

Abstract
Dengue is the world's most common mosquito-borne viral infection and a leading cause of morbidity throughout the tropics and subtropics. Viruses are known to evade the establishment of an antiviral state by regulating the activation of interferon regulatory factor 3 (IRF3), a critical transcription factor in the alpha/beta interferon induction pathway. Here, we show that dengue virus (DENV) circumvents the induction of the retinoic acid-inducible gene I-like receptor (RLR) pathway during infection by blocking serine 386 phosphorylation and nuclear translocation of IRF3. This effect is associated with the expression of nonstructural 2B/3 protein (NS2B/3) protease in human cells. Using interaction assays, we found that NS2B/3 interacts with the cellular IκB kinase ε (IKKε). Docking computational analysis revealed that in this interaction, NS2B/3 masks the kinase domain of IKKε and potentially affects its functionality. This observation is supported by the DENV-associated inhibition of the kinase activity of IKKε. Our data identify IKKε as a novel target of DENV NS2B/3 protease.
INTRODUCTION
Dengue virus (DENV) is a member of the genus Flavivirus in the family Flaviviridae. DENV causes dengue with or without warning signs or severe dengue that can result in shock or death (1). The World Health Organization reports that >2.5 billion people are at risk of dengue infection, and the number of cases continues to increase. This translates to 50 million infections annually, with 500,000 to 1 million cases of those being severe dengue and around 22,000 cases leading to death, mainly among children (1, 2). The disease is induced by four different viral serotypes, and although infection with one serotype confers protective immunity to that serotype, it does not protect the host from infection by other serotypes (3).
DENV is an enveloped single-stranded RNA (ssRNA) virus with positive polarity. Translation of the DENV genome produces a polyprotein, which upon cleavage by host and virus proteases yields three structural (C, E, and prM) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins. The viral protease NS2B/3 is composed of two subunits, NS3 and NS2B, which control the proteolytic activity of the enzyme (4, 5).
The innate immune response is the first line of defense against invading pathogens, and it is activated in mammalian hosts immediately after viral infection. This response is triggered when cell pattern recognition receptors (PRRs) detect pathogen-associated molecular patterns (PAMPs), leading to the activation of multiple signaling pathways and transcription factors (6). Toll-like receptor 3 (TLR3) and the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are important PRRs that recognize the viral replication intermediate, double-stranded RNA (dsRNA) (7). The RLR family comprises cytoplasmic RNA helicases that share a homologous DExD/H box. The two principal members are RIG-I and melanoma differentiation-associated gene 5 (MDA5) (8). The binding of dsRNA to PRRs activates interferon regulatory factor 3 (IRF3), a key transcription factor that induces the production of alpha/beta interferon (IFN-α/β) (type I IFNs). Both types of dsRNA receptors utilize adaptor molecules to facilitate signaling: TLR3 uses Toll interleukin-1 (IL-1) receptor domain-containing adaptor inducing IFN-β (TRIF) and RLR signals through mitochondrial antiviral signaling protein (MAVS), also called IPS1, VISA, and CARDIF (6). TRIF and MAVS interact with TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) independently of the activated dsRNA receptor. Downstream, the kinases phosphorylate IRF3 at several serine/threonine residues, thereby promoting its dimerization and translocation into the nucleus (6, 9). This phosphorylation leads to the activation of multiple genes, including IFN-β, which is crucial to the establishment of the antiviral state that blocks viral replication.
Recently, another protein, known as stimulator of the interferon gene (STING) (also known as MITA, ERIS, or TMEM173), was also shown to be associated with the production of type I IFNs. STING is an endoplasmic reticulum transmembrane protein and was first described as a cytosolic DNA sensor, but different studies have shown that it is also triggered by viral RNA (10). The activation of this pathway induces the dimerization of STING, which is then phosphorylated and ubiquitinated to activate IRF3 (11).
Viruses have evolved strategies to evade the type I IFN response. Some of these include RNA viruses that are of clinical relevance to humans, such as influenza virus (by NS1 protein), Ebola virus (by VP35 protein), and Nipah virus (by W protein), among others (12–15). Members of the family Flaviviridae, like hepatitis C virus (HCV) and West Nile virus (WNV), also have evolved unique strategies to control the innate immune response in order to prevent viral clearance and promote viral replication (16). Dengue virus has been shown to inhibit the type I IFN signaling cascade by targeting different proteins of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway (17, 18). In particular, the nonstructural DENV proteins NS2A, NS4A, NS4B, and NS5 have been involved in that inhibitory effect (19–22). Different studies have demonstrated that the HCV serine protease NS3/4A cleaves the adaptors TRIF and MAVS (23, 24). Other viruses use their proteases to disrupt the IFN pathways; for example, the picornavirus 3Cpro cleaves RIG-I and abrogates its activity (25). Recently, it was shown that DENV NS2B/3 inhibits the type I IFN response in human dendritic cells by cleaving STING (26). However, the precise mechanism by which DENV protease inhibits the induction pathway is still unclear.
Here, we investigated in vitro the interactions of DENV with the TLR3 and RLR pathways and the effects of the DENV NS2B/3 serine protease on the IKKε kinase. Our results show that DENV interrupts the RIG-I signaling pathway, blocking the nuclear translocation and S386 phosphorylation of IRF3 by a direct interaction of NS2B/3 with IKKε that allows for masking of the protein kinase domain.
MATERIALS AND METHODS
Cell culture and viruses.
HEK293 cells stably expressing TLR3 (293/TLR3) (InvivoGen, San Diego, CA) were propagated as a confluent monolayer in tissue culture flasks. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% inactivated fetal bovine serum (FBS), 1% (vol/vol) penicillin and streptomycin, and 10 μg/ml blasticidin at 37°C in 5% CO2. DENV2 strain NGC-44 was propagated in C6/36 cells, and titers were determined on Vero cells by a plaque assay.
Infection and stimulation.
The 293/TLR3 cells were seeded onto 24-well plates at 4.5 × 105 cells/well and after 24 h were infected with DENV2 at an multiplicity of infection (MOI) of 4; 12 h later, the cells were mock stimulated or stimulated to activate TLR3 with 100 μg/ml poly(I·C) or RLRs with 10 μg/ml poly(I·C) (low molecular weight [LMW])/LyoVec. Poly(I·C)/LyoVec is a synthetic dsRNA polymer complexed with the transfection reagent LyoVec (catalog no. tlrl-picwlv; InvivoGen, San Diego, CA). Unlike naked poly(I·C), which is recognized by TLR3, transfected poly(I·C) is sensed by RLRs. At 24 h postinfection (12 h poststimulation), the cells were lysed and stored at −80°C for subsequent Western blot analysis.
Transfection of 293/TLR3 cells.
The 293/TLR3 cells were transfected with hemagglutinin (HA)-tagged plasmids encoding DENV2 protease NS2B/3 or NS2B/3-S135A with impaired protease activity (kindly provided by Adolfo García-Sastre of the Mount Sinai School of Medicine, NY). Transfection was done using 1 μg of plasmid DNA with Lipofectamine 2000 (Invitrogen) at a density of 3.6 × 105 cells/ml. The cells were then cultured in 24-well dishes and after 12 h were stimulated with 10 μg/ml poly(I·C)/LyoVec. Twenty-four hours posttransfection (12 h poststimulation), the cells were collected, lysed, and stored at −80°C for subsequent Western blot analysis.
IKKε cleavage and functional evaluation.
For cleavage evaluation, 1 μg of Flag-tagged IKKε (kindly provided by K. Fitzgerald, University of Massachusetts Medical School, MA) and 1 μg of HA-tagged NS2B/3 or NS2B/NS3-S135A plasmid were transfected into 293/TLR3 cells, and the cells were collected and lysed after 48 h. For cleavage evaluation of endogenous IKKε after infection, the cells were infected with DENV2 at an MOI of 4 for 24 h, and the cells were then collected and lysed. For IKKε functional evaluation, the cells were transfected with 1 μg of Flag-tagged IKKε plasmid for 24 h, followed by infection with DENV2 at an MOI of 4 for 24 h, after which the cells were collected and lysed. The samples were analyzed by Western blotting using equivalent amounts of protein.
Western blot analysis.
The cells were lysed in RIPA buffer (50 mM Tris HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 2 mM EDTA, 1% sodium dodecyl sulfate) containing protease inhibitors. Equivalent amounts of protein from 20 to 40 μg (determined by the Micro BCA protein assay; Pierce, Rockford, IL) were loaded onto a 12% SDS-polyacrylamide gel. After electrophoresis, the gel was transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA) using a Trans-Blot semidry transfer cell (Bio-Rad) apparatus. After incubation with blocking solution (5% dry milk and 0.1% Tween 20 in Tris-buffered saline [TBST]) at room temperature for 1 h, the membranes were incubated overnight with the respective antibodies. The following monoclonal or polyclonal antibodies were used to probe the blots: mouse anti-IRF3 (R&D Systems, Minneapolis, MN), rabbit anti-phosphoserine 386 IRF-3 (Abcam, Cambridge, MA), rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam), rabbit anti-DENV-NS5 (kindly provided by Adolfo García-Sastre of Mount Sinai School of Medicine, NY), rabbit anti-IKKε (Cell Signaling Technology, Danvers, MA), mouse anti-Flag (Sigma-Aldrich), and rabbit anti-HA (Sigma-Aldrich) antibodies. The blots were washed three times for 5 min with TBST, incubated for 2 h with peroxidase-conjugated secondary goat anti-rabbit or anti-mouse (Bio-Rad) antibody, and washed again three times. SuperSignal West Pico chemiluminescent detection methods (Pierce) were used to visualize the proteins according to the manufacturer's instructions and analyzed using the VersaDoc imaging system model 4000 (Bio-Rad).
Immunofluorescence staining.
The cells were grown overnight to 50% confluence on microscope coverslips and then fixed and permeabilized in cold acetone-methanol (1:1). The cell nuclei were permeabilized for 5 min in 0.5% Nonidet P-40. Rabbit anti-IRF3 or anti-phosphoserine 386 IRF-3 (Abcam) and mouse anti-flavivirus E protein (4G2) (ATCC) were used as primary antibodies. Texas Red- and fluorescein isothiocyanate-conjugated anti-rabbit or anti-mouse antibodies were used as secondary antibodies (Jackson ImmunoResearch). Nuclear chromatin staining was performed by incubation in a phosphate-buffered saline solution containing 0.5 mg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Bio-Rad) and then mounted using ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA). The images were observed with a Zeiss Observer Z1 confocal laser-scanning microscope coupled to a Zeiss LSM 510 Meta EC. The system was controlled using Zeiss ZEN 2009 software.
Coimmunoprecipitation.
The 293/TLR3 cells were cotransfected as described above with Flag-IKKε (1 μg of DNA) and HA-NS2B/3, HA-NS2B/3-S135A, or empty plasmid (1 μg of DNA). After 48 h posttransfection, the cells were collected and lysed. The cell lysates (200 μl) were precleared for 3 h with 20 μl of agarose beads. The coimmunoprecipitation procedures were conducted according to the manufacturer's instructions (Pierce HA tag IP/Co-IP kit), including using a kit-supplied HA-tagged positive control to verify correct immunoprecipitation. Bound and unbound fractions were analyzed by Western blotting using anti-Flag (Sigma-Aldrich) and anti-HA (Sigma-Aldrich) antibodies.
Immunostaining by in situ proximity ligation assay.
The 293/TLR3 cells were transfected as described above, grown to 50% confluence on BD BioCoat poly-d-lysine 8-well culture slides (BD Biosciences), and after 48 h were fixed and permeabilized in cold acetone-methanol (1:1). The cell nuclei were permeabilized for 5 min in 0.5% Nonidet P-40. Protein-protein interactions were detected using a Duolink II proximity ligation assay red kit composed of anti-rabbit proximity ligation assay (PLA) probe Plus, anti-mouse PLA probe Minus (Olink Bioscience, Uppsala, Sweden) and antibodies against HA-tag (Sigma-Aldrich), IKKε (Cell Signaling Technologies), and IRF3 (Abcam). Single detections were analyzed using anti-rabbit PLA Plus and Minus probes for IKKε or IRF3 and anti-mouse PLA Plus and Minus probes for HA-tag proteins. The images were observed with a Zeiss Observer Z1 confocal laser-scanning microscope coupled to a Zeiss LSM 510 Meta EC. The system was controlled using Zeiss ZEN 2009 software.
Computational analysis.
The properties of the NS2B/3 cleavage site and preference sequences were searched for in the literature (4, 28). The ScanProsite databases were employed to predict putative cleavage sites in human IKKε (GenBank accession no. AAF45307.1). We used the pattern search tool of Prosite with the motif [KR] (2,3)-[SGA], which represents two or three residues of Lys and/or Arg followed by any of Ser, Gly, or Ala.
The protein-protein model of IKKε and NS2B/3 was examined using the automatic docking ClusPro 2.0 server (29). Since an experimentally determined three-dimensional structure for IKKε was not available, we used homology modeling to predict its three-dimensional (3D) structure using the Phyre2 server (protein homology/analogy recognition engine version 2.0) based on the resolved 3D structure of human TBK1 (Protein Data Bank ID, 4IM2). Docking was performed using the Protein Data Bank (PDB) homology model of IKKε and DENV NS2B/3 (Protein Data Bank ID, 2FOM). The highest-ranked model was selected and visualized using the PyMOL program.
Statistical analysis.
Data were analyzed by using unpaired two-tailed t tests or by one-way analysis of variance (ANOVA) followed by a Tukey multiple-comparison test. The results were expressed as the mean ± standard error of the mean (SEM). Differences were considered significant with P values of <0.05 or <0.01. All statistical tests were performed with the GraphPad Instat software version 3.06 and Prism 5.
RESULTS
DENV blocks IRF3 phosphorylation of serine 386.
Upon viral infection, TLR3 and RLRs are the major sensors that trigger IRF3 activation in response to stimulation. IKKε and TBK1 activate IRF3 by the phosphorylation of specific residues in its C terminus. The roles of the different phosphorylation sites have remained controversial, but the main phosphorylated residues are serine 386 (S386) and serine 396 (S396) (30). In this study, we tested IRF3 phosphorylation at S386, which has been suggested to be critical for IRF3 activation (31). Poly(I·C), a synthetic dsRNA, is a stimulator of TLR3; however, when poly(I·C) is transfected into the cells, it functions as an RLR stimulator (32). We showed that in both cases (dsRNA uptake and transfection), poly(I·C) stimulated IRF3 phosphorylation at S386 (Fig. 1A and B).
FIG 1.
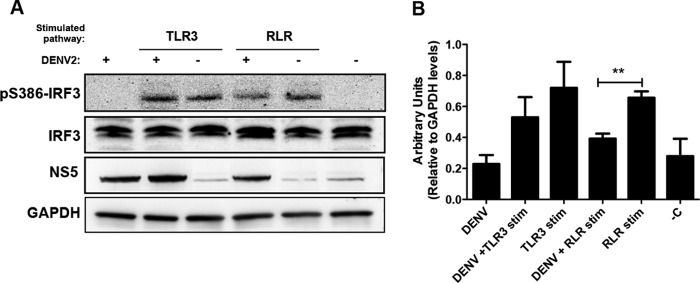
Interferon regulatory factor 3 (IRF3) phosphorylation state at residue S386 after infection and/or stimulation of TLR3 or RLR pathways. (A) Levels of phosphorylated IRF3 (pS386-IRF3) and IRF3 on a Western blot prepared with 20 μg of whole-cell extract recovered from cells treated as indicated and collected at 24 h postinfection and 12 h poststimulation. GAPDH was detected in these same cell lysates as a loading control and DENV NS5 protein as evidence of viral replication. (B) Densitometric analysis of pS386-IRF3 protein levels normalized to GAPDH are shown in arbitrary units. **, significant difference between RLR-stimulated cells that were DENV infected and mock infected (P < 0.01, two-tailed Student's t test) (n = 3). Each bar represents the mean ± SEM. −C, negative control.
Human embryonic kidney cells with stable expression of TLR3 (293/TLR3) and that are responsive to RLR ligands were infected with DENV and stimulated for TLR3 or RLR 12 h later. To analyze the ability of DENV to block S386 phosphorylation of IRF3 (pS386-IRF3), cell lysates were subjected to immunoblotting 24 h postinfection (12 h poststimulation) (Fig. 1A) and the signals were evaluated by densitometry (Fig. 1B). Cells that were infected but not stimulated showed no S386-IRF3 phosphorylation (Fig. 1A and B). However, the stimulation of noninfected 293/TLR3 cells for TLR3 or RLR resulted in S386-IRF3 phosphorylation. When such cells were also infected with DENV, they exhibited a moderate reduction in the level of pS386-IRF3 after stimulation of TLR3, compared to the noninfected stimulated cells (P > 0.05). Furthermore, the virus significantly diminished pS386-IRF3 levels after RLR stimulation (P < 0.01) (Fig. 1B). Immunoblotting against NS5 confirmed DENV replication in 293/TLR3 cells.
Because laboratory strains may accumulate adaptive mutations through passages, in addition to analyzing the DENV2 laboratory strain NGC and the DENV1 Western Pacific 74 strain to detect S386-IRF3 phosphorylation, we also analyzed primary DENV2 isolated from patients in Puerto Rico. The inhibition of IRF3 phosphorylation after in vitro DENV infection was confirmed (data not shown). This suggests that DENV-induced inhibition of S386-IRF3 phosphorylation is widely associated with different DENV serotypes and strains.
DENV infection results in inhibition of RLR-induced IRF3 nuclear translocation.
Previous works confirmed that following cellular stimulation of a PRR, the IKKε and TBK1 kinases phosphorylate IRF3, which then homodimerizes and rapidly translocates to the nucleus (6, 33). To analyze DENV-associated nuclear translocation of IRF3, we compared DENV- and mock-infected 293/TLR3 cells by using immunofluorescence confocal microscopy. The results demonstrate nuclear translocation of IRF3 after TLR3 or RLR stimulation occurs in mock-infected cells (Fig. 2B and C) compared to unstimulated control cells (Fig. 2A). Similarly, in the DENV-infected/unstimulated cells, nuclear translocation of IRF3 did not occur (Fig. 2D). However, in the DENV-infected/stimulated cells, RLR-induced IRF3 nuclear translocation was blocked (Fig. 2F) and there was moderate inhibition of TLR3-induced IRF3 nuclear translocation (Fig. 2E). To determine if the observed decrease in nuclear translocation is related to DENV-associated suppression of IRF3 activation, we performed the same experiments but used a specific pS386-IRF3 antibody. Control cells that were mock infected and unstimulated showed no presence of pS386-IRF3 (Fig. 2G), whereas both TLR3 and RLR stimulation induced S386-IRF3 phosphorylation and nuclear translocation in mock-infected cells (Fig. 2H and I). The phosphorylation of S386-IRF3 was not detected in either the cytoplasm or nucleus of DENV-infected/unstimulated cells (Fig. 2J). However, although the virus failed to block TLR3-induced IRF3 phosphorylation and nuclear translocation (Fig. 2K), it strongly antagonized RLR-induced S386-IRF3 phosphorylation and nuclear localization (Fig. 2L). This is in agreement with the results obtained with the anti-IRF3 antibody (Fig. 2A to F). Because of the robust and significant blockage of RLR-induced S386-IRF3 phosphorylation and nuclear translocation by the DENV virus, this pathway was chosen for study in all further experiments.
FIG 2.
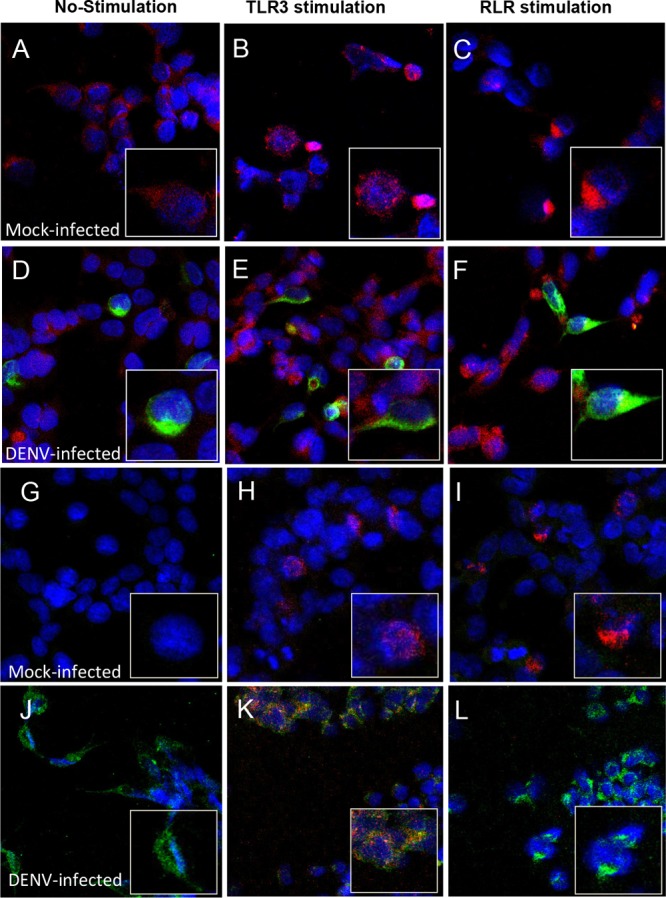
Cellular localization of IRF3 and pS386-IRF3 after DENV infection. The 293/TLR3 cells were infected or mock infected with DENV and stimulated or not for TLR3 or RLR as indicated. The cells were dually labeled for DENV and IRF3 (A to C, mock infected; D to F, DENV infected) or pS386-IRF3 (G to I, mock infected; J to L, DENV infected). Cells were stained with fluorescent, conjugated secondary antibodies and observed by confocal microscopy. Red indicates IRF3 or pS386-IRF3, green indicates DENV infection, and blue indicates nuclei stained with DAPI.
DENV NS2B/3 protease inhibits S386-IRF3 phosphorylation.
Recently, it was demonstrated that the DENV catalytically active protease complex NS2B/3 antagonizes the type I IFN response in human cells (34). To further address the role of the catalytic activity of DENV NS2B/3 in suppressing RLR-induced S386-IRF3 phosphorylation in 293/TLR3 cells, we performed immunoblotting of cells transfected with plasmids coding for either a wild-type DENV protease or a mutant variant with a single amino acid substitution, Ser135 replaced with Ala (S135A), which eliminates the protease activity (35) (Fig. 3A). A densitometric analysis of a pS386-IRF3 immunoblot of lysate from RLR-stimulated cells transfected with an empty plasmid showed a 3.1-fold level of induction compared with negative-control cells (Fig. 3B). However, in lysates of NS2B/3-transfected cells stimulated for RLR, the induction of pS386-IRF3 was significantly reduced compared to the positive-control (+C) cells that received only stimulation (P < 0.05) (Fig. 3B). Of note, cells transfected with NS2B/3-S135A were found to have lower levels of RLR-induced pS386-IRF3, but the degree of reduction was not significant compared to that of the +C cells.
FIG 3.
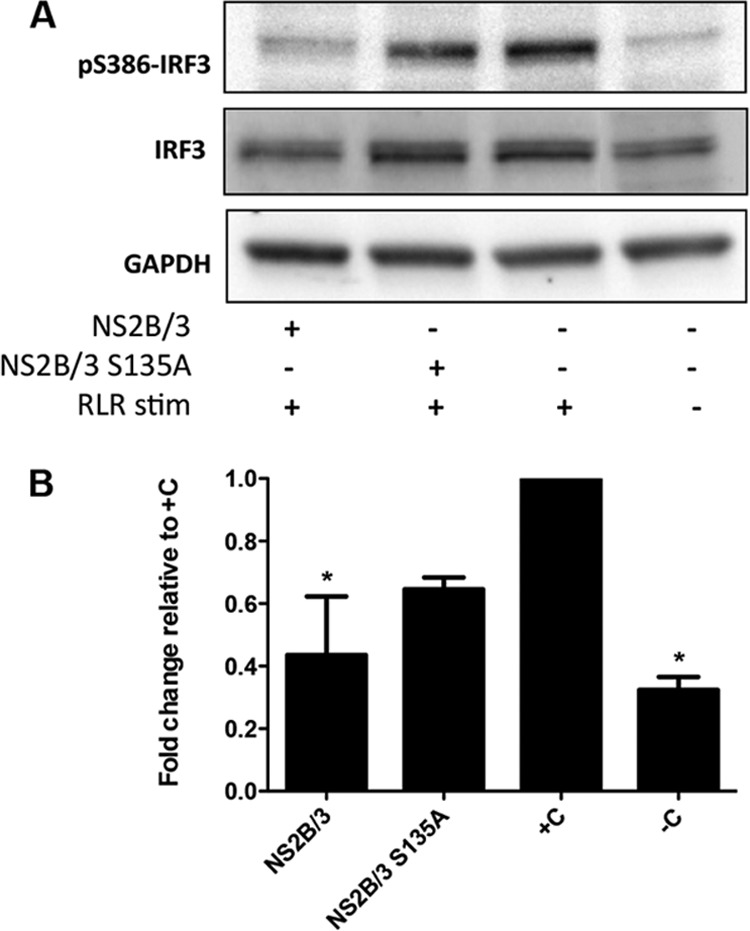
NS2B/3 inhibits RLR-associated induction of pS386-IRF3 phosphorylation. (A) The cells were transfected with NS2B/3- or NS2B/3-S135A-encoding plasmids, and 36 h after transfection, they were stimulated for RLR pathways for another 12 h and then cell lysate (20 μg) was analyzed by Western blotting using an anti-pS386-IRF3 antibody. (B) Densitometric analyses of pS386-IRF3 protein levels normalized to GAPDH are shown as fold change compared to the positive control (+C). *, significant difference compared to the +C (P < 0.05, one-way ANOVA) (n = 2). Each bar represents the mean ± SEM.
IKKε interacts with both the catalytically active and inactive forms of DENV protease.
The NS2B/3-associated inhibition of IRF3 phosphorylation establishes the relevance of studying the interplay between the protease and kinases that directly activate IRF3. To further address this, we performed interaction assays between NS2B/3 and IKKε, which is a kinase involved in phosphorylating IRF3. First, we used the in situ proximity ligation assay (PLA), a technique based on immunodetection that identifies any interaction between two proteins in close proximity by generating fluorescence. The cells were transfected with plasmids coding for HA-tagged NS2B/3 or NS2B/3-S135A or were cotransfected with Flag-tagged IKKε (Fig. 4). Using the same method, we were able to detect single proteins as evidence of the individual expression of IRF3, IKKε, HA-tagged NS2B/3, and NS2B/3-S135A (Fig. 4A, panels a to d). Once expression of the single proteins was confirmed, we tested whether any protein-protein interactions could be detected. As a positive control for interacting proteins, we used the cyclic AMP response element-binding (CREB)-binding protein/p300 (CBP/p300) and IRF3, which are well-described protein-protein partners (36). We first looked at cells that were transfected with any form of the protease and analyzed them for interactions between the protease and endogenous IKKε (Fig. 4A, panels e and f). Imaging of IKKε in transfected cells expressing the active protease NS2B/3 revealed no red fluorescence (Fig. 4A, panel e). However, cells transfected with the catalytically inactive protease showed positive protein-protein interactions with endogenous IKKε (Fig. 4A, panel f). Cells transfected with plasmids coding for IKKε and either the active protease NS2B/3 or the catalytically inactive protease NS2B/3-S135A were positive for protein-protein interactions in both cases compared with controls (Fig. 4A, panels g and h). However, the interaction of IKKε with NS2B/3-S135A resulted in a stronger signal than the interaction with catalytically active protease.
FIG 4.

DENV protease interacts with human IKKε. (A) 293/TLR3 cells were transfected with HA-NS2B/3 or HA-NS2B/3-S135A and Flag-IKKε or empty plasmid and at 48 h posttransfection were evaluated by proximity ligation assay (PLA). Single detection of proteins as controls for expression: IRF3 (a), IKKε (b), HA-NS2B/3 (c), and HA-NS2B/3-S135A (d). Red fluorescence indicates positive expression of these proteins and blue indicates nuclear staining by DAPI. (e to j) Double detection of protein-protein interactions. HA-tagged proteins were stained with a primary anti-HA antibody and a secondary PLA Minus probe, and IKKε was stained using a primary anti-IKKε antibody and a secondary PLA Plus probe. When the proteins are in close proximity, fluorescence is generated. The absence of red fluorescence reflects no protein-protein interaction. Shown are the endogenous expression of IKKε (e and f) and IKKε overexpression (g and h). (B) Cells were lysed at 48 h posttransfection and extracts coimmunoprecipitated with antibody against HA, and the bound and unbound fractions were analyzed by immunoblotting for IKKε.
To validate the results obtained with the PLA, we transfected cells with the same plasmids coding for the proteases and the kinase. Then, we performed coimmunoprecipitation (coIP) assays with either NS2B/3 or NS2B/3-S135A (Fig. 4B, lanes 1 and 2, respectively) using an antibody against HA and analyzed the immunoprecipitates by immunoblotting for Flag-IKKε. Consistent with the PLA results, we detected a stronger interaction for IKKε with NS2B/3-S135A than with the catalytically active protease NS2B/3. This effect was observed in the unbound fraction, where a portion of the expressed Flag-IKKε was detected in NS2B/3-transfected cells (Fig. 4B, lane 1). As expected, Flag-IKKε was detected in the unbound fraction from control cells that were transfected with HA-tag empty plasmid and Flag-IKKε (Fig. 4B, lane 3).
Protein expression of IKKε is not affected by NS2B/3 or NS2B/3-S135A, but DENV infection restricts IKKε kinase activity.
To determine if IKKε is a substrate of DENV NS2B/3 protease, we conducted a computational analysis to identify putative protease cleavage sites within the IKKε sequence. We identified two putative cleavage sites, one in the N terminus of the protein, specifically in the ATP-binding region of the kinase domain between residues 30 and 31, and the other in the C terminus between residues 559 and 560 (Fig. 5A). We then evaluated if IKKε expression was regulated or showed cleavage products after NS2B/3 transfection or DENV infection. We performed Western blot analysis of the cells cotransfected with plasmids coding for N-terminally Flag-tagged IKKε and HA-tagged NS2B/3 or NS2B/3-S135A and used two different antibodies to increase the chance of detecting the potential cleavage products of IKKε. The antibodies used were an anti-Flag antibody that can recognize the transfected protein and any N-terminal cleavage product and a monoclonal anti-IKKε antibody that recognizes residues surrounding Val345 and that will therefore detect cleavage products containing those residues. The results show that IKKε was equally expressed in cells transfected with NS2B/3 or NS2B/3-S135A. Furthermore, we detected only unique specific bands with no presence of cleavage products using two different antibodies (Fig. 5B). This effect was also observed in DENV-infected cells, in which neither IKKε endogenous expression was regulated nor its cleavage products detected after DENV infection (Fig. 5C).
FIG 5.

Analysis of IKKε putative cleavage by DENV protease NS2B/3 and IKKε functionality after DENV infection. (A) Representation of IKKε structure, including two putative target sites for NS2B/3 protease. (B and C) IKKε cleavage evaluation by Western blotting. (B) 293/TLR3 cells were transfected with Flag-IKKε- and NS2B/3- or NS2B/3-S135A-encoding plasmids for 48 h, and then the cell lysate (20 μg) was analyzed using an anti-Flag or anti-IKKε antibody. (C) Mock- or DENV-infected cells after 24 h postinfection analyzed for IKKε endogenous expression in 40 μg of whole-cell extract using an anti-IKKε antibody. (D) IKKε functionality evaluation. In the top blot, the cells were transfected with IKKε for 24 h, and then infected with DENV-2 for another 24 h to evaluate the ability of DENV to affect IKKε-associated phosphorylation of S386-IRF3. (E) Densitometric analysis of pS386-IRF3 protein levels. The results were normalized to GAPDH. The asterisks represent a significant fold change compared to IKKε-transfected cells using one-way ANOVA: *, P < 0.05; **, P < 0.01 (n = 2). Each bar represents the mean ± SEM.
Because the expression of the protease did not affect IKKε expression, we wanted to ascertain if IKKε functionality was affected by viral infection. For this, we overexpressed IKKε in 293/TLR3 cells to evaluate by immunoblotting whether DENV antagonizes specific IKKε-induced S386-IRF3 phosphorylation activity (Fig. 5D). A densitometric analysis of the cells that were infected with DENV only showed a significantly lower level of S386-IRF3 phosphorylation than cells transfected with IKKε (P < 0.001) (Fig. 5E). However, when lysates of the infected/transfected cells were compared with lysates from cells that were transfected only, DENV infection caused a significant reduction in the IKKε-associated kinase activity that phosphorylates S386-IRF3 (P < 0.05).
NS2B/3 restricts the kinase domain of IKKε.
To examine the potential effect of NS2B/3 structure on IKKε functionality, we employed a computational docking approach that allows for the prediction of protein-protein interactions. As no crystal structure data are currently available for IKKε, we first generated a predicted structure for IKKε using the protein fold recognition server Phyre2. This software modeled the three-dimensional (3D) structure of IKKε using as a template the crystal structure of the kinase TBK1 (PDB ID, 4IM2) available in the Protein Data Bank. The software modeled 610 residues of IKKε with 100.0% confidence by the single highest-scoring template and generated the secondary structure prediction. TBK1 and IKKε are homologous proteins with similarly organized kinase domains and that share >70% amino acid sequence identity (37–39). The output of the 3D structure of IKKε was used to run an in silico analysis of the protein-protein interactions using the fully automatic ClusPro 2.0 docking server.
The modeled structure shows the domains and functional regions of the two interacting proteins (Fig. 6). The side view shows that the NS2B/3 protease complex potentially binds the N-terminal region of IKKε, specifically masking part of the kinase domain. The top view shows more clearly the binding of NS2B/3 to part of the IKKε ATP-binding region (Fig. 6, middle panels). The close-up panels show docking of the viral protease at the ATP-binding region of the kinase and how this interaction masks the accessibility of the ATP-binding Lys38 residue and the proton acceptor Asp135.
FIG 6.

Interaction model of human IKKε with the NS2B/3 protein complex. Shown are the molecular surfaces for human IKKε and NS2B/3 (PDB ID, 2FOM) separated and docked with a side view (upper panel) and a top view (lower panel). The yellow residues represent the IKKε protein kinase domain, residues in orange represent the ATP-binding region, and the remaining residues are shown in gray. Blue residues represent the NS2B/3 complex, and magenta represents the catalytic triad (His51, Asp5, and Ser135). The close-up panels (bottom) illustrate in red two important residues in the kinase domain, Lys38 (ATP-binding) and Asp135 (proton acceptor). The model was generated using the fully automatic ClusPro 2.0 protein-protein docking server and visualized using PyMOL.
DISCUSSION
Type I IFNs have an important role in antiviral host defense, but their responses are not completely understood in DENV infection. To date, most studies have focused on IFN signaling and described the mechanisms of immune evasion of DENV involving the JAK-STAT pathway. For example, the transfection of cells with NS4B partially blocks the activation of STAT1 and IFN-β-stimulated gene expression (40). Similarly, the viral polymerase NS5 binds STAT2 to induce its proteasomal degradation and thereby inhibits IFN responses (22, 41). Less is known about evasion of DENV of the IFN induction pathway, but recently, the DENV NS2B/3 protease was identified as a relevant viral protein in that process (26, 42). In the present study, we performed a series of in vitro and computational experiments to better understand the strategies that DENV employs to inhibit S386-IRF3 phosphorylation during infection and how the NS2B/3 protease modulates IKKε, one of the main kinases associated with the IFN induction pathway.
Regardless of which PRR is stimulated after viral infection, in most cell types, IKKε and TBK1 are the kinases responsible for IRF3 phosphorylation, and the phosphorylation of serine residues in its C-terminal regulatory region is the most critical step in IRF3 activation (6). The roles of specific phosphorylation sites of IRF3 are a subject of some controversy; however, it is clear that IRF3 has two important phosphorylation sites: site 1, which includes S385 and S386, and site 2, which principally includes S396 and S398 (30). The data support a two-step phosphorylation model where phosphorylation at S396 results in IRF3 dimerization and the removal of an autoinhibitory structure that allows the dimer to interact with the coactivator CBP/p300, which in turn facilitates phosphorylation at S385 or S386 (43). In this study, we sought to determine whether DENV regulates IRF3 phosphorylation at S386, which has been suggested to be a major determinant of IRF3 activation (31). Western blot analysis demonstrated that after DENV infection of 293/TLR3 cells, the virus has the ability to actively block RLR-induced IRF3 activation by inhibiting S386-IRF3 phosphorylation. We also observed a moderate impact on S386 phosphorylation when the TLR3 pathway was stimulated (Fig. 1). These results are supported by previous studies showing DENV inhibition of IRF3 phosphorylation at S398 (44). Taken together, these results suggest that DENV can inhibit IRF3 phosphorylation of at least two different serines of the IRF3 phosphoserine cluster necessary for its activation. The impact of HCV infection on the phosphorylation of IRF3 at different serine residues has also been assessed and was similarly found to inhibit the phosphorylation of both serines (45).
The immunofluorescence analysis used to evaluate the nuclear translocation of IRF3 supports the results of the Western blot assays. DENV infection blocks RLR-induced nuclear translocation of IRF3 and moderately reduces TLR3-induced translocation (Fig. 2). Moreover, DENV completely abolished the detection of cytoplasmic and nuclear pS386-IRF3 after infection or dual treatment with infection and RLR-associated stimulation. The virus was unable to inhibit pS386-IRF3 translocation after TLR3-associated stimulation. The immunofluorescence results demonstrated the ability of the virus to actively block IRF3 and pS386-IRF3 nuclear translocation.
In our studies, we observed that DENV delays the early innate immune response more effectively by blocking the RLR pathway than the TLR3 pathway (Fig. 1 and 2). The relevance of the RLR pathway in DENV has been assessed and demonstrated delayed activation of RIG-I compared to MDA5 after DENV infection of HUH-7 cells up to 48 h postinfection (46). Furthermore, Ubol et al. (47) showed that DENV causes reduced RIG-I expression at 12 h postinfection, without any effect on MDA5. Studies have demonstrated the relevance of the length of dsRNA to the activation of the innate immune response and identified long poly(I·C) as being essential for MDA5-mediated IFN production and short poly(I·C) as necessary for activation of the RIG-I pathway (32). Considering that we transfected cells with low-molecular-weight poly(I·C), we can argue that the RIG-I pathway is probably the major target in our experiments and also the major target of the innate immune response for DENV.
Recent studies identified DENV protease as an important viral protein related to the subversion of IRF3 phosphorylation at the S396 residue (42). Here, we showed that 293/TLR3 cells transfected with the enzymatically active protease (NS2B/3) exhibited a significant reduction in the level of RLR-induced S386-IRF3 phosphorylation (Fig. 3), which is in agreement with previous studies that identified the requirement of the catalytically active viral protease complex for reducing type I IFN production (34). Interestingly, we showed that the catalytically inactive protease moderately inhibited IRF3 activation. Studies that evaluated DENV-mediated inhibition of Japanese encephalitis virus-induced S396-IRF3 phosphorylation produced similar results, showing the partial inhibition of IRF3 phosphorylation when NS2B/3-S135A was expressed in A549 cells (42). Recently, studies of HCV demonstrated that the NS2 protease inhibits Sendai virus-induced expression of the IFN-β promoter regardless of the proteolytic activity of NS2 (48). Moreover, engagement of the arenavirus nucleoprotein seems to sequester IKKε into an inactive complex without any proteolytic processing (49). These results support a model in which DENV protease, like the proteases of other viruses, can operate by catalysis-dependent and -independent mechanisms to inhibit the IFN induction pathway.
We studied IKKε, one of the two kinases associated with IRF3 phosphorylation. First, we sought to determine whether viral protease physically interacts with this kinase. Interaction experiments using PLA and coIP demonstrated that both NS2B/3 and NS2B/3-S135A are able to interact with the kinase, but the IKKε-NS2B/3-S135A interaction seems to be stronger than the interaction with the active protease (Fig. 4). At this point, we have no experimental evidence to explain this phenomenon, but our results suggest that the IKKε-NS2B/3 interaction is mediated by a catalysis-independent mechanism, and it is possible that catalysis-dependent activity modulates the stability of this interaction, which would be consistent with the IRF3 phosphorylation results (Fig. 3).
Our observation is supported by recent findings demonstrating that the DENV NS2B/3 protease physically interacts with STING, and the interaction induces the cleavage of STING by a catalysis-dependent mechanism (26, 42). Furthermore, these studies showed that both the catalytically active and inactive forms of the protease coimmunoprecipitated with STING, which is in agreement with our observations. Those data and our results support that the viral protease in both its active and inactive forms interacts with at least two different proteins of the IFN induction pathway, namely, STING and IKKε. Studies that characterized the role of STING in the multiprotein complex that induces type I IFN revealed that STING dimerizes and acts as a scaffold protein to recruit IKKε/TBK1 and IRF3 (50, 51). In this scenario, the DENV protease-mediated cleavage of STING alters the assembly of the complexes required for IRF3 activation and can thus explain the reduced IKKε-NS2B/3 interaction that we observed. However, in the absence of cleavage, the complex will still be stable, allowing a stronger IKKε-NS2B/3-S135A interaction. Nevertheless, understanding the dynamics of multiprotein complexes is challenging and will require more studies to decipher the component functions.
The interaction between IKKε and NS2B/3 can be explained by two different events. One possible mechanism involves protease binding to IKKε to induce its proteolytic cleavage. Although the kinase has two putative cleavage sites, our assay did not detect any IKKε cleavage products (Fig. 5A to C). The second mechanism may be that the association of the viral protease with the cellular kinase interferes with the IKKε activity domains. Here, we confirmed the impact of this IKKε-NS2B/3 interaction on kinase functionality by showing that infection with DENV significantly decreases IKKε kinase activity (Fig. 5D). This result strongly suggests that DENV negatively alters the kinase activity of IKKε.
In order to better understand the IKKε-NS2B/3 interaction, we conducted a docking computational analysis to model the structural basis of the interaction. We obtained evidence showing that the NS2B/3 protease interacts with IKKε, specifically with the kinase domain (Fig. 6). The adopted conformation masks the protein kinase and ATP-binding domains by steric hindrance, which supports the previous observation of an interaction between the two proteins that is independent of the proteolytic activity of NS2B/3. This interaction model supports the experimental observation of a DENV-associated inhibition of IKKε kinase function (Fig. 5D). Our structural analyses are relevant because the interaction of NS2B/3 with IKKε may not only affect the physical interaction of IRF3 with the kinase domain of IKKε, but it may also interfere with the access of other important proteins of the multiprotein complex that induces type I IFN transcription. A similar mechanism of inhibition has been described in another positive-stranded RNA virus: the papain-like protease of coronavirus antagonizes the innate immunity by disrupting the assembly of the STING-MAVS-IKKε/TBK1 complex (11). Thus, further studies with IKKε may reveal the mechanistic details that modulate this innate immune response.
In summary, this study describes a new unrecognized function of DENV NS2B/3. DENV protease counteracts the IFN induction pathway through direct interaction with IKKε by catalysis-dependent and -independent mechanisms interfering with the kinase activity of IKKε. DENV NS2B/3 plays a role in DENV pathogenicity, and therefore the knowledge gained by characterizing the mechanisms through which DENV evades innate immunity might provide insights that will spark the development of novel therapeutic strategies against this virus.
ACKNOWLEDGMENTS
We thank Jorge Muñoz-Jordán, Adolfo García-Sastre, and Katherine Fitzgerald for providing us with protocols and reagents, Gilberto Santiago for his critical review of the manuscript, and Teresa Arana and Roberto Medina for their technical support. We also thank the Associate Deanship of Biomedical Sciences at the University of Puerto Rico School of Medicine.
We also acknowledge the contributions to this study by grants ISI0 RR-13705-01, DBI-0923132, 2G12-RR003051, and 8G12-MD007600. This work was funded by the University of Puerto Rico MBRS-RISE program R25GM061838 (to Y.I.A.R), and partially by NIH/NIAID U24OD01042 and U42OD011128 (to C.A.S).
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.WHO and TDR 2009. Dengue: guidelines for diagnosis, treatment, prevention and control: new edition. World Health Organization and the Special Programme for Research and Training in Tropical Diseases (TDR), Geneva, Switzerland [Google Scholar]
- 2.Gubler DJ. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10:100–103. 10.1016/S0966-842X(01)02288-0 [DOI] [PubMed] [Google Scholar]
- 3.Fink J, Gu F, Ling L, Tolfvenstam T, Olfat F, Chin KC, Aw P, George J, Kuznetsov VA, Schreiber M, Vasudevan SG, Hibberd ML. 2007. Host gene expression profiling of dengue virus infection in cell lines and patients. PLoS Negl. Trop. Dis. 1:e86. 10.1371/journal.pntd.0000086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falgout B, Pethel M, Zhang YM, Lai CJ. 1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 65:2467–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Lim SP, Beer D, Patel V, Wen D, Tumanut C, Tully DC, Williams JA, Jiricek J, Priestle JP, Harris JL, Vasudevan SG. 2005. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 280:28766–28774. 10.1074/jbc.M500588200 [DOI] [PubMed] [Google Scholar]
- 6.Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644–658. 10.1038/nri1900 [DOI] [PubMed] [Google Scholar]
- 7.Meylan E, Tschopp J. 2006. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell 22:561–569. 10.1016/j.molcel.2006.05.012 [DOI] [PubMed] [Google Scholar]
- 8.Yoneyama M, Fujita T. 2007. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 18:545–551. 10.1016/j.cytogfr.2007.06.023 [DOI] [PubMed] [Google Scholar]
- 9.Noyce RS, Collins SE, Mossman KL. 2009. Differential modification of interferon regulatory factor 3 following virus particle entry. J. Virol. 83:4013–4022. 10.1128/JVI.02069-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. 2009. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. U. S. A. 106:8653–8658. 10.1073/pnas.0900850106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, Clementz MA, Banach BS, Li K, Baker SC, Chen Z. 2012. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7:e30802. 10.1371/journal.pone.0030802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donelan NR, Dauber B, Wang X, Basler CF, Wolff T, García-Sastre A. 2004. The N- and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 78:11574–11582. 10.1128/JVI.78.21.11574-11582.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, García-Sastre A. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 74:7989–7996. 10.1128/JVI.74.17.7989-7996.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Mühlberger E, Bray M, Klenk HD, Palese P, García-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77:7945–7956. 10.1128/JVI.77.14.7945-7956.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw ML, Cardenas WB, Zamarin D, Palese P, Basler CF. 2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and Toll-like receptor 3-triggered signaling pathways. J. Virol. 79:6078–6088. 10.1128/JVI.79.10.6078-6088.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keller BC, Johnson CL, Erickson AK, Gale M., Jr 2007. Innate immune evasion by hepatitis C virus and West Nile virus. Cytokine Growth Factor Rev. 18:535–544. 10.1016/j.cytogfr.2007.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho LJ, Hung LF, Weng CY, Wu WL, Chou P, Lin YL, Chang DM, Tai TY, Lai JH. 2005. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J. Immunol. 174:8163–8172 [DOI] [PubMed] [Google Scholar]
- 18.Tsai YT, Chen YH, Chang DM, Chen PC, Lai JH. 2011. Janus kinase/signal transducer and activator of transcription 3 signaling pathway is crucial in chemokine production from hepatocytes infected by dengue virus. Exp. Biol. Med. (Maywood) 236:1156–1165. 10.1258/ebm.2011.011060 [DOI] [PubMed] [Google Scholar]
- 19.Muñoz-Jordán JL. 2010. Subversion of interferon by dengue virus. Curr. Top. Microbiol. Immunol. 338:35–44. 10.1007/978-3-642-02215-9_3 [DOI] [PubMed] [Google Scholar]
- 20.Muñoz-Jordán JL, Fredericksen BL. 2010. How flaviviruses activate and suppress the interferon response. Viruses 2:676–691. 10.3390/v2020676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muñoz-Jordán JL, Sánchez-Burgos GG, Laurent-Rolle M, García-Sastre A. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U. S. A. 100:14333–14338. 10.1073/pnas.2335168100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashour J, Laurent-Rolle M, Shi PY, García-Sastre A. 2009. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 83:5408–5418. 10.1128/JVI.02188-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meylan E, Tschopp J, Karin M. 2006. Intracellular pattern recognition receptors in the host response. Nature 442:39–44. 10.1038/nature04946 [DOI] [PubMed] [Google Scholar]
- 24.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102:2992–2997. 10.1073/pnas.0408824102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, Akira S, Mechti N. 2009. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393:311–318. 10.1016/j.virol.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 26.Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A. 2012. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 8:e1002934. 10.1371/journal.ppat.1002934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reference deleted.
- 28.Falgout B, Miller RH, Lai CJ. 1993. Deletion analysis of dengue virus type 4 nonstructural protein NS2B: identification of a domain required for NS2B-NS3 protease activity. J. Virol. 67:2034–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. 2004. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50. 10.1093/bioinformatics/btg371 [DOI] [PubMed] [Google Scholar]
- 30.Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE, Jr, Lin K. 2008. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J. Mol. Biol. 379:251–260. 10.1016/j.jmb.2008.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. 2004. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 279:9698–9702. 10.1074/jbc.M310616200 [DOI] [PubMed] [Google Scholar]
- 32.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610. 10.1084/jem.20080091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A. 2010. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol. 84:9760–9774. 10.1128/JVI.01051-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khumthong R, Angsuthanasombat C, Panyim S, Katzenmeier G. 2002. In vitro determination of dengue virus type 2 NS2B-NS3 protease activity with fluorescent peptide substrates. J. Biochem. Mol. Biol. 35:206–212. 10.5483/BMBRep.2002.35.2.206 [DOI] [PubMed] [Google Scholar]
- 36.Yang H, Lin CH, Ma G, Orr M, Baffi MO, Wathelet MG. 2002. Transcriptional activity of interferon regulatory factor (IRF)-3 depends on multiple protein-protein interactions. Eur. J. Biochem. 269:6142–6151. 10.1046/j.1432-1033.2002.03330.x [DOI] [PubMed] [Google Scholar]
- 37.Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18:6694–6704. 10.1093/emboj/18.23.6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC, Lin R, Hiscott J. 2004. Activation of TBK1 and IKKepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 78:10636–10649. 10.1128/JVI.78.19.10636-10649.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu D, Zhu Z, Zhou AY, Yun CH, Lee KE, Toms AV, Li Y, Dunn GP, Chan E, Thai T, Yang S, Ficarro SB, Marto JA, Jeon H, Hahn WC, Barbie DA, Eck MJ. 2013. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 3:747–758. 10.1016/j.celrep.2013.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, Martínez-Sobrido L, Ashok M, Lipkin WI, García-Sastre A. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004–8013. 10.1128/JVI.79.13.8004-8013.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazzon M, Jones M, Davidson A, Chain B, Jacobs M. 2009. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 200:1261–1270. 10.1086/605847 [DOI] [PubMed] [Google Scholar]
- 42.Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL. 2012. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 8:e1002780. 10.1371/journal.ppat.1002780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panne D, McWhirter SM, Maniatis T, Harrison SC. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282:22816–22822. 10.1074/jbc.M703019200 [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Madoz JR, Bernal-Rubio D, Kaminski D, Boyd K, Fernandez-Sesma A. 2010. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J. Virol. 84:4845–4850. 10.1128/JVI.02514-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U. S. A. 102:2986–2991. 10.1073/pnas.0408707102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nasirudeen AM, Wong HH, Thien P, Xu S, Lam KP, Liu DX. 2011. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 5:e926. 10.1371/journal.pntd.0000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ubol S, Phuklia W, Kalayanarooj S, Modhiran N. 2010. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J. Infect. Dis. 201:923–935. 10.1086/651018 [DOI] [PubMed] [Google Scholar]
- 48.Kaukinen P, Sillanpää M, Nousiainen L, Melen K, Julkunen I. 2013. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J. Med. Virol. 85:71–82. 10.1002/jmv.23442 [DOI] [PubMed] [Google Scholar]
- 49.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martínez-Sobrido L, de la Torre JC, Kunz S. 2012. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKϵ. J. Virol. 86:7728–7738. 10.1128/JVI.00187-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5:ra20. 10.1126/scisignal.2002521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burdette DL, Vance RE. 2013. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 14:19–26. 10.1038/ni.2491 [DOI] [PubMed] [Google Scholar]