

Abstract
Therapy for hepatitis C virus (HCV) infection has advanced with the recent approval of direct-acting antivirals in combination with peginterferon and ribavirin. New antivirals with novel targets are still needed to further improve the treatment of hepatitis C. Previously reported screening methods for HCV inhibitors either are limited to a virus-specific function or apply a screening method at a single dose, which usually leads to high false-positive or -negative rates. We developed a quantitative high-throughput screening (qHTS) assay platform with a cell-based HCV infection system. This highly sensitive assay can be miniaturized to a 1,536-well format for screening of large chemical libraries. All candidates are screened over a 7-concentration dose range to give EC50s (compound concentrations at 50% efficacy) and dose-response curves. Using this assay format, we screened a library of pharmacologically active compounds (LOPAC). Based on the profile of dose-dependent curves of HCV inhibition and cytotoxicity, 22 compounds with adequate curves and EC50s of <10 μM were selected for validation. In two additional independent assays, 17 of them demonstrated specific inhibition of HCV infection. Ten potential candidates with efficacies of >70% and CC50s (compound concentrations at 50% cytotoxicity) of <30 μM from these validated hits were characterized for their target stages in the HCV replication cycle. In this screen, we identified both known and novel hits with diverse structural and functional features targeting various stages of the HCV replication cycle. The pilot screen demonstrates that this assay system is highly robust and effective in identifying novel HCV inhibitors and that it can be readily applied to large-scale screening of small-molecule libraries.
INTRODUCTION
Hepatitis C virus (HCV) infection affects approximately 200 million people worldwide (1). The majority of HCV-infected patients fail to clear the virus, and many progress to chronic liver diseases, including cirrhosis with a risk of developing hepatocellular carcinoma. Recent development of direct-acting antivirals (DAAs) against HCV, such as telaprevir and daclatasvir, still requires combination with peginterferon and ribavirin for maximal efficacy (2). New agents are needed to develop therapy that is not based on peginterferon because of its various limitations.
Most of the assay development to screen for anti-HCV compounds has focused on enzymatic functions of virus-encoded proteins, such as viral protease and polymerase. Screening based on various enzymatic assays has led to the discovery of HCV-specific DAAs (3). However, targeting a specific viral protein can be associated with rapid emergence of drug-resistant viral mutations, as shown by studies of monotherapy with these DAAs (4). Cell-based screening of anti-HCV molecules has also been conducted with the HCV replicon system (5, 6), which involves only the RNA replication step of the viral replication cycle and cannot target other viral infection steps, including viral entry, processing, assembly, and secretion (7, 8). Phenotypic screening with a cell-based infectious HCV system would cover potential drug targets at all stages of the HCV replication cycle. Targeting multiple key steps in the viral replication cycle not only increases antiviral efficacy, but also reduces the emergence of drug resistance (9).
Previously, we and others developed a highly robust cell culture system for infectious HCV (HCVcc) (10). Further studies demonstrated that reporter genes can be genetically engineered into certain locations of the HCV genome without impairing the viability of the virus (11). Several groups have reported cell-based HCV infection systems for the screening of HCV inhibitors in various assay formats (12–15). Gastaminza et al. developed a colorimetric assay measuring HCV E2 protein produced by HCV-infected cells in a 96-well plate format (12). Using this method, the group identified compounds that target multiple aspects of the HCV replication cycle in the screen of the NIH Clinical Collection library, composed of 446 clinically approved molecules. Chockalingam et al. developed a method that reports HCV infection via a cell death phenotype by introducing an HCV NS3-4A protease-cleavable derivative of the proapoptotic factor Bid, mBid, into the HCV-permissive cell line Huh7.5 (14). In the system, an HCV inhibitor could rescue the cells from the HCV-induced cytopathic effect. Based on the ability of the HCV NS3 protease to cleave synthetic peptides containing the enzyme's natural viral cleavage sites, Yu et al. applied a cell-based hepatitis C virus infection fluorescence resonance energy transfer (FRET) assay for antiviral-compound screening (13). Using a luciferase reporter that is directly inserted into the HCV genome, Wichroski et al. adapted the infection system in a 384-well format for the screening of HCV inhibitors (15).
Because of the limitations of their respective reporter activities, all the above-mentioned systems have limited throughput for compound screens. They may be useful for screening small compound collections but are not feasible for high-throughput screening (HTS) of large compound libraries with millions of compounds.
In this study, we developed a cell-based HCV infection system with a highly sensitive Gaussia luciferase (Gluc) reporter for detection of viral infection. This unbiased phenotypic approach captures the entire HCV replication cycle that is amenable to HTS. This cell-based HCV infection assay has been miniaturized into 1,536-well plates and validated in a pilot screen of a commercially available chemical library.
MATERIALS AND METHODS
Cells and chemicals.
The HCV-permissive human hepatoma cell line Huh7.5.1 and other Huh7-derived cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) with 10% fetal bovine serum (FBS) and antibiotics in 5% CO2 at 37°C. Cyclosporine, 2′-C-methylcytidine, ribavirin, and the library of pharmacologically active compounds (LOPAC) containing 1,280 compounds were purchased from Sigma-Aldrich. All chemicals were dissolved in dimethyl sulfoxide (DMSO) at 10 mM as a stock solution.
Generation of a Cre-Lox-based reporter system for the detection of HCV infection.
A Cre-Lox-based reporter platform was established to detect HCV infection in cell culture. The platform is composed of two parts: the HCV-Cre virus and the LoxPGluc cell line (Fig. 1). HCV-Cre was constructed by inserting the Cre DNA recombinase gene into the HCV genome (J6/JFH1 clone) between the NS5A and NS5B genes. The HCV NS3 serine protease cleavage signal sequence (TTVCCSMSY) was engineered to flank both ends of the Cre gene. First, a two-step PCR was employed to introduce an MluI and a BglII restriction site with two flanking HCV NS3 cleavage signal sequences, one on the 5′ side of MluI and the other on the 3′ side of BglII, into the juncture between NS5A and NS5B. In the first step, two pairs of primers, NS5A-7358-F (5′-ATATCAGAAGCCCTCCAGCA-3′)/NS5A-BglII-MluI-5AB-R (5′-CGTCTCAGATCTGGACGCGTCATAAGACATGGAGCAGCACACGGT-3′) and NS5B-MluI-BglII-5AB-F (5′-TCTTATGACGCGTCCAGATCTGAGACGACCGTGTGCTGCTCCATG-3′)/NS5B-8569-R (5′-CGCTAGGGCTTTCACATAGC-3′), were used to produce two fragments. Then, a second overlap PCR was performed using the two amplified fragments as templates and NS5A-7358-F and NS5B-8569-R as primers. The final PCR product was digested with RsrII and SnaBI and cloned back into the original pJ6/JFH1 plasmid. A Cre fragment (1,032 bp) with MluI and BglII restriction sites attached on both ends was then inserted into the newly created pJ6/JFH1 to obtain pJ6/JFH1-NS5A/B-Cre (pHCV-Cre), in which a Cre recombinase gene was flanked on both ends by authentic NS3 cleavage signal sequences, DTTVCCSMSY at the 5′ end and ETTVCCSMSY at the 3′ end (Fig. 1A). The LoxPGluc cell line was produced by transfecting a Gluc expression vector (pCAG-LoxPGluc) into HCV-permissive Huh7.5.1 cells and selected by G418 resistance. The Gluc expression vector was constructed by cloning the Gluc reporter gene into a Cre/LoxP-controlled conditional expression cassette, pCALNL5 (16). pCALNL5 contains a CAG promoter, a synthetic promoter consisting of the cytomegalovirus (CMV) early enhancer and a chicken β-actin promoter (17), and an intervening sequence flanked by two LoxP sites after the promoter. The intervening sequence contains signals for transcriptional termination and has been used for the regulation of gene expression of both mammalian cell lines and transgenic mice (18, 19). The Gluc gene was obtained from pCMV-Gluc (New England Biolabs) by SacI and NotI digestion and cloned into the SacI and SmaI sites of pCALNL5 to generate pCAG-LoxPGluc, in which Gluc expression is driven by the CAG promoter and regulated by LoxP (Fig. 1A). After transfection of pCAG-LoxPGluc into the cells, individual clones were selected with G418 (100 μg/ml) and characterized by HCV core immunofluorescent staining and luciferase reporter activity upon HCV-Cre infection. The clone supporting the highest level of HCV infection (determined by HCV core positivity) and luciferase activity was amplified and stocked for HTS assay.
FIG 1.

Cre-Lox HCV reporter platform for cell-based HTS assay. (A) Schematic representation of HCV-Cre and pCAG-LoxPGluc constructs. The junctional sequences containing two NS3-NS4A cleavage sites (arrows) flanking the Cre gene are shown. (B) Diagram of HCV-Cre infection and Gluc production in Huh7.5.1.LoxPGLuc cells.
After infection of the Huh7.5.1.LoxPGLuc cells with HCV-Cre, Cre recombinase specifically recognizes the LoxP sequence and efficiently removes the intervening stop sequence (18). Gluc is then produced and secreted under the control of the CAG promoter (Fig. 1B). Thus, the luciferase activity of the HCV-Cre-infected cell reflects the level of HCV infection and replication.
Production of HCV and pseudoviral particles.
Plasmid cDNA encoding a wild-type (WT) HCV J6/JFH1 clone (HCV-WT), HCV with the Renilla luciferase gene at the 3′ end of p7 (HCV-Luc), or HCV-Cre was linearized with XbaI and in vitro transcribed with T7 RNA polymerase. The production of HCV-WT, HCV-Luc, and HCV-Cre was conducted by transfecting 20 μg of in vitro-transcribed mRNA encompassing the whole HCV genome into 2 × 106 Huh7.5.1 cells in 10-cm dishes using the transfection reagent DMRIE-C (Invitrogen). The cells were passaged every other day from day 3 after transfection. The culture media containing virus were collected daily, filtered through 0.22-μm low-protein-binding filters, and aliquoted; had their titers determined; and were stored at −80°C until use. An envelope-deficient retroviral HIV vector carrying a luciferase reporter gene (pNL4-3.Luc.R-E-) and the plasmid pHEF-VSVG encoding envelope proteins of vesicular stomatitis virus (VSV)-G were obtained from the NIH AIDS Reagent Program (Division of AIDS, NIAID, NIH). pNL4-3.Luc.R-E- was originally from Nathaniel Landau (20). pHEF-VSVG was from Lung-Ji Chang (21). Plasmids encoding HCV genotype 1a envelope proteins were kindly provided by Stuart Ray (Johns Hopkins University). Pseudotyped viruses (VSV-Gpp and HCVpp-1a) were produced by cotransfection of DNA vectors encoding the respective envelope glycoproteins with pNL4-3.Luc.R-E- at a 1:1 ratio into 293T cells, as described previously (22, 23).
Compound library preparation and quantitative HTS (qHTS) assay.
A LOPAC consisting of 1,280 known compounds was serially diluted in DMSO at a 1:5 ratio to give seven stock concentrations ranging from 0.64 μM to 10 mM in 384-well plates. Then, these compounds were reformatted at 7 μl/well into 1,536-well plates as compound source plates.
The cell-based HCV infection assay was miniaturized in the 1,536-well plate format for HTS. A qHTS using the cell-based HCV infection assay was performed as described previously using a fully automated robotic screening system (Kalypsys, San Diego, CA) (24). Briefly, LoxPGluc cells were plated in white cell plates at a density of 750 cells/well in 3 μl of medium and cultured for 16 h. The LOPAC compounds were added to the cell plates at 23 nl/well using a Pintool station (Waco, San Diego, CA), followed by addition of 2.5 μl/well HCV-Cre-containing medium (at a multiplicity of infection [MOI] of ∼0.05). The final compound concentrations incubated with cells ranged from 2.68 nM to 42 μM. A serial dilution of the known HCV inhibitor cyclosporine was included in each plate as a positive control. Two days after compound treatment and viral infection, cells were harvested for detection of HCV infection by luciferase assay, and 4.5 μl of premixed assay buffer with substrate (Renilla luciferase assay kit; Promega) was added directly to the cell culture medium of each well. After 10 min of incubation at room temperature, the luciferase signal was detected in a luminescence plate reader (PerkinElmer). The signal-to-basal (S/B) ratio and Z′ factor (a measure to evaluate the suitability of an assay for HTS) were calculated from means and standard deviations of the positive and negative controls (SDp and SDn, respectively) as follows: Z′ = 1 − 3 × (meanp + meann)/|SDp − SDn|.
Secondary confirmation assays of primary hits.
Primary hits of LOPAC screening were selected and confirmed by HCV-Luc and 2-part HCV-WT infection assays. Huh7.5.1 cells were seeded in 96-well plates (104 cells/well) and cultured overnight, followed by infection with HCV-Luc, together with 10 μM each compound. Inhibition of HCV was analyzed with the luciferase assay kit 48 h after HCV-Luc infection. The 2-part HCV infection assay was performed as described previously (25). Briefly, Huh7.5.1 cells were plated in 384-well plates (3,000 cells/well) and infected with HCV-WT (part 1). After 48 h, the medium was replica plated onto a new plate with Huh7.5.1 cells for infection as the part 2 experiment. The cells from part 1 were then fixed with 4% paraformaldehyde and stained with anti-HCV core 6G7 monoclonal antibody, followed by an anti-mouse Alexa Fluor 488 secondary antibody (Invitrogen). The cell nuclei were counterstained with Hoechst dye (Invitrogen). The cells were imaged with a fluorescent cell imager and analyzed with Metamorph Cell Scoring software (Molecular Devices Inc.). Forty-eight hours after the transfer of the part 1 supernatant, the cells of part 2 plates were stained for HCV core expression and imaged as in part 1.
HCV entry and replication assays.
The HCV entry assay was performed with HCVpp-1a and VSV-Gpp infection. Huh7.5.1 cells seeded in 96-well plates (104 cells/well) were cultured overnight. Then, the cells were pretreated with 10 μM the compounds or DMSO (negative control) in 5 replicates for 10 min, followed by infection of HCVpp-1a and VSV-Gpp for 4 h. The cells were then washed and cultured in medium for 48 h, followed by a luciferase assay to detect HCV entry into cells.
HCV replicon genotype 2a with a Renilla luciferase reporter was tested for HCV replication against the selected compounds. Huh7.5.1 cells were plated into a 96-well plate (104 cells/well) and incubated overnight. Then, the cells were transiently transfected with the replicon mRNA with DMRIE-C for 4 h. After removing the transfection reagent, the cells were incubated with DMEM containing 10 μM each compound or DMSO control in 5 replicates. The cells were harvested for luciferase assay after 48-h compound treatment.
HCV single-cycle infection assay.
A single-round infectious defective HCV particle (HCVsc) generated by a replicon trans-packaging system was used for the HCV single-cycle infection assay (26). HCVsc can infect and replicate but does not assemble new virions. Thus, the system allows the distinction of viral entry/replication from assembly/secretion. The infectious HCVsc was generated by cotransfecting pHH/SGR-Luc, a bicistronic subgenomic reporter replicon with a Pol I promoter/terminator, and a HCV-core-NS2 expression plasmid (both plasmids were provided by Tetsuro Suzuki). The assay was performed in 96-well plates with cultured Huh7.5.1 cells. The cells were inoculated with the infectious HCVsc, together with the tested compounds. The luciferase activity of the cells was measured 48 h after the compound treatment.
Cytotoxicity assay.
Since general cytotoxicity of compounds can interfere with cell-based assays, we carried out a parallel ATP content assay (ATPlite assay kit; PerkinElmer) under the same conditions as the luciferase assay to monitor the cytotoxicity of the compounds. Briefly, after the cells were incubated with the compounds, an equal volume of ATP content assay mixture containing cell lysis buffer, firefly luciferase, and luciferase substrate (5 μl/well for 1,536-well plates and 100 μl/well for 96-well plates) was added to each well. After 15 min of incubation at room temperature, the plates were read in a luminescence plate reader.
Data analysis.
Primary screen data and curve fitting were analyzed with customized software developed internally (27). The maximal response (100% activity) was set as the response to 10 μM cyclosporine and the basal response (0% activity) as the response to the DMSO control. The efficacy of a compound refers to the highest response standardized to the maximal response to cyclosporine (as defined above) and is derived from the concentration-response curve where the inhibition reaches the plateau of maximal response. The EC50 (compound concentration at 50% efficacy) and CC50 (compound concentration at 50% cytotoxicity) values of compounds in the confirmation assays were calculated from the dose-response curves by nonlinear regression analysis using Prism software (GraphPad Software, San Diego, CA).
RESULTS
HCV-Cre induces robust Gluc production in reporter cells suitable for the HTS format.
To generate a reporter system that is simple and highly sensitive with minimal assay steps, we chose the secretive Gaussia luciferase (Gluc) as a reporter, because it produces a higher luminescence signal than Renilla luciferase. This Gluc reporter is a secreted form of luciferase that can be detected easily without cell lysis. Initially, a number of recombinant HCVs with genetically engineered insertion of the Gluc gene in various locations of the viral genome (the 5′ end, between p7 and NS2, or between NS5A and NS5B) were generated. However, none of them produced sufficient luminescence signals for HTS assay in 1,536-well plates (data not shown). We then developed a binary system for highly sensitive detection of HCV infection using the Cre-Lox system (18). A Gluc-containing Huh7.5.1 cell line was generated in which Gluc expression was silenced, due to the intervening sequence flanked by the LoxP sites (Fig. 1). Upon infection by the recombinant HCV-Cre, the intervening sequence was removed by the Cre recombinase, and Gluc was expressed and detected in the medium.
Based on our previous experience, we inserted the Cre gene between NS5A and NS5B in the HCV genome. To demonstrate that insertion of the Cre gene in this location does not have a major deleterious effect on HCV replication and infectivity, viral replication and production of infectious virus were compared between HCV-Cre and HCV-WT (Fig. 2A to C). Following transfection of the HCV RNAs, the levels of HCV secreted into the medium were similar in HCV-Cre and HCV-WT (Fig. 2A). Reinfection of naive Huh7.5.1 cells with the same medium also demonstrated that HCV-Cre produced amounts of intracellular and extracellular HCV RNAs similar to those of HCV-WT (Fig. 2B).
FIG 2.

HCV-Cre and HCV-WT replicate and infect similarly in Huh7.5.1 cells. (A) HCV RNA levels from culture media collected at various times following transfection of in vitro-transcribed HCV-Cre or HCV-WT RNA were quantified by real-time PCR analysis. The amount of HCV in the media is expressed as copy numbers of HCV RNA/ml. (B) Naive Huh7.5.1 cells plated in 12-well plate were infected with 100 μl of HCV-Cre and HCV-WT media collected at various times following transfection. Total RNAs were isolated from both the media and cells at 48 h after infection. The HCV RNA was quantified by real-time PCR analysis. (C) Naive Huh7.5.1 cells plated on 96-well plates were inoculated with serial dilutions of culture supernatant collected at various times following HCV RNA transfection, as indicated. After 3 days, the cells were processed for HCV core immunofluorescent staining (examples are shown below). HCV core-positive foci were counted, and the number of FFU of each virus was calculated as FFU/ml. (D) Induction of Gluc activity in LoxPGluc cells following HCV-Cre infection. Huh7.5.1.LoxPGLuc cells plated on 96-well plates were inoculated with HCV-Cre-containing medium (10 μl/well) harvested at various times following HCV-Cre mRNA transfection. The Gluc activity, defined as relative luciferase units (RLU), of the cultured medium was measured at 48 h after HCV-Cre infection. The data at each time point are means and standard errors of the mean (SEM) from 5 replicates. (E) Correlation between the multiplicity of infection (MOI) of HCV-Cre and Gluc activities. Huh7.5.1.LoxPGLuc cells plated on 96-well plates were inoculated with various titers (MOI) of HCV-Cre as indicated. Forty-eight hours after HCV-Cre infection, 10 μl of culture medium was transferred to an assay plate, and the Gluc activity was measured. The data are means ± SEM from 5 replicates.
Following reinfection of naive Huh7.5.1 cells with the virus-containing medium, viral infectivity was analyzed by HCV core immunofluorescent staining. HCV-Cre and HCV-WT had similar levels of infectious viral particles, as reflected by the number of focus-forming units (FFU)/ml (Fig. 2C). These results suggest that HCV-Cre replicated and produced infectious virus similarly to HCV-WT. Therefore, HCV appears to tolerate insertion of a foreign gene between NS5A and NS5B, which is consistent with previous publications (28).
To determine if the luminescence signals of the HCV-Cre–LoxPGluc system are suitable for HTS, HCV-Cre harvested at various days posttransfection was tested for the ability to induce Gluc production in the LoxPGluc cells. The Gluc activity in the medium of LoxPGluc cells at 48 h after HCV-Cre infection in 96-well plates was measured (Fig. 2D). HCV-Cre harvested at 2 days posttransfection started to show Gluc induction activity in the LoxPGluc cells. HCV-Cre harvested between day 4 and day 7 produced the highest levels of Gluc induction. The Gluc activity started to decline later and remained at a measurable level for HCV-Cre harvested up to day 10 posttransfection. The results indicate that HCV-Cre harvested between days 4 and 10 is optimal to infect LoxPGluc cells for robust Gluc reporter activity. Furthermore, we demonstrated that the Gluc signals correlate well with the infectious titers of HCV-Cre (Fig. 2E).
Assay development and miniaturization.
As shown above, HCV-Cre efficiently infects Huh7.5.1.LoxPGLuc cells and produces a high level of Gluc reporter activity in 96-well plates. The assay system was then miniaturized into the 1,536-well plate format for HTS. To optimize the assay conditions in 1,536-well plates, various cell densities (500, 750, 1,000, and 1,250 cells/well) and incubation times (0 h, 4 h, 8 h, and 16 h after plating the cells) prior to viral infection were examined. We found that cells seeded at a density of 750 cells/well and cultured overnight before viral infection yielded the most consistent and highest Gluc signal (data not shown).
To determine the robustness of this assay in the 1,536-well format, the S/B ratio and Z′ factor were examined. In multiple repeated experiments, the S/B ratios were typically above 40 and the Z′ factors were above 0.6 (Fig. 3A). The reproducible dose-response curves of two known HCV inhibitors, cyclosporine (29, 30) and 2′-C-methylcytidine (31), were generated in the 1,536-well plate format (Fig. 3B). Together, the results indicate that this assay is robust in the 1,536-well plate format and thus suitable for HTS.
FIG 3.
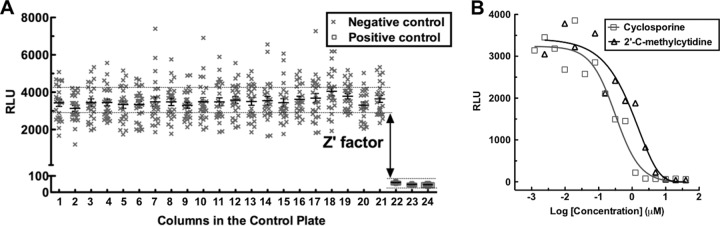
The HCV-Cre–LoxPGluc system is suitable for qHTS. (A) Huh7.5.1.LoxPGLuc cells were seeded in a 1,536-well plate at a density of 750 cells/well and infected with 2.5 μl of HCV-Cre (MOI, 0.05). Gluc activity was measured 2 days after viral infection. The horizontal lines and error bars represent means ± SEM. Columns 1 to 21 are HCV-infected cells only (treated with DMSO control). In columns 22 to 24, HCV-infected cells were treated with 3 known HCV inhibitors, ribavirin (100 μg/ml), 2′-C-methylcytidine (10 μM), and cyclosporine (10 μM). The dotted lines indicate the approximate range of Gluc signals within the standard errors of all the columns. (B) Dose-dependent response of HCV inhibition by 2′-C-methylcytidine and cyclosporine in 1,536-well plates. The cells were treated with 1:2 serial dilutions of the compounds starting at 20 μM. The results shown are representative of 3 independent experiments.
Pilot qHTS of LOPAC.
To further validate the HTS assay system, we performed a quantitative HTS on the LOPAC. All the compounds were assayed in 7 concentrations (2.68 nM to 42 μM) at a 1:5 serial dilution, and each assay plate contained a dose-response curve of the control inhibitor cyclosporine. The S/B ratio and Z′ value were consistent and robust in all plates, with minimal plate-to-plate variation. The primary screen data were analyzed to generate concentration-response curves with four curve classes (24). Class 1 curves are well fitted (r2 ≥ 0.9) and have both upper and lower asymptotes with either complete response (efficacy, >80%) or partial response (30 to 80%). Class 2 curves are incomplete, with only one asymptote. Class 3 curves display activity only at the highest tested concentration and have an efficacy of >30%. Class 4 curves have low (efficacy, <30%) or no response and were deemed inactive (24).
Based on the curve classes, 123 compounds with concentration-dependent curves of class 1 or 2 in the luciferase assay (active in HCV inhibition) and class 3 or 4 in the ATPlite assay (low or no toxicity) were selected as the primary hits (see Table S1 in the supplemental material). The primary hits were used as an initial pool for further selection of potential candidates for lead compounds according to compound potency and the maximal inhibitory response obtained from the concentration-response curves. Among them, the 22 most active compounds with EC50s under 10 μM were selected for the secondary confirmation and validation screens (Table 1 and Fig. 4).
TABLE 1.
Summary of hits from primary screen and secondary validation assays
| Primary hit | Primary screen |
Secondary validation screen |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Gluc assay |
ATPlite assay |
HCV-Luc assayc | ATPlite assayc | 2-Part HCV infection core-staining assay |
|||||
| Curve class | EC50 (μM) | Efficacy (%) | Curve class | CC50 (μM) | Part 1 (EC50 [μM]) | Part 2 (EC50 [μM]) | |||
| Amperozide hydrochloride | 2 | 7.31 | 92.2 | 4 | >42.0 | 11.9 ± 5.38 | 92.3 ± 3.09 | 1.70 | 2.49 |
| Anthraquinoneb | 1 | 3.66 | 83.0 | 3 | 25.9 | 0.310 ± 0.0900 | 74.5 ± 13.8 | >30.0 | >30.0 |
| Benztropine mesylate | 2 | 3.66 | 55.7 | 4 | >42.0 | 12.6 ± 2.63 | 82.0 ± 6.16 | 2.55 | 2.65 |
| BIO | 1 | 2.06 | 70.1 | 4 | >42.0 | 1.29 ± 0.610 | 88.1 ± 15.5 | >30.0 | 6.48 |
| cis-(Z)-flupenthixol dihydrochloride | 2 | 2.06 | 73.9 | 3 | 20.6 | 4.77 ± 1.49 | 97.8 ± 10.9 | 1.23 | 0.319 |
| Cyclosporine | 1 | 1.16 | 85.6 | 4 | >42.0 | 0.512 ± 0.0600 | 95.6 ± 13.6 | 0.321 | 0.301 |
| Diacylglycerol kinase inhibitor I | 2 | 5.80 | 84.7 | 4 | >42.0 | 35.4 ± 16.3 | 102 ± 8.87 | 12.6 | 0.541 |
| Dilazep hydrochloride | 2 | 8.20 | 116 | 4 | >42.0 | 5.49 ± 2.97 | 80.3 ± 9.86 | 4.56 | 4.38 |
| Ellipticinea | 1 | 3.26 | 90.3 | 4 | >42.0 | 0.440 ± 0.140 | 0.250 ± 0.0600 | NAd | NA |
| Emetine dihydrochloride hydratea | 1 | 0.210 | 85.6 | 3 | 25.9 | 0.360 ± 0.0150 | 35.6 ± 2.12 | NA | NA |
| Fiduxosin hydrochloride | 2 | 9.20 | 103 | 3 | 25.9 | 1.18 ± 0.850 | 100 ± 2.70 | 2.87 | 1.15 |
| Flunarizine dihydrochloride | 1 | 1.64 | 73.5 | 3 | 41.1 | 27.6 ± 9.41 | 82.0 ± 8.59 | 0.613 | 0.270 |
| GR 127935 hydrochloride hydrate | 1 | 2.59 | 86.7 | 3 | 16.4 | 0.780 ± 0.690 | 78.8 ± 10.8 | 0.444 | 0.463 |
| GW2974 | 2 | 9.20 | 94.8 | 4 | >42.0 | 7.68 ± 3.76 | 91.3 ± 6.99 | 17.0 | 19.5 |
| Perphenazine | 2 | 7.31 | 113 | 3 | 16.4 | 0.810 ± 0.400 | 73.6 ± 6.65 | 4.56 | 4.31 |
| Quinacrine dihydrochloride | 2 | 5.80 | 94.9 | 3 | 20.6 | 0.510 ± 0.230 | 78.5 ± 1.20 | 2.93 | 2.81 |
| Ritanserin | 1 | 1.84 | 93.3 | 4 | >42.0 | 1.46 ± 1.09 | 91.5 ± 14.1 | 4.84 | 2.95 |
| Roscovitine | 2 | 8.20 | 90.5 | 4 | >42.0 | 27.5 ± 11.8 | 97.2 ± 10.3 | 24.6 | 19.0 |
| SB 242084 dihydrochloride hydrate | 2 | 9.20 | 93.5 | 3 | 32.6 | 32.0 ± 23.2 | 84.8 ± 15.0 | 10.8 | 4.25 |
| Thioridazine hydrochloride | 2 | 6.51 | 83.2 | 3 | 20.6 | 29.1 ± 8.39 | 98.4 ± 9.97 | 2.16 | 1.96 |
| Trifluoperazine dihydrochlorideb | 2 | 4.11 | 85.2 | 3 | 16.4 | 2.91 ± 2.02 | 99.2 ± 3.62 | 29.7 | 19.1 |
| U-73343a | 1 | 1.46 | 74.8 | 3 | >42.0 | 0.940 ± 1.22 | 0.860 ± 0.960 | NA | NA |
Significant cytotoxicity in the ATPlite assay of the validation screen at a concentration of 10 μM.
Discrepant results between the Luc-based assay and HCV core-staining assay.
Compounds were tested at 10-μM concentration. The numbers (means ± SD) are percentages of the DMSO control from 5 replicates.
NA, not applicable.
FIG 4.

Dose-dependent curves of primary-hit compounds with EC50s of <10 μM, efficacies of >70%, and CC50s of >30 μM from qHTS of the LOPAC. HCV inhibition (symbols and lines in black) was determined by Gluc activity and cytotoxicity (symbols and lines in gray) by ATPlite assay. The figures were plotted based on the results of nine 1,536-well plates of cultured LoxPGLuc cells infected with HCV-Cre and 7 serial dilutions (from 2.56 to 40 μM) of LOPAC compounds as described in Materials and Methods. Two sets of identically treated plates were used in the analyses, one for Gluc activity and the other for cytotoxicity by ATPlite assay.
Confirmation of the primary hits.
The 22 most active compounds selected from the primary screen were examined in two secondary assays, including the HCV-Luc infection assay with 10 μM compounds in a 96-well plate format (Table 1) and a parallel cytotoxicity assay. Three of these compounds produced significant cytotoxicity and were excluded from further follow-up assays. The other 19 compounds were confirmed to inhibit R-Luc activities in the HCV-Luc assay without significant cytotoxicity (Table 1).
A second compound confirmation assay was then performed using the two-part HCV infection with HCV core immunofluorescent staining (25). Because the wild-type HCV was used to infect cells instead of the recombinant HCV, the assay helped to eliminate the false-positive compounds that might inhibit the luciferase reporter nonspecifically. The two-part HCV infection assay can also help determine whether the inhibitor is targeting the early or late stage of the HCV replication cycle (25), as an early-stage inhibitor exhibits dose-dependent inhibition in both parts of the assay and a late-stage inhibitor typically shows more potent inhibition in the part 2 assay. Two hits were eliminated because of minor inhibitions in both parts of the infection assay. Overall, 17 compounds out of the 22 primary hits were confirmed in the above-mentioned secondary assays, with a confirmation rate of 78% (Table 1). Two compounds, BIO [(2′Z,3′E)-6-bromoindirubin-3′-oxime] and diacylglycerol (DAG) kinase inhibitor I, showed significantly greater inhibitory effects in part 2 and minimal effect in part 1 (Fig. 5), suggesting that they target the late stage of the HCV replication cycle. A few other compounds, like ritanserin, also showed a modestly more potent effect in part 2 than in part 1 and are discussed below.
FIG 5.

A two-part HCV infection assay shows distinct patterns between HCV early-stage and late-stage inhibitors. Huh7.5.1 cells cultured in 96-well plates were infected with HCV-WT (MOI, 0.05), together with compounds in 1:3 serial dilutions starting at 30 μM (part 1). Culture media were transferred to a replica plate with fresh cells after 48 h of culture (part 2). Cells from both parts were processed for HCV core immunofluorescent staining. Cell nuclei were counterstained with Hoechst dye, and percentages of HCV core-positive cells were calculated and normalized against the DMSO control and plotted as dose-dependent curves. Representatives of 2 early-stage inhibitors (flunarizine and cyclosporine) and late-stage HCV inhibitors (BIO and DAG kinase inhibitor I) are shown. The data are means ± SEM from 8 replicates.
Characterization of validated hits by HCV replication cycle assays.
In order to select potential lead compounds for further consideration, the following criteria were applied: an EC50 of <10 μM, an efficacy of ≥70%, and a CC50 of >30 μM. A subset of 10 compounds, including cyclosporine (0.78% of the LOPAC), were selected for further characterization (Table 2). Their structures are shown in Fig. 6. The HCV pseudoviral particle (HCVpp) resembles HCV, has the same envelope proteins as HCV, and presumably enters the cell via a similar mechanism (32). Using HCVpp, we studied these 10 compounds for their effects on HCV entry. Dilazep hydrochloride and roscovitine inhibited HCVpp infection (<50% of the DMSO control) at 10 μM concentration (Table 2) and were classified as HCV entry inhibitors. Using the HCV subgenomic replicon assay, we tested the effects of these 10 compounds on HCV replication. We found that SB242084 and cyclosporine significantly inhibited HCV replicon activities (Table 2).
TABLE 2.
Characterization of lead hits in the HCV replication cycle
| Primary hita | Entryb |
Replicationb (replicon) | Single cycleb (HCVsc) | Target stagec | |
|---|---|---|---|---|---|
| HCVpp | VSV-Gpp | ||||
| Amperozide hydrochloride | 107 ± 29.9 | 105 ± 33.6 | 94.3 ± 40.9 | 11.0 ± 8.05 | Undetermined |
| BIO | 104 ± 31.9 | 115 ± 17.2 | 88.3 ± 34.8 | 55.3 ± 0.360 | Assembly/secretion |
| Cyclosporine | 47.5 ± 21.5 | 87.0 ± 14.0 | 0.750 ± 2.06 | 0.750 ± 0.310 | Replication |
| Diacylglycerol kinase inhibitor I | 80.2 ± 19.1 | 73.8 ± 19.7 | 110 ± 26.4 | 50.1 ± 8.87 | Assembly/secretion |
| Dilazep hydrochloride | 38.5 ± 15.5 | 87.9 ± 9.60 | 61.0 ± 6.12 | 2.87 ± 2.67 | Entry |
| Flunarizine dihydrochloride | 124 ± 30.4 | 88.3 ± 13.1 | 94.4 ± 14.6 | 14.9 ± 7.60 | Undetermined |
| GW2974 | 65.7 ± 32.5 | 94.0 ± 20.2 | 123 ± 54.3 | 36.5 ± 2.48 | Entry? |
| Ritanserin | 87.9 ± 23.3 | 76.4 ± 29.7 | 68.9 ± 24.0 | 56.4 ± 2.91 | Assembly/secretion |
| Roscovitine | 45.5 ± 23.3 | 82.9 ± 21.0 | 109 ± 33.7 | 27.0 ± 19.0 | Entry |
| SB 242084 dihydrochloride hydrate | 119 ± 32.5 | 79.4 ± 22.9 | 30.7 ± 21.3 | 12.4 ± 1.22 | Replication |
All the compounds were tested at 10 μM concentration.
The numbers (means ± SD) are percentages of the DMSO control from 5 replicates.
Determination of the specific target stage of the HCV replication cycle is based on various virologic assays (<50% of the control was considered significant).
FIG 6.

Chemical structures of lead compounds selected for characterization of their targeting stages in the HCV replication cycle.
In the HCV single-cycle infection assay, the virus lacks the ability to assemble into new infectious virions after going through entry and replication steps (26). As expected, the two late-stage inhibitors (BIO and DAG kinase inhibitor I) identified from the two-part HCV infection assay showed minimal effects in this assay and also had no significant effect on either HCVpp infection or replicon activity (Table 2). Due to structure similarity (Fig. 6), ritanserin and DAG kinase inhibitor I probably target the same step of the viral replication cycle, i.e., assembly/secretion. Our data are consistent with ritanserin acting on a late stage of the HCV replication cycle (Tables 1 and 2), but it probably also has some effect on the early stage of the HCV replication cycle. Amperozide hydrochloride and flunarizine dihydrochloride could not be specifically assigned to a single stage of the HCV replication cycle because they appeared to inhibit multiple stages of the HCV replication cycle.
DISCUSSION
In this study, we report the development of a novel quantitative HTS assay platform using a cell-based HCV infection system for identification of novel HCV inhibitors. A pilot screen of LOPAC using this assay platform not only yielded active compounds targeting cellular pathways involved in the HCV replication cycle, but also identified novel inhibitors whose mechanism of action has yet to be elucidated. Through systematic characterization of these compounds, we have demonstrated that some of the confirmed inhibitors target various stages of the HCV replication cycle. Therefore, this novel assay platform provides a phenotypic approach for HTS to identify antivirals against HCV.
Recently, several groups have reported cell-based HCV infection systems for the screening of HCV inhibitors in various assay formats (12–15). Traditionally, HTS was performed at a single fixed concentration of each candidate compound, which is prone to false positives and false negatives and requires extensive follow-up studies. It becomes particularly cumbersome for screening libraries exceeding 100,000 compounds, where thousands of primary hits may need to be selected for confirmation after primary screens. The qHTS approach, with a dose-response curve for each compound in the primary screen, produces potency and maximal-response data for the primary hits that allow structural grouping and early structure-activity analysis of the active compounds (24). The hit selection for the confirmation experiments is based on more logical criteria and a small set of selected hits, which can accelerate the hit-to-lead discovery process. Our new HCV assay can be performed in a 1,536-well plate format that is suitable for qHTS of large compound collections.
To generate a highly sensitive HCV reporter system that meets the requirement for qHTS, we utilized the Cre-LoxP system to further amplify the reporter signal, where Cre was inserted into the HCV genome and the LoxPGluc reporter gene was permanently introduced into the HCV-permissive Huh7.5.1 cells (Fig. 1). Conceivably, a few HCV-Cre enzymes can result in the generation of large numbers of Gluc molecules, thus amplifying the reporter signal. As expected, the system greatly increased the sensitivity of detection and allowed us to perform the experiment in 1,536-well plates.
In the system, it is crucial that HCV-Cre is robust in both infection and replication. It is worth noting that insertion of Cre between NS5A and NS5B in the HCV genome does not significantly alter the infectivity and replication of HCV in Huh7.5.1 cells (Fig. 2A), consistent with previous publications (28). The small size of the Cre gene (∼1 kbp) is also an advantage.
Because of the high sensitivity of the reporter activity, the HCV-Cre–LoxPGluc system has a high S/B ratio that is routinely over 40 in the 1,536-well plate format. The wide dynamic range more than overcomes the usually high variation associated with cell-based assays. This feature also contributes to the high Z′ factor of the assay (consistently above 0.6). By using known HCV inhibitors, cyclosporine and 2′-C-methylcytidine, we demonstrated that the platform is very robust and sensitive for the HTS of HCV inhibitors. In addition, a typical dose-response curve can be generated in the system, and therefore, it is suitable for the qHTS format.
Our pilot screen with the Sigma LOPAC containing 1,280 compounds with known pharmacological activities further demonstrated the feasibility and validity of our system for qHTS assay. In compounds exhibiting profiles of robust HCV inhibition and low cytotoxicity based on the dose-dependent curves, we selected 22 primary hits with EC50s of <10 μM for confirmation. Except for 3 compounds showing substantial toxicity and 2 possibly nonspecific inhibitors of the Luc reporter, 17 of them were validated with two independent assay systems. Additionally, 10 of the compounds also met the criteria of an efficacy of >70% and a CC50 of <30 μM, more stringent criteria for the selection of lead compound candidates. These results with the pilot screen suggest that the HCV-Cre–LoxPGluc system is not only quantitatively efficient, but also highly specific.
One of the validated hits was cyclosporine, a known inhibitor of HCV replication, and its derivative is currently under clinical development (33). Several compounds, such as DAG kinase inhibitor I and GW 2974, target cellular pathways that have been implicated in the HCV replication cycle. DAG kinase is an important factor involved in lipid droplet biogenesis (34, 35). Lipid formation plays a crucial role in HCV replication and assembly (36, 37). GW 2974 is an inhibitor of epidermal growth factor receptor (EGFR). EGFR has been implicated in the HCV entry pathway (38). Ritanserin and SB 242084 are 5-HT2 receptor antagonists that are used for the treatment of neurological disorders (39–41). Flunarizine is a nonselective calcium entry blocker with histamine H1 receptor-blocking activity (42), and amperozide is an atypical antipsychotic with high affinity for 5-HT2 receptors (43). These compounds have not been shown to target any steps of the HCV replication cycle and therefore may be explored as new HCV therapeutic agents with potentially novel mechanisms.
Several other compounds possess highly inhibitory activities against HCV (EC50, <10 μM) but have unacceptable toxicity (a CC50 of <30 μM or a CC50/EC50 selectivity of <30). One of them is fluoxetine hydrochloride, which is a serotonin reuptake inhibitor used clinically as an antidepressant (44). Prochlorperazine dimaleate, used clinically as an antiemetic, is an inhibitor of actin polymerization and has been shown to block HCV infection (12). Rottlerin, an inhibitor of Na+/H+ exchange and macropinocytosis, has been shown to be important in the entry processes of several viruses but so far not for HCV (45). Chloroquine, a lysosomotropic agent known to inhibit HCV entry by interfering with clathrin-mediated endocytosis (46) and HCV replication by targeting virus-associated autophagy (47), was not included in our validation list due to its relatively low efficacy (EC50 = 23.11 μM) in our primary screen. This pilot screen demonstrated that this HCV infection assay system can be applied to identify novel HCV inhibitors and is suitable for qHTS.
This cell-based HCV infection assay encompasses the entire HCV replication cycle and therefore is suitable for identification of compounds targeting either host factors or the virus at multiple stages of the HCV replication cycle. With various tertiary assays to characterize the potential lead compound hits from the LOPAC, we showed that the targets of these anti-HCV compounds include all stages of the HCV replication cycle. Therefore, the screening platform developed here provides an unbiased screen of compounds that cover the entire viral replication cycle and should greatly enhance the likelihood of identifying novel HCV inhibitors from large-scale screens of small-molecule libraries.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marissa Winkler and Baihua Zhang for their excellent technical work and Sam Michael and Mike Balcom for assisting in robotically controlled compound administration and plate reading.
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Center for Advancing Translational Sciences, National Institutes of Health.
Footnotes
Published ahead of print 25 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02094-13.
REFERENCES
- 1.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. 2000. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 132:296–305. 10.7326/0003-4819-132-4-200002150-00008 [DOI] [PubMed] [Google Scholar]
- 2.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N. Engl. J. Med. 368:1907–1917. 10.1056/NEJMra1213651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enomoto M, Tamori A, Kawada N. 2009. Emerging antiviral drugs for hepatitis C virus. Rev. Recent Clin. Trials 4:179–184. 10.2174/157488709789957628 [DOI] [PubMed] [Google Scholar]
- 4.Schiffer JT, Scott J, Corey L. 2011. A siege of hepatitis: fighting a defiant virus. Nat. Med. 17:253–254. 10.1038/nm0311-253 [DOI] [PubMed] [Google Scholar]
- 5.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110 [DOI] [PubMed] [Google Scholar]
- 6.Kim SS, Peng LF, Lin W, Choe WH, Sakamoto N, Kato N, Ikeda M, Schreiber SL, Chung RT. 2007. A cell-based, high-throughput screen for small molecule regulators of hepatitis C virus replication. Gastroenterology 132:311–320. 10.1053/j.gastro.2006.10.032 [DOI] [PubMed] [Google Scholar]
- 7.Lapidot A, Berchanski A, Borkow G. 2008. Insight into the mechanisms of aminoglycoside derivatives interaction with HIV-1 entry steps and viral gene transcription. FEBS J. 275:5236–5257. 10.1111/j.1742-4658.2008.06657.x [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharya S, Osman H. 2009. Novel targets for anti-retroviral therapy. J. Infect. 59:377–386. 10.1016/j.jinf.2009.09.014 [DOI] [PubMed] [Google Scholar]
- 9.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462. 10.1053/j.gastro.2009.11.055 [DOI] [PubMed] [Google Scholar]
- 10.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 80:5308–5320. 10.1128/JVI.02460-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gastaminza P, Whitten-Bauer C, Chisari FV. 2010. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. U. S. A. 107:291–296. 10.1073/pnas.0912966107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu X, Sainz B, Jr, Petukhov PA, Uprichard SL. 2012. Identification of hepatitis C virus inhibitors targeting different aspects of infection using a cell-based assay. Antimicrob. Agents Chemother. 56:6109–6120. 10.1128/AAC.01413-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chockalingam K, Simeon RL, Rice CM, Chen Z. 2010. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc. Natl. Acad. Sci. U. S. A. 107:3764–3769. 10.1073/pnas.0915117107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wichroski MJ, Fang J, Eggers BJ, Rose RE, Mazzucco CE, Pokornowski KA, Baldick CJ, Anthony MN, Dowling CJ, Barber LE, Leet JE, Beno BR, Gerritz SW, Agler ML, Cockett MI, Tenney DJ. 2012. High-throughput screening and rapid inhibitor triage using an infectious chimeric Hepatitis C virus. PLoS One 7:e42609. 10.1371/journal.pone.0042609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanegae Y, Lee G, Sato Y, Tanaka M, Nakai M, Sakaki T, Sugano S, Saito I. 1995. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site-specific cre recombinase. Nucleic Acids Res. 23:3816–3821. 10.1093/nar/23.19.3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. 10.1016/0378-1119(91)90434-D [DOI] [PubMed] [Google Scholar]
- 18.Sauer B, Henderson N. 1988. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. U. S. A. 85:5166–5170. 10.1073/pnas.85.14.5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orban PC, Chui D, Marth JD. 1992. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 89:6861–6865. 10.1073/pnas.89.15.6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He JL, Choe S, Walker R, Dimarzio P, Morgan DO, Landau NR. 1995. Human immunodeficiency virus type-1 viral protein r (vpr) arrests cells in the g(2) phase of the cell cycle by inhibiting p34(cdc2) activity. J. Virol. 69:6705–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang LJ, Urlacher V, Iwakuma T, Cui Y, Zucali J. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 6:715–728. 10.1038/sj.gt.3300895 [DOI] [PubMed] [Google Scholar]
- 22.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276. 10.1073/pnas.0832180100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowd KA, Hershow RC, Yawetz S, Larussa P, Diaz C, Landesman SH, Paul ME, Read JS, Lu M, Thomas DL, Netski DM, Ray SC. 2008. Maternal neutralizing antibody and transmission of hepatitis C virus to infants. J. Infect. Dis. 198:1651–1655. 10.1086/593067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. 2006. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U. S. A. 103:11473–11478. 10.1073/pnas.0604348103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. 2009. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U. S. A. 106:16410–16415. 10.1073/pnas.0907439106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masaki T, Suzuki R, Saeed M, Mori K, Matsuda M, Aizaki H, Ishii K, Maki N, Miyamura T, Matsuura Y, Wakita T, Suzuki T. 2010. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. J. Virol. 84:5824–5835. 10.1128/JVI.02397-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Jadhav A, Southal N, Huang R, Nguyen DT. 2010. A grid algorithm for high throughput fitting of dose-response curve data. Curr. Chem. Genomics 4:57–66. 10.2174/1875397301004010057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horwitz JA, Dorner M, Friling T, Donovan BM, Vogt A, Loureiro J, Oh T, Rice CM, Ploss A. 2013. Expression of heterologous proteins flanked by NS3-4A cleavage sites within the hepatitis C virus polyprotein. Virology 439:23–33. 10.1016/j.virol.2013.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa M, Sakamoto N, Enomoto N, Tanabe Y, Kanazawa N, Koyama T, Kurosaki M, Maekawa S, Yamashiro T, Chen CH, Itsui Y, Kakinuma S, Watanabe M. 2004. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 313:42–47. 10.1016/j.bbrc.2003.11.080 [DOI] [PubMed] [Google Scholar]
- 30.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. 2003. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38:1282–1288. 10.1053/jhep.2003.50449 [DOI] [PubMed] [Google Scholar]
- 31.Bassit L, Grier J, Bennett M, Schinazi RF. 2008. Combinations of 2′-C-methylcytidine analogues with interferon-alpha2b and triple combination with ribavirin in the hepatitis C virus replicon system. Antivir. Chem. Chemother. 19:25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartosch B, Cosset FL. 2009. Studying HCV cell entry with HCV pseudoparticles (HCVpp). Methods Mol. Biol. 510:279–293. 10.1007/978-1-59745-394-3_21 [DOI] [PubMed] [Google Scholar]
- 33.Fischer G, Gallay P, Hopkins S. 2010. Cyclophilin inhibitors for the treatment of HCV infection. Curr. Opin. Investig. Drugs 11:911–918 [PubMed] [Google Scholar]
- 34.Turro S, Ingelmo-Torres M, Estanyol JM, Tebar F, Fernandez MA, Albor CV, Gaus K, Grewal T, Enrich C, Pol A. 2006. Identification and characterization of associated with [sic] lipid droplet protein 1: a novel membrane-associated protein that resides on hepatic lipid droplets. Traffic 7:1254–1269. 10.1111/j.1600-0854.2006.00465.x [DOI] [PubMed] [Google Scholar]
- 35.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Jr, Ott M. 2010. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16:1295–1298. 10.1038/nm.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 19:95–103. 10.1016/j.tim.2010.11.005 [DOI] [PubMed] [Google Scholar]
- 37.Li Q, Pene V, Krishnamurthy S, Cha H, Liang TJ. 2013. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat. Med. 19:722–729. 10.1038/nm.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17:589–595. 10.1038/nm.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nappi G, Sandrini G, Granella F, Ruiz L, Cerutti G, Facchinetti F, Blandini F, Manzoni GC. 1990. A new 5-HT2 antagonist (ritanserin) in the treatment of chronic headache with depression. A double-blind study vs amitriptyline. Headache 30:439–444 [DOI] [PubMed] [Google Scholar]
- 40.Bromidge SM, Duckworth M, Forbes IT, Ham P, King FD, Thewlis KM, Blaney FE, Naylor CB, Blackburn TP, Kennett GA, Wood MD, Clarke SE. 1997. 6-Chloro-5-methyl-1-[[2-[(2-methyl-3-pyridyl)oxy]-5-pyridyl]carbamoyl]-indoline (SB-242084): the first selective and brain penetrant 5-HT2C receptor antagonist. J. Med. Chem. 40:3494–3496. 10.1021/jm970424c [DOI] [PubMed] [Google Scholar]
- 41.Kennett GA, Wood MD, Bright F, Trail B, Riley G, Holland V, Avenell KY, Stean T, Upton N, Bromidge S, Forbes IT, Brown AM, Middlemiss DN, Blackburn TP. 1997. SB 242084, a selective and brain penetrant 5-HT2C receptor antagonist. Neuropharmacology 36:609–620. 10.1016/S0028-3908(97)00038-5 [DOI] [PubMed] [Google Scholar]
- 42.Amery WK. 1983. Flunarizine, a calcium channel blocker: a new prophylactic drug in migraine. Headache 23:70–74. 10.1111/j.1526-4610.1983.hed2302070.x [DOI] [PubMed] [Google Scholar]
- 43.Axelsson R, Nilsson A, Christensson E, Bjork A. 1991. Effects of amperozide in schizophrenia. An open study of a potent 5-HT2 receptor antagonist. Psychopharmacology 104:287–292 [DOI] [PubMed] [Google Scholar]
- 44.Chen J, Gao K, Kemp DE. 2011. Second-generation antipsychotics in major depressive disorder: update and clinical perspective. Curr. Opin. Psychiatry 24:10–17. 10.1097/YCO.0b013e3283413505 [DOI] [PubMed] [Google Scholar]
- 45.Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 83:4895–4911. 10.1128/JVI.02498-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80:6964–6972. 10.1128/JVI.00024-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, Ikejima K, Kitamura T, Enomoto N, Sakai T, Kominami E, Watanabe S. 2010. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 45:195–203. 10.1007/s00535-009-0132-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.