

Abstract
Alpha-toxin (AT) is a major virulence factor in the disease pathogenesis of Staphylococcus aureus. We previously identified a monoclonal antibody (MAb) against AT that reduced disease severity in a mouse dermonecrosis model. Here, we evaluate the activity of an affinity-optimized variant, LC10, in a mouse model of S. aureus pneumonia. Passive immunization with LC10 increased survival and reduced bacterial numbers in the lungs and kidneys of infected mice and showed protection against diverse S. aureus clinical isolates. The lungs of S. aureus-infected mice exhibited bacterial pneumonia, including widespread inflammation, whereas the lungs of mice that received LC10 exhibited minimal inflammation and retained healthy architecture. Consistent with reduced immune cell infiltration, LC10-treated animals had significantly lower (P < 0.05) proinflammatory cytokine and chemokine levels in the bronchoalveolar lavage fluid than did those of the control animals. This reduction in inflammation and damage to the LC10-treated animals resulted in reduced vascular protein leakage and CO2 levels in the blood. LC10 was also assessed for its therapeutic activity in combination with vancomycin or linezolid. Treatment with a combination of LC10 and vancomycin or linezolid resulted in a significant increase (P < 0.05) in survival relative to the monotherapies and was deemed additive to synergistic by isobologram analysis. Consistent with improved survival, the lungs of animals treated with antibiotic plus LC10 exhibited less inflammatory tissue damage than those that received monotherapy. These data provide insight into the mechanisms of protection provided by AT inhibition and support AT as a promising target for immunoprophylaxis or adjunctive therapy against S. aureus pneumonia.
INTRODUCTION
Staphylococcus aureus is a leading cause of pneumonia in hospitalized patients and increasingly in healthy individuals in the community (1, 2). S. aureus pneumonia is a life-threatening disease, with mortality rates reported to be as high as 60% (2). Treatment of these infections is complicated by the fact that ∼50% of S. aureus isolates from patients with pneumonia are methicillin-resistant S. aureus (MRSA), thereby reducing safe and effective treatment options (1). While vancomycin (VAN) is the primary therapy for MRSA pneumonia, the mortality rate in pneumonia patients treated with vancomycin remains high, and the emergence of intermediate resistance to glycopeptides potentially limits the usefulness of this class of antibiotics (3–5). Linezolid (LZD) is currently the only other drug with anti-MRSA activity that is approved for the treatment of nosocomial pneumonia in the United States and Europe (4, 6). A consistent evolution toward antibiotic resistance, along with the scarcity of new agents, has led to the exploration of alternate methods of prophylaxis and therapy against various bacterial pathogens (7–10). One such approach is the development of monoclonal antibodies (MAbs) targeting S. aureus and its virulence determinants, which may be used in prophylaxis or perhaps in adjunctive therapy with antibiotics (11–15).
Mice infected intranasally (i.n.) with S. aureus exhibit an early increase in proinflammatory mediators (e.g., interleukin-1 beta [IL-1β], keratinocyte chemoattractant [KC], and macrophage inflammatory protein 2 [MIP-2]), leading to increased lung protein levels, polymorphonuclear leukocyte (PMN) influx, necrosis, and ultimately a consolidating pneumonia similar to that seen in humans (16–18). A key virulence determinant involved in the pathogenesis of murine S. aureus pneumonia is the pore-forming toxin, alpha-toxin (AT). AT is secreted as a 33-kDa soluble monomer that binds to the recently identified receptor, ADAM-10, on target cell membranes (19). After binding, AT undergoes a conformational change resulting in the formation of a heptameric transmembrane β-barrel, leading to cell lysis. At sublytic concentrations, AT has also been demonstrated to exert significant cytotoxic effects (20). AT binding and oligomerization on macrophage membranes activate the NLRP3 inflammasome that along with other staphylococcal pathogen-associated molecular patterns (PAMPs) induces IL-1β secretion and promotes cell death (21, 22). AT also activates ADAM-10-mediated proteolysis of E-cadherin present in cell-cell adhesive contacts, leading to a disruption in epithelial and endothelial integrity and contributing to the epithelial damage typically seen in pneumonia (23–27). Consistent with the inhibition of these activities, active and passive immunization directed against AT has been demonstrated to limit the severity of S. aureus pneumonia in mice (12, 13, 28).
We previously identified the monoclonal antibody (MAb) 2A3, which neutralizes AT and promotes a robust host immune response leading to reduced disease severity in a mouse dermonecrosis model (11, 29). Here, we evaluated the efficacy and the mechanism of action of LC10, an affinity-optimized 2A3 variant, in a murine S. aureus pneumonia model. We demonstrate that LC10 prophylaxis results in improved survival and a reduction in the hyperinflammatory response and lung damage associated with S. aureus pneumonia. Additionally, the therapeutic administration of LC10 in combination with either of two frontline antibiotics, vancomycin or linezolid, resulted in reduced lung damage and improved survival relative to use of the antibiotics alone. Taken together, these results provide support for the continued development of an anti-AT approach for the prevention or treatment of S. aureus pneumonia.
MATERIALS AND METHODS
Bacterial strains and chemicals.
S. aureus strains NRS382 (pulsed-field gel electrophoresis [PFGE] type USA100, clonal complex 5 [CC5]) and NRS261 (CC30) were obtained from the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) (Chantilly, VA). S. aureus strain FPR3757 (ATCC BAA-1556, PFGE type USA300) was obtained from the American Type Culture Collection, and S. aureus strain SF8300 (PFGE type USA300) was generously provided by Binh An Diep (University of California at San Francisco).
The S. aureus strains were streaked onto Trypticase soy agar (TSA) plates (BBL, Becton, Dickinson Laboratories, Franklin Lakes, NJ) and grown overnight at 37°C in Trypticase soy broth (TSB) (BBL). The bacteria were diluted 1:100 in TSB and grown to an optical density at 600 nm (OD600) of ∼0.8. The culture was centrifuged and the bacterial pellet resuspended in phosphate-buffered saline (PBS) (pH 7.2) (Invitrogen, Carlsbad, CA) with 10% glycerol to ∼1 × 1010 CFU/ml; the aliquots were stored at −80°C.
Vancomycin (VAN) (Sigma-Aldrich, St. Louis, MO) was prepared in 5% dextrose, and linezolid (LZD) (Tecoland Corporation, Edison, NJ) was dissolved in 5% aqueous hydroxypropyl-beta-cyclodextrin (Sigma-Aldrich, St. Louis, MO). Anti-AT MAb LC10 and isotype control R347 were diluted in sterile PBS (pH 7.2).
In vitro susceptibility testing.
MICs were determined by the broth microdilution method, according to the CLSI guidelines (11). The MIC was defined as the lowest concentration of antibiotic that prevented visible growth after an incubation period of 16 to 20 h.
In vivo models.
All animal studies were approved by the MedImmune Institutional Animal Care and Usage Committee and were conducted in an Association for Accreditation and Assessment of Laboratory Animal Care (AAALAC)-accredited facility in compliance with U.S. regulations governing the housing and use of animals.
Murine pneumonia model.
Specific-pathogen-free 7- to 9-week-old female C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were anesthetized with 3% isoflurane (Butler Schein Animal Health, Dublin, OH) and maintained with oxygen at 3 liters/min before inoculation with 50 μl of an S. aureus suspension (1 × 108 to 2 × 108 CFU) into the left and right nares. The animals were placed into a cage in a supine position for recovery. The mice were either sacrificed at predetermined time points or assessed for survival for 7 days following bacterial challenge. In prophylaxis, LC10 and the isotype control R347 were administered intraperitoneally (i.p.) 24 h prior to the bacterial challenge or 1 h postinfection. For therapeutic administration, the mice were dosed subcutaneously 1 h postinfection. Vancomycin dosing was continued at 12, 24, 36, 48, and 60 h postinfection. Linezolid treatment was discontinued after 36 h. LC10 was administered i.p. as a single therapeutic dose 1 h postinfection. To evaluate the synergy between LC10 and the antibiotics, vancomycin was titrated from 200 mg/kg of body weight/day to 25 mg/kg/day, with LC10 at doses from 45 mg/kg/day to 5 mg/kg/day. Linezolid was dosed at concentrations from 1.25 mg/kg/day to 20 mg/kg/day, and LC10 was titrated from 5 mg/kg/day to 45 mg/kg/day. The isobole exponents were determined using the isobologram equation (30).
CFU determination in lungs and kidneys.
Lung and kidney samples were harvested into 1.5-ml Lysing Matrix A tubes (MP Biomedicals, Solon, OH) and homogenized for 30 to 60 s using a FastPrep-24 homogenizer (MP Biomedicals). Organ homogenates were then serially diluted in PBS and plated on TSA plates for CFU enumeration.
Histopathology.
The infected mice (n = 5) were euthanized and the lungs were removed, inflated, and fixed in 10% neutral buffered formalin (VWR, Radnor, PA). The lungs were processed and embedded in a frontal plane in molten paraffin wax at approximately 60°C, and the sections were cut at 4 μm on a rotary microtome. All sections were allowed to dry and placed in a 60°C oven for ≥15 min prior to staining with Gill's hematoxylin (Mercedes Medical, Sarasota, FL) and eosin (Surgipath, Richmond, IL). All stained sections were analyzed using a Nikon 80i microscope with 10× and 40× objectives and reviewed by a pathologist who was blinded to the treatment groups.
Bronchoalveolar lavage.
Bronchoalveolar lavage (BAL) was performed at indicated time points. Each animal was euthanized with CO2, the chest was opened, and the trachea was exposed. One milliliter sterile PBS was flushed into the lungs, and the BAL fluid was harvested using a 1-ml syringe. The samples were centrifuged at 8,000 rpm for 5 min, and the supernatants were aliquoted for storage at −80°C. BAL fluid was assayed for total protein concentration using a modification of the Bradford and Lowry assay methods (Bio-Rad Laboratories, Richmond, CA), according to the manufacturer's specifications.
Cytokine analysis.
BAL fluid aliquots were thawed on ice and the cytokine and chemokine levels were analyzed using a Meso Scale proinflammatory cytokine kit (Meso Scale Discovery, Gaithersburg, MD) or MILLIPLEX MAP mouse cytokine/chemokine kit (Millipore), according to the instructions of each manufacturer. The data were expressed in pg/ml.
Arterial blood gas analyses.
Blood was drawn from a tail artery into a heparinized syringe using a mouse restraint device. Blood gas analysis was immediately performed using a VetScan i-STAT 1 analyzer (Abaxis, Union City, CA).
Statistical analyses.
The data were analyzed using t tests, Mann-Whitney tests, analysis of variance (ANOVA) followed by Dunnett's test, Kruskal-Wallis followed by Dunn's test, or log-rank test where appropriate. All statistical analyses were performed using GraphPad Prism version 5.0. A P value of <0.05 was considered statistically significant.
RESULTS
Anti-AT significantly increases survival in a murine pneumonia model.
We previously reported the identification of a potent AT-neutralizing MAb, 2A3, which significantly reduced lesion size when delivered prophylactically in a murine dermonecrosis model and its activity correlated directly with affinity (11). 2A3 was affinity optimized by systematic mutation of its complementarity determining regions (CDRs), followed by competition-based selections. This led to the generation of the 2A3 derivative, LC10, which exhibited a ∼10-fold-higher affinity for AT (kD, 89 pM). Although its affinity was increased, the potency of LC10 remained similar to that of 2A3 in vitro and in vivo (see Fig. S1 in the supplemental material). To assess the activity of LC10 in S. aureus-induced pneumonia, 7-week-old C57BL/6J mice were passively immunized with LC10 (at 20, 12, 6, 3, 1.5, or 0.5 mg/kg) or isotype control R347 (15 mg/kg) 24 h prior to intranasal (i.n.) infection with 3 × 108 CFU S. aureus FPR3757 (USA300), and survival was monitored for 7 days. LC10 prophylaxis resulted in a dose-dependent increase in survival relative to an isotype-matched control MAb, R347 (P = 0.0005 at a dose of 0.5 mg/kg) (Fig. 1A). To determine if LC10 protected from severe disease caused by diverse clinical isolates, mice (n = 10) were passively immunized with LC10 (at 45, 15, or 5 mg/kg) or R347 (45 mg/kg) 24 h prior to intranasal infection with ∼2 × 108 CFU of NRS382 (USA100, CC5), NRS261 (CC30), or SF8300 (USA300). In each case, LC10 prophylaxis resulted in a significant increase in survival (P < 0.005) over the 7-day experiment (Fig. 1B to D), indicating that LC10 can provide protection against diverse S. aureus clinical isolates.
FIG 1.

Anti-AT MAbs prevent death in murine pneumonia model. C57BL/6J mice (n = 10) were injected i.p. with anti-AT MAb or R347 as a negative control. The animals were i.n. challenged 24 h later with 3 × 108 CFU of S. aureus USA300 (A), NRS261 (B), NRS382 (C) or SF8300 (D). Survival was monitored for 6 days postinfection. The asterisks indicate statistical significance versus the R347-treated group using the log-rank test.
Fc-γ receptor binding in vivo does not play a significant role in LC10 activity.
The binding of anthrax toxin-neutralizing IgG to Fc-γ receptors on phagocytic cells has been reported to be important for complete anthrax toxin neutralization in vitro and in vivo (31, 32). It is unknown if this is also an important attribute for effective AT neutralization in vivo. Therefore, LC10 harboring the N297Q Fc mutation (LC10N297Q) was constructed to determine if binding to Fc-γ receptors was required for AT neutralization in a murine pneumonia model. N297Q renders IgG heavy chains aglycosylated and defective for Fc gamma receptor (Fc-γR) binding (33–35). LC10N297Q Fc was confirmed to be aglycosylated and to be defective for binding to a murine Fc-γ receptor (see Fig. S2 and S3 in the supplemental material). Prophylaxis with LC10N297Q resulted in survival rates in S. aureus pneumonia similar to those of LC10 (Fig. 2), indicating that binding to Fc-γ receptors does not play a significant role in AT neutralization by LC10 and protection from death in a murine pneumonia model.
FIG 2.

Fc function is not required for LC10 improved survival. C57BL/6 mice (n = 10) were i.p. injected with LC10, LC10N297Q, or R347 as a negative control. The animals were challenged i.n. 24 h later with 3 × 108 CFU of S. aureus USA300. Survival was monitored for 6 days postinfection.
LC10 reduces bacterial load.
To better understand the effect of LC10 on the pathogenesis of S. aureus pneumonia, the bacterial numbers in the lungs and kidneys of the infected mice were assessed 48 h postinfection. The animals passively immunized with LC10 (at a dose of 15 mg/kg) exhibited a significant reduction (P < 0.011) in bacterial CFU in the lungs of infected mice relative to the R347 group (Fig. 3A). Although not significant, LC10 prophylaxis also reduced hematogenous bacterial spread to the kidneys to a level at which the bacteria were detectable in only 2/10 animals compared with 7/10 in the R347-treated group (Fig. 3B). AT neutralization by LC10 during pneumonia therefore reduces bacterial load in the lungs and reduces bacterial dissemination.
FIG 3.

Anti-AT MAbs reduce bacterial numbers in murine pneumonia model. C57BL/6J (n = 10) mice were injected i.p. with LC10 or negative control, R347 (15 mg/kg). The animals were challenged i.n. 24 h later with 1 × 108 CFU SF8300. The lungs (A) and kidneys (B) were harvested 24 h postinfection and processed for CFU enumeration.
LC10 prophylaxis reduces inflammation and pulmonary damage associated with S. aureus pneumonia.
S. aureus pneumonia is characterized by neutrophilic inflammation leading to alveolar consolidation and a loss of alveolar structure (16, 17, 36, 37). Therefore, the effect of LC10 prophylaxis on lung pathology during S. aureus pneumonia was examined 14 h postinfection. Histologically, there was a clear difference in the lungs of LC10-treated mice compared to the lungs of the mice that received R347 (Fig. 4A). Consistent with S. aureus pneumonia, lungs from the mice that were passively immunized with R347 exhibited areas of severe alveolar inflammation characterized by prominent accumulations of bacteria admixed with abundant neutrophils with cellular debris, necrosis, edema, and hemorrhage. In contrast, the lungs of mice that received LC10 exhibited mild to moderate alveolar inflammation with alveolar and perivascular neutrophil infiltrates. There was no evidence of necrosis or damage to the alveolar architecture in the LC10-treated animals (Fig. 4B). To gain insight into the effect of AT neutralization on the local inflammatory response, proinflammatory cytokine and chemokine levels in the BAL fluid of S. aureus-infected mice passively immunized with LC10 or R347 were quantified. Consistent with the histopathology results, the animals immunized with LC10 exhibited a significant decrease (P < 0.05) in the levels of 16 proinflammatory cytokines and chemokines, such as IL-6, IL-1β, and tumor necrosis factor (TNF), relative to those that received R347 (Fig. 5). These results indicate that LC10 prophylaxis blunts an AT-mediated hyperinflammatory cytokine response typically seen during S. aureus pneumonia, resulting in reduced immune cell infiltration and pathophysiology.
FIG 4.
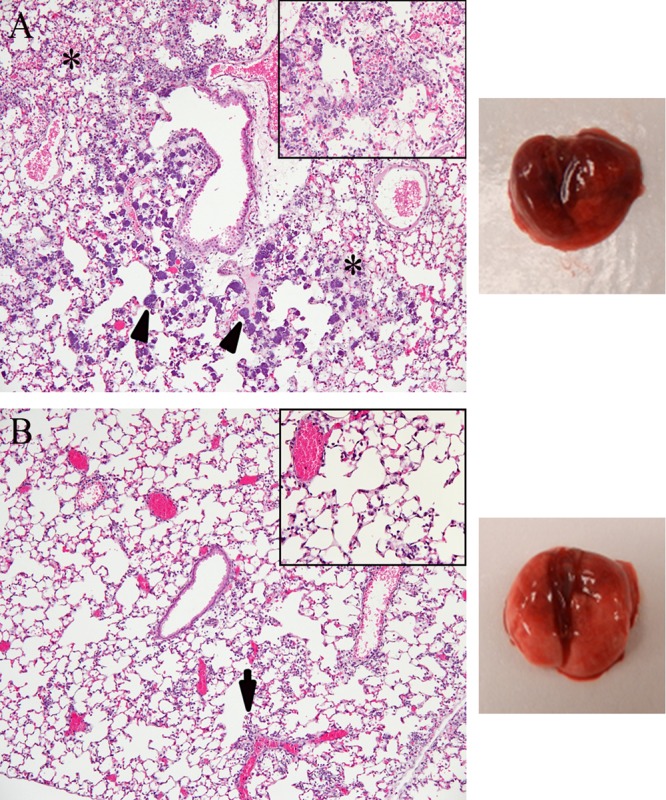
Anti-AT MAbs reduce pulmonary damage in prophylaxis. Mice received 15 mg/kg LC10 or R347 i.p. 24 h prior to infection with SF8300 (1.5 × 108 CFU). The mouse lungs were harvested 14 h after infection, sectioned, stained, and examined microscopically. (A) In the R347 group, there was marked to severe alveolar inflammation with neutrophils, areas of necrosis, edema, and hemorrhage (denoted by asterisks) admixed with bacterial colonies (arrowhead) (10×); inset, bacteria admixed with abundant neutrophils in alveolar spaces (40×). (B) In the LC10 group, there was mild inflammation of neutrophils in alveolar spaces and mild perivascular cuffing (arrow) (10×); inset, few neutrophils in alveolar spaces (40×).
FIG 5.

Proinflammatory cytokine and chemokine response in BAL fluids. The animals (n = 10) were i.n. challenged with SF8300 24 h after i.p. injection with anti-AT MAb LC10 or irrelevant human IgG1 R347. Fourteen hours postinfection, BAL fluids were harvested and stored at −80°C. Cytokine/chemokine levels were measured by Luminex. The data were analyzed by the t test, and the asterisks indicate statistical difference compared with the R347 group (P < 0.05). G-CSF, granulocyte colony-stimulating factor; IP10, inducible protein 10; LIF, leukemia inhibitory factor; LIX, lipopolysaccharide-induced CXC chemokine.
LC10 prophylaxis preserves lung integrity and improves lung function.
Two hallmarks of the pulmonary inflammation and damage seen during bacterial pneumonia are vascular leakage, resulting in increased lung protein levels, and reduced lung function/gas exchange (16, 36, 38–40). To determine if LC10 prophylaxis had an impact on these measures of lung integrity and pulmonary function, BAL fluid and arterial blood samples were collected from infected mice passively immunized with LC10 or R347 to measure protein leakage into the lungs and the carbon dioxide tension (PaCO2) in the arterial blood, respectively. The mice that were passively immunized with LC10 had significantly reduced total protein levels (P = 0.005) as determined by the Bradford protein assay (Fig. 6A). Furthermore, mice that received LC10 had significantly lower arterial PaCO2 (P < 0.0001) (Fig. 6B) than the R347-treated mice, which is indicative of improved lung function and gas exchange following prophylaxis with LC10. These results provide further evidence for the protective effect of LC10 on lung integrity and lung function.
FIG 6.

Total protein concentration and arterial blood gas. The animals (n = 10) were challenged i.n. with SF8300 24 h after i.p. injection with anti-AT MAb LC10 or irrelevant human IgG1 R347. (A) Fourteen hours postinfection, BAL fluids were harvested, and the total protein concentration was analyzed using Bradford and Lowry assay methods. The data were analyzed by the t test. (B) Fourteen hours postinfection, blood was drawn from a tail artery into a heparinized syringe. Blood gas analysis was performed using a VetScan i-STAT 1 analyzer. The data were analyzed by the t test.
LC10 exhibits synergy when administered with vancomycin or linezolid.
The results above indicate that LC10 prophylaxis reduces S. aureus pneumonia disease severity. S. aureus infections are difficult to treat even when the infecting strains are susceptible to antibiotics (41–43). Foletti et al. (13) reported on the benefit of the therapeutic administration of an anti-AT MAb after S. aureus infection in a murine pneumonia model. Additionally, they demonstrated that treatment with their anti-AT in combination with the protein synthesis inhibitor linezolid resulted in an improved outcome relative to treatment with either monotherapy. We took this further and evaluated different therapeutic dose combinations of LC10 (intraperitoneal) with vancomycin or linezolid (subcutaneous) and conducted isobologram analyses using the lethal pneumonia model to quantify the interaction between LC10 and vancomycin or linezolid. All molecules were delivered 1 h postinfection. An isobologram model adequately described the effects of vancomycin and LC10 combinations with an isobole exponent of 0.72, indicating a moderate synergistic effect (Fig. 7A). This is illustrated in Fig. 7B, in which the treatment of S. aureus-infected mice with subtherapeutic concentrations of LC10 (15 mg/kg) plus vancomycin (40 mg/kg/day) resulted in a significant increase (P < 0.026) in survival relative to treatment with either monotherapy. These subtherapeutic concentrations were determined empirically by dose-ranging experiments. Isobologram analysis of LC10 in combination with linezolid revealed a linezolid-dose-dependent isobole exponent. The isobole exponent increased from 0.48 (synergy) at a linezolid dose of 1.25 mg/kg/day to 1.0 (additive) at doses of 5 to 10 mg/kg/day (Fig. 7C). In other words, the synergy between LC10 and linezolid decreased as the linezolid dose increased. This response, shown in Fig. 7D, likely results from linezolid-mediated inhibition of all bacterial protein synthesis, including AT. Overall, the results presented here indicate that S. aureus pneumonia may be easier to treat with antibiotics when LC10 is present, with the effects of LC10 treatment being additive or synergistic depending on the type and dose of antibiotic administered.
FIG 7.

The addition of LC10 to vancomycin or linezolid treatment enhances survival. (A) Groups of mice were infected and treated 1 h postinfection with VAN at concentrations ranging from 5 mg/kg/day to 200 mg/kg/day and with LC10 at 5 mg/kg to 100 mg/kg. (B) LZD doses ranged from 1.25 mg/kg/day to 10 mg/kg/day. The LC10 concentration was fixed at 15 mg/kg. Survival was monitored for 7 days. The closed and open circles represent the observed % survival. The solid lines represent the predicted % survival. (C) Groups of mice were infected and treated as described above with VAN (40 mg/kg/day, filled green squares), VAN plus LC10 (40 mg/kg/day plus 15 mg/kg, open green squares), LC10 (15 mg/kg/day, filled purple diamonds), or R347 (15 mg/kg/day, filled black circles). The addition of LC10 to vancomycin was synergistic and significantly improved survival compared to vancomycin monotherapy (P < 0.026, log-rank test). (D) Filled blue circles, LZD (5 mg/kg/day); open blue circles, LZD plus LC10 (5 mg/kg/day plus 15 mg/kg); filled teal triangles, LZD (2.5 mg/kg/day); open teal triangles, LZD plus LC10 (2.5 mg/kg/day plus 15 mg/kg); filled purple diamonds, LC10 (15 mg/kg/day); filled black circles, R347 (15 mg/kg/day). A dose of LZD at 5 mg/kg/day was additive when added to LC10 (P < 0.027, log-rank test). LZD at 2.5 mg/kg/day was synergistic with linezolid and enhanced survival relative to linezolid monotherapy (P < 0.0001, log-rank test).
LC10 in combination with vancomycin or linezolid improves lung pathology and reduces bacterial load.
In prophylaxis, LC10 improved survival and reduced immune cell infiltration and damage to infected lungs. To determine if the survival benefit afforded by the combination of LC10 plus antibiotic therapy resulted in reduced lung damage, lung histopathology was also examined following treatment with antibiotic monotherapy or antibiotic plus LC10 combination therapy. To mimic the treatment of an infection with a bacterium with reduced susceptibility or inadequate exposure, the mice were dosed 1 h postinfection with concentrations of LC10 (intraperitoneal) and antibiotics (subcutaneous) that resulted in 30 to 40% survival when administered alone. These concentrations were determined empirically by dose-ranging experiments (data not shown). Lung sections from the mice infected with S. aureus and treated 1 h postinfection with vancomycin, linezolid, LC10, or LC10 plus antibiotic combinations were examined microscopically following hematoxylin and eosin (H&E) staining (Fig. 8). Twenty-four hours postinfection, the lungs from the mice that were treated with R347 exhibited severe multifocal alveolar inflammation with necrosis and abundant bacterial colonies. The lungs of the animals that were treated with subtherapeutic doses of vancomycin (40 mg/kg/day) or LC10 (15 mg/kg/day) exhibited areas of marked neutrophilic inflammation, necrosis, and scattered bacterial colonies, along with areas of alveolar necrosis, hemorrhage, and loss of alveolar architecture. In contrast, mice that received the vancomycin plus LC10 combination therapy had areas of mild to moderate inflammation, with occasional bacterial colonies in the alveolar spaces and little or no evidence of necrosis, hemorrhage, or damage to the alveolar architecture. The lungs of mice treated with a subtherapeutic linezolid dose (5 mg/kg/day) exhibited scattered bacterial colonies with areas of edema and necrosis, whereas in the lung sections of mice treated with a linezolid-plus-LC10 combination, there was moderate inflammation, and scattered bacterial colonies were apparent with no evidence of edema or necrosis. Consistent with the histopathology results, the bacterial numbers were also found to be reduced in the lungs of mice that received the combination therapies compared with those that received one of the monotherapies (Table 1). These results support the survival data presented above and provide further support for the hypothesis that S. aureus pneumonia is easier to treat with antibiotics when LC10 is present.
FIG 8.
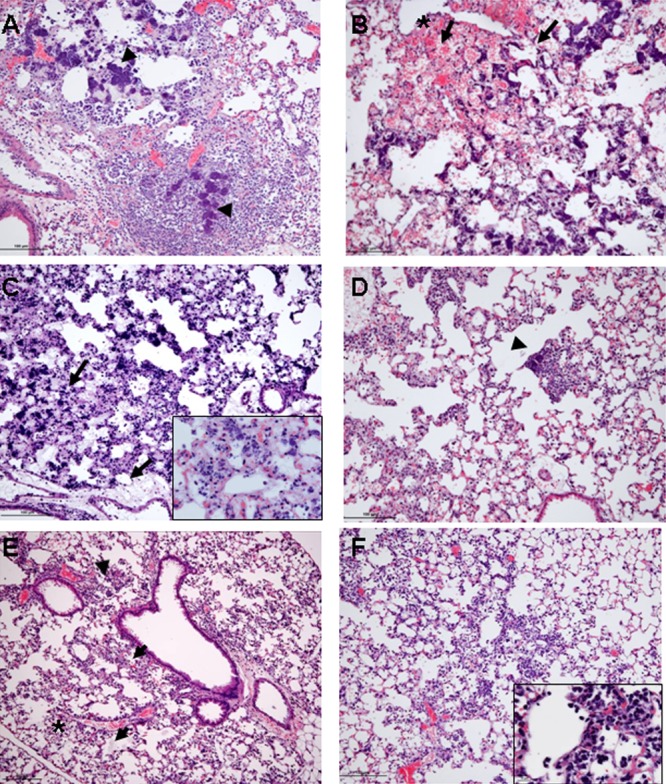
Antibody plus antibiotic combination therapy reduces pulmonary damage. The histopathology of mouse lungs was evaluated 24 h postinfection with BAA-1556 and treatment 1 h postinfection as described above with the following: (A) R347, marked to severe inflammation with edema and necrosis and bacterial colonies (arrowhead); (B) LC10, severe inflammation with prominent bacterial colonies, necrosis, edema, hemorrhage (denoted by asterisk), and multifocal loss of alveolar architecture; (C) vancomycin, scattered bacterial colonies with areas of edema and necrosis (inset showing edema, proteinaceous material in alveolar space 40×); (D) vancomycin plus LC10, moderate inflammation with some bacterial colonies (arrowhead); (E) linezolid, mild to moderate neutrophilic inflammation with areas of necrosis (denoted by asterisk) and scattered bacterial colonies (arrowhead); (F) linezolid plus LC10, mild inflammation with isolated bacteria (inset showing bacterial colonies 40×). All photomicrographs are at ×10 magnification.
TABLE 1.
CFU recovered from lung homogenates of C57BL/6J mice infected with MRSA and treated with various therapies
| Treatment | Log10 CFU/ml | SEM |
|---|---|---|
| VAN | 8.88 | 0.22 |
| VAN + LC10 | 6.79a | 1.64 |
| LZD | 7.50 | 0.64 |
| LZD + LC10 | 6.28 | 1.23 |
| LC10 | 9.12 | 1.09 |
| R347 | 8.62 | 0.28 |
Significantly different from VAN monotherapy (P < 0.0.03, t test).
DISCUSSION
Alpha-toxin (AT) is a pore-forming toxin produced by a majority of S. aureus clinical isolates, and it has been demonstrated to be a key virulence determinant in numerous S. aureus infection models (e.g., dermonecrosis, pneumonia, sepsis, endocarditis, and keratitis) using S. aureus mutants defective for AT expression or methods of passive and active immunization (11–13, 28, 44–50). As a consequence, AT is actively under investigation as a target for immunoprophylactic methods against S. aureus disease.
We previously identified a high-affinity AT-neutralizing MAb, 2A3, and reported on its efficacy in a murine dermonecrosis model (11). Here, we report on LC10, an affinity-optimized version of 2A3, and characterize its mechanism of action when administered both in prophylactic and in adjunctive therapy with antibiotics in an acute S. aureus pneumonia model. LC10 prophylaxis protected mice from lethal intranasal challenges with diverse clinical isolates (FPR3757 [USA300], NRS261 [CC30], NRS382 [USA100], and SF8300 [USA300]) and exhibited a 90% effective dose (ED90) and 50% effective dose (ED50) of 14.3 mg/kg and 4.3 mg/kg, respectively, against S. aureus FPR3757 (USA300). Prophylaxis with LC10 also resulted in a significant decrease in bacterial burden in the lungs and reduced bacterial spread to the kidneys in the infected mice. These results are consistent with published results, demonstrating that prophylaxis with an anti-AT MAb prior to intranasal challenge with S. aureus reduces disease severity and promotes survival (12).
An intense inflammatory response leading to damaging immune cell infiltration is thought to play a major role in the pathogenesis of bacterial pneumonia and acute lung injury in humans (51, 52). S. aureus pneumonia is similarly characterized by a robust inflammatory response and immune cell infiltration leading to localized alveolar consolidation, necrosis, and the destruction of the lung alveolar architecture (16, 17). AT is a key virulence determinant in S. aureus pneumonia and plays a role in both the hyperinflammatory response and lung damage (20–23, 53). AT has been reported to activate the NLRP3 inflammasome in vitro and in vivo, resulting in IL-1β production and increased disease severity in a mouse pneumonia model (21, 22). IL-1β has been reported to induce the production of a variety of chemokines, such as MIP-2, MIP-1α, monocyte chemotactic protein 1 (MCP-1), and IL-8 (KC), which are involved in cellular recruitment (52). Supporting a role for AT in IL-1β expression and the inflammatory response, Bartlett et al. (18) showed that mice challenged intranasally with an AT knockout mutant exhibited a significant reduction in IL-1β, KC, and MIP-2, which correlated with a reduction in neutrophil influx into the lungs. Similarly, active immunization targeting AT has been shown to reduce IL-1β levels in mice with S. aureus pneumonia (28). To further characterize the effect of AT neutralization on the damaging inflammatory response associated with S. aureus pneumonia, a panel of cytokines and chemokines was measured in bronchoalveolar lavage (BAL) fluid samples of mice passively immunized with either the isotype control IgG R347 or LC10. Not only did AT neutralization with LC10 reduce KC, MIP-2, and IL-1β levels in the lungs of mice 24 h postinfection, it significantly reduced the levels of 13 other cytokines and chemokines (e.g., granulocyte-macrophage colony-stimulating factor [GM-CSF], MIP-1α, and IL-17) involved in the inflammatory response and cellular recruitment (Fig. 5). These results are supported by a reduction in the cellular infiltration and lung damage seen upon histologic analysis of the infected lungs (Fig. 4A). AT neutralization with LC10 therefore acts to dampen the damaging inflammatory response that is characteristic of pneumonia, supporting a role for AT in the overall dysregulation of the immune response and providing a rationale for targeting AT in prophylactic methods and therapy against S. aureus pneumonia.
In addition to its role in inciting a damaging immune response during S. aureus pneumonia, AT has also been shown to promote lung damage resulting in pulmonary edema and decreased lung function (22, 23). AT does this by directly lysing lung epithelial cells and at sublytic concentrations by activating the metalloprotease activity of its receptor, ADAM-10. This results in the cleavage of E-cadherin in tight cell junctions and leads to fluid influx and pulmonary edema (23). Consistent with these reports, the neutralization of AT in animals passively immunized with LC10 prior to i.n. infection with S. aureus exhibited limited to no damage to the alveolar architecture and reduced levels of pulmonary edema as measured by protein levels in the bronchoalveolar lavage fluid. The LC10-treated mice also exhibited reduced CO2 levels in the blood, consistent with improved lung function relative to the mice that received R347. These results indicate that prophylaxis with a high-affinity AT-neutralizing MAb prior to infection reduces much of the damage associated with S. aureus pneumonia; this results in improved lung integrity and pulmonary function, which ultimately promotes bacterial clearance and survival. Taken together, the data presented here suggest that a positive disease outcome is dependent on both a reduction in bacterial CFU and a reduction in the inflammation and damage associated with S. aureus pneumonia.
Although antibacterial MAbs have been primarily explored for their value in the prevention of disease, prior to the antibiotic era, antibodies, and in particular antitoxins, in the form of immune serum from an exposed individual or animal, were used to treat bacterial infections with some success (54). Given their complementary mechanisms of action, lack of drug-drug interactions, and extended half-lives, antibacterial antibodies may provide a benefit when used in combination with antibiotics to treat bacterial infections. In fact, recent reports demonstrate the benefit of treating Pseudomonas aeruginosa and S. aureus infections with a MAb-antibiotic combination over treating with antibiotics alone (13, 55). For example, DiGiandomenico et al. (56) demonstrated that the combination of a novel anti-P. aeruginosa bispecific antibody with either ciprofloxacin, meropenem, or tobramycin resulted in synergy over treatment with either antibody or with antibiotic alone. Similarly, Foletti et al. (13) reported that the coadministration of an anti-AT MAb with linezolid provided benefit over treatment with either the MAb or linezolid; however, synergy was not examined in that study. We took this observation further and titrated LC10 in combination with either linezolid or vancomycin over a range of concentrations and conducted an isobologram analysis. Our results indicate that a combination of LC10 with vancomycin was mildly synergistic over a range of concentrations, whereas a combination of LC10 with linezolid was synergistic at subtherapeutic doses but additive as the concentration of linezolid increased (Fig. 7). This difference between vancomycin and linezolid may be explained based on their mechanisms of action. Linezolid is a protein synthesis inhibitor, and part of its utility in treating S. aureus infections likely results from its inhibition of toxin expression (57). Therefore, in the presence of linezolid, LC10 neutralizes the small amount of AT present, resulting in an additive effect over using linezolid alone. However, as linezolid concentrations decrease as dictated by its pharmacokinetics (PK), or if the infecting organism exhibits an elevated MIC to linezolid, more AT is made, which is neutralized by LC10, resulting in a synergistic interaction between LC10 and linezolid. On the other hand, vancomycin, which interferes with cell wall biosynthesis, does not reduce AT levels and may even increase AT levels by releasing cytosolic AT upon bacterial cell lysis. As vancomycin kills the bacteria, LC10 neutralizes the AT that is already present or released by the lysed bacteria, thereby reducing tissue damage and dampening the inflammatory response, resulting in synergy with vancomycin. These results indicate that treatment with LC10 in combination with antibiotics provides benefit over antibiotic therapy alone and may be a useful addition to the current antibacterial armamentarium to treat S. aureus pneumonia.
The results presented here support previous reports by demonstrating that immunoprophylaxis against AT reduces disease severity in an S. aureus pneumonia model. In addition, our results provide insight into the mechanism by which AT neutralization improves disease outcome by suppressing the damaging hyperinflammatory response, preserving lung integrity, and improving lung function, leading to bacterial clearance and improved survival. Similarly, the therapeutic administration of LC10 in combination with vancomycin or linezolid results in improved survival relative to the monotherapies by reducing inflammation and lung damage. Taken together, these results indicate that an anti-AT MAb, such as LC10, may be a valuable addition to the methods that are currently available for the prevention and treatment of serious S. aureus disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jose Martinez and Diana Pao for technical assistance.
Footnotes
Published ahead of print 2 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02190-13.
REFERENCES
- 1.Kollef MH, Shorr A, Tabak YP, Gupta V, Liu LZ, Johannes RS. 2005. Epidemiology and outcomes of health-care-associated pneumonia: results from a large US database of culture-positive pneumonia. Chest 128:3854–3862. 10.1378/chest.128.6.3854 [DOI] [PubMed] [Google Scholar]
- 2.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687. 10.1128/CMR.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Docobo-Pérez F, López-Rojas R, Domínguez-Herrera J, Jiménez-Mejias ME, Pichardo C, Ibáñez-Martínez J, Pachón J. 2012. Efficacy of linezolid versus a pharmacodynamically optimized vancomycin therapy in an experimental pneumonia model caused by methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 67:1961–1967. 10.1093/jac/dks142 [DOI] [PubMed] [Google Scholar]
- 4.Pletz MW, Burkhardt O, Welte T. 2010. Nosocomial methicillin-resistant Staphylococcus aureus (MRSA) pneumonia: linezolid or vancomycin? Comparison of pharmacology and clinical efficacy. Eur. J. Med. Res. 15:507–513. 10.1186/2047-783X-15-12-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walkey AJ, O'Donnell MR, Wiener RS. 2011. Linezolid vs glycopeptide antibiotics for the treatment of suspected methicillin-resistant Staphylococcus aureus nosocomial pneumonia: a meta-analysis of randomized controlled trials. Chest 139:1148–1155. 10.1378/chest.10-1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, J Rybak M, Talan DA, Chambers HF. 2011. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin. Infect. Dis. 52:285–292. 10.1093/cid/cir034 [DOI] [PubMed] [Google Scholar]
- 7.Digiandomenico A, Warrener P, Hamilton M, Guillard S, Ravn P, Minter R, Camara MM, Venkatraman V, Macgill RS, Lin J, Wang Q, Keller AE, Bonnell JC, Tomich M, Jermutus L, McCarthy MP, Melnick DA, Suzich JA, Stover CK. 2012. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J. Exp. Med. 209:1273–1287. 10.1084/jem.20120033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 362:197–205. 10.1056/NEJMoa0907635 [DOI] [PubMed] [Google Scholar]
- 9.François B, Luyt CE, Dugard A, Wolff M, Diehl JL, Jaber S, Forel JM, Garot D, Kipnis E, Mebazaa A, Misset B, Andremont A, Ploy MC, Jacobs A, Yarranton G, Pearce T, Fagon JY, Chastre J. 2012. Safety and pharmacokinetics of an anti-PcrV PEGylated monoclonal antibody fragment in mechanically ventilated patients colonized with Pseudomonas aeruginosa: a randomized, double-blind, placebo-controlled trial. Crit. Care Med. 40:2320–2326. 10.1097/CCM.0b013e31825334f6 [DOI] [PubMed] [Google Scholar]
- 10.Projan SJ, Shlaes DM. 2004. Antibacterial drug discovery: is it all downhill from here? Clin. Microbiol. Infect. 10(Suppl 4):18–22. 10.1111/j.1465-0691.2004.1006.x [DOI] [PubMed] [Google Scholar]
- 11.Tkaczyk C, Hua L, Varkey R, Shi Y, Dettinger L, Woods R, Barnes A, MacGill RS, Wilson S, Chowdhury P, Stover CK, Sellman BR. 2012. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 19:377–385. 10.1128/CVI.05589-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ragle BE, Bubeck Wardenburg J. 2009. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 77:2712–2718. 10.1128/IAI.00115-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foletti D, Strop P, Shaughnessy L, Hasa-Moreno A, Casas MG, Russell M, Bee C, Wu S, Pham A, Zeng Z, Pons J, Rajpal A, Shelton D. 2013. Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus α-hemolysin. J. Mol. Biol. 425:1641–1654. 10.1016/j.jmb.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 14.DeJonge M, Burchfield D, Bloom B, Duenas M, Walker W, Polak M, Jung E, Millard D, Schelonka R, Eyal F, Morris A, Kapik B, Roberson D, Kesler K, Patti J, Hetherington S. 2007. Clinical trial of safety and efficacy of INH-A21 for the prevention of nosocomial staphylococcal bloodstream infection in premature infants. J. Pediatr. 151:260–265, 265e1. 10.1016/j.jpeds.2007.04.060 [DOI] [PubMed] [Google Scholar]
- 15.Cheung GY, Otto M. 2012. The potential use of toxin antibodies as a strategy for controlling acute Staphylococcus aureus infections. Expert Opin. Ther. Targets 16:601–612. 10.1517/14728222.2012.682573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ventura CL, Higdon R, Hohmann L, Martin D, Kolker E, Liggitt HD, Skerrett SJ, Rubens CE. 2008. Staphylococcus aureus elicits marked alterations in the airway proteome during early pneumonia. Infect. Immun. 76:5862–5872. 10.1128/IAI.00865-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bubeck Wardenburg J, Patel RJ, Schneewind O. 2007. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 75:1040–1044. 10.1128/IAI.01313-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartlett AH, Foster TJ, Hayashida A, Park PW. 2008. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J. Infect. Dis. 198:1529–1535. 10.1086/592758 [DOI] [PubMed] [Google Scholar]
- 19.Wilke GA, Bubeck Wardenburg J. 2010. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. U. S. A. 107:13473–13478. 10.1073/pnas.1001815107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berube BJ, Bubeck Wardenburg J. 2013. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel) 5:1140–1166. 10.3390/toxins5061140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA. 2009. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One 4:e7446. 10.1371/journal.pone.0007446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, Jania C, Doerschuk CM, Tilley SL, Duncan JA. 2012. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 205:807–817. 10.1093/infdis/jir846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. 2011. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17:1310–1314. 10.1038/nm.2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gumbiner BM. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84:345–357. 10.1016/S0092-8674(00)81279-9 [DOI] [PubMed] [Google Scholar]
- 25.Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, de Strooper B, Hartmann D, Saftig P. 2005. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. U. S. A. 102:9182–9187. 10.1073/pnas.0500918102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoshima N, Wang Y, Bubeck Wardenburg J. 2012. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J. Invest. Dermatol. 132:1513–1516. 10.1038/jid.2011.462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. 2012. ADAM10 mediates vascular injury induced by Staphylococcus aureus α-hemolysin. J. Infect. Dis. 206:352–356. 10.1093/infdis/jis192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bubeck Wardenburg J, Schneewind O. 2008. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205:287–294. 10.1084/jem.20072208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tkaczyk C, Hamilton MM, Datta V, Yang XP, Hilliard JJ, Stephens GL, Sadowska A, Hua L, O'Day T, Suzich J, Stover CK, Sellman BR. 2013. Staphylococcus aureus alpha toxin suppresses effective innate and adaptive immune responses in a murine dermonecrosis model. PLoS One 8:e75103. 10.1371/journal.pone.0075103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Machado SG, Robinson GA. 1994. A direct, general approach based on isobolograms for assessing the joint action of drugs in pre-clinical experiments. Stat. Med. 13:2289–2309. 10.1002/sim.4780132202 [DOI] [PubMed] [Google Scholar]
- 31.Verma A, Ngundi MM, Meade BD, De Pascalis R, Elkins KL, Burns DL. 2009. Analysis of the Fc gamma receptor-dependent component of neutralization measured by anthrax toxin neutralization assays. Clin. Vaccine Immunol. 16:1405–1412. 10.1128/CVI.00194-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vitale L, Blanset D, Lowy I, O'Neill T, Goldstein J, Little SF, Andrews GP, Dorough G, Taylor RK, Keler T. 2006. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect. Immun. 74:5840–5847. 10.1128/IAI.00712-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, Parren PW. 2012. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 189:3430–3438. 10.4049/jimmunol.1200356 [DOI] [PubMed] [Google Scholar]
- 34.Balsitis SJ, Williams KL, Lachica R, Flores D, Kyle JL, Mehlhop E, Johnson S, Diamond MS, Beatty PR, Harris E. 2010. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 6:e1000790. 10.1371/journal.ppat.1000790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao MH, Morrison SL. 1989. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143:2595–2601 [PubMed] [Google Scholar]
- 36.Diep BA, Chan L, Tattevin P, Kajikawa O, Martin TR, Basuino L, Mai TT, Marbach H, Braughton KR, Whitney AR, Gardner DJ, Fan X, Tseng CW, Liu GY, Badiou C, Etienne J, Lina G, Matthay MA, DeLeo FR, Chambers HF. 2010. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. U. S. A. 107:5587–5592. 10.1073/pnas.0912403107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker D, Prince A. 2012. Immunopathogenesis of Staphylococcus aureus pulmonary infection. Semin. Immunopathol. 34:281–297. 10.1007/s00281-011-0291-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parker JC, Townsley MI. 2004. Evaluation of lung injury in rats and mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L231–L246. 10.1152/ajplung.00049.2003 [DOI] [PubMed] [Google Scholar]
- 39.Kenyon NJ, van der Vliet A, Schock BC, Okamoto T, McGrew GM, Last JA. 2002. Susceptibility to ozone-induced acute lung injury in iNOS-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L540–L545. 10.1152/ajplung.00297.2001 [DOI] [PubMed] [Google Scholar]
- 40.Yamauchi T, Shima M, Kuwaki T, Ando M, Ohmichi M, Fukuda Y, Adachi M. 2002. Acute effects of ozone exposure on lung function in mice sensitized to ovalbumin. Toxicology 172:69–78. 10.1016/S0300-483X(01)00588-1 [DOI] [PubMed] [Google Scholar]
- 41.Fowler VG, Jr, Olsen MK, Corey GR, Woods CW, Cabell CH, Reller LB, Cheng AC, Dudley T, Oddone EZ. 2003. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch. Intern. Med. 163:2066–2072. 10.1001/archinte.163.17.2066 [DOI] [PubMed] [Google Scholar]
- 42.de Kraker ME, Wolkewitz M, Davey PG, Koller W, Berger J, Nagler J, Icket C, Kalenic S, Horvatic J, Seifert H, Kaasch AJ, Paniara O, Argyropoulou A, Bompola M, Smyth E, Skally M, Raglio A, Dumpis U, Kelmere AM, Borg M, Xuereb D, Ghita MC, Noble M, Kolman J, Grabljevec S, Turner D, Lansbury L, Grundmann H, BURDEN Study Group 2011. Clinical impact of antimicrobial resistance in European hospitals: excess mortality and length of hospital stay related to methicillin-resistant Staphylococcus aureus bloodstream infections. Antimicrob. Agents Chemother. 55:1598–1605. 10.1128/AAC.01157-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JL, Chen SY, Wang JT, Wu GH, Chiang WC, Hsueh PR, Chen YC, Chang SC. 2008. Comparison of both clinical features and mortality risk associated with bacteremia due to community-acquired methicillin-resistant Staphylococcus aureus and methicillin-susceptible S. aureus. Clin. Infect. Dis. 46:799–806. 10.1086/527389 [DOI] [PubMed] [Google Scholar]
- 44.Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. 2007. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13:1405–1406. 10.1038/nm1207-1405 [DOI] [PubMed] [Google Scholar]
- 45.Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202:1050–1058. 10.1086/656043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rauch S, DeDent AC, Kim HK, Bubeck Wardenburg J, Missiakas DM, Schneewind O. 2012. Abscess formation and alpha-hemolysin induced toxicity in a mouse model of Staphylococcus aureus peritoneal infection. Infect. Immun. 80:3721–3732. 10.1128/IAI.00442-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kernodle DS, Voladri RK, Menzies BE, Hager CC, Edwards KM. 1997. Expression of an antisense hla fragment in Staphylococcus aureus reduces alpha-toxin production in vitro and attenuates lethal activity in a murine model. Infect. Immun. 65:179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, Bubeck Wardenburg J, Schneewind O, Otto M, Deleo FR. 2011. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis. 204:937–941. 10.1093/infdis/jir441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dajcs JJ, Thibodeaux BA, Girgis DO, O'Callaghan RJ. 2002. Corneal virulence of Staphylococcus aureus in an experimental model of keratitis. DNA Cell Biol. 21:375–382. 10.1089/10445490260099656 [DOI] [PubMed] [Google Scholar]
- 50.Bayer AS, Ramos MD, Menzies BE, Yeaman MR, Shen AJ, Cheung AL. 1997. Hyperproduction of alpha-toxin by Staphylococcus aureus results in paradoxically reduced virulence in experimental endocarditis: a host defense role for platelet microbicidal proteins. Infect. Immun. 65:4652–4660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rastogi D, Ratner AJ, Prince A. 2001. Host-bacterial interactions in the initiation of inflammation. Paediatr. Respir. Rev. 2:245–252. 10.1053/prrv.2001.0147 [DOI] [PubMed] [Google Scholar]
- 52.Goodman RB, Pugin J, Lee JS, Matthay MA. 2003. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 14:523–535. 10.1016/S1359-6101(03)00059-5 [DOI] [PubMed] [Google Scholar]
- 53.Frank KM, Zhou T, Moreno-Vinasco L, Hollett B, Garcia JG, Bubeck Wardenburg J. 2012. Host response signature to Staphylococcus aureus alpha-hemolysin implicates pulmonary Th17 response. Infect. Immun. 80:3161–3169. 10.1128/IAI.00191-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pope CG. 1963. Development of knowledge of antitoxins. Br. Med. Bull. 19:230–234 [DOI] [PubMed] [Google Scholar]
- 55.Song Y, Baer M, Srinivasan R, Lima J, Yarranton G, Bebbington C, Lynch SV. 2012. PcrV antibody-antibiotic combination improves survival in Pseudomonas aeruginosa-infected mice. Eur. J. Clin. Microbiol. Infect. Dis.. 31:1837–1845. 10.1007/s10096-011-1509-2 [DOI] [PubMed] [Google Scholar]
- 56.DiGiandomenico A, Keller A, Gao C, Hilliard JJ, Camara M, Warrener P, Rainey G, Gao C, Dimasi N, Sellman B, Stover CK. 2012. Abstr. B-1734. Controlling Pseudomonas aeruginosa infections using a novel anti-Psl/PcrV multispecific antibody approach. ICAAC. http://www.icaaconline.com/php/icaac2013abstracts/data/papers/2012/B/2012_B-1734.htm [Google Scholar]
- 57.Diep BA, Afasizheva A, Le HN, Kajikawa O, Matute-Bello G, Tkaczyk C, Sellman B, Badiou C, Lina G, Chambers HF. 2013. Effects of linezolid on suppressing in vivo production of staphylococcal toxins and improving survival outcomes in a rabbit model of methicillin-resistant Staphylococcus aureus necrotizing pneumonia. J. Infect. Dis. 208:75–82. 10.1093/infdis/jit129 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.