

Abstract
The cysteine protease cruzipain is essential for the viability, infectivity, and virulence of Trypanosoma cruzi, the causative agent of Chagas disease. Thus, inhibitors of cruzipain are considered promising anti-T. cruzi chemotherapeutic agents. Reversible cruzipain inhibitors containing a nitrile “warhead” were prepared and demonstrated 50% inhibitory concentrations (IC50s) as potent as 1 nM in baculovirus-generated cruzipain enzyme assays. In epimastigote and intracellular amastigote in vitro assays, the most potent compounds demonstrated antiparasitic behavior in the 5 to 10 μM IC50 range; however, trypomastigote production from the amastigote form was ∼90 to 95% inhibited at 2 μM. Two key compounds, Cz007 and Cz008, with IC50s of 1.1 and 1.8 nM, respectively, against the recombinant enzyme were tested in a murine model of acute T. cruzi infection, with oral dosing in chow for 28 days at doses from 3 to 50 mg/kg of body weight. At 3 mg/kg of Cz007 and 3 mg/kg of Cz008, the blood parasitemia areas under the concentration-time curves were 16% and 25% of the untreated group, respectively. At sacrifice, 24 days after immunosuppression with cyclophosphamide, parasite presence in blood, heart, and esophagus was evaluated. Based on negative quantitative PCR results in all three tissues, cure rates in surviving animals were 90% for Cz007 at 3 mg/kg, 78% for Cz008 at 3 mg/kg, and 71% for benznidazole, the control compound, at 50 mg/kg.
INTRODUCTION
Trypanosoma cruzi is the causative agent of Chagas disease, which is endemic from the southwest United States to Patagonia. Historically, Chagas disease was a disease of poor and rural populations in Central and South America, where it was transmitted primarily by triatomine insect vectors (e.g., Triatoma infestans, Rhodnius prolixus) (1). Although vector-based transmission has been controlled in several regions through massive public health programs (2), T. cruzi can also be transmitted by transfusion, transplantation, and contaminated foods and vertically from mother to child. In many parts of the Americas where vector-driven transmission has been controlled, transfusion has become the major route of transmission (3). Furthermore, mother-to-child transmission can maintain Chagas disease in populations long after vectorial transmission has ceased. Although congenital transmission is thought to occur in <5% of children born of positive mothers, family clustering and multigenerational transmission have been reported (4). Furthermore, due to international migrations, Chagas disease is now considered an emerging disease in several nonendemic areas in the world, including the United States, Europe, Japan, and Australia, where nonvectorial transmission of the parasite is becoming a serious public health threat (5).
Chemotherapy options are limited, with only two trypanocidal drugs available: nifurtimox and benznidazole. However, only benznidazole is in common use, due to the risk of serious central nervous system and peripheral neurotoxicity with nifurtimox (6). Although some authorities feel that the risks of benznidazole-associated side effects have been overemphasized (7), this drug has been reported to cause significant neurotoxicity (seizures), peripheral neurotoxicity, serious dermatologic conditions, leukopenia, and thrombocytopenia (7). Treatment in acute (<6 months since infection) and congenital infection is generally effective (>95% “cure”) (6). The efficacy rate is slightly lower in “early” infection (i.e., 76% cure in children >6 months but <14 years of age), but there is no adequate treatment for Chagas disease once the patient has entered the indeterminate or chronic phase of infection (<6% cure) (8, 9). Thus, once the disease has entered the chronic phase, the benefit of benznidazole treatment on parasite load and clinical evolution is controversial (10), yet the majority of people currently infected with T. cruzi fall into this last category. Chagas disease is receiving more attention in drug discovery and drug development, and promising new therapies for Chagas disease have recently entered clinical trials, with several others in the pipeline (10, 11, 12, 13).
The cysteine protease cruzipain, a member of the papain family, is essential for the viability and virulence of T. cruzi, and thus inhibitors of cruzipain are considered promising agents for Chagas disease therapies (14, 15, 16, 17, 18, 19). Studies with irreversible cruzipain inhibitors have shown the biochemical impact of inhibiting cruzipain on T. cruzi. In particular, prototype K777 (also known as K11777) has been efficacious in preclinical models of T. cruzi infection, including immunocompetant and immunodeficient mice (20, 21) and dogs (22). An α-ketone irreversible cruzipain inhibitor has also recently shown efficacy in a murine model (23). These studies show the potential of cruzipain inhibitors as anti-T. cruzi therapies.
Cruzipain is closely related to the human cysteine protease family of cathepsins. Recently, the discovery of reversible cathepsin K inhibitors containing a nitrile “warhead” have been reported (24). One of these compounds, MK-0822 (odanacatib) Fig. 1), is currently in phase III development for postmenopausal osteoporosis (25–27). The structural similarities between cruzipain and cathepsin K suggested that reversible cruzipain inhibitors could be successful anti-T. cruzi agents (28), and a drug discovery effort was mounted to identify nitrile-containing cruzipain inhibitors (29). This paper describes the in vitro and in vivo efficacy data for the resulting compounds and demonstrates that these reversible cruzipain inhibitors are orally bioavailable, well tolerated, and highly efficacious in a preclinical model of acute Chagas disease.
FIG 1.

Structures of K777, odanacatib, and the nitrile-containing cruzipain inhibitors used in this study (20, 29, 30).
MATERIALS AND METHODS
General.
Cruzipain inhibitors were synthesized at the Merck Frosst Canada & Co. laboratories (Kirkland, Quebec, Canada) according to published procedures (20, 29, 30). Reagents were obtained from standard commercial sources unless specified below. The Brazil strain of T. cruzi was used (31) for all in vitro and in vivo studies. All animal studies were reviewed and approved by the Merck Frosst and McGill University IACUC committees.
Enzyme expression for screening.
A recombinant cruzipain enzyme preparation was generated in a baculovirus system by using a construct from amino acids (aa) −104 to 212 (construct 334), which is identical to the full-length cruzipain-1 sequence described by Eakin et al. and is herein referred to as cruzipain-ΔC (32). It comprises the prodomain but not the C terminus, and a 6-aa His tag was added. The supernatant produced after 72 h demonstrated enough activity to be used without further purification.
Cruzipain enzyme activity.
Approximately 900 proprietary cysteine protease inhibitors (20, 29, 30) were screened by a 3-point titration at 0.06, 0.3, and 1.5 μM against cruzipain-ΔC. Definitive 50% inhibitory concentrations (IC50s) for selected compounds were determined by using 11-point serial dilutions (1/3) at final concentrations of 0.0085 to 500 μM, or as appropriate. Two microliters of 50× dimethyl sulfoxide (DMSO) stock solutions were added to 100 μl of cruzipain-ΔC (500 ng/ml) in the assay buffer solution (50 mM NaOAc [pH 5.5], 5 mM dithiothreitol, and 10% [vol/vol] DMSO). The assay solutions were mixed for 5 to 10 s on a shaker plate and incubated for 15 min at room temperature. Ten microliters of 20 μM Z-Phe-Arg-AMC buffer was added to the assay solutions. Hydrolysis of the coumarin leaving group (AMC) was followed by spectrofluorometry (excitation wavelength, 350 nm; emission wavelength, 460 nm) for 10 min. Percent inhibitions were calculated by fitting experimental values to a standard mathematical model for dose-response curves.
Cathepsin enzyme assays.
Assays for human cathepsins B, F, K, L, S, and V and mouse cathepsins B and S were performed as described previously (30, 33, 34).
In vitro effects of cruzipain inhibitors on epimastigotes.
A [3H]thymidine incorporation assay was used to assess compound activity, and it was performed in two ways; (i) parasites were treated with compounds for 48 h, the compounds were removed by washing, and the [3H]thymidine assay was performed for 48 h; (ii) parasites were treated with the compound for 48 h, the compound was removed by washing, fresh medium wash was added, the parasites were incubated for an additional 4 days (recovery experiment), and then the [3H]thymidine assay was performed for 48 h. Specifically, the epimastigote form of T. cruzi (Brazil strain [31]) was grown in a 25-ml flask in 10 ml liver infusion tryptose (LIT) broth at 28°C with agitation at 80 rpm (orbital shaker; Forma Scientific, CA). The LIT medium contained 20 g LIT broth (Difco Laboratories, Detroit, MI), 5 g tryptose (Difco), 4 g NaCl, 0.4 g KCl, 8 g Na2HPO4, and 2 g of dextrose (Gibco, Burlington, ON, Canada) in 1 liter of distilled water (pH 7.4) and was filter sterilized. Heat-inactivated 10% fetal bovine serum (FBS; Wisent, Inc., St. Bruno, Quebec, Canada), 1% penicillin-streptomycin (Wisent, Inc.), and 25 mg L21 hemin (Sigma, St. Louis, MO) were dissolved in 2.5 ml of water and 2.5 ml (50/50 [vol/vol]) of triethanolamine (Sigma, Oakville, ON, Canada) and added to the LIT medium. Test compounds at final concentrations of 0.1, 1, 10, and 100 μM were incubated with 107 epimastigotes/ml in 24-well flat-bottom plates (Costar, Tewksbury, MA), and the final volume was 1 ml/well. The plates were incubated in a shaker at 80 rpm (orbital shaker; Forma Scientific) for 48 h at 28°C. Parasites were harvested in Eppendorf tubes and washed three times with 1 M phosphate-buffered saline (PBS; pH 7.4) by centrifugation at 850 × g for 15 min at 4°C to remove the compounds. Epimastigote replication was measured in a [3H]thymidine incorporation assay, in which epimastigotes were seeded in duplicate into 96-well plates with LIT medium (final volume, 200 μl), and 1 μCi per well of [3H]thymidine was added. Parasites were incubated at 28°C for 48 h and were then subjected to one freeze-thaw cycle before cellular DNA was harvested onto glass fiber filters. [3H]thymidine incorporation was measured by using a beta counter (Microbeta, Wallac, Finland). The results are expressed as mean counts per minute (cpm) of duplicate samples (35). For the 4-day recovery experiment, after washing to remove the compound, the parasites were incubated for 4 days in medium, as described above, prior to initiating the [3H]thymidine incorporation assay.
Antiamastigote activity.
Cytotoxicity of compounds was tested in U937 cells (data not shown) and J774 cells (see the description of our preliminary work with these cells in the supplemental material), and the latter were selected for the antiamastigote activity assay. Trypomastigotes were first prepared in a 10-day procedure involving infection of Vero cells with epimastigotes at a 1:10 ratio on day 3, as described by Berrizbeitia et al. (36). On day 10, trypomastigotes were harvested and used to infect the J774 cells, which were seeded in 24-well microplates with round-bottom coverslips. Trypomastigotes of the Brazil strain (31) were added at a parasite-to-cell ratio of 10:1 for 24 h at 37°C. Extracellular parasites were removed by 3 washes with PBS, pH 7.2. The test compounds were diluted in fresh medium at 2, 5, 10, and 20 μM and refreshed every 24 h for 72 h. The experiment was done in triplicate. Coverslips were fixed and stained with May Grunwald Giemsa stain (Sigma). The average number of amastigotes per macrophage in 100 macrophages was determined by light microscopy. For Cz005, some cytotoxicity was observed at 10 and 100 μM, and fewer than 100 macrophages were counted. The percent antiamastigote activity was determined as follows: [1 − (number of amastigotes/100 cells in treated group)/(number of amastigotes/100 cells in control group)] × 100.
Antitrypomastigote activity.
Viable trypomastigotes in the medium were harvested at 72 h from the antiamastigote activity assay mixture described above. Trypomastigotes were resuspended in trypan blue solution (0.4%; Sigma), and live parasites were counted in a Neubauer chamber.
Pharmacokinetic (PK) studies of cruzipain inhibitors.
Male CD-1 mice weighing approximately 25 g were dosed orally by gavage with Cz007 (20 mg/kg) or Cz005 (10 mg/kg) as a suspension in 0.5% methocellulose (10-ml/kg dose volume), respectively. Blood samples were collected from the tail vein at 15 and 30 min and 1, 2, 4, 6, 8, and 24 h and placed into 0.1 M trisodium citrate buffer for analysis according to standard procedures (37). Mice were dosed intravenously with Cz007 (5 mg/kg) or Cz005 (2 mg/kg) in 60% polyethylene glycol (5-ml/kg dose volume) by direct injection into the jugular vein of conscious mice. Blood samples were taken at 5 and 30 min and 1, 2, 4, 6, 8, and 24 h and treated as described above.
Murine tolerability and tissue distribution studies for Cz007 and Cz008.
Tolerability studies were conducted using male CD-1 mice weighing ∼35 g that were individually housed. Mice were exposed to a 12-h light/dark cycle with lights on at 6 a.m. and were preconditioned with blank chow for 10 days. Cz007 and Cz008 were then provided in chow formulation (see below) for 5 days in preweighed food jars (product number 910018; Diets, Inc., Bethlehem, PA) that were changed daily, with weighing, to determine food consumption. Mice were examined and weighed daily. Ten microliters of blood for PK was collected by tail bleed and placed into 0.1 M trisodium citrate buffer for analysis (37). Blood samples collected at sacrifice were also assessed for clinical chemistry markers (aspartate transaminase, alanine aminotransferase, lactate dehydrogenase, creatinine kinase, alkaline phosphatase, and total bilirubin). Heart, esophagus, and liver were harvested at sacrifice for determination of Cz007 and Cz008 concentrations.
Chow preparation with Cz007 and Cz008.
Test articles were prepared for dosing as a mixture in powdered chow (2018M; Harland Teklad). For both Cz007 and Cz008, chow prepared at 22, 90, and 425 ppm was considered to provide doses of 3, 10, and 50 mg/kg, since the consumption of 5 g of food per 35-g mouse per day was established by weighing the food jars during the 5-day tolerability study. After milling to a particle size of <10 μM in a Fritsch mortar grinder pulverisette 2 (Fritsch GmbH, Germany), the total amounts of drug needed for the studies were systematically mixed with chow by serial dilution as detailed in the supplemental material.
Benznidazole dosing.
Benznidazole was provided in water containing 0.0075% Tween 20 at a concentration of 0.25 mg/ml. Given a water consumption rate of 5 ml/day per 25-g mouse, this corresponds to ∼50 mg/kg. Water consumption would be correspondingly higher for a larger mouse.
Bioanalysis.
Standard curves for each tissue type were prepared with corresponding blank tissue homogenates. All compounds were analyzed by quantitative liquid chromatography-mass spectrometry (LC-MS) by using a SCIEX 2000 apparatus (Toronto, Canada) with positive ion electron spray ionization. LC separation was achieved by using a Luna C18 3-μm (50- by 2-mm) LC column (Phenomenex) with a mobile-phase gradient consisting of acetonitrile and 0.1% formic acid. Analysis of Cz007 was done using a 3-min gradient of acetonitrile from 35 to 95%, with MS ionization at +30 eV. For Cz005 and Cz008, a 2-min gradient of acetonitrile from 10 to 95% acetonitrile was used with ionization at +40 eV. Benznidazole was analyzed on the same system with a gradient of 20 to 95% acetonitrile and ionization of +30 eV.
Parasite culture for inoculation.
Epimastigotes were collected during the logarithmic growth phase, as previously described, and washed three times in 1 M PBS (pH 7.4) by centrifugation at 850 × g for 15 min at 4°C. The trypomastigotes for infection were then prepared as previously described (38) by infecting Vero cell monolayers (ATCC CCL-81) with the epimastigotes, harvesting the supernatant after 7 days, purifying the trypomastigotes in a column, and counting them in a Neubauer chamber.
In vivo murine efficacy studies.
Two separate efficacy studies were conducted, and the experimental designs of both are summarized in Table 1. Mice were housed 5 per cage and were inoculated intraperitoneally with 104 trypomastigotes of the Brazil strain (31) of T. cruzi. Treatment was started 2 days postinfection. For parasitemia assessments, 10-μl blood samples were taken from the tail vein, and parasitemia was determined by manual counting using a Neubauer chamber. The limit of detection was 1 parasite in 20 microscopic fields, which corresponds to 2.5 × 105 parasites per ml of blood (39). Mice were also evaluated for weight and body temperature 3 times a week throughout the studies. Body temperatures were measured using implantable programmable temperature transponders (Bio Medic Data Systems, Inc., Seaford, DE).
TABLE 1.
Experimental design of in vivo efficacy studies
| Compound or control | Dose (mg/kg) | Infected? | Expt 1 (no. of mice) |
Expt 2 (no. of mice) | |
|---|---|---|---|---|---|
| Endpoint on day 28 | Endpoint on day 90 | Endpoint on day 113 | |||
| Cz007 | 3 | Yes | 10 | ||
| 10 | Yes | 10 | |||
| 50 | Yes | 10 | |||
| 50 | 5 | ||||
| Cz008 | 3 | Yes | 10 | ||
| 10 | Yes | 10 | 10 | 10 | |
| 50 | Yes | 10 | 10 | ||
| 50 | 10 | 10 | |||
| Benznidazole | 50 | Yes | 10 | 10 | 10 |
| Infected, untreated | Yes | 10 | 10 | 10 | |
| Mock infected, untreated | 5 | 5 | 5 | ||
In the first study (experiment 1), eight groups of 10 male CD-1 mice, 6 to 8 weeks old and weighing 37 g, were used. Only Cz008 was evaluated in this first experiment, at doses of 10 and 50 mg/kg, from day 2 to day 28 in a food formulation, while benznidazole was provided in drinking water, as described above. Control mice consisted of untreated infected mice, untreated mock-infected mice, and Cz008 (50 mg/kg)-treated mock-infected groups. One cohort of each treatment group was sacrificed at day 28 for an interim analysis, and the remaining animals were sacrificed at day 90. Blood levels of benznidazole and Cz008 were determined at day 28, and qualitative PCR was determined in blood at day 90.
In the second murine efficacy study (experiment 2), the initial 30 days were identical with respect to inoculation and monitoring. Mice were treated for 28 days with Cz008 at 3 or 10 mg/kg, Cz007 at 3, 10, or 50 mg/kg, both dosed in chow, or benznidazole at 50 mg/kg, provided in drinking water. Control mice consisted of untreated infected mice, untreated mock-infected mice, and Cz007 (50 mg/kg)-treated mock-infected groups. At day 30, blood samples were taken by tail bleed (37) for compound level determinations. From day 30 to the end of the study, mice were treatment-free, receiving regular chow and drinking water. On day 90, immunosuppression was induced by 200 mg/kg cyclophosphamide (Sigma) via intraperitoneal injection in 0.9% saline (40). Body weight, temperature, and parasitemia were determined daily until sacrifice of all surviving animals by CO2 asphyxiation at day 113. At sacrifice, blood, heart, and esophagus were collected for real-time quantitative PCR (qPCR) and were frozen at −20°C until analysis.
Qualitative PCR.
From each animal, 200 μl of blood and 10 mg of tissues were collected for DNA purification by use of a Qiagen minikit (Germantown, MD) following the manufacturer's instructions. PCR primers used were TCZ-3, 5′-TGCACTCGGCTGATCGTTT-3′, and TCZ-4, 5′-ATTCCTCCAAGCAGCGGATA-3′ (41). Each 50-μl reaction mixture contained 2 U of Taq DNA polymerase (Promega, Madison, WI), 200 μM deoxynucleoside triphosphates (Promega, Madison, WI), 2 mM MgCl2, 1 μM primers, and 20 μl of DNA sample.
Quantitative PCR.
DNA was extracted from blood, heart, and esophagus by using a QIAamp DNA minikit from Qiagen. The primers used were TCZ-3, 5′-TGCACTCGGCTGATCGTTT-3′, and TCZ-4, 5′-ATTCCTCCAAGCAGCGGATA-3′ (41). Hybridization probes were designed and purchased from TIB Molbiol, Berlin, Germany (T. cruzi LC640, 5′-LC640-TGATGCAGCAGCCGCTCGAA-PH, and T. cruzi FL, 5′-CGAAACAAAAATTTGGACCACAAGT-FL). Real-time PCR and subsequent melting curve analyses were performed on a Light Cycler 1.5 instrument (Roche Diagnostics, Germany). The final volume for the master mix was 20 μl, and it contained 1 μM each primer, 0.4 μM probes, and 15 μl 1× Hybprobe (Light Cycler; Roche, Germany). The limit of detection was 5 parasites/ml.
Data analysis.
IC50s for enzyme potency were calculated using standard 4-point-fit parameters (SoftmaxPro; Molecular Devices). Pharmacokinetics data were analyzed by using an in-house analysis software with standard formulae for bioavailability, area under the concentration-time curve (AUC), half-life, and volume of distribution. Parasite experimental data were analyzed, and statistics were determined using unpaired t tests as calculated using GraphPad Prism version 5.
RESULTS
Chemistry and enzyme assays.
As described by Beaulieu et al. (29), the 900-compound library of nitrile-containing inhibitors demonstrated a distinct structure-activity relationship (SAR) against cruzipain-ΔC, and several analogs were found to have low, nanomolar potency. Based on this SAR, new compounds were specifically prepared to optimize for potency and pharmacokinetic properties. From these compounds, a small panel comprising a weak (Cz003; 3.7 μM), a moderate (Cz001; 170 nM), and two potent inhibitors (Cz002 and Cz005; 1 to 2 nM) were chosen for in vitro testing against the parasite (Fig. 1 and Table 2). Of these two potent compounds, Cz002 was neutral and Cz005 was basic. All compounds were also assessed for their inhibition of key off-target human cathepsins, and in most cases, the compounds showed little selectivity for cruzipain over the human cathepsins. The irreversible and basic inhibitor K777, which was used as a reference compound, had no selectivity over human cathepsins either. Inhibitors Cz007 and Cz008, which were used for murine in vivo efficacy studies, had IC50s of 1 to 2 nM against cruzipain-ΔC. Cz007 did not demonstrate any appreciable selectivity versus human cathepsin enzymes. Cz008 had modest selectivity over cathepsins B, F, L, and S but was a potent inhibitor of cathepsin K (Table 2).
TABLE 2.
Activities of compounds against cruzipain-ΔC and human cathepsin enzymes
| Compound | Mean ± SDa IC50 (n) |
Reference | ||||||
|---|---|---|---|---|---|---|---|---|
| Cruzipain-ΔC | CatB | CatF | CatK | CatL | CatS | CatV | ||
| Cz003 (weak, neutral) | 3,700 (2) | —b | — | 1 ± 0.3 (3) | 5,990 ± 1,380 (3) | — | — | |
| Cz001 (moderate, neutral) | 170 (2) | — | — | <0.3 (2) | 2,253 (2) | — | — | |
| Cz002 (potent, neutral) | 1.2 (2) | 692 ± 177 (3) | 8 ± 2 (3) | 0.7 ± 0.7 (2) | 104 ± 52 (4) | 40 ± 12 (4) | — | |
| Cz005 (potent, basic) | 2.4 (2) | 12 ± 4 (4) | — | <0.2 (7) | 62 ± 25 (5) | 16 ± 2 (4) | — | |
| K777 (irreversible) | 3.5 (2) | 9 (2) | 3 (2) | 1.8 (2) | <0.2 (3) | <0.2 (2) | — | |
| Cz007 (potent, neutral) | 1.1 (2) | 9 ± 2 (9) | 4 ± 1 (8) | <0.5 (1) | 49 ± 19 (9) | 1.6 ± 0.2 (8) | 2 (1) | 29 |
| Cz008c (potent, basic) | 1.8 (2) | 584 ± 304 (4) | 53 ± 21 (3) | <0.005d (1) | 109 ± 11 (4) | 414 ± 141 (4) | — | 30 |
| Odanacatib (cathepsin K) | — | 1,034 | — | 0.2 | 2,995 | 60 | — | 27 |
Each entry is the mean concentration ± the standard deviation (with n in parentheses), unless otherwise indicated. Values may be slightly different than those reported by Beaulieu et al. (29) and Black et al. (30) due to additional testing (higher n values).
—, not tested.
Cz008 is the single diastereomer of Cz005 with the trifluoromethyl side chain stereochemistry resolved.
For a description of the low protein assay, see Black et al. (30).
In vitro antiparasite assays.
In the epimastigote assay using [3H]thymidine incorporation as an indicator of anti-T. cruzi activity, the rank order of compound potency was the same as that observed versus the cruzipain-ΔC enzyme (Tables 2 and 3 and Fig. 2A). However, there was a systematic ∼1,000-fold shift between the enzyme potency and the whole-cell parasitic assay results (Table 3) such that the potent neutral and basic inhibitors Cz002 and Cz005 (1 to 2 nM), were active against the parasites, with IC50s of 1 to 10 μM. The less potent compounds, Cz001 and Cz003, did not show substantial activity against whole cells.
TABLE 3.
Potencies of compounds in the cruzipain-ΔC epimastigote, amastigote, and trypomastigote assaya
| Compound | IC50 (μM) |
||||
|---|---|---|---|---|---|
| Cruzipain-ΔC | Epimastigote 48 h | Epimastigote 48 h + 4-day recovery | Amastigote 72 h | Trypomastigote 72 h | |
| Cz003 | 3.7 | >100 | >100 | >20 | ∼10 |
| Cz001 | 0.17 | >100 | ∼100 | >20 | ∼10 |
| Cz002 | 0.0011 | 1–10 | 1–10 | 5–10 | <2 |
| Cz005b | 0.0018 | 1–10 | 1–10 | 5–10 | <2 |
| K777 | 0.0035 | 10–100 | ∼10 | ∼10 | <2 |
FIG 2.
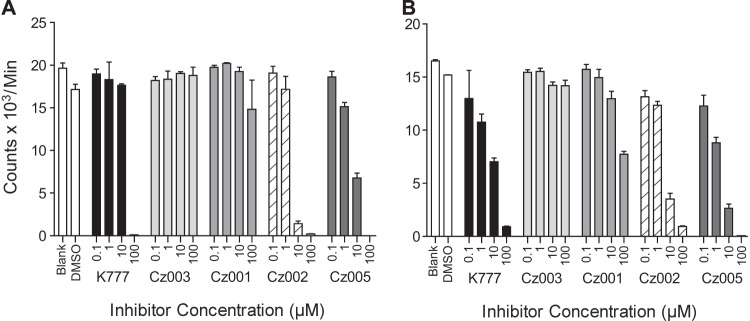
Activities of key compounds in epimastigote assays as measured by [3H]thymidine incorporation. (A) Epimastigotes were treated with test compounds for 48 h. After removal of test compound with 3 washes of PBS, the parasites were incubated with [3H]thymidine for 48 h. (B) After the treatment, the test compounds were removed by 3 PBS washes, and a 4-day recovery time was allowed before measurement of thymidine incorporation. Error bars represent standard deviations, and concentrations of compound are in micromolar units.
A 4-day recovery experiment was designed to provide a robust assessment of the reversible inhibitors. Standard assays use a readout which comprises 48 h of [3H]thymidine incorporation immediately following compound treatment, which is appropriate for irreversible inhibitors. Given that the cruzipain inhibitors used in the current study are reversible, the standard assay might overstate the effect of the inhibitor, since the off-rate is not taken into account. By removing the reversible inhibitor after 2 days of treatment and allowing the parasites to recover prior to thymidine incorporation, a better indication of the true potency of the inhibitors can be obtained. Low levels of thymidine incorporation after the 4-day washout period suggested killing activity of the inhibitors rather than a mechanism such as life cycle arrest, which is likely to be reversible after compound removal. In this assay, Cz002 and Cz005 gave apparent IC50s of 1 to 10 μM (Fig. 2A and B), and the moderate inhibitor, Cz001, showed some activity at 100 μM, whereas the inactive Cz003 showed no activity up to 100 μM. The behavior and potency of the two potent reversible inhibitors, Cz002 and Cz005, were comparable to the those of the irreversible inhibitor K777, except that the epimastigotes treated with Cz005 were round and showed no movement at the end of the experiment, whereas those treated with Cz002 and K777 were elongated and slow moving. This suggests that Cz005 had completely killed the parasites.
Prior to evaluating the compounds in the amastigote assay, the absence of cytotoxicity in J774 cells was tested up to 100 μM. The basic compounds, Cz005 and K777, showed some evidence of toxicity at 100 μM, although they were well tolerated at 10 μM (see the supplemental material). The reduction of intracellular amastigote numbers was similar for Cz002, Cz005, and K777 at 5 μM, and the IC50s for all 3 were estimated to be between 5 and 10 μM (Fig. 3A and B and Table 3). The less potent compounds Cz003 and Cz001 showed very little antiamastigote activity up to 20 μM. The number of trypomastigotes released into the medium was reduced by 97%, 89%, and 96% for Cz002, Cz005, and K777, respectively, at 2 μM (Fig. 3C). The less potent compounds, Cz001 and Cz003, had IC50s of 10 to 20 μM for trypomastigote release.
FIG 3.

Activities of key compounds against intracellular T. cruzi amastigotes. J774 cells were exposed to trypomastigotes, and after 48 h they were treated with the test compounds, each 24 h for 72 h. (A) Average number of amastigotes per macrophage, determined using Geisma staining and visual counting. (B) The calculated percentage of antiamastigote activity {[1 − (number of amastigotes/100 treated macrophages)/(number of amastigotes/100 untreated macrophages)] × 100}. (C) The number of trypomastigotes measured in the medium at 72 h based on trypan blue staining and counting in a Neubauer chamber. *, some toxicity of J774 cells was observed with this treatment, since a total of 100 J774 cells could not be reached in the counting procedure.
In vivo dosing paradigms.
For the in vivo efficacy studies, benznidazole was selected as the comparator, since it is the current standard of care in clinical treatment (5 mg/kg for 60 days). It was administered to mice in drinking water to deliver a dose of ∼50 mg/kg, since it had been reported that 100 mg/kg caused toxicity in mice (42). In vivo efficacy studies were conducted with analogs of the compounds used in the in vitro studies. Cz002 was found to have a bioavailability of only 4%, making it unsuitable for in vivo experiments. Cz007, which demonstrates similar cruzipain inhibition (IC50, 1.1 nM) and a bioavailability of 45%, was selected instead. For in vivo experiments, the basic compound Cz005 was replaced by its single diastereomer, Cz008, in which the trifluoromethyl side chain stereochemistry has the S configuration required for activity (Fig. 1). Although both Cz007 and Cz008 are ∼45 to 50% orally bioavailable, they showed suboptimal pharmacokinetic properties after once-a-day oral gavage, due to their short half-lives (Table 4). An effective and convenient regimen was developed that involved formulation in chow. Since mice feed regularly, they ingest a steady amount of test compound through a large part of the 24-hour day, which maintains relatively constant drug levels. Using formulated chow also reduces the technical complexity of the study and reduces handling of the animals.
TABLE 4.
Summary of pharmacokinetic parameters in CD-1 male mice for Cz007 and the diastereomeric mixture of Cz008 (Cz005)a
| Compound | Route | Vehicle | Dose (mg/kg) | Cmax (nM) | Tmax (h) | AUC (nM · h) | Clearance (ml/min/kg) | t1/2 (h) | Vd (liter/kg) | %F |
|---|---|---|---|---|---|---|---|---|---|---|
| Cz007 | p.o. | 0.5% Methocel | 20 | 1,350 | 1.5 | 6,400 | ||||
| i.v. | 60% PEG 200 | 5 | 3,600 | 37 | 1.1 | 3.7 | 45 | |||
| Cz005b | p.o. | 1% Methocel | 10 | 500 | 2 | 6,600 | ||||
| i.v. | 60% PEG 200 | 2 | 2,700 | 25 | 4.3 | 7.0 | 51 |
Animal were evaluated in groups of 2 (for Cz007) or 3 (for Cz005). Mice weighed approximately 25 g. Abbreviations: p.o., per os; i.v., intravenous; PEG 200, polyethylene glycol 200; Cmax, maximum concentration of drug in blood; Tmax, time to maximum concentation of drug in blood; t1/2, half-life; Vd, volume of distribution; %F, percent bioavailability.
Cz005 is a mixture of Cz008 and its diastereomer at the trifluoromethyl side chain position.
In 5-day feeding studies to test food consumption, tolerability, and blood and tissue levels at doses of 3, 10, or 50 mg/kg for Cz007 and 10 or 50 mg/kg for Cz008, mice showed consistent food consumption of ∼5 g per day and no weight loss, indicating that there was no test article taste aversion. Clinical chemistry results were within normal ranges (data not shown), and no adverse effects were observed, indicating good tolerability. Blood analysis at sacrifice indicated dose-dependent drug concentrations for both Cz007 and Cz008, and all doses gave blood and tissue levels that were at least 10-fold in excess of the IC50s against cruzipain-ΔC (Table 5). Cz007 and Cz008 concentrations measured in liver, heart, and jejunum at the end of the dosing regimen indicated dose-dependent concentrations in these tissues that were in general greater than those observed in blood, consistent with the volumes of distribution (Table 4).
TABLE 5.
Concentrations of Cz007 and Cz008 in blood and tissues at sacrifice after dosing in chow for 5 daysa
| Compound | Dose (mg/kg) | Food (ppm)b | Concn (nm) in body compartment (mean ± SD) |
|||
|---|---|---|---|---|---|---|
| Blood | Liver | Heart | Jejunum | |||
| Cz007 | 3 | 22 | 15 ± 8 | 754 ± 238 | 23 ± 8 | 448 ± 240 |
| 10 | 90 | 77 ± 33 | 4,534 ± 1,863 | 149 ± 94 | 2,712 ± 1,184 | |
| 50 | 425 | 159 ± 40 | 5,984 ± 1,071 | 266 ± 52 | 5,978 ± 3,310 | |
| Cz008 | 10 | 90 | 23 ± 6 | 1,985 ± 706 | 420 ± 228 | 1,955 ± 590 |
| 50 | 425 | 46 ± 20 | 12,605 ± 1.987 | 2.443 ± 1,175 | 15,502 ± 6,913 | |
There were five mice per treatment group, and animals were housed one per cage.
Test articles were mixed into powdered chow at concentrations (in ppm) predetermined to result in the target doses (in mg/kg).
In vivo efficacy study: experiment 1.
The chosen inoculation conditions produced a robust T. cruzi infection but did not cause mortality (see the supplemental material). The AUC of parasitemia was 10% and 35% of the vehicle control for 10 and 50 mg/kg of Cz008, respectively (Fig. 4A and C and Table 6). The AUC of parasitemia indicated that the 50-mg/kg Cz008 treatment was comparable to the benznidazole results at 50 mg/kg (P = 0.56), whereas the 10-mg/kg Cz008 dose group showed a significantly better response (P < 0.05), indicating an inverse dose-response relationship (see Discussion). There was no recrudescence of parasitemia after cessation of dosing. In this study, three mice died, one in each of the untreated (at day 63), benznidazole (at day 70), and Cz008 at 50 mg/kg (at day 49) groups. Qualitative PCR of blood at day 90 gave positive results for 5/9, 2/9, 1/10, and 2/9 mice for the vehicle, benznidazole, 10-mg/kg Cz008, and 50-mg/kg Cz008 groups, respectively. Day 28 average drug blood concentrations were 4.3 μM for benznidazole, 120 nM for Cz008 at 10 mg/kg, and 596 nM for Cz008 at 50 mg/kg. The mock-infected mice treated with 50 mg/kg Cz008 had a drug blood concentration of 411 nM.
FIG 4.

In vivo efficacy results in T cruzi-infected CD-1 mice (n = 10 per treatment group). (A and B) The blood parasitemia of mice inoculated with 104 trypomastigotes and treated from day 2 to day 30 with the cruzipain inhibitors, in their chow, at the indicated doses. Benznidazole in drinking water was used as a positive control at a dose of 50 mg/kg. In the experiment shown in panel B, mice were also treated with 200 mg/kg of cyclophosphamide at day 90 to cause immunosuppression. (C) The total AUC of blood parasitemia from day 0 to sacrifice in both studies. (D) The blood levels (nM) of benznidazole and the two cruzipain inhibitors on the last day of dosing in each experiment.
TABLE 6.
Blood parasitemia AUCs in the murine in vivo efficacy experiments
| Treatment | Dose (mg/kg) | % parasitemia AUC relative to vehicle control (mean ± SD) |
|
|---|---|---|---|
| Expt 1 | Expt 2 | ||
| Untreated | 0 | 100 ± 33 | 100 ± 21 |
| Benznidazole | 50 | 32 ± 23 | 44 ± 9 |
| Cz007 | 3 | NDa | 16 ± 3 |
| 10 | ND | 44 ± 10 | |
| 50 | ND | 46 ± 8 | |
| Cz008 | 3 | ND | 23 ± 6 |
| 10 | 10 ± 2 | 25 ± 7 | |
| 50 | 35 ± 30 | ND | |
ND, not determined.
In vivo efficacy study: experiment 2.
In experiment 2, both Cz007 and Cz008 were evaluated, a low dose of 3 mg/kg was added to the study design, cyclophosphamide treatment was introduced at day 90 to suppress the immune system to allow recrudescence if the animal had not been cured (40), and qPCR analyses of blood, heart, and esophagus were conducted at sacrifice to establish parasite loads and cure. The AUCs of parasitemia showed that the 10- and 50-mg/kg Cz007 treatments were as effective as benznidazole at 50 mg/kg, since all AUCs were in the range of 44 to 46% of the vehicle group (Fig. 4B and C and Table 6). Cz007 at 3 mg/kg and Cz008 at both 3 and 10 mg/kg showed even greater efficacy, with parasitemia AUCs ranging from 16 to 25% of the vehicle group. These were all statistically lower than benznidazole at 50 mg/kg (P < 0.0001 for each) and confirmed the inverse dose-response observed in experiment 1. Mouse deaths were observed in nearly all treatment groups, especially post-cyclophosphamide treatment, with the notable exception of the Cz007 3-mg/kg group (see Table S1 and Fig. S2 in the supplemental material).
Blood samples collected on day 30 were analyzed for the test articles (Fig. 4D). The average benznidazole blood concentration was 1.5 μM (Fig. 4D). The Cz007 blood levels for the 3-, 10-, and 50-mg/kg infected groups were 16, 57, and 254 nM, respectively, which were comparable to the 5-day tolerability study data of 37, 77, and 159 nM (Table 5). A mock-infected group dosed with 50 mg/kg Cz007 gave blood drug levels of 272 nM. For Cz008, the final blood drug levels for the 3- and 10-mg/kg groups were <2 and 3 nM, respectively, much lower than had been observed in the tolerability study and experiment 1, where blood concentrations of 23 nM and 120 nM, respectively, had been measured for 10-mg/kg dose groups.
Real-time qPCR from blood, heart, and esophagus demonstrated a marked reduction in parasite burden for all treatment groups in all tissues (Fig. 5). Prior to cyclophosphamide treatment (day 90), parasites were detected in the blood of some mice of all groups except for the Cz007 3-mg/kg group, in which none were detected (Fig. 5A). At sacrifice on day 113, the Cz007 3-mg/kg group still showed no detectable T. cruzi DNA in blood (Fig. 5B), with no deaths. In the heart tissues (Fig. 5C), a substantial reduction of parasite burden (≥3-log10 drop) was observed in all dose groups relative to the vehicle control group. In the Cz007 3-mg/kg dose group, only 1 of the 10 mice was positive, with a reading at the limit of detection of 5 parasites/ml (≥5-log10 drop). Finally, in the esophagus, the findings were similar, with the Cz007 3-mg/kg group having no mice test positive for T. cruzi (Fig. 5D).
FIG 5.

qPCR data in various tissues from in vivo efficacy experiment 2. (A) Data for blood collected at day 90, prior to cyclophosphamide treatment. (B) Data for blood collected at day 113, which was 24 days after cyclophosphamide treatment. (C and D) Heart and esophagus data, collected at sacrifice on day 113. In the fractions, the numerator is the number of mice positive by qPCR and the denominator is the number of mice that survived in the dose group. For tissue samples in which no parasites were detected (<5 parasites/ml), a value of 1 parasite/ml was plotted.
In Table 7, a summary of results is provided to show the numbers of animals with negative qPCR results in all 3 tissues at sacrifice, which was defined as parasitological cure. Across the treatment groups, the percentages of animals with cures ranged from 40 to 90%. The 3-mg/kg Cz007 dose group gave the best result, with 90% cure and all animals surviving to sacrifice (in this group, the only positive finding was a single mouse with a parasite signal of 5 parasites/ml in the heart), followed by Cz008 at 3 mg/kg (70% cure) and Cz007 at 50 mg/kg and Cz008 at 10 mg/kg (60% cure each), while benznidazole at 50 mg/kg afforded only a 50% cure. Of the mice that died between days 90 and 113, most had negative parasitemia and qPCR results prior to cyclophosphamide treatment (see Table S1 in the supplemental material). Since the animals were immunocompromised, death from an alternate infection cannot be ruled out.
TABLE 7.
Summary of negative qPCR findings for blood, heart, and esophagus samples from efficacy experiment 2, along with survival rates pre- and post-cyclophosphamide treatment
| Measurement | Vehicle | qPCR result for treatment groupa |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Benznidazole 50 mg/kg | Cz007 |
Cz008 |
Uninfected, untreated | Uninfected, Cz007 at 50 mg/kg | |||||
| 3 mg/kg | 10 mg/kg | 50 mg/kg | 3 mg/kg | 10 mg/kg | |||||
| Initial no. of mice | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 5 | 5 |
| Negative qPCR result(s) | |||||||||
| Day 90, blood | 3/7 (43) | 8/10 (80) | 10/10 (100) | 7/10 (70) | 8/10 (80) | 9/10 (90) | 9/10 (90) | 5/5 (100) | 5/5 (100) |
| Day 113, blood | 1/6 (17) | 6/7 (86) | 10/10 (100) | 6/7 (86) | 8/9 (89) | 9/9 (100) | 7/8 (88) | 5/5 (100) | 5/5 (100) |
| Day 113, heart | 0/6 (0) | 5/7 (71) | 9/10 (90) | 4/7 (57) | 6/9 (67) | 8/9 (89) | 6/8 (75) | 5/5 (100) | 5/5 (100) |
| Day113, esophagus | 1/6 (17) | 6/7 (86) | 10/10 (100) | 5/7 (71) | 7/9 (78) | 8/9 (89) | 7/8 (88) | 5/5 (100) | 5/5 (100) |
| All 4 measures | 0/6 (0) | 5/7 (71) | 9/10 (90) | 4/7 (57) | 6/9 (67) | 7/9 (78) | 6/8 (75) | 5/5 (100) | 5/5 (100) |
| All 4 measures including all mice | 0/10 (0) | 5/10 (50) | 9/10 (90) | 4/10 (40) | 6/10 (60) | 7/10 (70) | 6/10 (60) | 5/5 (100) | 5/5 (100) |
| Survival pre-cyclophosphamide | 7/10 (70) | 10/10 (100) | 10/10 (100) | 10/10 (100) | 10/10 (100) | 10/10 (100) | 10/10 (100) | 5/5 (100) | 5/5 (100) |
| Survival post-cyclophosphamide | 6/7 (86) | 7/10 (70) | 10/10 (100) | 7/10 (70) | 9/10 (90) | 9/10 (90) | 9/10 (90) | 5/5 (100) | 5/5 (100) |
| Total survival | 6/10 (60) | 7/10 (70) | 10/10 (100) | 7/10 (70) | 9/10 (90) | 9/10 (90) | 9/10 (90) | 5/5 (100) | 5/5 (100) |
The qPCR results were considered positive when ≥5 parasites/ml were detected. In the fractions, the numerator is the number of PCR-negative mice and the denominator is the number of surviving mice; the corresponding percentage is shown in parentheses.
DISCUSSION
The objective of this research was to establish whether reversible and orally bioavailable cruzipain inhibitors, exemplified by Cz007 and Cz008, are active against T. cruzi. In vitro and in vivo testing was conducted to establish whether these compounds are viable as potential treatments for Chagas disease. Successful efficacy studies with irreversible cruzipain inhibitors, such as K777 (20, 21, 22) and an α-ketone (23), have established proof of concept. The way is paved for second-generation compounds, such as reversible cruzipain inhibitors, which are expected to offer both efficacy and a superior toxicity profile, particularly if selectivity over human cathepsins can be achieved (15, 24, 43). The nitrile group has been characterized as an effective warhead for cysteine protease inhibitors, since it forms a covalent, but reversible, thioimidate complex with the enzyme active site sulfur. As an example, studies with odanacatib, a nitrile-containing cathepsin K inhibitor, demonstrated an enzyme off-rate half-life of approximately 14 min (27). Odanacatib is in late-stage clinical studies for postmenopausal osteoporosis (25, 26). It is generally well tolerated and has pharmacokinetic properties that allow for once-weekly dosing.
Odanacatib itself would not be expected to show potent cruzipain-ΔC activity, because the cyclopropyl group at P1 is too small to capitalize on binding in the S1 cruzipain enzyme pocket. The analogs Cz003 and Cz001, which also have a cyclopropyl at P1, illustrate this SAR. However, based on the promise of odanacatib, a cruzipain inhibitor project was initiated to explore analogs of odanacatib as possible therapies for Chagas disease.
The rank order of potency observed in both the epimastigote and amastigote assays correlated with the potencies observed in the cruzipain enzyme assay, although there was a shift of 3 orders of magnitude between the enzyme potency and the inhibitory effect on whole parasites (Table 3). This shift of enzyme potency and antiparasite activity with cruzipain inhibitors has been reported by others (44, 45, 46, 47, 48). The cause has not been elucidated but may be due to limited drug penetration to the intracellular parasites or a mechanism of action that is not adequately tested under the conditions of the assay.
In addition to its involvement in infectivity, cruzipain is involved in the life cycle of T. cruzi (49). Intracellular amastigote cell number reduction IC50s were in the 5 to 10 μM range, whereas the trypomastigote production was ∼90 to 95% inhibited at 2 μM. This assay could not distinguish between viable and dead intracellular amastigotes, and the highly reduced number of trypomastigotes could have been due to killing amastigotes or arrest of the life cycle.
The compounds Cz007 and Cz008 were not tested against the Brazil strain of T. cruzi due to constrained resources. However, subsequent studies in the Cl Td Tom strain (40) have demonstrated IC50s of 0.19, 0.17, and 0.76 μM for Cz007, Cz008, and benznidazole, respectively (H. J. Park and R. L. Tarleton, unpublished data).
In in vivo experiment 1 with the basic compound Cz008, the lack of recrudescence after treatment cessation and 10 to 22% positive PCR rate in the treatments groups (versus 55% in the untreated group), although encouraging, were not considered proof of cure, and an inverse dose response was suggested. These data indicated the need for a follow-up study with lower doses and a more convincing method for detection of residual parasite burden. Thus, in experiment 2, in which both Cz007 and Cz008 were evaluated, a lower dose of 3 mg/kg was added to the study design, cyclophosphamide treatment was introduced at day 90, and qPCR analyses of blood, heart, and esophagus were conducted to establish parasite burdens. This gave a more robust study from which rigorous cure rates could be established.
For both compounds, measured drug blood concentrations at the most efficacious doses were in the nanomolar range (Fig. 4D). Based on the IC50s observed in the whole-cell assays, these levels are too low for direct antiparasitic activity. While a slow killing mechanism may be possible at these lower concentrations due to impairment of the parasite (20, 50), one could also invoke mechanisms of action that are only possible in vivo, such as inhibition of direct tissue and macrophage invasion (49, 51), reduction of parasite-evoked inflammatory edema that may enable persistent parasitism (52, 53, 54), and abrogation of host immune evasion (55, 56). These biological effects are believed to be the result of cruzipain, which is tethered, secreted, or present in secluded sites of the host-parasite interface (16, 17, 19, 28, 51, 53) and would be readily accessible to cruzipain inhibitors in the blood compartment. The 1 to 2 nM IC50s of these compounds versus cruzipain and their nanomolar drug blood concentrations suggest that the efficacy observed in vivo is significantly driven by inhibition of extracellular cruzipain. However, the high volumes of distribution of both inhibitors, and particularly the basic compound Cz008, are reflected in their elevated tissue levels. For Cz008, the micromolar levels of drug present in the heart tissue could have contributed directly to antiparasitic activity. For Cz007, the heart drug concentrations were 10-fold lower than plasma drug concentrations, yet heart parasites were virtually absent at the 3-mg/kg dose, supporting the hypothesis of extracellular cruzipain inhibition.
T. cruzi parasites have acidic reservosomes, where cruzipain is implicated in protein processing and nutrient production. A basic cruzipain inhibitor would be expected to concentrate in these reservosomes, whereas as a neutral compound would not. If inhibition of the reservosome-localized cruzipain drives efficacy, a basic inhibitor should show superior activity (28, 57). If the inhibition of extracellular cruzipain inhibition drives efficacy, neutral and basic inhibitors should show similar efficacies. Basic compounds will accumulate in tissues rich in lysosomes (24), which may be advantageous for anti-T. cruzi agents due to favorable distribution to tissues that harbor the amastigotes, as well as concentration of the compound in the parasite reservosomes (57). However, basic cruzipain inhibitors would need to have exquisite selectivity profiles versus host cathepsins to be well tolerated (24, 43). Both neutral and basic compounds were evaluated in this study, and no clear differentiation was observed in either the in vitro or the in vivo assays. In the in vivo model, both compounds were equally effective at 3 mg/kg, with similar bioavailabilities. This finding is consistent with the role of extracellular cruzipain as the main target for the efficacy of these compounds.
A curious inverse dose-response curve was observed in both efficacy studies with both compounds. The poor selectivity of Cz007 versus other cathepsins (Table 2) and likely lysosomal concentration of basic Cz008 may have led to off-target inhibition of murine cathepsins. Potent IC50s against mouse cathepsin B (Cat B; 5.1 nM) and Cat S (<0.17 nM) were measured for Cz007. Cathepsins S, L, F, and/or V, which are known to be responsible for antigen processing (43), and their inhibition could have led to an impaired immune response at the higher doses. Unfortunately, attempts to demonstrate this in spleen tissues that were collected at sacrifice using published procedures (33, 58) gave inconclusive results (Wanda Cromlish, unpublished data). This immunosuppression hypothesis suggests that a cruzipain inhibitor that is highly selective over cathepsins would be efficacious at low doses in this mouse model and would exhibit a normal dose response. A potent, nonbasic cruzipain inhibitor, Cz009, with an excellent selectivity profile has recently been identified (29), and studies are in progress.
In summary, Cz007 and Cz008 are the first reported orally active, reversible cruzipain inhibitors capable of curing T. cruzi infection in an in vivo model. Both compounds were well tolerated and worked most effectively at doses of 3 mg/kg. Cz007 gave a 90% cure rate at 3 mg/kg with no animal deaths, even following immunosuppression with cyclophosphamide. Efficacy occurred with nanomolar drug blood levels, similar to their cruzipain-ΔC IC50s. The efficacy shown in these T. cruzi murine studies suggests that nitrile-containing cruzipain inhibitors show promise as a viable approach for a safe and effective treatment of Chagas disease.
Supplementary Material
ACKNOWLEDGMENTS
The Brazil T. cruzi strain was a kind gift of Herbert B. Tanowitz (Albert Einstein College of Medicine) and was used with permission for this research. Former Merck Frosst scientists who supported this work directly or indirectly are all acknowledged. In particular, we thank Kathleen Metters, Nancy Kelly, Alex Popovic, and Allegra Mascisch, who helped to make this work possible, the Laboratory Animal Resources group for generating clinical data during mouse tolerability studies, Wanda Cromlish for attempting to understand the inverse dose-response curve, and Hea Jin Park for allowing unpublished data to be included in this publication. Julio A. Urbina is gratefully acknowledged for extremely helpful discussions during the preparation of the manuscript.
Footnotes
Published ahead of print 9 December 2013
This paper is dedicated to the memory of the Merck Frosst Centre for Therapeutic Research, which was closed in October 2010.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01855-13.
REFERENCES
- 1.Beard CB, Pye G, Steurer FJ, Rodriguez R, Campman R, Peterson AT, Ramsey J, Wirtz RA, Robinson LE. 2003. Chagas disease in a domestic transmission cycle, southern Texas, USA. Emerg. Infect. Dis. 9:103–105. 10.3201/eid0901.020217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coura JR, Vinas PA. 2010. Chagas disease: a new worldwide challenge. Nature 465:S6–S7. 10.1038/nature09221 [DOI] [PubMed] [Google Scholar]
- 3.Moncayo A. 2003. Chagas disease: current epidemiological trends after the interruption of vectorial and transfusional transmission in the Southern Cone countries. Mem. Inst. Oswaldo Cruz 98:577–591. 10.1590/S0074-02762003000500001 [DOI] [PubMed] [Google Scholar]
- 4.Negrette OS, Mora MC, Basombrio MA. 2005. High prevalence of congenital Trypanosoma cruzi infection and family clustering in Salta, Argentina. Pediatrics 115:e668–e672. 10.1542/peds.2004-1732 [DOI] [PubMed] [Google Scholar]
- 5.Gascon J, Bern C, Pinazo MJ. 2010. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 115:22–27. 10.1016/j.actatropica.2009.07.019 [DOI] [PubMed] [Google Scholar]
- 6.Guedes PM, Silva GK, Gutierrez FR, Silva JS. 2011. Current status of Chagas disease chemotherapy. Expert Rev. Anti Infect. Ther. 9:609–620. 10.1586/eri.11.31 [DOI] [PubMed] [Google Scholar]
- 7.Viotti R, Vigliano C, Lococo B, Alvarez MG, Petti M, Bertocchi G, Armenti A. 2009. Side effects of benznidazole as treatment in chronic Chagas disease: fears and realities. Expert Rev. Anti Infect. Ther. 7:157–163. 10.1586/14787210.7.2.157 [DOI] [PubMed] [Google Scholar]
- 8.Perez-Molina JA, Perez-Ayala A, Moreno S, Fernandez-Gonzalez MC, Zamora J, Lopez-Velez R. 2009. Use of benznidazole to treat chronic Chagas' disease: a systematic review with a meta-analysis. J. Antimicrob. Chemother. 64:1139–1147. 10.1093/jac/dkp357 [DOI] [PubMed] [Google Scholar]
- 9.Bern C. 2011. Antitrypanosomal therapy for chronic Chagas' disease. N. Engl. J. Med. 364:2527–2534. 10.1056/NEJMct11014204 [DOI] [PubMed] [Google Scholar]
- 10.Urbina JA. 2010. New insights in Chagas' disease treatment. Drugs Future 35:409–419. 10.1358/dof.2010.035.05.1484391 [DOI] [Google Scholar]
- 11.Apt W. 2010. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des. Devel. Ther. 4:243–253 http://dx.doi.org/10.2147/DDDT.58338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribeiro I, Sevcsik AM, Alves F, Diap G, Don R, Harhay MO, Chang S, Pecoul B. 2009. New, improved treatments for Chagas disease: from the R&D pipeline to the patients. PLoS Negl. Trop. Dis. 3(7):e484. 10.1371/journal.pntd.0000484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahia MT, de Andrade IM, Martins TA, do Nascimento AF, Diniz LF, Caldas IS, Talvani A, Trunz BB, Torreele E, Ribeiro I. 2012. Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl. Trop. Dis. 6(11):e1870. 10.1371/journal.pntd.0001870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urbina JA. 2002. Chemotherapy of Chagas disease. Curr. Pharm. Des. 8:287–295. 10.2174/1381612023396177 [DOI] [PubMed] [Google Scholar]
- 15.McKerrow JH. 1999. Development of cysteine protease inhibitors as chemotherapy for parasitic diseases: insights on safety, target validation, and mechanism of action. Int. J. Parasitol. 29:833–837 [DOI] [PubMed] [Google Scholar]
- 16.Cazzulo JJ. 2002. Proteinases of Trypanosoma cruzi: patential targets for the chemotherapy of Chagas desease. Curr. Top. Med. Chem. 2:1261–1271. 10.2174/1568026023392995 [DOI] [PubMed] [Google Scholar]
- 17.Alvarez VE, Niemirowicz GT, Cazzulo JJ. 2012. The peptidases of Trypanosoma cruzi: digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochim. Biophys. Acta 1824:195–206. 10.1016/j.bbapap.2011.05.011 [DOI] [PubMed] [Google Scholar]
- 18.Sajid M, Robertson SA, Brinen LS, McKerrow JH. 2011. Cruzain: the path from target validation to the clinic. Adv. Exp. Med. Biol. 712:100–115. 10.1007/978-1-4419-8414-2_7 [DOI] [PubMed] [Google Scholar]
- 19.Caffrey CR, Lima AP, Steverding D. 2011. Cysteine peptidases of kinetoplastid parasites. Adv. Exp. Med. Biol. 712:84–99. 10.1007/978-1-4419-8414-2_6 [DOI] [PubMed] [Google Scholar]
- 20.Engel JC, Doyle PS, Hsieh I, McKerrow JH. 1998. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 188:725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doyle PS, Zhou YM, Engel JC, McKerrow JH. 2007. A cysteine protease inhibitor cures Chagas' disease in an immunodeficient-mouse model of infection. Antimicrob. Agents Chemother. 51:3932–3939. 10.1128/AAC.00436-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barr SC, Warner KL, Kornreic BG, Piscitelli J, Wolfe A, Benet L, McKerrow JH. 2005. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob. Agents Chemother. 49:5160–5161. 10.1128/AAC.49.12.5160-5161.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brak K, Kerr ID, Barrett KT, Fuchi N, Debnath M, Ang K, Engel JC, McKerrow JH, Doyle PS, Brinen LS, Ellman JA. 2010. Nonpeptidic tetrafluorophenoxymethyl ketone cruzain inhibitors as promising new leads for Chagas disease chemotherapy. J. Med. Chem. 53:1763–1773. 10.1021/jm901633v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Black WC, Percival MD. 2006. The consequences of lysosomotropism on the design of selective cathepsin K inhibitors. Chembiochem 7:1525–1535. 10.1002/cbic.200600149 [DOI] [PubMed] [Google Scholar]
- 25.Stoch SA, Zajic S, Stone J, Miller DL, Van Dyck K, Gutierrez MJ, De Decker M, Liu L, Liu Q, Scott BB, Panebianco D, Jin B, Duong LT, Gottesdiener K, Wagner JA. 2009. Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double-blind, randomized, placebo-controlled phase I studies. Clin. Pharmacol. Ther. 86:175–182. 10.1038/clpt.2009.60 [DOI] [PubMed] [Google Scholar]
- 26.Stoch SA, Zajic S, Stone JA, Miller DL, van Bortel L, Lasseter KC, Pramanik B, Cilissen C, Liu Q, Liu L, Scott BB, Panebianco D, Ding Y, Gottesdiener K, Wagner JA. 2012. Odanacatib, a selective cathepsin K inhibitor to treat osteoporosis: safety, tolerability, pharmacokinetics and pharmacodynamics. Results from single oral dose studies in healthy volunteers. Br. J. Clin. Pharmacol. 75:1240–1254. 10.1111/j.1365-2125.2012.04471.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gauthier JY, Chauret N, Cromlish W, Desmarais S, l, Duong T, Falgueyret JP, Kimmel DB, Lamontagne S, Leger S, LeRiche T, Li CS, Masse F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmer JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Therien M, Truong VL, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC. 2008. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 18:923–928. 10.1016/j.bmcl.2007.12.047 [DOI] [PubMed] [Google Scholar]
- 28.Nicoll-Griffith DA. 2012. Use of cysteine-reactive small molecules in drug discovery for trypanosomal disease. Expert Opin. Drug Discov. 7:353–366. 10.1517/17460441.2012.668520 [DOI] [PubMed] [Google Scholar]
- 29.Beaulieu C, Isabel E, Fortier A, Masse F, Mellon C, Methot N, Ndao M, Nicoll-Griffith D, Lee D, Park H, Black WC. 2010. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg. Med. Chem. Lett. 20:7444–7449. 10.1016/j.bmcl.2010.10.015 [DOI] [PubMed] [Google Scholar]
- 30.Black WC, Bayly CI, Davis DE, Desmarais S, Falgueyret JP, Leger S, Li CS, Masse F, McKay DJ, Palmer JT, Percival MD, Robichaud J, Tsou N, Zamboni R. 2005. Trifluoroethylamines as amide isosteres in inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 15:4741–4744 http://dx.doi.org/10.1016/j.bmcl.2005.07.071 [DOI] [PubMed] [Google Scholar]
- 31.Baum SG, Wittner M, Nadler JP, Horwitz SB, Dennis JE, Schiff PB, Tanowitz HB. 1981. Taxol, a microtubule stabilizing agent, blocks the replication of Trypanosoma cruzi. Proc. Natl. Acad. Sci. U. S. A. 78:4571–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eakin AE, Mills AA, Harth G, McKerrow JH, Craik CS. 1992. The sequence, organization, and expression of the major cysteine protease (cruzain) from Trypanosoma cruzi. J. Biol. Chem. 267:7411–7420 [PubMed] [Google Scholar]
- 33.Falgueyret JP, Desmarais S, Oballa R, Black WC, Cromlish W, Khougaz K, Lamontagne S, Masse F, Riendeau D, Toulmond S, Percival MD. 2005. Lysosomotropism of basic cathepsin K inhibitors contributes to increased cellular potencies against off-target cathepsins and reduced functional selectivity. J. Med. Chem. 48:7535–7543. 10.1021/jm0504961 [DOI] [PubMed] [Google Scholar]
- 34.Falgueyret JP, Black WC, Cromlish W, Desmarais S, Lamontagne S, Mellon C, Riendeau D, Rodan S, Tawa P, Wesolowski G, Bass KE, Venkatraman S, Percival MD. 2004. An activity-based probe for the determination of cysteine cathepsin protease activities in whole cells. Anal. Biochem. 335:218–227 http://dx.doi.org/10.1016/j.ab.2004.09.005 [DOI] [PubMed] [Google Scholar]
- 35.Piacenza L, Peluffo G, Radi R. 2001. L-arginine-dependent suppression of apoptosis in Trypanosoma cruzi: contribution of the nitric oxide and polyamine pathways. Proc. Natl. Acad. Sci. U. S. A. 98:7301–7306. 10.1073/pnas.121520398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berrizbeitia M, Ndao M, Bubis J, Gottschalk M, Ache A, Lacouture S, Medina M, Ward BJ. 2006. Purified excreted-secreted antigens from Trypanosoma cruzi trypomastigotes as tools for diagnosis of Chagas' disease. J. Clin. Microbiol. 44:291–296. 10.1128/JCM.44.2.291-296.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bateman KP, Castonguay G, Xu L, Rowland S, Nicoll-Griffith DA, Kelly N, Chan CC. 2001. Reduction of animal usage by serial bleeding of mice for pharmacokinetic studies: application of robotic sample preparation and fast liquid chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 754:245–251 [DOI] [PubMed] [Google Scholar]
- 38.Berrizbietia M, Ndao M, Gottschalk M, Ache A, Vasquez F, Lacouture S, Medina M, Ward BJ. 2004. Development and comparison of enzyme immunoassays for diagnosis of Chagas' disease using fixed forms of Trypanosoma cruzi (epimastigotes, amastigotes, and trypomastigotes) and assessment of antigen stability for the three assays. J. Clin. Microbiol. 42:1766–1769. 10.1128/JCM.42.4.1766-1769.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herbert WJ, Lumsden WH. 1976. Trypanosoma brucei: a rapid “matching” method for estimating the host's parasitemia. Exp. Parasitol. 40:427–431 [DOI] [PubMed] [Google Scholar]
- 40.Bustamante JM, Bixby LM, Tarleton RL. 2008. Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nat. Med. 14:542–550. 10.1038/nm1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ndao M, Kelly N, Normandin D, Maclean JD, Whiteman A, Kokoskin E, Arevalo I, Ward BJ. 2000. Trypanosoma cruzi infection of squirrel monkeys: comparison of blood smear examination, commercial enzyme-linked immunosorbent assay, and polymerase chain reaction analysis as screening tests for evaluation of monkey-related injuries. Comp. Med. 50:658–665 [PubMed] [Google Scholar]
- 42.Zaidenberg A, Luong T, Lirussi D, Bleiz J, Del Buono MB, Quijano G, Drut R, Kozubsky L, Marron A, Buschiazzo H. 2006. Treatment of experimental chronic chagas disease with trifluralin. Basic Clin. Pharmacol. Toxicol. 98:351–356. 10.1111/j.1742-7843.2006.pto_253.x [DOI] [PubMed] [Google Scholar]
- 43.Kometani M, Nonomura K, Tomoo T, Niwa S. 2010. Hurdles in the drug discovery of cathepsin K inhibitors. Curr. Top. Med. Chem. 10:733–744. 10.2174/156802610791113478 [DOI] [PubMed] [Google Scholar]
- 44.Choe Y, Brinen LS, Price MS, Engel JC, Lange M, Grisostomi C, Weston SG, Pallai PV, Cheng H, Hardy LW, Hartsough DS, McMakin M, Tilton RF, Baldino CM, Craik CS. 2005. Development of alpha-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg. Med. Chem. 13:2141–2156 http://dx.doi.org/10.1016/j.bmc.2004.12.053 [DOI] [PubMed] [Google Scholar]
- 45.Fujii N, Mallari JP, Hansell EJ, Mackey Z, Doyle P, Zhou YM, Gut J, Rosenthal PJ, McKerrow JH, Guy RK. 2005. Discovery of potent thiosemicarbazone inhibitors of rhodesain and cruzain. Bioorg. Med. Chem. Lett. 15:121–123 http://dx.doi.org/10.1016/j.bmcl.2004.10.023 [DOI] [PubMed] [Google Scholar]
- 46.Greenbaum DC, Mackey Z, Hansell E, Doyle P, Gut J, Caffrey CR, Lehrman J, Rosenthal PJ, McKerrow JH, Chibale K. 2004. Synthesis and structure-activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 47:3212–3219. 10.1021/jm030549j [DOI] [PubMed] [Google Scholar]
- 47.Bryant C, Kerr ID, Debnath M, Ang KK, Ratnam J, Ferreira RS, Jaishankar P, Zhao D, Arkin MR, McKerrow JH, Brinen LS, Renslo AR. 2009. Novel non-peptidic vinylsulfones targeting the S2 and S3 subsites of parasite cysteine proteases. Bioorg. Med. Chem. Lett. 19:6218–6221. 10.1016/j.bmcl.2009.08.098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ang KK, Ratnam J, Gut J, Legac J, Hansell E, Mackey ZB, Skrzypczynska KM, Debnath A, Engel JC, Rosenthal PJ, McKerrow JH, Arkin MR, Renslo AR. 2011. Mining a cathepsin inhibitor library for new antiparasitic drug leads. PLoS Negl. Trop. Dis. 5(5):e1023. 10.1371/journal.pntd.001023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meirelles MN, Juliano L, Carmona E, Silva SG, Costa EM, Murta AC, Scharfstein J. 1992. Inhibitors of the major cysteinyl proteinase (GP57/51) impair host cell invasion and arrest the intracellular development of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol. 52:175–184 [DOI] [PubMed] [Google Scholar]
- 50.Engel JC, Doyle PS, Palmer J, Hsieh I, Bainton DF, McKerrow JH. 1998. Cysteine protease inhibitors alter Golgi complex ultrastructure and function in Trypanosoma cruzi. J. Cell Sci. 111:597–606 [DOI] [PubMed] [Google Scholar]
- 51.Aparicio IM, Scharfstein J, Lima AP. 2004. A new cruzipain-mediated pathway of human cell invasion by Trypanosoma cruzi requires trypomastigote membranes. Infect. Immun. 72:5892–5902. 10.1128/IAI.72.10.5892-5902.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benitez-Hernandez I, Mendez-Enriquez E, Ostoa P, Fortoul T, Ramirez JA, Stempin C, Cerban F, Soldevila G, Garcia-Zepeda EA. 2010. Proteolytic cleavage of chemokines by Trypanosoma cruzi's cruzipain inhibits chemokine functions by promoting the generation of antagonists. Immunobiology 215:413–426. 10.1016/j.imbio.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 53.Andrade D, Serra R, Svensjo E, Lima AP, Ramos ES, Jr, Fortes FS, Morandini AC, Morandi V, Soeiro MN, Tanowitz HB, Scharfstein J. 2012. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: a converging pathway leading to chagasic vasculopathy. Br. J. Pharmacol. 165:1333–1347. 10.1111/j.1476-5381.2011.01609.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scharfstein J, Andrade D, Svensjo E, Oliveira AC, Nascimento CR. 2012. The kallikrein-kinin system in experimental Chagas disease: a paradigm to investigate the impact of inflammatory edema on GPCR-mediated pathways of host cell invasion by Trypanosoma cruzi. Front. Immunol. 3:396. 10.3389/fimmu.2012.00396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berasain P, Carmona C, Frangione B, Cazzulo JJ, Goni F. 2003. Specific cleavage sites on human IgG subclasses by cruzipain, the major cysteine proteinase from Trypanosoma cruzi. Mol. Biochem. Parasitol. 130:23–29 http://dx.doi.org/10.1016/S0166-68511(03)00139-7 [DOI] [PubMed] [Google Scholar]
- 56.Doyle PS, Zhou YM, Hsieh I, Greenbaum DC, McKerrow JH, Engel JC. 2011. The Trypanosoma cruzi protease cruzain mediates immune evasion. PLoS Pathog. 7(9):e1002139. 10.1371/journal.ppat.1002139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKerrow JH, Doyle PS, Engel JC, Podust LM, Robertson SA, Ferreira R, Saxton T, Arkin M, Kerr ID, Brinen LS, Craik CS. 2009. Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 104(Suppl 1):263–269 http://dx.doi.org/10.1590/S0074-02762009000900034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desmarais S, Black WC, Oballa R, Lamontagne S, Riendeau D, Tawa P, l, Duong T, Pickarski M, Percival MD. 2008. Effect of cathepsin k inhibitor basicity on in vivo off-target activities. Mol. Pharmacol. 73:147–156. 10.1124/mol.107.039511 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.