isolates.
P. aeruginosa clinical isolates from CF patients were obtained from the Microbiological Diagnostic Laboratory of Beaumont Hospital, Dublin, Ireland. Isolate identity was confirmed by the BBL DrySlide oxidase test (BD) and the C-390 Diatab disk test (Rosco Diagnostics, Germany). Isolates were labeled PABH01 to -04.
CF BAL fluid collection.
BAL fluid samples were collected from consenting CF patients. The protocol for collection was approved by the Beaumont Hospital Ethics Research Committee. Briefly, 30 ml of sterile 0.9% NaCl was instilled into the right or left subsegmental bronchus, collected immediately, and stored at 4°C. After centrifugation at 1,500 rpm for 10 min at 4°C, the supernatants were removed and stored in aliquots at −80°C until required.
Peptide synthesis.
Three peptide sequences were investigated, D-Bac8c2, 5 Leu (rlwvlwrr [lowercase letters indicate d-amino acids]), D-HB43 (fakllaklakkll), and D-P188 Leu (kwklfkklpkflhlakkf). Bac8c is an 8-residue bactenecin derivative of net charge +3 (32). HB43 is a 13-residue, salt-resistant peptide of net charge +5 (26). P18 is an 18-residue, salt-resistant hybrid sequence of magainin 2 and cecropin A of net charge +8 (33).
The parent sequences were assembled by automated solid-phase peptide synthesis on a 433A synthesizer (Applied Biosystems, United Kingdom) from 9-fluorenylmethoxy carbonyl (Fmoc)-protected d-amino acids (Merck Chemical, United Kingdom) according to the Fmoc-tBu strategy with HATU [O-(7-azabenzotriazole-1-yl)-N,N,N,N′-tetramethyluronium hexafluorophosphate]/DIEA (N,N-diisopropylethylamine) coupling chemistry from a Rink amide MBHA (4-methylbenzhydrylamine) resin (Sigma, Ireland). Single coupling cycles using a 10-fold excess of Fmoc-amino acids were employed, except for the leucine at position 8 in P188 Leu, where double coupling was required. For the prodrug candidates, the addition of the AAAG linker and four glutamic acids and N-terminal acetylation were carried out manually with l-amino acids in a syringe fitted with a Teflon frit and Kaiser Test monitoring. Peptides were deprotected and cleaved from the synthesis resin using a mixture of 80% trifluoroacetic acid, 5% water, 5% triisopropylsilane, 5% thioanisole, and 5% 1,2-ethanedithiol at room temperature (RT) for either 2 h (pro-HB43 and pro-P18) or 4 h (pro-Bac8c). They were then precipitated, washed three times with 10-ml portions of diethyl ether, dried, dissolved in distilled water, and lyophilized. Chromatographic analysis and purification were performed on a BioCAD Sprint perfusion chromatography workstation (PerSeptive Biosystems, United Kingdom) using Gemini columns (Phenomenex) (110 Å, 5 m, C18, 4.6-mm diameter, and 250-mm length or 100-mm diameter/250-mm length for the analytic or semipreparative columns, respectively). Buffers used were mobile-phase A (0.1% trifluoroacetic acid [TFA] in water) and mobile-phase B (0.1% TFA in acetonitrile) with a gradient of 5% to 65% buffer B in 18 column volumes (analytical) or 5 column volumes (semipreparative) with a flow rate of 1 ml/min (analytical) or 5 ml/min (semipreparative) and single-wavelength detection at 214 nm. Purified peptides were finally characterized by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry using an α-cyano-4-hydroxy-cinnamic acid matrix.
Enzymatic cleavage of pro-HDPs.
Each pro-HDP (250 μg/ml) was incubated with 5 μg/ml purified NE or with 5 or 10 μg/ml pseudolysin (PE) (Elastin Products Company) for 24 h at 37°C in PBS (pH 7.4). At various time points, samples were removed from the incubation mixture and analyzed by high-performance liquid chromatography (HPLC) and MALDI-TOF.
Neutrophil elastase determination.
Neutrophil elastase activity was determined in BAL fluids by measuring the cleavage of the NE substrate, AAPV-pNA (N-methoxysuccinyl-ala-ala-pro-val-P-nitroanilide) (Sigma, Ireland). Briefly, 1 mM AAPV-pNA was added to BAL fluids (diluted as appropriate) in buffer containing 0.05 M sodium acetate and 0.1 M sodium chloride (pH 7.5) in 96-well plates. The rate of AAPV-pNA cleavage was measured as the increase in absorbance at 405 nm over 5 min at 37°C using a Victor X3 2030 multilabel reader (PerkinElmer). Reaction mixtures containing 0.25 to 5 μg/ml purified NE (Elastin Products) were used to construct a standard curve from which the NE concentration of the BAL fluids was calculated.
Susceptibility testing.
MICs were determined using the broth microdilution method according to the guidelines of the Clinical and Laboratory Standards Institute (CLSI) (34), with modifications for cationic peptides as described by Wu and Hancock (35). Briefly, serial doubling dilutions of peptide were made in a sterile solution containing 0.2% (wt/vol) bovine serum albumin (BSA) and 0.01% (vol/vol) acetic acid. These were added to a 96-well microtiter plate with a 1.5 × 105 CFU/ml inoculum of P. aeruginosa reference strain PAO1 and clinical isolates in Mueller-Hinton broth (non-cation adjusted; Oxoid, United Kingdom). The lowest peptide concentration showing no visible growth was recorded as the MIC.
Bactericidal activity.
P. aeruginosa strain PAO1 and clinical isolates were grown overnight at 37°C on Mueller-Hinton (MH) agar. Suspensions were prepared from isolated colonies to a density of a 1.0 McFarland standard (bioMérieux, Ireland) and were further diluted 1/100 in potassium phosphate buffer (pH 7.4) containing 0.2% BSA. Assays were carried out in microcentrifuge tubes and contained 0.78 to 25 μg/ml peptides, 10% (vol/vol) P. aeruginosa suspension (approximately 1.5 × 106 CFU/ml), and 10 mM potassium phosphate buffer (pH 7.4) containing 0.2% BSA. To allow adequate contact time, reaction mixtures were incubated at 37°C and 200 rpm in a shaker incubator (Gallenkamp, United Kingdom) for 1 h and then diluted 1/10 with 0.95% (wt/vol) NaCl. After vortex mixing for 30 s was performed, a 100-μl aliquot was spread onto MH agar and incubated overnight at 37°C. Percent killing activity was calculated from viable counts (CFU/ml) from assays containing peptides compared to control assays not containing peptide. The assay allowed easy modification of conditions, and the effects of the addition of purified NE (5 to 20 μg/ml), CF BAL fluid (25% [vol/vol]), and NaCl (300 to 600 mM) on killing activity were determined by the addition of these to the assays and the inclusion of appropriate controls. Statistical analyses of the data were carried out using Graphpad Prism software and the two-tailed unpaired t test.
Cell culture.
Cystic fibrosis bronchial epithelial (CFBE41o-) cells were cultured in minimal essential medium, supplemented with 10% (vol/vol) fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, at 37°C in a humidified atmosphere with 5% CO2.
Toxicology assay.
Cells were seeded on 96-well plates at a density of 3 × 105 cells/ml and incubated for 24 h at 37°C. The cells were then treated with 0.2 to 300 μM peptides and their prodrugs in serum-free media. After incubation, the medium was removed and the cells were incubated with 500 μg/ml of MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (Sigma, Ireland) for 4 h. The MTT solution was then removed, and 100 μl of dimethyl sulfoxide was added to each well, mixing by shaking was performed, and the absorbance at 560 nm was recorded.
RESULTS
Pro-HDPs are cleaved by purified NE.
Cleavage of pro-HDPs using 5 μg/ml NE was shown after 1 h of incubation. Cleavage mainly occurred between the first and second alanines of the linker group and, to a lesser extent, between the second and third alanines. This cleavage pattern (i.e., AAG-HDP and AG-HDP) was found for pro-Bac8c and pro-P18. Pro-HB43 yielded predominantly AG-HB43. These cleavage products were termed “fragment HDPs” and were synthesized as controls. The AAAG linker was not labile with respect to pseudolysin, as indicated by the detection of only pro-HDPs after incubation with this enzyme for up to 24 h.
Pro-HDPs have greater MIC values than parent and fragment HDPs.
The MIC values for both parent and fragment HDPs were comparable (Table 1). The additional glycine and alanine residues increased the MIC from 4 to 8 μg/ml for the HB43 series, and there was no increase for the P18 or Bac8c series. In all cases, the pro-HDPs had MIC values greater than or equal to the highest concentration tested (≥64 μg/ml). The MICs for both pro-HDPs and fragment HDPs toward PAO1 were comparable to those found for the four clinical isolates tested (Table 1).
TABLE 1.
MICs for parent HDPs, fragment HDPs, and pro-HDPs versus P. aeruginosa PAO1 and clinical isolates
| Peptide | Sequencea | MIC (μg/ml) vs P. aeruginosa strains |
||||
|---|---|---|---|---|---|---|
| PAO1 | PABH01 | PABH02 | PABH03 | PABH04 | ||
| Bac8c | rlwvlwrr-NH2 | 4 | NTb | NT | NT | NT |
| AAG-Bac8c | AAGrlwvlwrr-NH2 | 4 | 8 | 8 | 16 | 8 |
| Pro-Bac8c | Ac-EEEEAAAGrlwvlwrr-NH2 | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 |
| P18 | kwklfkklpkfhlhlakkf-NH2 | 2 | NT | NT | NT | NT |
| AAG-P18 | AAGkwklfkklpkfhlhlakkf-NH2 | 2 | 2 | 4 | 4 | 2 |
| Pro-P18 | Ac-EEEEAAAGkwklfkklpkfhlhlakkf-NH2 | ≥64 | 64 | 64 | ≥64 | 64 |
| HB43 | fakllaklakkll-NH2 | 4 | NT | NT | NT | NT |
| AG-HB43 | AGfakllaklakkll-NH2 | 8 | 4 | 8 | 4 | 4 |
| Pro-HB43 | Ac-EEEEAAAGfakllaklakkll-NH2 | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 |
Uppercase characters represent l-amino acids; lowercase characters represent d-amino acids.
NT, not tested.
Pro-HDPs are bactericidal toward PAO1 in the presence of NE.
The bactericidal activities of parent and fragment HDPs were comparable (Fig. 1) over the concentration range tested. As 100% killing of PAO1 was achieved at 6.25 μg/ml for parent and fragment HDPs, this concentration was selected for bactericidal testing of pro-HDPs in the presence of NE. Similar to the susceptibility determined on the basis of MIC values, bactericidal activity was negligible for pro-Bac8c and pro-HB43 in the absence of NE, but in contrast to the poor activity of pro-P18 determined on the basis of the MIC value, this pro-HDP was bactericidal (66.8% ± 11.7% killing activity) under the bactericidal assay conditions (Fig. 2). Bactericidal activity increased in the presence of NE for all pro-HDPs. The bactericidal activity with 5 μg/ml NE increased to 73.3% ± 0.2% for pro-Bac8c and 90.1% ± 5.8% for pro-HB43. The activity also increased for pro-P18, to 93.5% ± 4.7%, but this result was not statistically significant (P = 0.102). The level of bactericidal activity appeared to be independent of the NE concentration tested. No bactericidal effect was observed for NE alone at any of the concentrations investigated (data not shown).
FIG 1.

Bactericidal activity of parent and fragment HDPs against P. aeruginosa PAO1. Data shown are the means ± standard errors of the means (SEM) of the results of three independent assays carried out in duplicate.
FIG 2.

Effect of 5 to 20 μg/ml NE on the bactericidal activity of pro-HDPs against P. aeruginosa PAO1. NE was added to assays containing 6.25 μg/ml pro-HDPs. Killing activity levels shown are the means ± SEM of the results of three independent assays carried out in duplicate. Where no killing activity was observed, no bar is shown.
In the presence of NaCl, pro-HDPs are activated by CF BAL fluids.
Six CF BAL fluid samples were used to investigate pro-HDP activation. The NE concentrations in CF BAL fluid samples differed (range, 0 to 193.3 μg/ml) (Table 2). With the exception of CF005, which had significant bactericidal activity (at a 25% [vol/vol] final assay concentration) against PAO1 (74.5%), the killing activity of the CF BAL fluids against PAO1 was <20%.
TABLE 2.
Comparison of the neutrophil elastase concentration and bactericidal activity of different CF BAL fluid samples
| CF BAL fluid sample | NE concn (μg/ml) | % killing ± SEMa |
|---|---|---|
| CF001 | 35.7 | 0 |
| CF002 | NDb | 0 |
| CF003 | 136 | 7.8 ± 6.8 |
| CF004 | 46.4 | 10.4 ± 10.6 |
| CF005 | 193.3 | 74.5 ± 4.6 |
| CF006 | 62.8 | 15.6 ± 3.3 |
Data represent means ± SEM of the results of 3 separate determinations of killing activity versus P. aeruginosa PAO1 where 25% (vol/vol) BAL fluid was added to assays.
ND, not detected.
The bactericidal activities of the pro-HDPs were not restored by any of the CF BAL fluids investigated. Pro-P18 retained bactericidal activity in the absence of CF BAL fluid. However, after accounting for activity due to BAL alone, the bactericidal activity of pro-P18 was reduced by the addition of CF BAL fluid (Fig. 3C).
FIG 3.
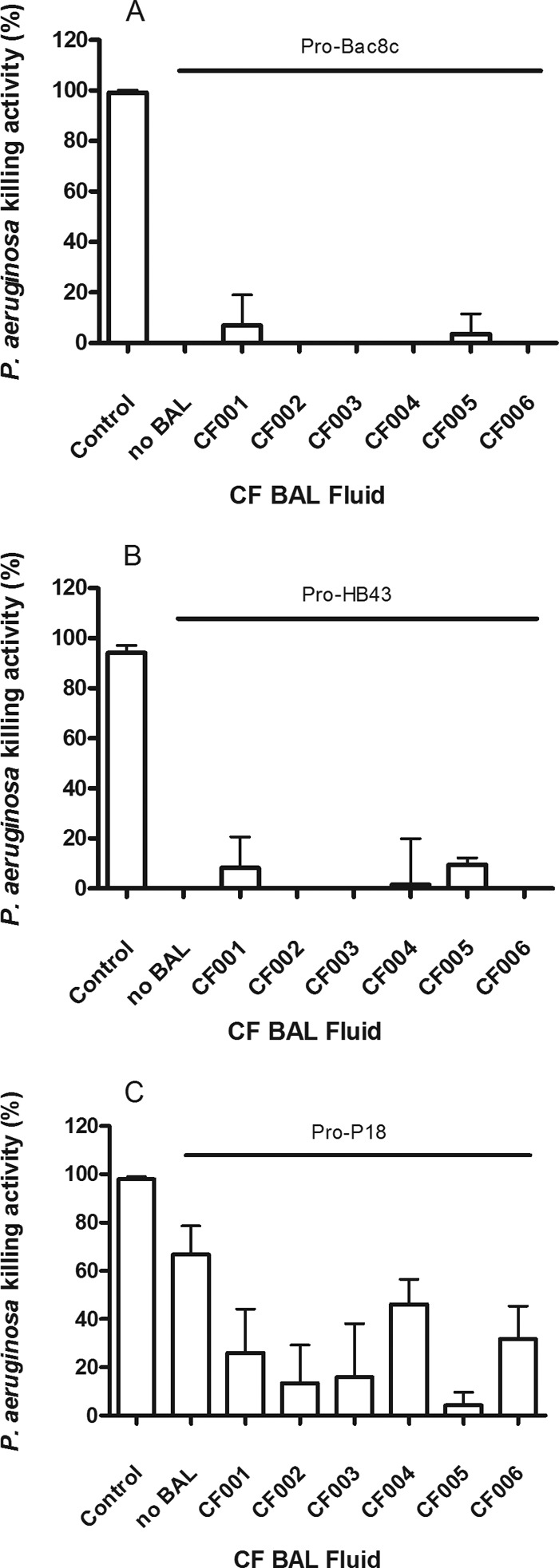
Effect of 25% (vol/vol) BAL fluid on the bactericidal activity of pro-Bac8c (A), pro-HB43 (B), and pro-P18 (C) against P. aeruginosa PAO1. BAL fluid (CF001 to CF006) was added to assays containing 6.25 μg/ml pro-HDPs. Killing activity levels shown are the means ± SEM of the results of three independent assays where activity due to CF BAL fluid alone was subtracted. Where no killing activity was observed, no bar is shown. Controls represent 6.25 μg/ml of the corresponding fragment HDP without BAL fluid, e.g., AAGBac8c for pro-Bac8c.
The addition of 300 mM NaCl (which reduces nonspecific electrostatic interaction with anionic components of CF BAL fluid) and an increase of the pro-HDP concentration to 25 μg/ml in the bactericidal assays resulted in the restoration of bactericidal activity of pro-HB43 and pro-P18 but not pro-Bac8c. This effect was shown for three selected CF BAL fluid samples with NE concentrations in the range of 46.4 to 136 μg/ml (Fig. 4). However, compared to that of the HB43 parent peptide, the bactericidal activity of Bac8c was reduced in the presence of NaCl (Fig. S1 in the supplemental material). NaCl alone did not increase the bactericidal activity of CF BAL fluid (see Fig. S2 in the supplemental material). Pro-P18 alone again showed bactericidal activity, although the addition of NaCl in this case appeared to reduce its bactericidal activity (39% ± 4.8%). Similar results were observed with 77.5% (vol/vol) BAL fluid (data not shown).
FIG 4.

Effect of 300 mM NaCl on the bactericidal activity of pro-HDPs (25 μg/ml) against P. aeruginosa PAO1 in the presence of 25% (vol/vol) CF BAL fluids. Values shown are the means ± SEM of the results of three independent assays where activity due to CF BAL fluid alone was subtracted. Where no killing activity was observed, no bar is shown.
Further increasing the NaCl concentration (to 450 mM and 600 mM) did not increase the bactericidal activity of pro-HB43 (BAL fluid CF004; see Fig. S3 in the supplemental material). While pro-HB43 did not have as high of a bactericidal activity against PAO1 as did pro-P18, the activity was significantly increased with 300 mM NaCl and CF004 BAL against the CF clinical isolates (Fig. 5). In the case of the PABH02 isolate, the activity increased from 0% (pro-HB43 with NaCl) to 99.2% ± 0.7% (pro-HB43 with NaCl and BAL fluid).
FIG 5.

Effect of 300 mM NaCl on the bactericidal activity of pro-HB43 (25 μg/ml) against P. aeruginosa clinical isolates from CF patients in the presence of 25% (vol/vol) CF004 BAL fluid. Values shown are the means ± SEM of the results of three independent assays where activity due to CF BAL fluid alone has been subtracted. Where no killing activity was observed, no bar is shown.
Pro-HDPs displayed lower cytotoxicity against CFBE41o- cells than fragment HDPs.
The 50% inhibitory concentrations (IC50s) were higher for the pro-HDPs than for the fragment HDPs (Table 3). The addition of the promoiety increased the IC50 from 38.3 μM to >300 μM for pro-Bac8c, from 3.6 μM to 50.8 μM for pro-HB43, and from 35.5 μM to 77.3 μM for pro-P18.
TABLE 3.
Comparison of the IC50s of pro-HDP and fragment HDP against CFBE41o- cells
| Peptide | Sequence | IC50 (μM)a |
|---|---|---|
| Pro-Bac8c | Ac-EEEEAAAGrlwvlwrr-NH2 | >300 |
| AAG-Bac8c | AAGrlwvlwrr-NH2 | 38.3 |
| Pro-HB43 | Ac-EEEEAAAGfakllaklakkll-NH2 | 50.8 |
| AG-HB43 | AGfakllaklakkll-NH2 | 3.6 |
| Pro-P18 | Ac-EEEEAAAGkwklfkklpkfhlhlakkf-NH2 | 77.3 |
| AAG-P18 | AAGkwklfkklpkfhlhlakkf-NH2 | 35.5 |
Data represent means of the results of 3 separate determinations.
DISCUSSION
Life expectancy in cystic fibrosis has been greatly increased by the use of antibiotics to treat lower-airway infections and delay their progression from acute to chronic (5). However, significant challenges remain to be overcome. Once a patient becomes chronically infected by P. aeruginosa, the current guidelines recommend long-term antibiotic therapy. This has led to the development of antibiotic resistance due to selective pressure and subtherapeutic local drug concentrations (1, 3, 6, 36, 37). In addition, P. aeruginosa can grow as a biofilm, which can result in further reduced susceptibility to certain antibiotics (1, 2, 6, 38). Currently used antibiotics cannot eradicate P. aeruginosa chronic infection, and the resulting exaggerated inflammatory response leads to progressive loss of lung function (21). The requirement is for novel therapeutic agents that are highly active against CF pathogens and less prone to resistance, with high local antibiotic concentrations deliverable in the endobronchial space without the accompanying systemic effects and toxicity. The latter requirement can be met by the use of inhaled delivery (3, 6, 7), while the former emphasizes the need for new therapeutic agents such as HDPs. Without novel antimicrobials, there is a risk that many of the gains made in CF treatment may be lost with increasing resistance.
Potential host toxicity is currently a limitation with respect to the development of HDPs as antimicrobial agents. Here we demonstrate that prodrug modification can mask HDP bactericidal activity and reduce host cytotoxic effects. Furthermore, the reversible conjugation of the promoiety can be targeted for cleavage by NE in a manner similar to that of the enzymatic activation of LL-37 by proteinase 3 in vivo (19), allowing for targeted restoration of bactericidal activity and limitation of cytotoxic effects. In addition, the recovery of activity for two of the pro-HDPs (pro-HB43 and pro-P18) by the use of NE has been shown under in vitro conditions that are representative of in vivo conditions in the CF lung through the use of CF BAL fluid. The differential activities found for these pro-HDPs under the assay conditions may be explained by the different sequences and net charges of the parent HDPs. The NE concentration required to cleave the pro-HDPs was within the range observed in CF BAL fluids. NE levels differed considerably in the CF BAL fluid samples in this study, and wide variations have been reported by others. For example, Rees et al. reported ranges of 0.47 to 18.5 μM (13.8 to 545.8 μg/ml) in the sputum (10). NE concentrations have also been reported to vary in CF patients over time (7). It has previously been estimated, based on the level of dilution of CF BAL fluid during collection, that the NE concentrations at the respiratory surface (as opposed to the CF BAL fluid itself) may be in the range of 92 to 185 μM (2.7 to 5.5 mg/ml) (39).
Although purified NE was shown to reactivate pro-HDPs in the present study, this effect was not initially apparent in CF BAL fluids containing NE, despite confirmation of NE concentrations that were high enough to catalyze the cleavage step. CF-BAL may contain several components that could inactivate HDPs. For example, LL-37 bactericidal activity is inhibited by proteolytic degradation by NE and cathepsin D in CF BAL fluid (31). Furthermore, cationic HDPs bind to anionic components of BAL and CF sputum, such as f-actin, extracellular DNA (27, 28), mucins (29), and glycosaminoglycans (31, 40). In addition, both extracellular DNA and mucins have been reported to increase the MIC values for synthetic HDPs such as HB43 (26). Inactivation by proteases in CF BAL fluid was unlikely in the present study due to their construction from d-amino acids and the demonstration of the stability of the parent HDPs in the presence of NE by the use of HPLC analysis (see the supplemental material). However, increased concentrations of the HDPs and the addition of NaCl restored the bactericidal activity of pro-HB43 and pro-P18 in CF BAL fluid, suggesting that anionic components of BAL fluid may prevent HDP activity and that the effect can be reversed by the addition of salt, similar to the liberation of LL-37 from BAL fluid components (31). The bactericidal activity of 25% (vol/vol) BAL fluid was found not to increase with 300 mM NaCl, indicating that increases in activity were due to the pro-HDPs. In addition, the stringent cleavage and recovery conditions demonstrated here for activation in the lung environment, such as the addition of NaCl to disrupt electrostatic interactions that may contribute to inactivation of HDPs, may potentially be met therapeutically by inhalation of saline solution. Should coadministration of NaCl be required for activity in vivo, a precedent already exists; inhaled hypertonic saline solution is in use at 1.2 M in CF treatment for the improvement of lung function (31, 41). The failure to achieve activation of pro-Bac8c in the presence of CF BAL fluid and NaCl, in contrast to pro-HB43 and pro-P18, may be related to the greater salt tolerance of the latter two HDPs which has been shown previously (26, 42).
We have demonstrated the effectiveness of the NE-targeted approach for two pro-HDPs here, but there is further scope for the development of this approach for other HDPs. Highly active HDPs, with various length, charge, activity, and cytotoxicity characteristics, have been described in the literature (26, 32, 43–47). However, the appropriate selection of the parent HDP requires consideration. In the present study, since the number of glutamic acids added to the pro-HDPs was limited to four, the final pro-HDPs were of different net charges (−1, 0, and +3 for pro-Bac8c, pro-HB43, and pro-P18, respectively). Therefore, the bactericidal activity and cytotoxicity of P18, which still retained a significant cationic net charge, were not masked as markedly as for the other candidates (Fig. 3). This residual activity of pro-P18 was not initially apparent where an MIC of ≥64 mg/ml was determined. However, discrepancies in effective antimicrobial concentrations between MIC and bactericidal assays for antimicrobial peptides have been previously observed (44, 48, 49). It is possible that the bactericidal activity was masked in the broth microdilution assay used to determine MIC, which is carried out on actively metabolizing cells, where the growth of residual surviving cells is supported. This was not the case in the bactericidal assay, where viable cell counts were measured following incubation of the pro-HDP with a fixed bacterial population. These results demonstrate that a high starting net charge may be undesirable for the modification described here. The net charge reduction, while being sufficient against Staphylococcus aureus (25), may be insufficient against Gram-negative organisms such as P. aeruginosa. Alternatively, other synthetic approaches may be useful for parent HDPs with a high net charge, such as the incorporation of a spacer group (e.g., β-alanine) to extend the oligoglutamic acid moiety (25).
The linker moiety of pro-HDPs may also be tailored to contain specific sequences cleavable by enzymes appropriate to the application or the disease. In the present study, the AAAG linker was labile with respect to human NE but not pseudolysin from P. aeruginosa, both of which are relevant to CF. It is possible that AAAG can be cleaved by other host/bacterial enzymes and that additional modification of the linker may lead to increased selectivity.
Suitable HDP candidates for pulmonary delivery as pro-HDPs for treatment of CF infections would ideally be short, highly active against P. aeruginosa, highly salt resistant, and of lower net charge. The issue of net charge has been observed previously by our group for cephalothin-Bac8c conjugates, where activity was incompletely masked (50). The use of d-amino acids, which have been shown to be resistant to proteolysis (51–53), may be necessary to avoid deactivation by the abundant proteases of the CF lung. In addition, any potential candidate would have to be compatible with local lung delivery such as is used with tobramycin and colistin in treatment of CF (3, 6, 54).
This prodrug approach potentially provides the means to deliver effective bactericidal HDPs to the CF lung in therapeutic concentrations while limiting cytotoxicity distal to the site of activation and infection (i.e., the endobronchial space). HDP prodrugs may provide novel anti-infective agents against acute bacterial infection in CF and alternative therapeutics in the prevention of chronic pseudomonal infections. Further development and testing of the best candidate pro-HDPs, evaluated using in vitro studies such as those described here, should be investigated in CF animal models to establish a proof of concept as a prerequisite to preclinical testing.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Higher Education Authority, Ireland, under the BioAT program in Cycle 5 of the Programme for Research in Third-Level Institutions and by the Science Foundation Ireland under Equipment Grant no. 06/RFP/CHO024/602 EC07 for the peptide synthesizer.
We thank Graeme Kelly, Łukasz Frankiewicz, and Aoife O'Connor, Department of Chemistry, RCSI, and Paul McKiernan and Bojana Mirkovic, Pulmonary Research Division, RCSI, for technical assistance.
Footnotes
Published ahead of print 25 November 2013
D.F.-H. and M.D. are joint senior authors.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01167-13.
REFERENCES
- 1.Gibson RL, Burns JL, Ramsey BW. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168:918–951. 10.1164/rccm.200304-505SO [DOI] [PubMed] [Google Scholar]
- 2.Callaghan M, McClean S. 2012. Bacterial host interactions in cystic fibrosis. Curr. Opin. Microbiol. 15:71–77. 10.1016/j.mib.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 3.Maiz L, Giron RM, Olveira C, Quintana E, Lamas A, Pastor D, Canton R, Mensa J. 16 April 2013. Inhaled antibiotics for the treatment of chronic bronchopulmonary Pseudomonas aeruginosa infection in cystic fibrosis: systematic review of randomised controlled trials. Expert Opin. Pharmacother. 10.1517/14656566.2013.790366 [DOI] [PubMed] [Google Scholar]
- 4.Ren CL, Konstan MW, Yegin A, Rasouliyan L, Trzaskoma B, Morgan WJ, Regelmann W. 2012. Multiple antibiotic-resistant Pseudomonas aeruginosa and lung function decline in patients with cystic fibrosis. J. Cyst. Fibros. 11:293–299. 10.1016/j.jcf.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain K, Smyth AR. 2012. Current dilemmas in antimicrobial therapy in cystic fibrosis. Expert Rev. Respir. Med. 6:407–422. 10.1586/ers.12.39 [DOI] [PubMed] [Google Scholar]
- 6.Greally P, Whitaker P, Peckham D. 2012. Challenges with current inhaled treatments for chronic Pseudomonas aeruginosa infection in patients with cystic fibrosis. Curr. Med. Res. Opin. 28:1059–1067. 10.1185/03007995.2012.674500 [DOI] [PubMed] [Google Scholar]
- 7.Kelly E, Greene CM, McElvaney NG. 2008. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 12:145–157. 10.1517/14728222.12.2.145 [DOI] [PubMed] [Google Scholar]
- 8.Voynow JA, Fischer BM, Zheng S. 2008. Proteases and cystic fibrosis. Int. J. Biochem. Cell Biol. 40:1238–1245. 10.1016/j.biocel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cosgrove S, Chotirmall SH, Greene CM, McElvaney NG. 2011. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/Toll-like receptor pathway. J. Biol. Chem. 286:7692–7704. 10.1074/jbc.M110.183863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rees DD, Brain JD, Wohl ME, Humes JL, Mumford RA. 1997. Inhibition of neutrophil elastase in CF sputum by L-658,758. J. Pharmacol. Exp. Ther. 283:1201–1206 [PubMed] [Google Scholar]
- 11.Hancock RE, Sahl HG. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551–1557. 10.1038/nbt1267 [DOI] [PubMed] [Google Scholar]
- 12.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395. 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- 13.Sadler K, Eom KD, Yang JL, Dimitrova Y, Tam JP. 2002. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry 41:14150–14157. 10.1021/bi026661l [DOI] [PubMed] [Google Scholar]
- 14.Westerhoff HV, Juretić D, Hendler RW, Zasloff M. 1989. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. U. S. A. 86:6597–6601. 10.1073/pnas.86.17.6597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeaman MR, Bayer AS, Koo SP, Foss W, Sullam PM. 1998. Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J. Clin. Invest. 101:178–187. 10.1172/JCI562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fjell CD, Hiss JA, Hancock RE, Schneider G. 2012. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11:37–51. 10.1038/nrd3591 [DOI] [PubMed] [Google Scholar]
- 17.Devocelle M. 2012. Targeted antimicrobial peptides. Front. Immunol. 3:309. 10.3389/fimmu.2012.00309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitzgerald-Hughes D, Devocelle M, Humphreys H. 2012. Beyond conventional antibiotics for the future treatment of methicillin-resistant Staphylococcus aureus infections: two novel alternatives. FEMS Immunol. Med. Microbiol. 65:399–412. 10.1111/j.1574-695X.2012.00954.x [DOI] [PubMed] [Google Scholar]
- 19.Giuliani A, Pirri G, Nicoletto S. 2007. Antimicrobial peptides: an overview of a promising class of therapeutics. Cent. Eur. J. Biol. 2:1–33. 10.2478/s11535-007-0010-5 [DOI] [Google Scholar]
- 20.Steinstraesser L, Kraneburg UM, Hirsch T, Kesting M, Steinau HU, Jacobsen F, Al-Benna S. 2009. Host defense peptides as effector molecules of the innate immune response: a sledgehammer for drug resistance? Int. J. Mol. Sci. 10:3951–3970. 10.3390/ijms10093951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Döring G, Flume P, Heijerman H, Elborn JS; Consensus Study Group. 2012. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J. Cyst. Fibros. 11:461–479. 10.1016/j.jcf.2012.10.004 [DOI] [PubMed] [Google Scholar]
- 22.Arnusch CJ, Pieters RJ, Breukink E. 2012. Enhanced membrane pore formation through high-affinity targeted antimicrobial peptides. PLoS One 7:e39768. 10.1371/journal.pone.0039768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckert R, He J, Yarbrough DK, Qi F, Anderson MH, Shi W. 2006. Targeted killing of Streptococcus mutans by a pheromone-guided “smart” antimicrobial peptide. Antimicrob. Agents Chemother. 50:3651–3657. 10.1128/AAC.00622-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franzman MR, Burnell KK, Dehkordi-Vakil FH, Guthmiller JM, Dawson DV, Brogden KA. 2009. Targeted antimicrobial activity of a specific IgG-SMAP28 conjugate against Porphyromonas gingivalis in a mixed culture. Int. J. Antimicrob. Agents 33:14–20. 10.1016/j.ijantimicag.2008.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desgranges S, Le Prieult F, Daly A, Lydon J, Brennan M, Rai DK, Subasinghage AP, Hewage CM, Cryan SA, Greene C, McElvaney NG, Smyth TP, Fitzgerald-Hughes D, Humphreys H, Devocelle M. 2011. In vitro activities of synthetic host defense propeptides processed by neutrophil elastase against cystic fibrosis pathogens. Antimicrob. Agents Chemother. 55:2487–2489. 10.1128/AAC.01384-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Parente J, Harris SM, Woods DE, Hancock RE, Falla TJ. 2005. Antimicrobial peptide therapeutics for cystic fibrosis. Antimicrob. Agents Chemother. 49:2921–2927. 10.1128/AAC.49.7.2921-2927.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiner DJ, Bucki R, Janmey PA. 2003. The antimicrobial activity of the cathelicidin LL-37 is inhibited by F-actin bundles and restored by gelsolin. Am. J. Respir. Cell Mol. Biol. 28:738–745. 10.1165/rcmb.2002-0191OC [DOI] [PubMed] [Google Scholar]
- 28.Bucki R, Byfield FJ, Janmey PA. 2007. Release of the antimicrobial peptide LL-37 from DNA/F-actin bundles in cystic fibrosis sputum. Eur. Respir. J. 29:624–632. 10.1183/09031936.00080806 [DOI] [PubMed] [Google Scholar]
- 29.Felgentreff K, Beisswenger C, Griese M, Gulder T, Bringmann G, BAL fluids R. 2006. The antimicrobial peptide cathelicidin interacts with airway mucus. Peptides 27:3100–3106. 10.1016/j.peptides.2006.07.018 [DOI] [PubMed] [Google Scholar]
- 30.Baranska-Rybak W, Sonesson A, Nowicki R, Schmidtchen A. 2005. Glycosaminoglycans inhibit the antibacterial action of LL-37 in biological fluids. J. Investig. Dermatol. 125:A70–A70. 10.1093/jac/dki460 [DOI] [PubMed] [Google Scholar]
- 31.Bergsson G, Reeves EP, McNally P, Chotirmall SH, Greene CM, Greally P, Murphy P, O'Neill SJ, McElvaney NG. 2009. LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J. Immunol. 183:543–551. 10.4049/jimmunol.0803959 [DOI] [PubMed] [Google Scholar]
- 32.Hilpert K, Volkmer-Engert R, Walter T, Hancock REW. 2005. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotech. 23:1008–1012. 10.1038/nbt1113 [DOI] [PubMed] [Google Scholar]
- 33.Shin SY, Kang JH, Hahm KS. 1999. Structure-antibacterial, antitumor and hemolytic activity relationships of cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J. Pept. Res. 53:82–90. 10.1111/j.1399-3011.1999.tb01620.x [DOI] [PubMed] [Google Scholar]
- 34.CLSI 2010. Performance standards for antimicrobial susceptibility testing, 20th informational supplement, vol M100-S12 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 35.Wu M, Hancock RE. 1999. Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 274:29–35. 10.1074/jbc.274.1.29 [DOI] [PubMed] [Google Scholar]
- 36.Livermore DM. 2009. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 64(Suppl 1):i29–36. 10.1093/jac/dkp255 [DOI] [PubMed] [Google Scholar]
- 37.Septimus EJ, Kuper KM. 2009. Clinical challenges in addressing resistance to antimicrobial drugs in the twenty-first century. Clin. Pharmacol. Ther. 86:336–339. 10.1038/clpt.2009.122 [DOI] [PubMed] [Google Scholar]
- 38.Overhage J, Campisano A, Bains M, Torfs EC, Rehm BH, Hancock RE. 2008. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 76:4176–4182. 10.1128/IAI.00318-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greene CM, Carroll TP, Smith SG, Taggart CC, Devaney J, Griffin S, O'Neill SJ, McElvaney NG. 2005. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 174:1638–1646 [DOI] [PubMed] [Google Scholar]
- 40.Barańska-Rybak W, Sonesson A, Nowicki R, Schmidtchen A. 2006. Glycosaminoglycans inhibit the antibacterial activity of LL-37 in biological fluids. J. Antimicrob. Chemother. 57:260–265. 10.1093/jac/dki460 [DOI] [PubMed] [Google Scholar]
- 41.Kellett F, Robert NM. 2011. Nebulised 7% hypertonic saline improves lung function and quality of life in bronchiectasis. Respir. Med. 105:1831–1835. 10.1016/j.rmed.2011.07.019 [DOI] [PubMed] [Google Scholar]
- 42.Shin SY, Yang ST, Park EJ, Eom SH, Song WK, Kim Y, Hahm KS, Kim JI. 2002. Salt resistance and synergistic effect with vancomycin of alpha-helical antimicrobial peptide P18. Biochem. Biophys. Res. Commun. 290:558–562. 10.1006/bbrc.2001.6234 [DOI] [PubMed] [Google Scholar]
- 43.Mai J, Tian XL, Gallant JW, Merkley N, Biswas Z, Syvitski R, Douglas SE, Ling J, Li YH. 2011. A novel target-specific, salt-resistant antimicrobial peptide against the cariogenic pathogen Streptococcus mutans. Antimicrob. Agents Chemother. 55:5205–5213. 10.1128/AAC.05175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scudiero O, Galdiero S, Nigro E, Del Vecchio L, Di Noto R, Cantisani M, Colavita I, Galdiero M, Cassiman JJ, Daniele A, Pedone C, Salvatore F. 2013. Chimeric beta-defensin analogs, including the novel 3NI analog, display salt-resistant antimicrobial activity and lack toxicity in human epithelial cell lines. Antimicrob. Agents Chemother. 57:1701–1708. 10.1128/AAC.00934-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Wong ES, Whitley JC, Li J, Stringer JM, Short KR, Renfree MB, Belov K, Cocks BG. 2011. Ancient antimicrobial peptides kill antibiotic-resistant pathogens: Australian mammals provide new options. PLoS One 6:e24030. 10.1371/journal.pone.0024030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott MG, Yan H, Hancock RE. 1999. Biological properties of structurally related alpha-helical cationic antimicrobial peptides. Infect. Immun. 67:2005–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobsen F, Mohammadi-Tabrisi A, Hirsch T, Mittler D, Mygind PH, Sonksen CP, Raventos D, Kristensen HH, Gatermann S, Lehnhardt M, Daigeler A, Steinau HU, Steinstraesser L. 2007. Antimicrobial activity of the recombinant designer host defence peptide P-novispirin GIO in infected full-thickness wounds of porcine skin. J. Antimicrob. Chemother. 59:493–498. 10.1093/jac/dkl513 [DOI] [PubMed] [Google Scholar]
- 48.Scudiero O, Galdiero S, Cantisani M, Di Noto R, Vitiello M, Galdiero M, Naclerio G, Cassiman JJ, Pedone C, Castaldo G, Salvatore F. 2010. Novel synthetic, salt-resistant analogs of human beta-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob. Agents Chemother. 54:2312–2322. 10.1128/AAC.01550-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC. 2013. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob. Agents Chemother. 57:2511-2521. 10.1128/AAC.02218-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desgranges S, Ruddle CC, Burke LP, McFadden TM, O'Brien JE, Fitzgerald-Hughes D, Humphreys H, Smyth TP, Devocelle M. 2012. β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. RSC Adv. 2:2480–2492. 10.1039/c2ra01351g [DOI] [Google Scholar]
- 51.Strömstedt AA, Pasupuleti M, Schmidtchen A, Malmsten M. 2009. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob. Agents Chemother. 53:593–602. 10.1128/AAC.00477-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bessalle R, Kapitkovsky A, Gorea A, Shalit I, Fridkin M. 1990. All-D-magainin: chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 274:151–155. 10.1016/0014-5793(90)81351-N [DOI] [PubMed] [Google Scholar]
- 53.Hong SY, Oh JE, Lee KH. 1999. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 58:1775–1780. 10.1016/S0006-2952(99)00259-2 [DOI] [PubMed] [Google Scholar]
- 54.Proesmans M, Vermeulen F, Boulanger L, Verhaegen J, De Boeck K. 2013. Comparison of two treatment regimens for eradication of Pseudomonas aeruginosa infection in children with cystic fibrosis. J. Cyst. Fibros. 12:29–34. 10.1016/j.jcf.2012.06.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.