

Abstract
The antiviral efficacy of stavudine depends on the trough concentration of its intracellular metabolite, stavudine-triphosphate (d4T-TP), while the degree of stavudine's mitochondrial toxicity depends on its peak concentration. Rates of mitochondrial toxicity are high when stavudine is used at the current standard pediatric dose (1 mg/kg twice daily [BID]). Evidence from adult work suggests that half of the original standard adult dose (i.e., 20 mg BID) may be equally effective, with markedly less mitochondrial toxicity. We present a population pharmacokinetic model to predict intracellular d4T-TP concentrations in pediatric HIV-infected patients administered a dose of 0.5 mg/kg BID. Our model predicted that the reduced pediatric dose would result in a trough intracellular d4T-TP concentration above that of the reduced 20-mg adult dose and a peak concentration below that of the 20-mg adult dose. The simulated pediatric intracellular d4T-TP at 0.5 mg/kg BID resulted in median peak and trough values of approximately 23.9 fmol/106 cells (95% prediction interval [PI], 14.2 to 41 fmol/106 cells) and 14.8 fmol/106 cells (95% PI, 7.2 to 31 fmol/106 cells), respectively. The peak and trough concentrations resulting from a 20-mg BID adult dose were 28.4 fmol/106 cells (95% PI, 17.3 to 45.5 fmol/106 cells) and 13 fmol/106 cells (95% PI, 6.8 to 28.6 fmol/106 cells), respectively. Halving the current standard pediatric dose should therefore not compromise antiviral efficacy, while markedly reducing mitochondrial toxicity.
INTRODUCTION
Pediatric HIV is a common and neglected disease (1). Among other obstacles, pediatric clinicians in the developing world are severely hampered by the lack of pediatric antiretroviral formulations appropriate for sub-Saharan Africa, where over 90% of the world's 3.4 million HIV-infected children live. Stavudine (also known as d4T) is a nucleoside reverse transcriptase inhibitor. At the standard pediatric dose, it may cause apoptosis of subcutaneous fat cells, a process known as lipoatrophy, in up to 37% of children (2). Here, the limbs and face become disfigured, potentially leading to stigmatization and reduced adherence to therapy (3). Stavudine remains one of the most commonly used antiretroviral drugs for children in sub-Saharan Africa due to its short-term safety profile and long shelf life. In 2010, abacavir replaced stavudine in South Africa's first-line antiretroviral therapy (ART) regimen for children; however, children already on stavudine continued if there were no signs of lipoatrophy. South Africa's 2013 ART guidelines have recently advised that all virologically suppressed children on stavudine be automatically switched to abacavir (4), and currently around 24,000 South African children remain on stavudine out of a total of 157,000 on ART (Corry van der Walt and Jaco Stokes, South African National Department of Health, personal communication, 15 October 2013). Recent data have questioned the efficacy of abacavir-based ART in children (5), which if verified, may lead to reintroduction of stavudine. Outside of South Africa, abacavir use is limited due to its high cost. Stavudine also remains an important second-line option in South Africa because of the gastrointestinal intolerance and bone marrow toxicity associated with zidovudine (particularly in the context of already high rates of chronic anemia) and the complexity of taking didanosine. Tenofovir is not widely licensed for use in children due to concerns about its renal and bone toxicities, which may be more pronounced in growing children than in adults.
There is strong evidence that lipoatrophy related to stavudine can be avoided with a smaller dose (6–9). The current standard pediatric dose of stavudine, 1 mg/kg twice daily (BID), was established by extrapolation from the plasma pharmacokinetic (PK) parameters of the adult dose by using data from a few small, well-controlled pediatric pharmacokinetic studies. Those studies showed that an oral dose of 1 mg/kg BID in children under 30 kg gives plasma exposure similar to that of 40 mg BID in adults weighing more than 60 kg and that an oral dose of 0.5 mg/kg BID in children gives plasma exposure similar to that of adults taking 20 mg BID (10–12). Preapproval adult dose-finding studies performed in the 1990s found that 20 mg BID and 40 mg BID had equivalent antiviral effects (13–16). The final recommended dose of 40 mg BID was chosen fairly arbitrarily after a large trial chose to test 40 mg BID rather than a lower dose, with stavudine having minimal short-term toxicity at the higher dose (17). This was despite the outcome of the much larger Parallel Track study, in which the 40-mg BID arm was stopped by the Data Safety Monitoring Board due to excessive rates of delayed peripheral neuropathy, all patients being switched to 20 mg BID (13). In 2007, an influential systematic review by Hill et al. showed that a reduced adult dose of either 20 or 30 mg BID significantly reduced the frequency of delayed toxicity (lipoatrophy, dyslipidemia, lactic acidosis, and peripheral neuropathy) while maintaining excellent antiviral efficacy (6). In response, the World Health Organization recommended that the adult dose be reduced (18). The children's dose, however, has not yet been reduced.
The plasma pharmacokinetics of stavudine has been characterized in adults and children (10–12, 19, 20). Stavudine is rapidly cleared from circulation after dosing (half-life [t1/2] of ∼1 h), and the magnitude of its antiviral and toxic effects depends on the intracellular concentration of its phosphorylated metabolite, stavudine triphosphate (d4T-TP), whose t1/2 is ∼7 h (20, 21). We hypothesized that the predicted trough intracellular concentration of d4T-TP in children on 0.5 mg/kg BID would be equivalent to the trough intracellular concentration of adults on 20 mg BID, and therefore that a dose of 0.5 mg/kg BID in children weighing less than 30 kg is likely to have an equivalent antiviral effect to 20 mg BID in adults weighing more than 60 kg.
MATERIALS AND METHODS
Data sets used in the model development.
The plasma concentration-time data of Aspen stavudine administered as capsule and suspension came from a bioequivalence study (22). The original bioequivalence study compared the exposure of Aspen stavudine to that of Cipla Stavir and opened versus intact capsule dosing methods. In the opened capsule dosing, the caregiver would disperse the capsule content in 5 ml water and then withdraw the required dose using a syringe. Fourteen HIV-seronegative adults fasted for 6 h prior to the dosing administration and 2 h after the dose was administered. Blood was drawn predose and at 15, 30, 45, 60, and 90 min and 2, 4, 6, and 8 h after dosing. The study had a randomized crossover design. Subjects received either a single dose of 30 mg stavudine as an intact capsule or the same dose in suspension using the opened capsule administration method on separate study days. Both dosing methods were bioequivalent. We undertook a population pharmacokinetic analysis, using only data from Aspen stavudine, treating the two dosing methods as different occasions to evaluate interoccasion variability (IOV).
Population pharmacokinetic models.
The population PK models were developed using a nonlinear mixed-effects modeling approach. The first-order conditional maximum likelihood estimation was performed with the NONMEM program (double precision, version 7.1.2; ICON Development Solutions, Ellicott City, MD); the subroutine within NONMEM was the linear mammillary model (ADVAN1 with TRANS2).
The one-compartment model with first-order elimination and zero-order absorption provided the best fit to the data set. The structural pharmacokinetic model for the one-compartment model consists of absorption duration (D), oral clearance (CL), and volume of the central compartment (V). The relative bioavailability parameter (F) assumed the value of 1. Exponential interindividual variability terms were included in the pharmacokinetic parameters. Interoccasion variability (IOV) was introduced to the model (equation 1):
| (1) |
TV represents the typical value. ηi and κij describe interindividual and interoccasion (within an individual) variability and are assumed to be independently, normally distributed parameters both with mean zero and variances and , respectively.
Once the base model was established, covariate screening was carried out using the stepwise covariate model (SCM) in PerlSpeaksNONMEM. The covariates tested included the demographic variables age, height, weight, body surface area, and creatinine clearance. The hypothesis testing to discriminate among alternative hierarchical structural models was based on the P values for the forward inclusion and backward elimination at 0.05 and 0.01, respectively. Both linear and power relationships were explored for a covariate relationship.
The allometric scaling based on body weight (BW) was applied to the pharmacokinetic parameter CL, as described by equation 2, once body weight was determined to be the only influential covariate:
| (2) |
θ1 is the typical value (TV) at the median body weight of the study population, and θ2 is the allometric scaling exponent for the body weight normalized by the median.
The accuracy and robustness of the final model were evaluated using a nonparametric bootstrap procedure. The resampling was repeated 1,000 times. The final population pharmacokinetic model was fitted to each of the bootstrap data sets, and a set of model parameters were determined for each run. The 95% confidence intervals (CI) were computed.
Model validation.
A degenerate visual predictive check was performed by the simulation of the parameter estimates of the final model to generate 1,000 individual profiles. The median and 95% prediction intervals (PI) for the concentration at each time points were plotted and compared to the original data. In the adult simulation, weight was simulated following a normal distribution with a mean ± standard deviation (SD) of 74 ± 21 kg, as in the original population with a median body weight of 70 kg. In the pediatric population, a minimum and maximum of 6 and 30 kg, respectively, were simulated for body weight. Their ages were between 0.5 and 9 years, with 16% within the ages of 0.5 and 2 years, 25% between 2 and 4 years, and 58.3% between 4 and 8 years. The relationship between body weight and age was based on the growth charts from the Centers for Disease Control and Prevention (23). The covariate equation for pediatric age and volume of distribution was defined by the relationship established by Jullien et al. (12):
| (3) |
Both age and body weight distributions were used in the simulation of 1,000 pediatric profiles.
Intracellular concentration of d4T-TP.
The intracellular concentrations of d4T-TP in adults weighing over 60 kg and those under 60 kg who were administered 40 mg or 30 mg stavudine BID, respectively, were obtained from the first figure from Becher et al. (20). The same cellular parameters in the study by Hurwitz and Schinazi (24) were used to transform the intracellular concentrations from fmol/106 cells to μM. The intracellular concentrations of d4T-TP were computed assuming a cell volume of 32 pl and 8% activated lymphocyte fraction (24–27). We assumed that stavudine rapidly reached equilibrium between extra- and intracellular concentrations, as it is known that transporters of nucleosides on the cell membranes of lymphocytes are efficient and that equilibrium is achieved within milliseconds (28, 29). Hurwitz and Schinazi also had shown that the intracellular d4T-TP and extracellular stavudine concentrations were proportional, suggesting that phosphorylation was not saturable at the clinically relevant concentration (24). This supported the assumption that rapid equilibrium is achieved between extracellular and intracellular concentrations. The typical parameter values, assuming a body weight of 70 kg, for stavudine pharmacokinetics were used to simulate the plasma concentration-time profile following a 40-mg administration; steady state with an interdosing interval of 12 h was assumed in the pharmacokinetic model. The first-order accumulation (kpp) and decay (kdp) rate constants of d4T-TP accumulation in the intracellular compartment were then determined according to equation 4
| (4) |
where Cp represents the plasma stavudine concentration in μM and the d4T-TP concentrations were converted to μM. No interindividual variability was incorporated to the rate parameters kpp and kdp, as we assumed that the data from Becher et al. (20) are representative of a typical adult with a body weight of 70 kg. The residual variability in the intracellular d4T-TP concentration was described by a proportional error model.
The parameters kpp and kdp obtained from the model fit to the reported data were then used to simulate the intracellular d4T-TP concentration-time profiles in 1,000 pediatric individuals, assuming weights between 6 and 30 kg and ages between 0.5 and 9 years. The dose used in the simulation was 0.5 mg/kg BID. The simulated profiles in the pediatric patients were compared to the simulated adult profiles administered 20 and 40 mg BID at steady state.
RESULTS
Population PK model.
A one-compartment model with zero-order absorption and first-order elimination was fitted to the stavudine concentration-time data of individuals administered 30 mg intact and open capsules on different occasions (22). Table 1 provides the base and final pharmacokinetic model with parameter estimates and 95% CI from a 1,000-bootstrap resampling. The absorption of stavudine was rapid, and a first-order absorption could not sufficiently characterize the absorption process. Thus, a zero-order absorption process was used, and the duration of absorption was estimated. The parameter values for the final structural pharmacokinetic model reported in Table 1 were 19.0 liters/h for CL, 37.7 liters for V, and 0.298 h for the zero-order absorption duration. Values for the parameter ωp, which represents the approximate coefficients of variation for the interindividual variability for CL, V, and D, were 8.11%, 23.5%, and 47.8%, respectively. The πp values, the approximate coefficients of variation for the interoccasion variability within an individual, were 10% and 16% for CL and V. The covariate analysis included weight as an influential covariate of CL. Weight, however, was not an influential covariate of V, and thus no relationship between weight and V was included in the final model. The addition of body weight to the CL parameter resulted in a significant decrease of 12 points in the objective function value (OFV) compared to the basic model. The final covariate model was
| (5) |
TABLE 1.
Population pharmacokinetic model parameters of the base and final models for stavudine
| Parametera | Result from: |
|||
|---|---|---|---|---|
| Base model |
Final model |
|||
| Mean | 95% CIb | Mean | 95% CIb | |
| Structural model | ||||
| CL, liters/h | 20.0 | 16.8–28.9 | 19.0 | 17.2–20.7 |
| V, liters | 38.4 | 32.2–45.9 | 37.7 | 29–45 |
| D, h | 0.292 | 0.25–0.45 | 0.298 | 0.25–0.49 |
| kpp, h−1 | 0.269 | 9.6 × 10−5c | ||
| kdp, h−1 | 0.151 | 2.3 × 10−5c | ||
| Interindividual variability | ||||
| ωCL | 24.9 | 11.9–34.0 | 8.11 | 1.7–18.4 |
| ωV | 23.3 | 10.8–52.0 | 23.5 | 8.0–50.4 |
| ωD | 54.0 | 33.4–64.6 | 47.8 | 33.0–68.9 |
| IOV | ||||
| πCL | 12.2 | 3.2–25.1 | 10.0 | 2.07–17.3 |
| πV | 13.1 | 4.1–37.2 | 16.1 | 4.20–36.7 |
| Residual variability | ||||
| Proportional residual error for plasma d4T (σ1) | 0.239 | 0.16–0.29 | 0.226 | 0.147–0.285 |
| Additive residual error for plasma d4T (σ1), IU/dl | 45.9 | 20.3–59.8 | 47.9 | 18.0–59.7 |
| Proportional residual error for intracellular d4T (σ3) | 0.49 | 3.5 × 10−4c | ||
| Covariates | ||||
| Allometric exponent for CL by wtd | 0.775 | 0.39–1.29 | ||
| OFV | 2,456.44 | 2,266–2,570 | 2,444.04 | 2,243–2,564 |
Abbreviations: CL, clearance; V, volume of central compartment; D, zero-order duration; kpp, intracellular d4T-TP accumulation rate; kdp, intracellular d4T-TP decay rate; ωCL, ωV, and ωD, interindividual variability percent coefficients of variation (% CV) for CL, V, and D, respectively; πCL and πV, interoccasion variability (IOV) % CV for CL and V, respectively; OFV, objective function value.
95% CI obtained from the bootstrap 2.5th and 97.5th percentiles.
Standard error of estimate reported instead of 95% CI of bootstrap.
The covariate equation for CL by weight (BW) is .
The parameter estimates of the final structural model were not very different from that of the base model. However, the interindividual variability in CL was reduced significantly to 8%. The 95% CI of the parameter estimates was determined from the bootstrap results.
The pharmacokinetic profiles after intact and opened capsule administration, represented as black filled circles and opened circles on Fig. 1, were not markedly different, allowing us to treat the two formulations as different occasions (Fig. 1, left panel). The accuracy of the final model was evaluated by a posterior visual predictive check obtained from simulation of 1,000 profiles from the adult population weighing 74 ± 21 kg, assuming a normal distribution (Fig. 1, left panel).
FIG 1.

Accuracy of the final model evaluated by visual predictive check obtained from 1,000 simulations in adult patients (left) and another 1,000 in pediatric patients (right) receiving 30 mg and 1.23 mg/kg stavudine, respectively. In the left panel, the filled circles and the open circles represent stavudine plasma concentrations in adult patients given intact capsules and opened capsules, respectively, while the data points in the right panel are from Jullien et al. (12). The shaded areas correspond to the 95% prediction interval, whereas the line represents the median of the simulated profiles.
As age is an independent predictor of volume of distribution for stavudine in prepubertal children (12), it was incorporated as a covariate of V in the pediatric simulation. Weights of children ranging from 6 to 30 kg and at ages from 0.5 to 9 years were then used to evaluate whether the predicted stavudine exposure using our model in this weight range was consistent with the pediatric data from Jullien et al. (12). The predictability of the model in pediatric patients was evaluated based on the simulation of a dose of 1.23 mg/kg superimposed over the stavudine plasma concentrations from 272 children who were administered the same dose (Fig. 1, right panel), as reported by Jullien et al. (12). Our 95% PI of stavudine plasma concentration in children encompassed the upper half of the data reported by Jullien et al. (12).
The accuracy of the simulation was considered satisfactory. This model was then used to extrapolate the plasma stavudine exposure and intracellular concentration of phosphorylated stavudine in children given 0.5 mg/kg BID.
Modeling and simulation of the intracellular concentration of d4T-TP.
The transformed data of the in vivo measurements of phosphorylated stavudine in the lymphocyte peripheral blood mononuclear cells from Hurwitz and Schinazi (24)—originally from Becher et al. (20)—were used to determine the kpp and kdp parameters using equation 4. The model in equation 4 along with the final pharmacokinetic model using the median parameters was fitted to the pooled d4T-TP concentration-time profiles. The model assumed that the pooled d4T-TP data were at steady state. The parameter estimates were 0.269 h−1 and 0.151 h−1 for kpp and kdp, respectively. The parameter estimates were markedly different from that reported by Hurwitz and Schinazi (24). The likely explanation for the discrepancy is that our pharmacokinetic model was a zero-order absorption model, which allowed for a rapid rise to peak concentration within approximately 0.3 h. In contrast, their model was originally from Panhard et al. (30) and had a slow first-order absorption rate constant with an estimated time of peak concentration of approximately 1.5 to 2 h (30).
To evaluate how well the model predicts the intracellular concentration in adults, 2,000 adult profiles with the population weight following a uniform distribution of 60 to 90 kg administered 20 and 40 mg BID were simulated, with 1,000 profiles per dosing regimen. A subsequent simulation of another 1,000 profiles in children with the same age and body weight distributions as the one used in the model evaluation (administered 0.5 mg/kg) was compared to the adult profiles. Figure 2 shows the simulations of intracellular d4T-TP in pediatric patients compared to that in adult patients. The 95% PI of the simulation in adults receiving 40 mg encompassed 70% of the data from Becher et al. (20), wherein adults weighing more than 60 kg were receiving 40 mg BID and adults weighing less than 60 kg were receiving 30 mg BID. Our approach assumes that the first-order accumulation and decay rate constants were identical for all individuals and the interindividual difference in the intracellular d4T-TP was driven by the pharmacokinetic difference in plasma stavudine concentrations. This approach was taken because the intracellular d4T-TP values were from a different source. The limitation of the present study is the lack of the individual's intracellular d4T-TP concentrations and can only be circumvented by utilizing data from the literature whose study population closely resembles that of the present study. The prediction also does not include the residual variability, which was relatively large at an approximately 50% coefficient of variation (CV) and of the same magnitude as that reported by Hurwitz and Schinazi (24).
FIG 2.
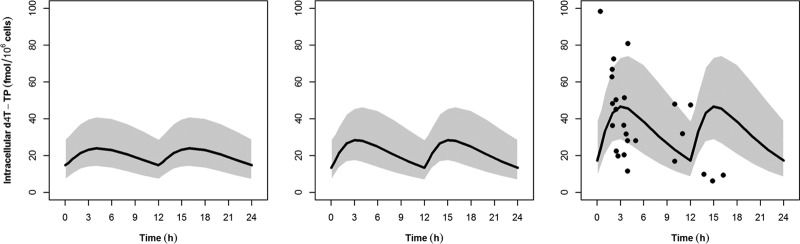
Intracellular stavudine triphosphate (d4T-TP) concentrations at steady state. Points represent actual data for compliant patients from Becher et al. (20). The shaded area and line represent the 95% prediction interval and median of the simulation in pediatric patients administered 0.5 mg/kg twice daily (left) and in adult patients administered 20 mg twice daily (center) and 40 mg twice daily (right).
Our model estimated a median trough intracellular d4T-TP concentration of 17.3 fmol/106 cells (95% PI, 8.4 to 39 fmol/106 cells) for the 40-mg BID dose, whereas 20 mg BID would result in a median trough concentration of 13 fmol/106 cells (95% PI, 6.8 to 28.6 fmol/106 cells). The peak concentrations of the 40-mg BID dose were significantly higher, with a median of 46.6 fmol/106 cells (95% PI, 28.6 to 73.3 fmol/106 cells), compared to 28.4 fmol/106 cells (95% PI, 17.3 to 45.5 fmol/106 cells) for the 20-mg BID dose. The predicted median peak concentration of intracellular d4T-TP in children taking 0.5 mg/kg BID was 23.9 fmol/106 cells (95% PI, 14.2 to 41 fmol/106 cells). This value was lower than the median peak in adults administered 20 mg BID. The median trough concentration in children taking 0.5 mg/kg BID was 14.8 fmol/106 cells (95% PI, 7.2 to 31 fmol/106 cells). This value lay above the median trough in adults administered 20 mg mg BID. The model estimated that the median intracellular levels of d4T-TP in children remained within the 95% PI range of concentrations of the adult 20-mg BID dose throughout the dosing interval but was below the adult median concentration for approximately 80% of the dosing interval.
DISCUSSION
The frequency and severity of stavudine's most common side effect in children, lipoatrophy, are strongly dose dependent. However, stavudine is rapidly cleared from the circulation, and the serum concentration-time curve is a poor predictor of the degree of mitochondrial toxicity and antiretroviral efficacy. Previous literature has revealed that the trough d4T-TP concentration determines the antiretroviral efficacy, while the peak d4T-TP concentration determines the degree of mitochondrial toxicity (31). A dose optimization should therefore aim to achieve an adequate d4T-TP trough (to retain efficacy) while reducing the peak d4T-TP concentration (to reduce toxicity). Our model predicted that a reduced pediatric dose of 0.5 mg/kg BID would result in a trough intracellular d4T-TP concentration above that of the reduced 20-mg adult dose and a peak concentration below that of the 20-mg adult dose, suggesting that 0.5 mg/kg BID should reduce mitochondrion-associated complications without compromising antiviral efficacy.
Until now there has been resistance to reducing the pediatric dose, with critics citing a lack of evidence showing that antiviral efficacy will be maintained at the lower dose. Our data suggest that a reduction in the pediatric dose to 0.5 mg/kg BID should not compromise antiviral efficacy, since it achieves intracellular d4T-TP levels similar to 20 mg BID in adults weighing more than 60 kg, which has excellent antiviral efficacy. In fact, due to the apparently longer half-life and flatter curve of d4T-TP in children, the predicted trough d4T-TP concentration in children taking 0.5 mg/kg BID lies between the median trough values of adults taking 20 and 40 mg BID, suggesting that a pediatric dose of 0.5 mg/kg BID in children under 30 kg may in fact have greater antiviral efficacy than 20 mg BID in adults over 60 kg. In addition, a substantial reduction in the standard pediatric dose should markedly reduce the frequency and severity of lipoatrophy and allow safer use of this highly effective, logistically simple, and cheap medication. The present model provides a means to predict intracellular d4T-TP exposure based on plasma exposure of the parent drug combined with a variety of cofactors. To our knowledge, this has not previously been done.
The population pharmacokinetic model for stavudine in pediatric patients, previously developed by Jullien et al. (12), was based on a one-compartment model with first-order elimination and zero-order absorption. Age, rather than weight, was a significant covariate of both CL/F and V/F in newborns through 16 years of age. The effect of weight may have been masked, because the majority of their pediatric patients were dosed by weight. We incorporated both weight and age as covariates in our simulated prepubertal population. Our weight-based simulation of stavudine pharmacokinetic profile at 1.23 mg/kg in pediatric patients covered the upper half of their data points. The interindividual variability in the present study was smaller than that in the study by Jullien et al. (12), most likely because our study population was more homogeneous (primarily healthy adult volunteers given 30 mg BID) and our simulation for the pediatric population was restricted to an age between 0.5 and 9 years with body weights between 6 and 30 kg, whereas Jullien et al. recruited from newborns to adolescents, using a more variable dosing regimen, depending on their age group: 0.61 mg/kg for the first 13 days of life, 1.23 mg/kg for children older than 13 days and weighing less than 30 kg, and 31.5 mg for children with body weights between 30 and 60 kg. Our model had a more rapid absorption, with the time of peak concentration at approximately 0.3 h, whereas their peak concentration occurred at approximately 1.72 h. This discrepancy may have been due to a difference in drug formulation, wherein the Aspen stavudine capsule may have a faster dissolution profile than the stavudine tablet that was used in 64% of their subjects. The median CL/F and V/F at 16.5 liters/h and 40.9 liters, respectively (12), were comparable in both studies, as were the peak concentrations.
The in vitro MIC (50% inhibitory concentration [IC50]) of d4T-TP against HIV-1 has been found to be between 0.009 and 6 μM, depending on the phenotypic and genotypic resistance to nucleoside reverse transcriptase inhibitors (32, 33). The standard adult doses achieved a median intracellular d4T-TP concentration in the midrange of the IC50s (approximately 2 to 3 μM) (24) yet produced an excellent virological response. The true in vivo IC50 of d4T-TP is difficult to determine. For that reason, we chose to compare the d4T-TP concentration of the reduced pediatric dose (0.5 mg/kg BID) to that of the clinically effective adult doses (20 and 40 mg BID).
Intracellular d4T-TP concentrations were not collected as part of our original data set (22). Rather, plasma data were used to develop the population pharmacokinetic model. An assumption was made that the intracellular d4T-TP data from Becher et al. (20) come from a representative 70-kg subject. Consequently, the pharmacodynamic parameters representing the intracellular accumulation and decay rate constants were identical for all simulated subjects. The true interindividual variability in the predicted peak and trough intracellular d4T-TP concentrations would probably be larger than our model predicted. However, the relative magnitudes between the dosing regimens evaluated by the model should be similar. This limitation should not significantly bias the comparison.
The estimated half-life of intracellular d4T-TP from our estimate based on the decay rate (kdp) was approximately 4.5 h, close to the 6.6 h reported in the literature (20, 21), whereas Hurwitz and Schinazi estimated a half-life of 1.3 h (24). It is likely that the population pharmacokinetic model from Panhard et al. (30) had a slower absorption than the model determined from the present study. Additionally, the analysis of Panhard et al. (30) did not demonstrate the covariate effect of body weight on the pharmacokinetic parameters, whereas the body weight was a significant covariate of clearance in the present study. This relationship between body weight and clearance allowed us to extrapolate the adult data to pediatric dosing.
The pediatric concentration-time profiles for stavudine are flatter than those in adults (12). The inclusion of an age effect on the volume of distribution and body weight on clearance may have contributed to the shape of the concentration-time profiles, which in turn drives the shape of the intracellular d4T-TP concentration-versus-time curves in pediatric patients. Even in the adult population, the model predicted that the range of intracellular d4T-TP concentrations is smaller in the group receiving 20 mg BID than in those receiving 40 mg BID (2.2-fold versus 2.8-fold difference between peak and trough concentrations). This lower peak may explain the observation that reduction of the stavudine dose from 40 mg to 30 mg in adults was associated with less mitochondrial DNA depletion in the peripheral blood mononuclear cells (9). To establish the relationship between the plasma concentration of stavudine and intracellular d4T-TP, we utilized a mass transfer form to determine the transfer from plasma to the intracellular compartment. With this modeling approach, we assumed that extracellular and intracellular stavudine concentrations equilibrate rapidly; this assumption is reasonable given that nucleoside analogs are subject to rapid transit into the intracellular side by transporters on the cell membranes of lymphocytes (28, 29).
Siccardi et al. used in silico simulation to show that the dose of efavirenz should be reduced in poor metabolizers of CYP2B6 to reduce central nervous system (CNS)-related toxicity (34). Their strategy of extrapolating their pharmacokinetic predicted information to the pharmacodynamic effect to argue for dose reduction of efavirenz is analogous to our argument for lowering the pediatric dose for stavudine.
With regard to adult dosing, our model predicted that the peak d4T-TP concentration in adults taking 40 mg BID was 64% higher than the peak in adults taking 20 mg BID. This translates into a substantially greater d4T-TP area under the curve in the 40-mg group, which may explain the markedly higher rate of mitochondrial toxicity associated with that dose in published literature. In contrast, the predicted trough d4T-TP concentration in the 20-mg group was only 25% lower than that of the 40-mg group, which may explain why the antiretroviral properties are retained when the adult dose is reduced from 40 to 20 mg BID.
Our work is in line with the first of five priority work areas in the World Health Organization and UNAIDS Treatment 2.0 strategy document (35), which aims to optimize the dosing of antiretroviral regimens available in low- to middle-income countries, particularly with regard to minimizing toxicities while retaining maximal efficacy. That strategy document reviews the history of antiretroviral scale-up, which required fast-tracking of drugs to reach the very large number of patients in desperate need of effective antiretroviral therapy. As a result of fast-tracking, many antiretroviral drugs may not have completed rigorous dose optimization evaluations prior to marketing and distribution. That dose optimization process is now being revisited, the most recent example being the Evaluation of Novel Concepts in Optimization of antiRetroviral Efficacy (ENCORE) trial, which showed noninferiority of 400 mg efavirenz compared to 600 mg (36). Similar trials are under way to evaluate reduced doses of zidovudine, stavudine, tenofovir, atazanavir, darunavir, and ritonavir (37, 38).
In conclusion, our model predicted that a reduced pediatric dose of 0.5 mg/kg BID would result in a trough intracellular d4T-TP concentration above that of the 20-mg adult dose and a peak concentration below that of the 20-mg adult dose, suggesting that a reduction in the current standard pediatric dose would not be expected to compromise antiviral efficacy while resulting in markedly less mitochondrial toxicity. However, clinical data of intracellular d4T-TP in children and adults on the reduced dose are needed to confirm this.
ACKNOWLEDGMENTS
We are indebted to Jaco Stokes, National Antiretroviral Monitor, South African National Department of Health, and Corry van der Walt, Senior Program Associate at Management Sciences for Health, for estimates of current national antiretroviral usage in South African children.
S.I. received support through a Fogarty International Clinical Research Fellowship grant (R24-TW007988-01), a pilot research grant (P30 AI036214-16, subaward 10304442) from the University of California San Diego Center for AIDS Research (UCSD CFAR), and a Southern Africa Consortium for Research Excellence (SACORE) grant (WTX055734) from the Wellcome Trust.
M.F.C. received grants from the National Institutes of Health (R01-AI 076199, 5R01HD069169-02, and R01-HD071664), from the National Institute of Allergy and Infectious Diseases (NIAID) through the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT) (5U01AI069521-01 to -04) and through the Comprehensive International Plan for Research in AIDS (CIPRA-SA) (1U19AI53217-01), from Social & Scientific Systems, Inc., through IMPAACT (BRS-IMPCT-S-11-000331-001458, BRS-IMPCT-S-11-000331-001552), and from USAID (674-A-00-09-00001-00), as well as a grant from the Centers for Disease Control and Prevention (2009-N-11094).
The recruits in this study were not coenrolled in any IMPAACT trial.
Footnotes
Published ahead of print 2 December 2013
REFERENCES
- 1.Lallemant M, Chang S, Cohen R, Pecoul B. 2011. Pediatric HIV—a neglected disease? N. Engl. J. Med. 365:581–583. 10.1056/NEJMp1107275 [DOI] [PubMed] [Google Scholar]
- 2.Innes S, Cotton MF, Haubrich R, Conradie MM, van Niekerk M, Edson C, Rabie H, Jain S, Sun X, Zollner W, Hough S, Browne SH. 2012. High prevalence of lipoatrophy in pre-pubertal South African children on antiretroviral therapy: a cross-sectional study. BMC Pediatr. 12:183. 10.1186/1471-2431-12-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Innes S, Levin L, Cotton M. 2009. Lipodystrophy syndrome in HIV-infected children on HAART. S. Afr. J. HIV Med. 10:76–80 http://www.sajhivmed.org.za/index.php/sajhivmed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.South African Department of Health 2013. The South African antiretroviral treatment guidelines. The South African National Department of Health, Pretoria, South Africa [Google Scholar]
- 5.Technau KG, Lazarus E, Kuhn L, Abrams EJ, Sorour G, Strehlau R, Reubenson G, Davies MA, Coovadia A. 2013. Poor early virologic performance and durability of abacavir-based first-line regimens for HIV-infected children. Pediatr. Infect. Dis. J. 32:851–855. 10.1097/INF.0b013e31828c3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill A, Ruxrungtham K, Hanvanich M, Katlama C, Wolf E, Soriano V, Milinkovic A, Gatell J, Ribera E. 2007. Systematic review of clinical trials evaluating low doses of stavudine as part of antiretroviral treatment. Expert Opin. Pharmacother. 8:679–688. 10.1517/14656566.8.5.679 [DOI] [PubMed] [Google Scholar]
- 7.McComsey GA, Lo Re V III, O'Riordan M, Walker UA, Lebrecht D, Baron E, Mounzer K, Frank I. 2008. Effect of reducing the dose of stavudine on body composition, bone density, and markers of mitochondrial toxicity in HIV-infected subjects: a randomized, controlled study. Clin. Infect. Dis. 46:1290–1296. 10.1086/529384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milinkovic A, Martinez E, Lopez S, de Lazzari E, Miro O, Vidal S, Blanco JL, Garrabou G, Laguno M, Arnaiz JA, Leon A, Larrousse M, Lonca M, Mallolas J, Gatell JM. 2007. The impact of reducing stavudine dose versus switching to tenofovir on plasma lipids, body composition and mitochondrial function in HIV-infected patients. Antivir. Ther. 12:407–415 http://www.intmedpress.com/serveFile.cfm?sUID=42389fbb-9eca-4a3a-a514-e0630ef9d42d [PubMed] [Google Scholar]
- 9.Sanchez-Conde M, de Mendoza C, Jimenez-Nacher I, Barreiro P, Gonzalez-Lahoz J, Soriano V. 2005. Reductions in stavudine dose might ameliorate mitochondrial-associated complications without compromising antiviral activity. HIV Clin. Trials 6:197–202. 10.1310/ED57-EU48-RK6A-E5U0 [DOI] [PubMed] [Google Scholar]
- 10.Kaul S, Kline MW, Church JA, Dunkle LM. 2001. Determination of dosing guidelines for stavudine (2′,3′-didehydro-3′-deoxythymidine) in children with human immunodeficiency virus infection. Antimicrob. Agents Chemother. 45:758–763. 10.1128/AAC.45.3.758-763.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kline MW, Dunkle LM, Church JA, Goldsmith JC, Harris AT, Federici ME, Schultze ME, Woods L, Loewen DF, Kaul S, et al. 1995. A phase I/II evaluation of stavudine (d4T) in children with human immunodeficiency virus infection. Pediatrics 96:247–252 [PubMed] [Google Scholar]
- 12.Jullien V, Rais A, Urien S, Dimet J, Delaugerre C, Bouillon-Pichault M, Rey E, Pons G, Blanche S, Treluyer JM. 2007. Age-related differences in the pharmacokinetics of stavudine in 272 children from birth to 16 years: a population analysis. Br. J. Clin. Pharmacol. 64:105–109. 10.1111/j.1365-2125.2007.02854.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson RE, Dunkle LM, Smaldone L, Adler M, Wirtz C, Kriesel D, Cross A, Martin RR. 1995. Design and implementation of the stavudine parallel-track program. J. Infect. Dis. 171(Suppl 2):S118–S122. 10.1093/infdis/171.Supplement_2.S118 [DOI] [PubMed] [Google Scholar]
- 14.Pollard RB, Peterson D, Hardy D, Pottage J, Murphy RL, Gathe J, Beall G, Rutkievicz V, Reynolds L, Cross AP, Dunkle LM. 1999. Safety and antiretroviral effects of combined didanosine and stavudine therapy in HIV-infected individuals with CD4 counts of 200 to 500 cells/mm3. J. Acquir. Immune Defic. Syndr. 22:39–48. 10.1097/00042560-199909010-00005 [DOI] [PubMed] [Google Scholar]
- 15.Petersen EA, Ramirez-Ronda CH, Hardy WD, Schwartz R, Sacks HS, Follansbee S, Peterson DM, Cross A, Anderson RE, Dunkle LM. 1995. Dose-related activity of stavudine in patients infected with human immunodeficiency virus. J. Infect. Dis. 171(Suppl 2):S131–S139. 10.1093/infdis/171.Supplement_2.S131 [DOI] [PubMed] [Google Scholar]
- 16.Ruxrungtham K, Kroon ED, Ungsedhapand C, Teeratakulpisarn S, Ubolyam S, Buranapraditkun S, van Leeuwen R, Weverling GJ, Kunanusont C, Lange JM, Cooper DA, Phanuphak P. 2000. A randomized, dose-finding study with didanosine plus stavudine versus didanosine alone in antiviral-naive, HIV-infected Thai patients. AIDS 14:1375–1382. 10.1097/00002030-200007070-00010 [DOI] [PubMed] [Google Scholar]
- 17.Spruance SL, Pavia AT, Mellors JW, Murphy R, Gathe J, Jr, Stool E, Jemsek JG, Dellamonica P, Cross A, Dunkle L. 1997. Clinical efficacy of monotherapy with stavudine compared with zidovudine in HIV-infected, zidovudine-experienced patients. A randomized, double-blind, controlled trial. Bristol-Myers Squibb Stavudine/019 Study Group. Ann. Intern. Med. 126:355–363 [DOI] [PubMed] [Google Scholar]
- 18.WHO 2007. Addendum to 2006 WHO guidelines on antiretroviral therapy for HIV infection in adults and adolescents. World Health Organization, Geneva, Switzerland [Google Scholar]
- 19.ter Hofstede HJ, Koopmans PP, Burger DM. 2008. Stavudine plasma concentrations and lipoatrophy. J. Antimicrob. Chemother. 61:933–938. 10.1093/jac/dkn041 [DOI] [PubMed] [Google Scholar]
- 20.Becher F, Landman R, Mboup S, Kane CN, Canestri A, Liegeois F, Vray M, Prevot MH, Leleu G, Benech H. 2004. Monitoring of didanosine and stavudine intracellular trisphosphorylated anabolite concentrations in HIV-infected patients. AIDS 18:181–187. 10.1097/00002030-200401230-00006 [DOI] [PubMed] [Google Scholar]
- 21.Dudley MN, Graham KK, Kaul S, Geletko S, Dunkle L, Browne M, Mayer K. 1992. Pharmacokinetics of stavudine in patients with AIDS or AIDS-related complex. J. Infect. Dis. 166:480–485. 10.1093/infdis/166.3.480 [DOI] [PubMed] [Google Scholar]
- 22.Innes S, Norman J, Smith P, Smuts M, Capparelli E, Rosenkranz B, Cotton M. 2011. Bioequivalence of dispersed stavudine: opened versus closed capsule dosing. Antivir. Ther. 16:1131–1134. 10.3851/IMP1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, Wei R, Curtin LR, Roche AF, Johnson CL. 2002. 2000 CDC growth charts for the United States: methods and development. Vital Health Stat. 11 11:1–190 [PubMed] [Google Scholar]
- 24.Hurwitz SJ, Schinazi RF. 2011. In silico study supports the efficacy of a reduced dose regimen for stavudine. Antivir. Ther. 92:372–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, Kost R, Hurley A, Weinberger L, Cesar D, Hellerstein MK, Ho DD. 2001. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 194:1277–1287. 10.1084/jem.194.9.1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 279:51545–51553. 10.1074/jbc.M408573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E, Schmidt D, Hoh R, Neese R, Macallan D, Deeks S, McCune JM. 1999. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat. Med. 5:83–89. 10.1038/4772 [DOI] [PubMed] [Google Scholar]
- 28.Balimane PV, Sinko PJ. 1999. Involvement of multiple transporters in the oral absorption of nucleoside analogues. Adv. Drug Deliv. Rev. 39:183–209. 10.1016/S0169-409X(99)00026-5 [DOI] [PubMed] [Google Scholar]
- 29.Plagemann PG, Wohlhueter RM, Woffendin C. 1988. Nucleoside and nucleobase transport in animal cells. Biochim. Biophys. Acta 947:405–443. 10.1016/0304-4157(88)90002-0 [DOI] [PubMed] [Google Scholar]
- 30.Panhard X, Legrand M, Taburet AM, Diquet B, Goujard C, Mentre F. 2007. Population pharmacokinetic analysis of lamivudine, stavudine and zidovudine in controlled HIV-infected patients on HAART. Eur. J. Clin. Pharmacol. 63:1019–1029. 10.1007/s00228-007-0337-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingo P, Cabeza MC, Pruvost A, Salazar J, Gutierrez Mdel M, Mateo MG, Domingo JC, Fernandez I, Villarroya F, Munoz J, Vidal F, Baiget M. 2010. Relationship between HIV/highly active antiretroviral therapy (HAART)-associated lipodystrophy syndrome and stavudine-triphosphate intracellular levels in patients with stavudine-based antiretroviral regimens. Clin. Infect. Dis. 50:1033–1040. 10.1086/651117 [DOI] [PubMed] [Google Scholar]
- 32.Calvez V, Costagliola D, Descamps D, Yvon A, Collin G, Cecile A, Delaugerre C, Damond F, Marcelin AG, Matheron S, Simon A, Valantin MA, Katlama C, Brun-Vezinet F. 2002. Impact of stavudine phenotype and thymidine analogues mutations on viral response to stavudine plus lamivudine in ALTIS 2 ANRS trial. Antivir. Ther. 7:211–218 http://www.intmedpress.com/serveFile.cfm?sUID=f8a7bf03-a413-46a2-acbd-5ac682945177 [PubMed] [Google Scholar]
- 33.Holguin A, Dietrich U, Immelmann A, Soriano V. 1998. Genotypic and phenotypic resistance to stavudine after long-term monotherapy. BMS-020 Spanish Study Group. Antivir. Ther. 3:183–186 [PubMed] [Google Scholar]
- 34.Siccardi M, Almond L, Schipani A, Csajka C, Marzolini C, Wyen C, Brockmeyer NH, Boffito M, Owen A, Back D. 2012. Pharmacokinetic and pharmacodynamic analysis of efavirenz dose reduction using an in vitro-in vivo extrapolation model. Clin. Pharmacol. Ther. 92:494–502. 10.1038/clpt.2012.61 [DOI] [PubMed] [Google Scholar]
- 35.WHO 2011. The treatment 2.0 framework for action: catalysing the next phase of treatment, care and support. World Health Organization, Geneva, Switzerland [Google Scholar]
- 36.Puls R, ENCORE1 Study Group 2013. 7th IAS Conf. HIV Pathog., Treat. Prevent., Kuala Lumpur, Malaysia, 30 June to 3 July 2013, abstr WELBB01 [Google Scholar]
- 37.Clayden P. 12 June 2012, posting date Retrofitting for purpose: treatment optimization. HIV-HCV-TB Pipeline Report. http://www.pipelinereport.org/TOC/treatment-optimization [Google Scholar]
- 38.Crawford KW, Ripin DH, Levin AD, Campbell JR, Flexner C. 2012. Optimising the manufacture, formulation, and dose of antiretroviral drugs for more cost-efficient delivery in resource-limited settings: a consensus statement. Lancet Infect. Dis. 12:550–560. 10.1016/S1473-3099(12)70134-2 [DOI] [PubMed] [Google Scholar]