

Abstract
Conventional therapy for human cytomegalovirus (CMV) relies on inhibition of the viral DNA polymerase. Ganciclovir (GCV) is the first-line therapy, but when GCV-resistant strains emerge, alternative therapies are extremely limited and are associated with significant toxicities. Combination of anti-CMV agents that act on different targets or stages of virus replication has not been well studied, mostly because of the limited number of anti-CMV agents. We report our investigation of combinations of agents that inhibit CMV by targeting the viral DNA polymerase, cellular kinases, or other cell/virus mechanisms yet to be discovered. The selected compounds differed by the slopes of their dose-response curve: compounds with a slope of 1 (GCV) representing one target or noncooperativity and compounds with high slopes indicating positive cooperativity. Analysis of anti-CMV drug combinations using the Bliss model (which accounts for the slope parameter) distinguished between combinations with synergistic, antagonistic, and additive activities. The combination of GCV and foscarnet was slightly synergistic; strong synergism was found when GCV was used with artemisinin-derived monomers or dimers or the MEK inhibitor U0126. The combination of GCV and cardiac glycosides (digoxin, digitoxin, and ouabain) was additive. The monomeric artemisinin artesunate was synergistic when combined with U0126 or the multikinase inhibitor sunitinib. However, the combination of artemisinin-derived dimers (molecular weights, 606 and 838) and U0126 or sunitinib was antagonistic. These results demonstrate that members of a specific drug class show similar patterns of combination with GCV and that the slope parameter plays an important role in the evaluation of drug combinations. Lastly, antagonism between different classes of CMV inhibitors may assist in target identification and improve the understanding of CMV inhibition by novel compounds.
INTRODUCTION
Cytomegalovirus (CMV) is the most common cause of congenitally acquired infection in the United States and is a major pathogen in solid organ transplant recipients and patients with AIDS (1–3). Anti-CMV compounds have been used with varied success in these patient populations, but the complexity of CMV disease and the need for prolonged courses of therapy for virus suppression result in serious side effects and the emergence of resistant viral mutants (4–8). The FDA-approved anti-CMV drugs ganciclovir (GCV), foscarnet (FOS), and cidofovir (CDV) belong to a single class of inhibitors, all targeting the viral DNA polymerase. The development and clinical evaluation of compounds that act on new viral targets, for example, the UL97 kinase inhibitor maribavir (9–11) and the terminase inhibitor AIC246 (12, 13), are under way. Cellular targets that could abrogate virus replication are also being studied as potential anti-CMV compounds (14). The role of anticellular antiviral inhibitors in CMV therapy is not defined as of yet; however, the potential use of such agents as either monotherapy (salvage therapy) or combination therapy with existing anti-CMV agents may be justified as their mechanisms of action against CMV replication become clear.
While combination therapy for cancer (chemotherapy) and some infectious diseases (tuberculosis, HIV infection, hepatitis C) has become the standard of care, a similar approach to CMV therapy is not a common practice, although combination of GCV and FOS has been reported in patients with CMV retinitis and is recommended for CMV encephalitis (15, 16). The lack of combination regimens is partially explained by the limited number of known anti-CMV agents with mechanisms of action different from those of the DNA polymerase inhibitors, insufficient in vitro data on the effect of combinations of anti-CMV agents on CMV replication, and a lack of standardization in analyzing the results obtained with drug combinations.
Previously reported in vitro combination studies were based on a plaque reduction assay or real-time PCR and investigated a small number of CMV inhibitors. The models used for analysis of combinations in vitro somewhat complicated data interpretation. For example, one study reported moderate synergism of GCV and FOS against the laboratory-adapted strain AD169 and several clinical isolates (17). The drug combination analysis used in that study was based on the fractional inhibitory concentration (FIC) value of the isobologram method, in which the effect of combinations of agents on CMV replication was evaluated by analysis of the changes of the drug concentrations leading to 50% virus inhibition (the 50% effective concentrations [EC50s]) of one compound in the presence of different concentrations of the other compound (17). Another study found the combination of GCV and FOS to be synergistic against the laboratory-adapted Towne strain and one of several clinical strains tested but not against AD169, on the basis of the mean combination index (CI) of the Chou-Talalay method (18, 19). The combination of GCV and maribavir (MBV) was antagonistic using the isobologram method, while FOS plus MBV and CDV plus MBV were additive (20). However, using a three-dimensional method (MacSynergy II), a strong synergism between FOS and MBV or CDV and MBV was found (21, 22). These discrepancies underscore the importance of identifying the factor(s) that may determine the appropriate model for analysis of drug combinations.
We reported that the CMV inhibitors artemisinins and cardiac glycosides acted earlier than GCV in CMV-infected cells, and limited combination studies indicated that GCV plus artemisinins had synergistic activities, while GCV plus digoxin showed only additive effects on CMV inhibition (23, 24). We now report a comprehensive analysis of multiple drug combinations. In this study, CMV inhibition was quantified in vitro using combinations of compounds that belong to one of three general classes: (i) compounds that target the viral DNA polymerase, (ii) compounds that target the viral DNA polymerase in combination with compounds targeting a known cellular protein(s), and (iii) compounds with cellular targets that are either defined or not yet known. We show that specific parameters included in a combination model can affect the interpretation of the outcome from the use of a drug combination and that in vitro combination studies may lead to targeted investigations into the mechanisms of action of novel anti-CMV compounds.
MATERIALS AND METHODS
Compounds.
The compounds used in this study and their mechanisms of action, when known, are listed in Table 1. Artemisinins (monomers and dimers), cardiac glycosides, and U0126 were reported to inhibit CMV replication and were therefore selected for this study (24–29). The multikinase inhibitor sunitinib has not previously been reported to inhibit CMV replication, but other kinase inhibitors have been shown to inhibit CMV in vitro (30). GCV was dissolved in distilled water; all other compounds were dissolved in dimethyl sulfoxide (DMSO). Stock solutions (10 mM) were stored at −80°C. The final concentration of DMSO in all experiments was less than 0.1%, a concentration at which no cytotoxicity was observed.
TABLE 1.
Compounds used for anti-CMV activity
| Compound (abbreviation or reference) | Source | Mechanism of action |
|---|---|---|
| Ganciclovir (GCV) | Sigma Chemical Co. | Inhibition of viral DNA polymerase |
| Foscarnet (FOS) | Sigma Chemical Co. | Inhibition of viral DNA polymerase |
| Artesunate (AS) | The Johns Hopkins University | NAa |
| Dimer 838 (37) | The Johns Hopkins University | NA |
| Dimer 606 (37) | The Johns Hopkins University | NA |
| Digoxin (DIG) | Sigma Chemical Co. | Cellular Na-K-ATPase inhibitor |
| Digitoxin | Sigma Chemical Co. | Cellular Na-K-ATPase inhibitor |
| Ouabain (OUA) | Sigma Chemical Co. | Cellular Na-K-ATPase inhibitor |
| Sunitinib | Sigma Chemical Co. | Cellular multikinase inhibitor |
| U0126 | Santa Cruz Biotechnology Inc. (Santa Cruz, CA) | Cellular MEK1/2 inhibitor |
NA, mechanism of action unknown.
Cell culture, virus infection, and antiviral assays.
Human foreskin fibroblasts (HFFs; CRL-2088; ATCC) from passages 12 to 16 were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA) in a 5% CO2 incubator at 37°C and used for infection with human CMV at a multiplicity of infection (MOI) of 1 PFU/cell. The luciferase reporter CMV system was used to quantify CMV inhibition by each drug individually and by different drug combinations. The pp28-luciferase Towne strain, which expresses luciferase under the control of the UL99 (pp28) late promoter, was generated by insertion of the reporter gene between the US9 and US10 open reading frames (ORFs) in the Towne genome (31). The expression of pp28-luciferase is strongly activated at 48 to 72 h postinfection (hpi), and this reporter system is sensitive and reproducible and highly correlates with plaque reduction (31). After 90 min adsorption, medium was removed and cells were washed with phosphate-buffered saline (PBS). DMEM with 4% FBS containing compounds was then added to each well. Infected and drug-treated HFFs were collected at 72 hpi, and lysates were assayed for luciferase activity using a luciferase assay kit (Promega, Madison, WI) on a GloMax-Multi detection system (Promega) according to the manufacturer's instructions. For plaque reduction assay, human embryonic lung fibroblasts were seeded into 12-well plates and incubated at 37°C 1 day prior to infection with the laboratory-adapted Towne CMV strain. The virus was diluted to a concentration which gave 50 to 60 plaques per well. Medium was aspirated from the wells, and virus suspension was added to each well in triplicate. Plates were incubated for 90 min with shaking every 10 min; thereafter, compounds were added and a methylcellulose overlay was applied to each well. Serial dilutions of the artemisinin-derived dimer with a molecular weight of 838 (dimer 838) and the MEK inhibitor U0126 were used. After incubation for 7 days, cells were stained with crystal violet. The stain was aspirated, wells were washed with phosphate-buffered saline, and plaques were counted. Infection with the TB40 endotheliotrophic strain (ATCC VR1578) at an MOI of 1 PFU/cell was also used for one representative drug combination in the synergistic and antagonistic categories.
Real-time PCR.
CMV DNA was extracted from supernatants of infected cells at 96 hpi using the automated BioRobot M48 instrument with a MagAttract Virus Mini-M48 kit and the Virus Mini-Protocol, version 1.1 (Qiagen, Valencia, CA). Cellular DNA was purified at 6 days postinfection from TB40-infected HFFs using a Wizard SV Genomic kit (Promega, Madison, WI). The quantitative CMV real-time PCR assay is based on detection of a 151-bp region from the highly conserved US17 gene (32). The limit of detection of the assay is 100 copies/ml (3.0 copies per reaction), and the measurable range is 2.4 to 8.0 log10 copies/ml. The PCR was performed using a total reaction volume of 50 μl. This included 25 μl of TaqMan 2× Universal PCR master mix (Applied Biosystems, Foster City, CA), 1.5 μl each of 10 μM primers, 1 μl of 10 μM 6-caraboxyfluorescein-labeled probe, 11 μl of distilled H2O, and 10 μl of template. Amplification was performed on a 7500 real-time PCR system (Applied Biosystems, Foster City, CA). PCR conditions were 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 15 s and 60°C for 60 s. Quantification standards were prepared by cloning the US17 amplicon in the pCR2.1-TOPO plasmid vector (Invitrogen, Carlsbad, CA). Serial 10-fold dilutions of plasmid from 7.0 to 1.0 log10 copies per reaction were included with each assay and used to establish a standard curve. Assay controls included quantified CMV AD169 DNA at 10 copies per reaction (Advanced Biotechnologies Inc., Columbia, MD) and quantified Towne CMV at 3.0 and 5.0 log10 copies/ml.
Cell viability.
Cell viability was determined by an 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT)-based colorimetric assay (Sigma-Aldrich, St. Louis, MO). A total of 1 × 106 cells per plate were seeded in 96-well microplates. The MTT assay was performed at the same time points of the antiviral assay with the same concentrations of compounds or combinations of compounds.
SDS-PAGE and immunoblot analysis.
Cells were serum starved for 3 days, followed by infection with CMV Towne (MOI = 1) and drug treatment. Cell lysates were collected at the indicated time points with 1× lysis buffer containing 10 mM NaF and 5 mM Na3VO4. Equivalent amounts of proteins were mixed with an equal volume of sample buffer (125 mM Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 5% β-mercaptoethanol) and boiled at 100°C for 10 min. Denatured proteins were resolved in Tris-glycine polyacrylamide gels (10%) and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA) by electroblotting. Membranes were incubated in blocking solution (5% bovine serum albumin and 0.1% Tween 20 in PBS [PBST]) for 1 h, washed with PBST, and incubated with antibody at 4°C overnight. Membranes were washed with PBST and incubated with horseradish peroxidase-conjugated secondary antibodies in PBST for 2 h at room temperature. Following washing with PBST, protein bands were visualized by chemiluminescence using SuperSignal West Dura and Pico reagents (Pierce Chemical, Rockford, IL). The following antibodies were used: rabbit phospho-p44/42 mitogen-activated protein kinase (extracellular signal-regulated kinase 1/2 [ERK1/2]; Cell Signaling Technology, Beverly, MA), rabbit phospho-MEK1/2 (Cell Signaling Technology, Beverly, MA), mouse anti-ERK1/2 (p44/p42; Millipore, Billerica, MA), mouse anti-β-actin (Millipore, Billerica, MA), horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (Cell Signaling Technology, Beverly, MA), and HRP-conjugated anti-mouse IgG (GE Healthcare, Waukesha, WI).
Dose-response curves.
The combined inhibitory effect of compounds on human CMV replication was determined in infected HFFs. A dose-response curve was first generated for each compound; the EC50 and slope were calculated. Dose-response curves were constructed following a classic description of dose-response relationships (33): log (fa/fu) = m log(D/EC50) or fa = 1/[1 + (EC50/D)m], where fa and fu (equal to 1 − f) are the fractions of viruses affected and unaffected by the drug, respectively; D is the drug concentration; EC50 is the drug concentration that causes 50% of the maximum inhibitory effect; and m is a slope parameter mathematically analogous to the Hill coefficient, a measure of cooperativity in the binding of multiple ligands to linked binding sites (33). A slope of 1 represents either one binding site (target) or a phenomenon of noncooperativity. Cooperative binding requires that the chemical compound have more than one binding site and cooperativity results from the interactions between binding sites. A positive cooperativity exists when binding at one site increases the affinity for ligand binding at another site (slope > 1). If binding at one site lowers the affinity for ligand at another site, the compound exhibits negative cooperativity (slope < 1). If the ligand binds at each site independently, the binding is noncooperative (slope = 1). An inhibitor that has one binding site cannot engage in cooperative binding. For drug combinations, the concentration of each drug was twice its EC50 with subsequent 2-fold serial dilutions.
Analysis of drug combination.
The methods used for quantifying the effect of two compounds on CMV replication were the isobologram method (34) and the Bliss model (35). In the isobologram method, the effects of two compounds are considered to be mutually exclusive (i.e., the compounds possess similar modes of action or compete for the same binding site). In the Bliss model, the effect of the drug combination represents the product of two probabilistically independent events, as described in the following equation (36):
where D is the drug concentration, m is the slope, EC50 is the effective concentration resulting in 50% virus inhibition, and 1 and 2 represent inhibitors 1 and 2, respectively. The combined effect of two inhibitors (fractional unaffected [FU1+2]) is computed as the product of the individual effects of the two inhibitors, FU1 and FU2. If the ratio of the observed fold inhibition divided by the expected fold inhibition is greater than 1, the compounds are synergistic. If the ratio is less than 1, the combination is considered antagonistic, and if it equals 1, the combination is additive.
RESULTS
Single-drug dose-response.
Traditionally, in vitro anti-CMV activities are described using the concentration resulting in 50% inhibition of virus replication (EC50). The anti-CMV activities and slopes of each compound used in this study were determined: the DNA polymerase inhibitors GCV and FOS; the artemisinin derivatives monomeric artesunate (AS), dimer primary alcohol (MW, 606 [dimer 606]), and dimer diphenyl phosphate (MW, 838 [dimer 838]); the cardiac glycosides digoxin, digitoxin, and ouabain; the multikinase inhibitor sunitinib; and the MEK inhibitor U0126 (Table 2). Artemisinin-derived dimers, highly effective inhibitors of CMV replication, have a dose-response curve characterized by a high slope (m ∼ 3) (37) compared to the slope of GCV and AS (m = 1).
TABLE 2.
EC50 and slope of each drug alone and combinations of compoundsa
| Combination | Drug 1 |
Drug 2 |
Bliss coefficient (Luc/PCR/plaque) | Effect | ||||
|---|---|---|---|---|---|---|---|---|
| Drug | EC50 (μM) | Slope (m) | Drug | EC50 (μM) | Slope (m) | |||
| Virus-virus | GCV | 1.15 ± 0.04 | 1.3 ± 0.06 | GCV | 1.29 ± 0.20 | 1.44 ± 0.29 | 1.05 | Ad |
| GCV | 1.14 ± 0.06 | 1.38 ± 0.09 | FOS | 74.2 ± 5.46 | 1.07 ± 0.16 | 1.26 | Sy | |
| Virus-cell | GCV | 1.23 ± 0.12 | 1.14 ± 0.14 | AS | 8.07 ± 0.26 | 1.43 ± 0.08 | 2.02 | Sy |
| GCV | 1.36 ± 0.03 | 1.35 ± 0.03 | 838 | 0.05 ± 0.00 | 3.32 ± 0.51 | 2.95 | Sy | |
| GCV | 1.29 ± 0.20 | 1.14 ± 0.14 | 606 | 0.11 ± 0.01 | 2.43 ± 0.23 | 1.77 | Sy | |
| GCV | 1.48 ± 0.07 | 1.40 ± 0.11 | DIG | 0.04 ± 0.00 | 2.78 ± 0.21 | 1.04 | Ad | |
| GCV | 1.57 ± 0.31 | 1.47 ± 0.50 | OUA | 0.01 ± 0.00 | 2.40 ± 0.59 | 1.09 | Ad | |
| GCV | 1.72 ± 0.19 | 1.49 ± 0.25 | Digitoxin | 0.03 ± 0.01 | 2.03 ± 0.58 | 1.04 | Ad | |
| GCV | 1.54 ± 0.03 | 1.24 ± 0.04 | U0126 | 43.74 ± 1.00 | 4.96 ± 0.43 | 2.11/2.16 | Sy | |
| Cell-cell | AS | 5.85 ± 0.97 | 1.3 ± 0.29 | 606 | 0.11 ± 0.00 | 2.67 ± 0.11 | 0.90 | Ad |
| AS | 9.64 ± 0.25 | 1.02 ± 0.03 | U0126 | 45.06 ± 1.17 | 4.76 ± 0.47 | 2.26 | Sy | |
| AS | 5.80 ± 0.67 | 1.35 ± 0.22 | 838 | 0.05 ± 0.00 | 3.58 ± 0.17 | 0.96 | Ad | |
| 606 | 0.11 ± 0.01 | 2.43 ± 0.23 | U0126 | 43.74 ± 1.00 | 4.96 ± 0.43 | 0.64/0.65 | An | |
| 838 | 0.04 ± 0.00 | 3.92 ± 0.17 | Sunitinib | 10.32 ± 0.87 | 1.94 ± 0.29 | 0.41 | An | |
| 838 | 0.04 ± 0.00 | 3.92 ± 0.17 | U0126 | 43.74 ± 1.00 | 4.96 ± 0.43 | 0.19/0.18/0.3 | An | |
| OUA | 0.02 ± 0.00 | 2.80 ± 0.35 | Sunitinib | 9.05 ± 0.83 | 2.61 ± 0.52 | 2.20 | Sy | |
| OUA | 0.01 ± 0.00 | 3.19 ± 1.02 | U0126 | 37.19 ± 3.40 | 2.9 ± 0.68 | 1.40 | Sy | |
For each combination experiment, the EC50 and slope of each compound were first determined. Inhibition of CMV replication was determined by luciferase expression, which is under the control of the late CMV pp28 gene promoter. Since the luciferase assay is highly sensitive, EC50s can show some variability; a change of 10 to 15% does not indicate a significant difference in the EC50. The Bliss coefficient was calculated on the basis of the observed/expected fold inhibition around the EC50 of the combination. A coefficient of >1 suggests synergism (Sy), a coefficient of <1 suggests antagonism (An), and a coefficient equal to 1 suggests an additive effect (Ad). The Bliss coefficient was determined on the basis of pp28-luciferase (Luc) expression at 72 hpi in cell lysates of CMV-infected compound-treated cells. For the drug combinations GCV plus U0126, 606 plus U0126, and 838 plus U0126, the Bliss coefficient was also calculated on the basis of real-time PCR performed at 96 hpi in supernatants of infected-treated cells. The combination of 838 and U0126 was also tested using a plaque reduction assay. DIG, digoxin; OUA, ouabain.
Analysis of drug combinations using the isobologram method and the Bliss model reflects an important role of the slope parameter.
CMV inhibition using combinations of compounds that have similar or different slopes was quantified. As a control, the combination of GCV and GCV (m = 1) was additive when analyzed using either the isobologram method or the Bliss model (data not shown and Table 2). The isobologram and Bliss methods correlated well when the combination of compounds possessing a slope of 1 was tested. However, when compounds with different slopes were used in combination, discordant findings were observed between the isobologram method and the Bliss model (Fig. 1; Table 2). The combination of GCV (m, ∼1) and dimer diphenyl phosphate 838 (m, >3) showed mild antagonism when the isobologram method was used but was highly synergistic when analyzed by the Bliss method. In previous combination studies, the slope of the tested compounds was mostly 1. Since the compounds used here for combination studies possess different slopes and because the isobologram method does not consider the slope of the dose-response curve, the Bliss model was selected for subsequent data analysis.
FIG 1.

Combination of artemisinin-derived dimer (dimer 838) and GCV analyzed using the isobologram method (left) or the Bliss model (right). HFFs were infected with pp28-luciferase CMV at an MOI of 1 PFU/cell and treated with dimer 838, GCV, and their combination. The concentrations of GCV and dimer 838 were started at 2× their EC50s, followed by 2-fold serial dilutions. The concentrations selected for the isobologram method (left) were started with different ratios of the GCV and dimer 838 combination, followed by 2-fold serial dilutions. The concentrations selected for the Bliss model (right) were started at a fixed ratio of the GCV and dimer 838 combination (1:1), followed by their 2-fold serial dilutions. Luciferase activity was measured in cell lysates collected at 72 hpi. Data represent mean values ± SDs of triplicate determinations from three independent experiments. FIC, fractional inhibitory concentration.
Specific drug combinations are additive, synergistic, or antagonistic in CMV inhibition.
Drug combinations consisting of compounds that have been reported to inhibit CMV replication were tested. Compounds generally belonged to classic CMV inhibitors (GCV, FOS), agents with known cellular targets (cardiac glycosides, U0126, sunitinib), and compounds with a yet unidentified target(s) (artemisinins). Using the Bliss model, we were able to distinguish between additive, synergistic, and antagonistic combinations. Combinations with an additive effect included GCV plus digoxin, digitoxin, or ouabain and AS plus dimer 838 or dimer 606, all of which had a Bliss coefficient (observed results by use of the Bliss model divided by the expected result by use of the Bliss model) of approximately 1 (Fig. 2; Table 2). The combination of GCV plus FOS showed mild synergy (Bliss coefficient, 1.26). Synergistic combinations were GCV plus AS, dimer 606, dimer 838, or U0126, AS plus U0126, and ouabain plus sunitinib, with Bliss coefficients of close to or >2. To ensure that synergistic inhibition was not a result of cellular toxicity, an MTT assay was performed. Cell viabilities for these combinations at their highest synergistic effect were 94%, 93%, 89%, 85%, 79%, and 77%, respectively, suggesting that enhanced CMV inhibition was likely achieved through inhibition of independent targets and not as a result of cellular toxicity. Ouabain plus U0126 also showed synergism, with a Bliss coefficient of 1.4 (Fig. 3; Table 2). The combination of GCV plus dimer 606 was also highly synergistic against the TB40 strain (see Table S1 in the supplemental material).
FIG 2.

Additive effect of AS plus dimer 838 and GCV plus digoxin. HFFs were infected with pp28-luciferase CMV (MOI = 1) and treated with each compound individually, followed by treatment with the drug combinations at various concentrations. Each compound was tested at 2× its EC50, followed by 2-fold serial dilutions. A fixed ratio of drug concentration was used, diluting each compound 2-fold in each test iteration. Luciferase activity was measured in cell lysates collected at 72 hpi. Data represent mean values ± SDs of triplicate determinations from three independent experiments. Solid line, theoretical (expected) Bliss dose-response curve; dotted line, observed Bliss curve. Additivity was determined on the basis of calculation of the Bliss coefficient at the EC50 of each drug combination.
FIG 3.

Synergistic activity of GCV and artemisinins. HFFs were infected with pp28-luciferase CMV and treated with each compound, followed by the combination, at various concentrations. The concentration of each compound alone was started at 2× its EC50, followed by 2-fold serial dilutions. The concentrations for the combination experiment were started at a fixed ratio, followed by 2-fold serial dilutions. Luciferase activity was measured in cell lysates collected at 72 hpi. Data represent mean values ± SDs of triplicate determinations from more than three independent experiments. Synergy was determined on the basis of calculation of the Bliss coefficient at the EC50 of each drug combination.
U0126 antagonizes the anti-CMV activities of artemisinin-derived dimers.
To understand the activities of artemisinins in CMV-infected cells, the combination of these compounds with the MEK inhibitor U0126 was tested. The Ras/Raf/MEK/ERK pathway has been suggested to play a role in the response of tumor cells to AS (38). Artemisinin-derived dimers likely represent a class of CMV inhibitors separate from the monomeric artemisinins (such as AS) on the basis of their different slopes (37). The combination of AS plus U0126 or the artemisinin dimers plus U0126 demonstrated different effects on CMV replication. The combination of dimer 838 plus U0126 or the upstream multikinase inhibitor sunitinib was highly antagonistic in CMV inhibition (Fig. 4A; Table 2), with a Bliss coefficient of <1. The antagonistic effect of U0126 plus dimer 838 was also demonstrated by real-time PCR from supernatants of infected cells and by a plaque reduction assay (Table 2). The antagonistic effect of dimer 838 plus U0126 is likely specific to the class of dimers because combination of the dimer primary alcohol (MW, 606) plus U0126 was also antagonistic when tested against the pp28-luciferase Towne and the TB40 strains (Table 2; see Table S1 in the supplemental material). In contrast, the combination of U0126 with GCV or AS was synergistic. U0126 plus ouabain showed mild synergy, and sunitinib plus ouabain was also synergistic (Table 2; Fig. 5). The most significant antagonism was observed when the compounds were added together at 6 hpi or when U0126 was added just after infection, followed by addition of dimer 838 at 6 hpi (Fig. 4B). The antagonistic effect of dimer 838 and U0126 was not a result of a chemical interaction between the two compounds because the same results were obtained when U0126 was added for 6 h, followed by removal of the medium, washing of the cells with PBS, and addition of dimer 838 in fresh medium (Fig. 4B). In addition, the levels of pERK and pMEK at 15 min postinfection and 8 hpi supported a lack of chemical interaction (Fig. 4C). The combination of dimer 838 plus U0126 reversed the effects of dimer 838 used alone on ERK phosphorylation. Since CMV inhibition with U0126 was achieved at concentrations that are higher than those required for inhibition of MEK1/2 activity and thus could represent off-target effects, the combination of U0126 with dimer 838 was also tested using U0126 at lower concentrations (0.1 μM and 1 μM) which inhibit MEK1/2 activity but do not result in CMV inhibition. Again, U0126 was able to offset the anti-CMV activities of dimer 838 (Fig. 4B and C).
FIG 4.

Antagonistic anti-CMV activity of U0126 plus dimer 838. (A) Antagonistic anti-CMV activity of dimer 838 plus sunitinib and dimer 838 plus U0126. HFFs were infected with pp28-luciferase CMV (MOI = 1) and treated with each compound individually, followed by the drug combination, as described in the legends to Fig. 2 and 3. Data represent mean values ± SDs of triplicate determinations from three independent experiments. (B) Effect of the drug combination applied at different time points on anti-CMV activity. (Left) HFFs were treated either before infection with U0126 (40 μM) or after infection, followed by compound removal, washing of the cells with PBS, and addition of dimer 838 (40 nM). Time points of testing of the combination of dimer 838 plus U0126 relative to the time of infection were, respectively, −2 and 0 h, −2 and 6 h, 0 and 0 h, 0 and 6 h, 6 and 0 h, and 6 and 6 h. (Right) HFFs were infected and treated with U0126 (100 nM), followed by its removal, washing of the cells with PBS, and addition of dimer 838 (40 nM). Time points of testing the combination relative to the time of infection were, respectively, 0 and 0 h, 0 and 6 h, 6 and 0 h, and 6 and 6 h. (C) Western blot of pMEK, pERK (p42/44), and total ERK at 15 min postinfection and 8 hpi. HFFs were serum starved for 72 h, followed by infection and treatment with dimer 838 (40 nM) plus U0126 (40 μM) (left) or dimer 838 (40 nM) plus U0126 (1 μM) (right). Infected-treated cells were harvested at the indicated time points, and lysates were subjected to Western blot analysis. Lanes NI, noninfected cells; lanes I, infected cells.
FIG 5.
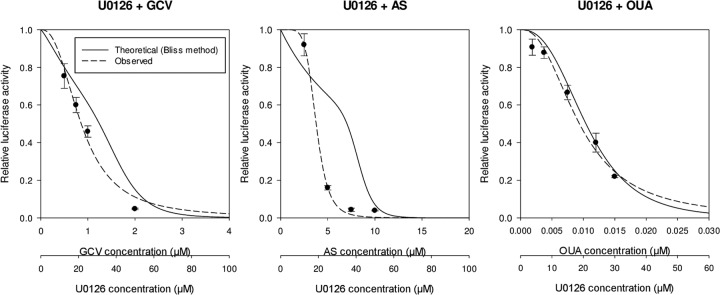
Synergistic combination of U0126 plus GCV, AS, or ouabain (OUA). HFFs were infected with pp28-luciferase CMV at an MOI of 1 PFU/cell and treated with each compound individually, followed by the combination, at various concentrations. The concentration of each compound was started at 2× its EC50, followed by 2-fold serial dilutions. A fixed ratio of drug concentration was used, diluting each compound 2-fold in each test iteration. Luciferase activity was measured in cell lysates collected at 72 hpi. Data represent mean values ± SDs of triplicate determinations from three independent experiments.
DISCUSSION
Specific anti-CMV drug combinations achieve additive, synergistic, or antagonistic effects on human CMV replication. The combination of the CMV DNA polymerase inhibitors GCV and FOS was mildly synergistic. The combination of GCV and all artemisinins (AS, dimer 838, dimer 606) demonstrated strong synergistic activity, and that of GCV and cardiac glycosides (digoxin, digitoxin, and ouabain) resulted in an additive effect. The two separate classes of CMV inhibitors, artemisinins and cardiac glycosides, were previously reported to have a higher slope than GCV, suggesting that these inhibitors belong to different classes of CMV inhibitors in which participation of multiple copies of the drug target may occur (24, 37). For other chronic viral infections, such as those with HIV, the slope was found to be an important factor not only in distinguishing between drug classes with a known mechanism of action but also in contributing to reduced antiviral activity even when the EC50 was unchanged in resistant virus mutants (39, 40). When the slope of the dose-response curve is higher than 1, small changes in drug concentration (D) can result in significant effects on virus replication. Since the slope of the dose-response curve is not accounted for in the isobologram method, analysis of anti-CMV drug combinations consisting of compounds with different slopes may be misinterpreted using this model. In addition, assuming that the tested compounds were active at different stages of virus replication, use of the Bliss model appeared to be the most appropriate method for data analysis of anti-CMV drug combinations.
Several methods have been used to calculate the expected dose-response relationship for combination therapy compared to that for monotherapy, of which the Loewe additivity (isobologram method) and Bliss independence methods have been commonly used (41). The Loewe additivity method assumes that two inhibitors act through a similar mechanism, and thus, the effects of each inhibitor and the inhibitor combination are related through equipotent dose ratios. In contrast, the Bliss independence method assumes that the two inhibitors act through independent mechanisms, leading to the concept of effect multiplication, in which combination therapy is represented as the union of two probabilistically independent events. Debate continues, however, as to which method performs better. Three-dimensional dose-response surfaces are also used to identify regions of robust synergistic behavior (21, 42). Using this methodology, an analysis of compounds acting late during virus replication was performed and revealed strong synergy between GCV plus FOS or CDV but antagonism between the viral DNA processing inhibitor BAY 38-4766 and GCV (22). The generation of dose-response surfaces requires an extensive checkerboard of inhibitor concentrations and does not consider the slope of the dose-response curve. On the basis of our results, determination of the slope of the dose-response curve may play an important role in choosing the model for analysis of drug combinations. The combination of GCV plus FOS (each has a slope of 1) showed a mild synergistic effect in CMV inhibition using either the isobologram or the Bliss method. However, the combination of GCV (m = 1) and artemisinin-derived dimers (m > 3) was synergistic by the Bliss method but mildly antagonistic by the isobologram method.
Most studies have focused on the discovery of synergy between compounds, but very few have identified the antagonistic effects of compounds (22). In the case of antagonism, the combined effect of two agents is less than what would be expected for the additive activities of the individual agents. Combination studies of cyclopropavir and GCV using real-time PCR showed antagonistic activity, consistent with the inhibition of the UL97 kinase by cyclopropavir (43). Similarly, maribavir antagonized the activity of GCV (20). Our results showed that the MEK1/2 inhibitor U0126 and the multikinase inhibitor sunitinib antagonize the anti-CMV activities of the dimer diphenyl phosphate (MW, 838). This antagonistic effect was also observed when dimer primary alcohol 606 was used in combination with U0126, suggesting that the antagonistic effect of U0126 is specific to the class of artemisinin-derived dimers. Antagonistic activity was proven by several antiviral assays, including late pp28-luciferase expression, virus DNA yield, and plaque reduction assays. These results suggest that inhibition of MEK activity may interfere with the anti-CMV activities of artemisinin-derived dimers but not those of the monomeric derivatives.
The antagonistic effect of U0126 on dimer 838 was achieved at low concentrations of U0126 that inhibit MEK1/2 activity and was not a result of chemical interaction, as proven by addition of the compounds at different time points and the expression level of pMEK and pERK. The antagonism of U0126 and dimer 838 may assist in identifying the specific cellular target of artemisinins, since the MEK1/2 inhibitor U0126 could abrogate the activities of dimers, suggesting that U0126 may interfere with the binding of the artemisinin dimer to its target.
Characterization of synergistic drug interactions is an active area of research which could result in the clinical use of combination chemotherapies for cancer and infectious diseases. The identification of new anti-CMV agents and the ability to screen for drug combinations may allow the development of new strategies for the treatment of CMV infection. Although our study was performed in vitro, the concentrations of several compounds used (GCV, AS, digitoxin, and U0126) correlate with concentrations achievable in serum. A combination of GCV with digitoxin (a cardiac glycoside used in Europe) may have clinical implications. While our data define a Bliss coefficient of >1 as synergy, the clinical significance of a specific drug combination must be determined in vivo. A mild in vitro synergy (such as that observed for GCV plus FOS) has shown beneficial effects in vivo. Combination therapy for CMV may reduce the frequency of resistant viral mutants and achieve efficacy with fewer side effects and enhanced potency by either additive or synergistic effects. Although the findings of in vitro studies may not always reflect the more complex in vivo activities of antimicrobial agents, they may be helpful in pointing to the mechanisms of action of antiviral compounds and in the future design of drug combinations for use in animal studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant 1R01AI093701, March of Dimes grant 6-FY11-268 (to R.A.-B.), and NIH grant AI 34885 (to G.H.P.).
Footnotes
Published ahead of print 25 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01972-13.
REFERENCES
- 1.Griffiths PD, Clark DA, Emery VC. 2000. Betaherpesviruses in transplant recipients. J. Antimicrob. Chemother. 45(Suppl T3):29–34. 10.1093/jac/45.suppl_4.29 [DOI] [PubMed] [Google Scholar]
- 2.Kovacs A, Schluchter M, Easley K, Demmler G, Shearer W, La RP, Pitt J, Cooper E, Goldfarb J, Hodes D, Kattan M, McIntosh K. 1999. Cytomegalovirus infection and HIV-1 disease progression in infants born to HIV-1-infected women. Pediatric Pulmonary and Cardiovascular Complications of Vertically Transmitted HIV Infection Study Group. N. Engl. J. Med. 341:77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demmler-Harrison GJ. 2009. Congenital cytomegalovirus: public health action towards awareness, prevention, and treatment. J. Clin. Virol. 46(Suppl 4):S1–S5. 10.1016/j.jcv.2009.10.007 [DOI] [PubMed] [Google Scholar]
- 4.Jabs DA, Martin BK, Forman MS, Dunn JP, Davis JL, Weinberg DV, Biron KK, Baldanti F, Hu H. 2001. Longitudinal observations on mutations conferring ganciclovir resistance in patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis: the Cytomegalovirus and Viral Resistance Study Group report number 8. Am. J. Ophthalmol. 132:700–710. 10.1016/S0002-9394(01)01161-8 [DOI] [PubMed] [Google Scholar]
- 5.Jabs DA, Martin BK, Forman MS. 2010. Mortality associated with resistant cytomegalovirus among patients with cytomegalovirus retinitis and AIDS. Ophthalmology 117:128–132. 10.1016/j.ophtha.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lurain NS, Chou S. 2010. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 23:689–712. 10.1128/CMR.00009-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou SW. 2001. Cytomegalovirus drug resistance and clinical implications. Transpl. Infect. Dis. 3(Suppl 2):20–24. 10.1034/j.1399-3062.2001.00004.x [DOI] [PubMed] [Google Scholar]
- 8.Chou S. 1999. Antiviral drug resistance in human cytomegalovirus. Transpl. Infect. Dis. 1:105–114. 10.1034/j.1399-3062.1999.010204.x [DOI] [PubMed] [Google Scholar]
- 9.Krosky PM, Baek MC, Coen DM. 2003. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 77:905–914. 10.1128/JVI.77.2.905-914.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams SL, Hartline CB, Kushner NL, Harden EA, Bidanset DJ, Drach JC, Townsend LB, Underwood MR, Biron KK, Kern ER. 2003. In vitro activities of benzimidazole d- and l-ribonucleosides against herpesviruses. Antimicrob. Agents Chemother. 47:2186–2192. 10.1128/AAC.47.7.2186-2192.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winston DJ, Saliba F, Blumberg E, Abouljoud M, Garcia-Diaz JB, Goss JA, Clough L, Avery R, Limaye AP, Ericzon BG, Navasa M, Troisi RI, Chen H, Villano SA, Uknis ME. 2012. Efficacy and safety of maribavir dosed at 100 mg orally twice daily for the prevention of cytomegalovirus disease in liver transplant recipients: a randomized, double-blind, multicenter controlled trial. Am. J. Transplant. 12:3021–3030. 10.1111/j.1600-6143.2012.04231.x [DOI] [PubMed] [Google Scholar]
- 12.Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, Ruebsamen-Schaeff H, Zimmermann H. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 54:1290–1297. 10.1128/AAC.01596-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaul DR, Stoelben S, Cober E, Ojo T, Sandusky E, Lischka P, Zimmermann H, Rubsamen-Schaeff H. 2011. First report of successful treatment of multidrug-resistant cytomegalovirus disease with the novel anti-CMV compound AIC246. Am. J. Transplant. 11:1079–1084. 10.1111/j.1600-6143.2011.03530.x [DOI] [PubMed] [Google Scholar]
- 14.Evers DL, Wang X, Huong SM, Andreoni KA, Huang ES. 2005. Inhibition of human cytomegalovirus signaling and replication by the immunosuppressant FK778. Antiviral Res. 65:1–12. 10.1016/j.antiviral.2004.03.007 [DOI] [PubMed] [Google Scholar]
- 15.Walton RC, Whitcup SM, Mueller BU, Lewis LL, Pizzo PA, Nussenblatt RB. 1995. Combined intravenous ganciclovir and foscarnet for children with recurrent cytomegalovirus retinitis. Ophthalmology 102:1865–1870. 10.1016/S0161-6420(95)30782-8 [DOI] [PubMed] [Google Scholar]
- 16.Tunkel AR, Glaser CA, Bloch KC, Sejvar JJ, Marra CM, Roos KL, Hartman BJ, Kaplan SL, Scheld WM, Whitley RJ. 2008. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin. Infect. Dis. 47:303–327. 10.1086/589747 [DOI] [PubMed] [Google Scholar]
- 17.Manischewitz JF, Quinnan GV, Jr, Lane HC, Wittek AE. 1990. Synergistic effect of ganciclovir and foscarnet on cytomegalovirus replication in vitro. Antimicrob. Agents Chemother. 34:373–375. 10.1128/AAC.34.2.373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27–55. 10.1016/0065-2571(84)90007-4 [DOI] [PubMed] [Google Scholar]
- 19.Manion DJ, Vibhagool A, Chou TC, Kaplan J, Caliendo A, Hirsch MS. 1996. Susceptibility of human cytomegalovirus to two-drug combinations in vitro. Antivir. Ther. 1:237–245 [PubMed] [Google Scholar]
- 20.Chou S, Marousek GI. 2006. Maribavir antagonizes the antiviral action of ganciclovir on human cytomegalovirus. Antimicrob. Agents Chemother. 50:3470–3472. 10.1128/AAC.00577-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prichard MN, Shipman C., Jr 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–205. 10.1016/0166-3542(90)90001-N [DOI] [PubMed] [Google Scholar]
- 22.Evers DL, Komazin G, Shin D, Hwang DD, Townsend LB, Drach JC. 2002. Interactions among antiviral drugs acting late in the replication cycle of human cytomegalovirus. Antiviral Res. 56:61–72. 10.1016/S0166-3542(02)00094-3 [DOI] [PubMed] [Google Scholar]
- 23.He R, Park K, Cai H, Kapoor A, Forman M, Mott B, Posner GH, Arav-Boger R. 2012. Artemisinin-derived dimer diphenyl phosphate is an irreversible inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 56:3508–3515. 10.1128/AAC.00519-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapoor A, Cai H, Forman M, He R, Shamay M, Arav-Boger R. 2012. Human cytomegalovirus inhibition by cardiac glycosides: evidence for involvement of the HERG gene. Antimicrob. Agents Chemother. 56:4891–4899. 10.1128/AAC.00898-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Efferth T, Marschall M, Wang X, Huong SM, Hauber I, Olbrich A, Kronschnabl M, Stamminger T, Huang ES. 2002. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 80:233–242. 10.1007/s00109-001-0300-8 [DOI] [PubMed] [Google Scholar]
- 26.Efferth T, Romero MR, Wolf DG, Stamminger T, Marin JJ, Marschall M. 2008. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 47:804–811. 10.1086/591195 [DOI] [PubMed] [Google Scholar]
- 27.Arav-Boger R, He R, Chiou CJ, Liu J, Woodard L, Rosenthal A, Jones-Brando L, Forman M, Posner GH. 2010. Artemisinin-derived dimers have greatly improved anti-cytomegalovirus activity compared to artemisinin monomers. PLoS One 5:e10370. 10.1371/journal.pone.0010370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartley C, Hartley M, Pardoe I, Knight A. 2006. Ionic contra-viral therapy (ICVT); a new approach to the treatment of DNA virus infections. Arch. Virol. 151:2495–2501. 10.1007/s00705-006-0824-x [DOI] [PubMed] [Google Scholar]
- 29.Johnson RA, Ma XL, Yurochko AD, Huang ES. 2001. The role of MKK1/2 kinase activity in human cytomegalovirus infection. J. Gen. Virol. 82:493–497 [DOI] [PubMed] [Google Scholar]
- 30.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. 10.1038/nature07209 [DOI] [PubMed] [Google Scholar]
- 31.He R, Sandford G, Hayward GS, Burns WH, Posner GH, Forman M, Arav-Boger R. 2011. Recombinant luciferase-expressing human cytomegalovirus (CMV) for evaluation of CMV inhibitors. Virol. J. 8:40. 10.1186/1743-422X-8-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka Y, Kanda Y, Kami M, Mori S, Hamaki T, Kusumi E, Miyakoshi S, Nannya Y, Chiba S, Arai Y, Mitani K, Hirai H, Mutou Y. 2002. Monitoring cytomegalovirus infection by antigenemia assay and two distinct plasma real-time PCR methods after hematopoietic stem cell transplantation. Bone Marrow Transplant. 30:315–319. 10.1038/sj.bmt.1703661 [DOI] [PubMed] [Google Scholar]
- 33.Hill AV. 1910. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curve. J. Physiol. 40:iv–vii [Google Scholar]
- 34.Loewe S. 1953. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 3:285–290 [PubMed] [Google Scholar]
- 35.Bliss CI. 1939. The toxicity of poisons applied jointly. Ann. Appl. Biol. 26:585–615. 10.1111/j.1744-7348.1939.tb06990.x [DOI] [Google Scholar]
- 36.Jilek BL, Zarr M, Sampah ME, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF. 2012. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 18:446–451. 10.1038/nm.2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He R, Mott BT, Rosenthal AS, Genna DT, Posner GH, Arav-Boger R. 2011. An artemisinin-derived dimer has highly potent anti-cytomegalovirus (CMV) and anti-cancer activities. PLoS One 6:e24334. 10.1371/journal.pone.0024334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konkimalla VB, McCubrey JA, Efferth T. 2009. The role of downstream signaling pathways of the epidermal growth factor receptor for artesunate's activity in cancer cells. Curr. Cancer Drug Targets 9:72–80. 10.2174/156800909787314020 [DOI] [PubMed] [Google Scholar]
- 39.Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, Siliciano RF. 2008. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14:762–766. 10.1038/nm1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sampah ME, Shen L, Jilek BL, Siliciano RF. 2011. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A. 108:7613–7618. 10.1073/pnas.1018360108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. 2006. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2:458–466. 10.1038/nchembio817 [DOI] [PubMed] [Google Scholar]
- 42.Greco WR, Bravo G, Parsons JC. 1995. The search for synergy: a critical review from a response surface perspective. Pharmacol. Rev. 47:331–385 [PubMed] [Google Scholar]
- 43.James SH, Hartline CB, Harden EA, Driebe EM, Schupp JM, Engelthaler DM, Keim PS, Bowlin TL, Kern ER, Prichard MN. 2011. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob. Agents Chemother. 55:4682–4691. 10.1128/AAC.00571-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.