

Abstract
Women showing normal cytology but diagnosed with a persistent high-risk human papillomavirus (HR-HPV) infection have a higher risk of developing high-grade cervical intraepithelial neoplasia and cervical cancer than noninfected women. As no therapeutic management other than surveillance is offered to these women, there is a major challenge to develop novel targeted therapies dedicated to the treatment of these patients. As such, E1 and E2 antigens, expressed early in the HPV life cycle, represent very interesting candidates. Both proteins are necessary for maintaining coordinated viral replication and gene synthesis during the differentiation process of the epithelium and are essential for the virus to complete its normal and propagative replication cycle. In the present study, we evaluated a new active targeted immunotherapeutic, a modified vaccinia virus Ankara (MVA) vector containing the E1 sequence of HPV16, aimed at inducing cellular immune responses with the potential to help and clear persistent HPV16-related infection. We carried out an extensive comparative time course analysis of the cellular immune responses induced by different schedules of immunization in C57BL/6 mice. We showed that multiple injections of MVA-E1 allowed sustained HPV16 E1-specific cellular immune responses in vaccinated mice and had no impact on the exhaustion phenotype of the generated HPV16 E1-specific CD8+ T cells, but they led to the differentiation of multifunctional effector T cells with high cytotoxic capacity. This study provides proof of concept that an MVA expressing HPV16 E1 can induce robust and long-lasting E1-specific responses and warrants further development of this candidate.
INTRODUCTION
Cervical cancer is the second most frequent cancer in women worldwide (1). It results from infection with high-risk human papillomaviruses (HR-HPV) through sexual contact (2). HPVs are small, nonenveloped, double-stranded DNA viruses. They are divided into cutaneous and mucosal subtypes and further classified as high risk (HR) or low risk, depending on the lesion they are associated with. Gene coding sequences are located on one DNA strand and designated early or late according to their expression during the viral life cycle. HPVs replicate in stratified squamous epithelia using the differentiation of the epithelium to regulate their replication (3). In the early stages of infection, HPV replicates its DNA to maintain the viral genome as a low-copy number nuclear episome. The E1 and E2 proteins are essential for viral replication. E1 displays DNA helicase activity and plays a role in viral replication and replication repression (3). E2 is a regulator of viral transcription and replication. It is necessary for efficient viral DNA replication, together with E1 (3), and controls the expression of E6 and E7 proteins, which allow the differentiation process to start (2). Expressions of late proteins L1 and L2 are initiated in the suprabasal layers, where viral particles are assembled and then released in the upper layers of the mucosal epithelium, ensuring viral propagation (2). One striking feature of HR-HPV infection is that its incidence far exceeds the number of individuals who develop HPV-associated malignancies, with 95% of HR-HPV infections of the cervix resolving spontaneously. In subjects with persistent infections, HR-HPV subtypes induce cervical intraepithelial neoplasia (CIN) histologically graded from 1 to 3. In the absence of therapeutic intervention, such dysplasias are likely to progress into in situ carcinoma, leading eventually to invasive cervical cancer. Viral integration in the host genome frequently occurs in malignant lesions, with a loss of function and expression of E1/E2 proteins. As a consequence, expression of the oncoproteins E6 and E7 is deregulated, which results in cellular transformation (2). This transformation occurs through the binding to, and inactivation of, p53 and retinoblastoma tumor suppressor genes by E6 and E7 proteins, respectively (2). The higher occurrence of HR-HPV-associated malignancies among immunocompromised individuals suggests that the immune system controls to some extent HPV infection. Two HR-HPV subtypes, HPV16 and HPV18, account for 70 to 80% of cervical cancers. Prophylactic vaccination for cervical cancer is now available for young women who are not yet sexually active, as two vaccines targeting the L1 viral capsid protein from HPV16 and HPV18 genotypes have been approved by authorities. From the onset of clinical use, these vaccines have together displayed more than 90% efficacy in the prevention of cervical lesion development (4). However, these prophylactic HPV vaccines do not show any therapeutic effect on preexisting HPV infection (5). Immunotherapeutic approaches targeting HPV-infected cells have been developed to induce efficient cellular immune responses in patients in whom HR-HPV infections do not resolve. Due to their permanent expression in HPV-transformed cells, E6 and E7 proteins were the prevailing targets of various delivery systems, including subunit vaccines, naked DNA, peptides, antigen-loaded dendritic cells, bacteria, and viral vectors (6).
For years, cervical cancer screening programs were based only on cytomorphological criteria. Nowadays, HPV DNA testing is being generalized and identifies a population for whom there is no available treatment. Indeed, women with normal cytology in whom HR-HPV genotypes are detected in two successive DNA tests 12 months apart are diagnosed as persistently infected. These women have a 100- to 200-fold greater risk of developing CIN2 than uninfected women (7). The development of a novel therapy dedicated to this population thus represents a major public health challenge. An innovative treatment could be an immunotherapy targeting E1 and E2 antigens, which are expressed early in the HPV life cycle, just following primary infection of the cervical epithelium, while the HPV genome is replicated as an episome (2). The therapeutic potential of HPV16 E1 as an immune target has already been described in the canine oral papillomavirus (COPV) (8) and cottontail rabbit papillomavirus (CRPV) models (9). Furthermore, a recent publication reported that E1 protein was dispensable for replication maintenance but not for the productive replication of HPV16 during keratinocyte differentiation, thus restricting the rationale for the development of E1 inhibitors, while strengthening that for the development of an immunotherapeutic approach (10).
Multiple clinical trials were conducted using modified vaccinia virus Ankara (MVA) as a vector for immunotherapy (11–14). MVA is a double-stranded DNA virus derived from a Turkish smallpox vaccine strain through more than 570 passages in primary chicken embryo fibroblasts (15). As a consequence, it is unable to complete its replication cycle in most mammalian cells. MVA has an excellent safety profile in humans (11, 15) and can contain large amounts of foreign DNA to induce, upon immunization, both humoral and cellular immune responses against the expressed foreign antigen (14, 16–18). Clinical immunotherapy protocols with MVA-based products have recently shown promise in settings where multiple injections over a relatively short period of time are applied (12, 13, 19). Yet, the impact of such closely administered injections of large quantities of viral material on the qualitative aspect of the immune response remains poorly studied. In the present study, we constructed a recombinant MVA vector carrying the sequence of the HPV16 E1 gene (MVA-E1) and characterized the cellular immunogenicity of this construct. We notably investigated the consequences of 1, 2, or 3 injections of MVA-E1 on the quality of the generated immune response in terms of recall capacities, immunological memory generation, CD4+ regulatory T (Treg) cell induction, multifunctionality, and in vivo killing capacities.
MATERIALS AND METHODS
Peptide identification.
We used the SYFPEITHI prediction algorithm (20) (see www.syfpeithi.de) to screen for putative epitopes in the HPV16 E1 sequence, which predicted the IAYKYAQL peptide (score, 30) to be H-2Kb restricted.
Mice.
Five-week-old C57BL/6 mice were purchased from Charles River laboratory (L'Arbresle, France). Animals were housed under specific pathogen-free conditions with water and food ad libitum. This study was conducted in compliance with European Union (EU) directive 2010/63/EU for animal experiments. An institutional ethical committee has approved the experiments performed in this study.
Recombinant MVA.
The sequence coding for the E1 gene from HPV16 was amplified by PCR using DNA extracted from CaSki cells (ATCC CRL-1550) as the template. E1 was mutated to generate a G482D modified protein which is devoid of its DNA replication function (21). The plasmid used for homologous recombination in the so-called deletion III corresponding site of the MVA genome was based on plasmid pTG1E (22). Flanking sequences (BRG3 and BRD3) surrounding the deletion III were amplified by PCR from MVATGN33 (a subclone isolated in our laboratory) DNA (23). The transfer plasmid also contained a fusion between the Aequorea victoria enhanced green fluorescent protein gene (eGFP gene, isolated from pEGP-C1; Clontech) and the E. coli xanthine-guanine phosphoribosyltransferase gene (gpt gene) under the control of the early/late vaccinia virus synthetic promoter p11K7.5 (kindly provided by R. Wittek, University of Lausanne). Synthesis of xanthine-guanine phosphoribosyltransferase enabled GPT+-recombinant MVA to form plaques in a selective medium containing mycophenolic acid, xanthine, and hypoxanthine. eGFP enables the visualization of recombinant MVA plaques. When the clonal selection was achieved, the selection marker eGFP–GPT, placed between two homologous sequences in the same orientation, was eliminated by several passages without selection. The modified HPV16 E1 gene was inserted downstream of the p7.5K promoter (24). Generation of MVA-E1 was performed by homologous recombination in primary chicken embryo fibroblasts (CEF). The transfer plasmid was transfected according to the standard calcium phosphate DNA precipitation method onto CEF previously infected with MVATGN33 at a multiplicity of infection (MOI) of 0.1 PFU/cell. Viral selection was performed by three rounds of plaque purification in the presence of a selective medium containing mycophenolic acid, xanthine, and hypoxanthine on CEF, and then the selection marker was eliminated by passages in nonselective medium. Absence of contamination by parental MVA was verified by PCR.
Western blot analysis.
Cell extracts from CEF infected with MVA-E1 or mock MVA were separated by SDS-PAGE and electrotransferred to a nitrocellulose membrane. E1 protein was detected using an in-house-made mouse polyclonal anti-HPV16 E1 serum diluted 1:5,000. Immune complexes were detected with horseradish peroxidase-conjugated goat anti-mouse immunoglobulins (Dako, P0047) and revealed with enhanced chemiluminescence (ECL) (Amersham).
Immunizations.
Prior to injection, MVA was diluted in specific buffer (10 mM Tris-HCl, 5% saccharose, 10 mM sodium glutamate, and 50 mM sodium chloride [pH 8]) under sterile conditions. Animals were injected subcutaneously (s.c.). Each injection consisted of 5 × 107 PFU in 100 μl. MVA was injected once, twice, or thrice with a 7-day interval between injections. Injections were scheduled so that in each tested group, day 21 represented the seventh day after the last MVA injection. Animals were boosted at the indicated days with MVA-E1. At the indicated time points, animals were sacrificed for immunogenicity testing.
ELISpot assay.
For detection of single gamma interferon (IFN-γ)-producing cells, 96-well polyvinylidene difluoride (PVDF) membranes (MultiScreen HTS; Millipore) were coated with a rat anti-mouse IFN-γ antibody (MabTech). Splenocytes (106/well) were cultured in these coated plates for 18 h at 37°C in a 5% CO2 atmosphere. The H2-Kb-restricted HPV16 E1-specific peptide (IAYKYAQL) was used at 1 μg/ml for in vitro stimulation. The irrelevant peptide RAHYNIVTF (HPV16E7 specific) and the MVA-derived peptide TSYKFESV were both used at 1 μg/ml as negative and positive controls, respectively. IFN-γ was detected with a biotinylated rat anti-mouse IFN-γ antibody coupled to alkaline phosphatase (Mabtech). Spots were revealed using 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium as the substrate and counted with an enzyme-linked immunosorbent spot (ELISpot) reader (Biosys 4000 series).
Flow cytometry.
Two million lymphocytes isolated from spleen were incubated for 10 min at room temperature with 2.5 μl of custom phycoerythrin (PE)-conjugated H-2Kb IAYKYAQL tetramer (Beckman Coulter). Lymphocytes were washed and then incubated for 30 min at 4°C with anti-mouse CD44, CD8, and CD3 antibodies (BD Biosciences). To analyze the memory phenotype of antigen-specific CD8+ T cells, tetramer-stained lymphocytes were incubated with anti-mouse CD8, CD3, CD62L (BD Biosciences), and CD127 (eBioscience) antibodies. The programmed death 1 (PD-1) phenotype was determined by the addition of an anti-mouse PD-1 antibody (R&D Systems) in the above-described setting.
For intracellular detection of cytokine production, splenocytes were stimulated for 16 h at 37°C with 1 μg/ml of the IAYKYAQL peptide in the presence of Golgistop (BD Biosciences). T lymphocytes were identified with anti-mouse CD3, CD8, and CD4 antibodies (BD Biosciences). Cells were then fixed and permeabilized 1 h in Fix/Perm buffer (eBioscience). Anti-mouse IFN-γ, interleukin-2 (IL-2), and tumor necrosis factor alpha (TNF-α) antibodies (BD Biosciences) were used to visualize intracellular cytokines. Flow cytometry was performed on an FACS Aria III (BD Biosciences).
In vivo cytotoxicity assay.
For in vivo measurement of CTL activity, 107 splenocytes from naive animals were pulsed for 1 h at 37°C with 10 μM IAYKYAQL peptide and labeled with 6.4 μM carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) for 10 min at 37°C. A second fraction of 107 unpulsed splenocytes was labeled with 0.4 μM CFSE. The two fractions were mixed in equal proportions for intravenous injection into recipient mice. Splenocytes were harvested 18 h later, prior to fluorescence-activated cell sorting (FACS) acquisition. The percentage of specific lysis was calculated using the formula percentage of specific lysis = 100 − (100 × [R in immunized group/mean R in naive group]), where the ratio (R) is the number of loaded cells/number of unpulsed cells.
Statistical analysis.
When indicated, statistical significance was determined by nonparametric Kruskal-Wallis and Dunn's multiple-comparison tests (GraphPad Software, Inc., CA). Differences were considered significant at P values of <0.05.
RESULTS
Expression of MVA-E1.
To verify that HPV16 E1 protein is effectively produced by infected cells, we incubated CEF with 0.2 PFU of MVA-E1 or mock MVA and performed the detection of HPV16 E1 by Western blot analysis 24 h later. HPV16 E1 protein could be specifically detected in MVA-E1-infected CEF (Fig. 1).
FIG 1.
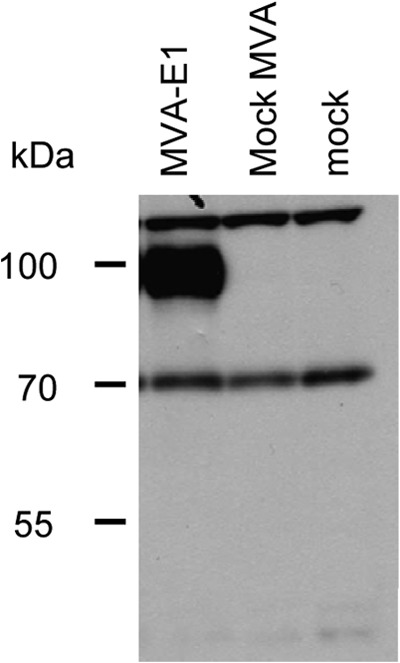
Expression of HPV16E1 protein in CEF infected with MVA-E1. CEF were infected with 0.2 PFU/cell of MVA-E1, with mock MVA, or without vector (mock). Cell extracts collected 24 h postinfection were analyzed by SDS-PAGE. HPV16E1 protein was detected by Western blot analysis using an in-house-made mouse polyclonal serum against HPV16E1.
Intensity and duration of the immune response.
To evaluate the impact of multiple injections of MVA-E1 on the robustness of the induced immune response, we immunized groups of C57BL/6 mice with one, two, or three injections of MVA-E1 and then looked at diverse immunological parameters.
We first performed a mouse IFN-γ ELISpot assay to analyze the level and duration of the cellular immune response. Upon in vitro stimulation with HPV16 E1 peptide IAYKYAQL, between 18,000 and 30,000 IFN-γ-secreting cells were found in the spleens of the animals vaccinated with MVA-E1 on day 21, whatever the number of MVA-E1 injections (Fig. 2A to C). The observed responses were specific for HPV16 E1, as no IFN-γ-secreting splenocytes were detected upon in vitro stimulation with an irrelevant peptide (data not shown) or in mice vaccinated with 3 injections of mock MVA (Fig. 2D). On day 28, the number of IFN-γ-secreting HPV16 E1-specific cells markedly decreased in all groups, except for those vaccinated with 3 injections of MVA-E1 (Fig. 2C). However, differences observed were not significant (Kruskal-Wallis test and Dunn's multiple-comparison test). Thereafter, in the spleens of the animals vaccinated with only one injection of MVA-E1 (Fig. 2A), <2,000 IFN-γ-secreting HPV16 E1-specific cells were detected, while in those vaccinated with 2 or 3 injections of MVA-E1, between 2,500 and 8,500 IFN-γ-secreting HPV16 E1-specific cells were detected until day 73 (Fig. 2B and C). All together, these results indicate that multiple injections of MVA-E1 induce sustained HPV16 E1 cellular immune responses in vaccinated mice.
FIG 2.

Time course analysis of the number of HPV16 E1-specific IFN-γ-producing cells (peptide IAYKYAQL) by ex vivo ELISpot assay. The y axis shows the total number of IFN-γ-secreting cells per spleen. Time of sampling is indicated on the x axis. The mean values ± the standard error of the mean (SEM) from 3 independent experiments (except for day 129, n = 1) are represented. In each experiment, five mice were sacrificed and their splenocytes were pooled at each time point. Individual values were calculated as the means of triplicate wells. (A, B, C) One (day 14), two (days 7 and 14), or three (days 0, 7, and 14) immunizations with MVA-E1, respectively. (D) Three immunizations with mock MVA vector (days 0, 7, and 14). Significance was evaluated by Kruskal-Wallis test and Dunn's multiple comparison test for each time point between groups receiving one, two, or three immunizations with MVA-E1. ns, not significant.
Impact of MVA-E1 boost immunization.
The anti-MVA cellular and humoral immune responses generated and detected after multiple immunizations with MVA vectors are important (data not shown). To investigate the impact of a booster injection with MVA-E1 on HPV16 E1-specific cellular immunity, groups of immunized mice were boosted 1, 2, or 4 months after the last MVA-E1 injection, and the intensity of HPV16 E1-specific cellular immune response was investigated 7 days later by IFN-γ ELISpot. In each immunized group, levels of recalled HPV16 E1-specific cellular immune response were equivalent to those of day 21, with the detection of 15,000 to 35,000 IFN-γ-secreting cells per spleen at each time point (Fig. 3A to C). These results indicate that in our experimental setting, the initial number of MVA-E1 injections had no noticeable impact on the intensity of the recalled HPV16 E1-specific cellular immunity. As expected, preexisting immunity to the MVA backbone prevented the induction of HPV16 E1-specific cellular immunity after MVA-E1 boosting (Fig. 2D and 3D).
FIG 3.

Impact of the MVA-E1 boost vaccination on the number of recalled HPV16 E1-specific IFN-γ-producing cells (peptide IAYKYAQL) by ex vivo ELISpot assay. Following initial injection schedules, mice from all groups were boosted with MVA-E1 at either day 46, 73, or 129, as indicated by the arrows. Immune responses were monitored 7 days after boost, at days 54, 80, and 136, respectively. The boost at day 129 was also monitored at day 158. The y axis shows the total number of IFN-γ-secreting cells per spleen. Time of sampling is indicated on the x axis. The mean values ± the SEM from two to three independent experiments are represented. In each experiment, five mice were sacrificed and their splenocytes were pooled at each time point. Individual values were calculated as the means of triplicate wells. (A, B, C) One (day 14), two (days 7 and 14), or three (days 0, 7, and 14) immunizations with MVA-E1, respectively. (D) Three immunizations with mock MVA vector (days 0, 7, and 14). Significance was evaluated by Kruskal-Wallis test and Dunn's multiple comparison test for each time point between groups receiving one, two, or three immunizations with MVA-E1. ns, not significant.
Memory and exhaustion phenotype of HPV16 E1-specific T cells.
Booster immunizations have important consequences on memory CD8+ T cell response quality (25, 26). While PD-1 is a marker of T cell exhaustion in chronic viral diseases (27–29) that has been shown to be induced by multiple immunizations (30), CD4+ CD25+ FoxP3+ regulatory T cells have the ability to suppress host immune responses and account for viral persistence and tissue damage during chronic viral infections (31). We analyzed the impact of multiple MVA-E1 immunizations as well as MVA-E1 boosting on these different parameters. Consistent with results obtained in IFN-γ ELISpot assays, percentages of H-2Kb-restricted IAYKYAQL-specific CD8 T cells observed in the spleen (detected by tetramer staining) were higher after multiple injections of MVA-E1 than after a single injection of MVA-E1 (Fig. 4A to C, left panels). These HPV16 E1-specific splenic CD8 T cells were analyzed for their expressions of CD62L and CD127, to discriminate between effector, effector memory, and central memory T cells (32). Whatever the time point considered, we did not observe marked differences in the proportions of HPV16 E1-specific effector, effector memory, and central memory T cells between the different groups in our experimental setup (Fig. 4A to C, middle panels). Effector memory T cells were prevailing on day 21. The proportion of effector T cells was high on day 73, but the effector memory phenotype became predominant again 7 days after boosting (day 80). On day 129, the tetramer-positive (Tet+) cells were mostly of the central memory phenotype but also with an important proportion of intermediate cells. Upon boost (day 136), the HPV16 E1-specific CD8+ T cells were mainly of the effector and effector memory phenotypes. One month later (day 158), the effector memory phenotype was again predominant. We also monitored PD-1 expression on these H-2Kb IAYKYAQL Tet+ CD8+ T cells. Again, no marked differences in the percentages of HPV16 E1-specific CD8+ T cells expressing PD-1 were seen between the different groups whatever the time point considered (Fig. 4A to C, right panels). Of note, the proportion of HPV16 E1-specific CD3+ CD8+ PD-1+ T cells was the highest on day 73 in all groups, but to a lesser extent in mice immunized with 3 injections of MVA-E1. Finally, we saw no particular trend concerning the numbers of CD4+ CD25+ FoxP3+ regulatory T cells among the different groups (1.3 × 105 to 5 × 105 regulatory T cells per spleen, data not shown).
FIG 4.

Time course analysis of memory and exhaustion phenotypes. Following initial immunizations, the HPV16 E1-specific immune response was monitored in the spleen by tetramer staining on 30,000 CD3+ CD8+ CD44high T cells (left panels). Boost immunizations with MVA-E1 were performed on either day 73 or 129, as indicated by the arrows. Boosted immune responses were monitored on days 80 and 136, respectively. Concerning the boost at day 129, an additional monitoring point was performed at day 158. Memory phenotypes of tetramer-positive cells were evaluated with anti-mouse CD62L and CD127 (middle panels). Stacked histograms show the relative proportions of central memory cells (CD62L+ CD127+, white), effector memory cells (CD62L− CD127+, light gray), effector cells (CD62L− CD127−, dark gray) and cells with intermediate phenotype (CD62L+ CD127−, black). Exhaustion phenotype of tetramer-positive cells was evaluated with anti-mouse PD-1 (right panels). Time of sampling is indicated on the x axis. The mean values ± the SEM from two independent experiments are represented. In each experiment, five mice were sacrificed and their splenocytes were pooled at each time point. (A, B, C) One (day 14), two (days 7 and 14), or three (days 0, 7, and 14) immunizations with MVA-E1, respectively.
All together, our results indicate that, in comparison with a single injection of MVA-E1, the administration of up to 3 injections of large quantities of MVA-E1 in a short period of time has no influence on the memory phenotype or on the PD-1 expression profiles of the generated HPV16 E1-specific CD8+ T cells (or on the number of CD4+ CD25+ FoxP3+ regulatory T cells). The same observations apply to secondary memory HPV16 E1-specific CD8+ T cells generated after booster injections.
Quality of the T cell response.
To fully characterize the quality of the immune response induced by our different immunization regimens, we performed intracellular cytokine staining following in vitro stimulation with H-2Kb restricted HPV16 E1 peptide (IAYKYAQL) and determined the percentages of HPV16 E1-specific T cells secreting IFN-γ, TNF-α, and IL-2 (Fig. 5A). Seven days after the last MVA-E1 injection, IAYKYAQL-specific CD8+ T cells were mainly characterized by the secretion of IFN-γ, TNF-α (Fig. 5A, upper panels, day 21), or both (Fig. 5A, lower panels, day 21). The highest frequencies of these cytokine-secreting CD8+ T cells were detected in animals that received more than one MVA-E1 injection. On days 73 and 129, IAYKYAQL-specific CD8+ T cells were essentially characterized by the secretion of IL-2, with no major differences in the frequencies observed among the different experimental groups. Cytokine secretion patterns were different depending on the time of MVA-E1 boosting. A booster injection on day 73 resulted mostly in the generation of IAYKYAQL-specific CD8+ T cells secreting IFN-γ, IL-2 (Fig. 5A, upper panels, d80), and IL-2 plus TNF-α (Fig. 5A, lower panels, day 80), whereas a booster injection on day 129 resulted in the generation of IAYKYAQL-specific CD8+ T cells secreting mainly IFN-γ (Fig. 5A, upper panels, day 136) and IFN-γ plus TNF-α (Fig. 5A, lower panels, day 136). There was a trend for a positive association between the number of initial MVA-E1 injections and the detected frequencies of cytokine-secreting CD8+ T cells on days 80 and 136, but differences were not significant (Kruskal-Wallis test and Dunn's multiple comparison test). In all groups, IAYKYAQL-specific CD8+ T cells secreting IFN-γ, TNF-α plus IL-2, IFN-γ plus TNF-α, IL-2 plus TNF-α, and IFN-γ plus IL-2 plus TNF-α were detected 1 month after the boost administered on day 129 (Fig. 5A, upper and lower panels, day 158). As specific T cells secreting more than one cytokine in response to a given antigen are considered to be more optimized for effector functions (33), we plotted in pie charts the percentages of CD8+ T cells secreting one, two, or three cytokines (Fig. 5B). On day 21, we observed a positive trend between the number of MVA-E1 injections and the proportion of multifunctional HPV16 E1-specific CD8+ T cells (Fig. 5B). The proportion of these multifunctional CD8+ T cells decreased with time in all groups on days 73 and 129, except on day 129 in the group that received 3 injections of MVA-E1. Booster injections on days 73 and 129 with MVA-E1 restored multifunctionality in all experimental groups (Fig. 5B, days 80 and 136). Frequencies of IAYKYAQL-specific cytokine-secreting CD8+ T cells were increased in all groups 1 month after the boost on day 129 (Fig. 5B, day 158) in comparison to frequencies on day 136. Moreover, at day 158, the majority of IAYKYAQL-specific CD8+ T cells were multiple cytokine-secreting CD8+ T cells.
FIG 5.

Time course analysis of cytokine production. Following immunization, the percentages of HPV16 E1-specific IFN-γ-, TNF-α-, and IL-2-producing CD3+ CD8+ lymphocytes were determined by intracellular cytokine staining. Boost immunizations with MVA-E1 were performed either on day 73 or 129. Boosted immune responses were monitored on days 80, 136, and 158. (A) The percentages of single cytokine-secreting cells (IL-2, TNF-α, and IFN-γ) are shown in the upper panels. The percentages of double and triple cytokine-secreting cells (IFN-γ plus IL-2 plus TNF-α, IL-2 plus TNF-α, TNF-α plus IFN-γ, and IFN-γ plus IL-2) are shown in the lower panels. (B) Pie charts indicate the proportions of single (white), double (gray), and triple (black) cytokine-secreting cells according to the data represented in panel A. The total frequencies of IAYKYAQL-specific cytokine-secreting CD3+ CD8+ lymphocytes are represented next to each pie. Backgrounds from mock MVA-immunized animals were subtracted from experimental values. The mean values ± the SEM from two independent experiments are represented. Five mice were sacrificed and their splenocytes were pooled for each time point.
All together, these results indicate that a multiple MVA-E1 injection regimen generates not only HPV16 E1-specific IFN-γ-secreting CD8+ T cells but also multifunctional effector cells, including IFN-γ plus TNF-α double cytokine-secreting HPV16 E1-specific CD8+ T cells.
Functional analysis: in vivo killing assay.
In order to associate these ex vivo observations with the in vivo functionality of the generated HPV16 E1-specific CD8+ T cells, we performed an in vivo killing assay using splenocytes loaded with H-2Kb-restricted HPV16 E1 peptide IAYKYAQL as targets. In all groups, very high percentages of specific killing of IAYKYAQL-loaded targets were observed 7 days after the last MVA-E1 injection, on day 21 (Fig. 6). Consistent with results obtained in the IFN-γ ELISpot assays and tetramer staining, low percentages of specific killing of IAYKYAQL-loaded targets were detected at day 73. Seven days after boost, on day 80, the percentages of specific killing were very high again. Whatever the time point considered, percentages of specific killing were significantly different between the MVA-E1-injected experimental groups and the mock MVA-injected control group (Fig. 6D), thus highlighting again the inhibitory effect of preexisting immunity to MVA backbone in the induction of an HPV16 E1-specific and functional cellular immunity. No significant differences were observed among the experimental groups (Kruskal-Wallis test and Dunn's multiple comparison test) (Fig. 6A to C).
FIG 6.

Time course analysis of in vivo cytotoxicity. Following immunization, cytotoxicity to cells loaded with HPV16 E1 peptide IAYKYAQL was monitored by in vivo killing assay. Time of sampling is indicated on the x axis. For all groups, boost immunization with MVA HPV16 E1 was performed on day 73, as indicated by the arrows. Boosted cytotoxicity was monitored on day 80. Bars indicate the percentage of specific killing. The mean values ± the SEM from 5 animals for each time point from two independent experiments are represented. (A, B, C) One (day 14), two (days 7 and 14), or three (days 0, 7, and 14) immunizations with MVA-E1, respectively. (D) Three immunizations with mock MVA vector (days 0, 7 and 14). Significance was evaluated by the Kruskal-Wallis test and Dunn's multiple comparison test for each time point between groups receiving one, two, or three immunizations with MVA-E1. ns, not significant.
These results thus indicate that immunization with multiple injections of MVA-E1 generates fully functional HPV16 E1-specific CD8+ T cells, with high cytotoxic capacity.
DISCUSSION
To evaluate a novel therapeutic strategy dedicated to women with normal cytology but persistent HR-HPV infection, we constructed a new MVA vector expressing HPV16 E1 protein. Some clinical immunotherapy protocols based on MVA comprise numerous injections in a relatively short time period (12, 13, 19). Such types of schedules are known to induce efficient immunogenicity in preclinical studies (34). However, little is known about the effects on long term immunogenicity, generation of memory cells, exhaustion of T cells, proliferation of regulatory T cells, and profiles of the cytokines secreted by specific T cells. We showed that the differences observed between 1 and 2 or 3 MVA-E1 injections are mainly quantitative. Indeed, there was a trend to detect more HPV16 E1-specific IFN-γ-secreting cells in the spleens of the animals that received more than one injection of MVA-E1, and results indicated that these cells tended to persist for longer periods of time. Our observations indicate that the number of initial MVA-E1 injections did not influence the memory phenotypes or the PD-1 expression profiles of HPV16 E1-specific CD3+ CD8+ T cells in the time course study that was performed here. Our results also demonstrated that multiple injections of large quantities of MVA-E1 had no impact on the proliferation of CD4+ Treg lymphocytes. Similar to other mouse models using lymphocytic choriomeningitis virus (LCMV) or vesicular stomatitis virus (VSV), HPV16 E1-specific CD3+ CD8+ T cells generated by MVA-E1 immunizations were secreting IFN-γ and IFN-γ plus TNF-α at the peak of the response (day 21), while later on (days 73 and 129), we detected HPV16 E1-specific CD3+ CD8+ T cells secreting IL-2 (33, 35, 36). Of note, a qualitative differences were observed in the patterns of cytokines secreted by HPV16 E1-specific CD8+T cells. It appeared that more than one injection of MVA-E1 was necessary to induce substantial frequencies of HPV16 E1-specific CD3+ CD8+ lymphocytes secreting IFN-γ plus TNF-α at day 21, suggesting that at least two injections of MVA-E1 are necessary to induce multifunctional HPV16 E1-specific CD3+ CD8+ T cells. Unfortunately, the in vivo killing assay that we performed was not discriminant enough to further support this observation, as no significant differences were observed between the MVA-E1 groups. Indeed, multifunctional CD3+ CD8+ T cells that secrete both IFN-γ and TNF-α have been shown to be endowed with higher cytolytic capacities in comparison to CD3+ CD8+ T cells that secrete IFN-γ only (33).
We analyzed the impact of multiple close immunizations with MVA-E1 on anamnestic responses to the HPV16 E1 antigen following booster immunizations with the same MVA-E1 vector. At the tested time points, the specific response against HPV16 E1 could be boosted by MVA-E1 injection to similar levels in all groups, regardless of the initial immunization schedule. Again, the initial number of MVA-E1 injections had no impact either on the memory phenotypes or on the PD-1 expression profiles of anamnestic HPV16 E1-specific CD8+ T cells generated. Rather, it appeared that the time of boost with MVA-E1 was of importance in regard to the quality of the anamnestic response. Indeed, recalled HPV16 E1-specific CD8+ T cells generated on day 80 secreted mainly IFN-γ and IL-2 plus TNF-α, whereas those generated on day 136 secreted predominantly IFN-γ and IFN-γ plus TNF-α. Moreover, the frequencies of multifunctional HPV16 E1-specific CD8+ T cells were elevated 1 month following the day-129 boost (day 158). Of note, HPV16 E1-specific CD8+ T cells were mostly of the effector/effector memory phenotype on day 73 and mainly PD-1 positive, while they were primarily of the central memory phenotype on day 129 and mostly PD-1 negative. Relative to the high and short-term duration of antigen expression by MVA vectors after s.c. immunization in comparison to other vectors (37), this might constitute a specific feature of MVA-based vaccination; boosting must be performed 4 months after priming to induce multifunctional and persistent antigen-specific CD8+ T cells.
In two representative animal models, CRPV and COPV, recombinant Listeria monocytogenes secreting the E1 viral protein and DNA plasmid encoding codon optimized E1 viral protein conferred prophylactic protection against respective viral challenges (8, 9). In the CRPV model, it was shown that a cell-mediated immune response was associated with the prophylactic protection observed, whereas in the COPV model, no detectable humoral response was associated with the prophylactic protection observed, thus leading the authors to suggest that the mechanism by which the protection was generated may be cell mediated. Furthermore, in the COPV model, protection was clearly linked to an in vitro detection of E1 protein expression (8). Upon MVA-E1 infection, chicken embryo fibroblasts produced substantial amounts of HPV16-E1, as detected by Western blot analysis.
In humans, the study of cervical T cell populations in detail remains complicated, as available cervical tissue is scarce and conventional methods for T cell isolation yield very low numbers of cells. What is known is that cervical T cells in normal mucosa have an effector memory phenotype (CD45RO+ CD62L− CCR7−) and are slightly activated (CD25low/CD69low) (38). Thus far, therapeutic vaccines targeting advanced CIN lesions have succeeded in establishing detectable systemic immunity to HPV16 E6 or E7 proteins, but clinical benefits remained below expectations (39). It is tempting to associate these observations with the inability of classical routes of immunization (intramuscular or subcutaneous) to generate effective mucosal immunity. Similarly to what has been described for gut homing of antigen-specific T cells, homing of primed T cells to the genital mucosa is thought to rely on the interaction of α4/Eβ7 integrin and CCR9 expressed on their cell surface with their respective ligands MAdCAM1 and CCL25 (39). In that sense, it has been shown that 30% of mucosal CD3+ lymphocytes expressed β7 integrins and CCR9 (40). In addition, a positive correlation between high numbers of αEβ7+ intraepithelial lymphocytes and the rate of spontaneous regression of cervical intraepithelial neoplasia has been observed in women (40).
Taken together with our demonstration that the MVA-E1 vector induces efficient and multifunctional HPV16 E1-specific CD8 T cell responses of the effector and effector memory phenotype and that multiple close injections of MVA-E1 can be performed without risk of developing immunomodulatory mechanisms, this information makes us confident that MVA-E1 would be therapeutically efficient in light of the recent findings concerning the immunity of the female genital mucosa. Further development of MVA-E1 as a novel immunotherapeutic to treat persistently HPV16-infected women having a normal cytology would need to address the generation of efficient genital mucosa immunity to HPV16 E1.
ACKNOWLEDGMENTS
This work is a contribution to ADNA (Advanced Diagnostics for New Therapeutic Approaches), a program dedicated to personalized medicine, coordinated by Institut Mérieux and supported and partially funded by the French public agency BPI (Banque Publique d'Investissement).
We thank G. Inchauspé for critical reading of the manuscript.
Footnotes
Published ahead of print 4 December 2013
REFERENCES
- 1.Jenkins M, Chiriva-Internati M, Mirandola L, Tonroy C, Tedjarati SS, Davis N, D'Cunha N, Tijani L, Hardwick F, Nguyen D, Kast WM, Cobos E. 2012. Perspective for prophylaxis and treatment of cervical cancer: an immunological approach. Int. Rev. Immunol. 31:3–21. 10.3109/08830185.2011.637254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer 2:342–350. 10.1038/nrc798 [DOI] [PubMed] [Google Scholar]
- 3.Sanclemente G, Gill DK. 2002. Human papillomavirus molecular biology and pathogenesis. J. Eur. Acad. Dermatol. Venereol. 16:231–240. 10.1046/j.1473-2165.2002.00419.x [DOI] [PubMed] [Google Scholar]
- 4.Rowhani-Rahbar A, Mao C, Hughes JP, Alvarez FB, Bryan JT, Hawes SE, Weiss NS, Koutsky LA. 2009. Longer term efficacy of a prophylactic monovalent human papillomavirus type 16 vaccine. Vaccine 27:5612–5619. 10.1016/j.vaccine.2009.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hildesheim A, Herrero R. 2007. Human papillomavirus vaccine should be given before sexual debut for maximum benefit. J. Infect. Dis. 196:1431–1432. 10.1086/522869 [DOI] [PubMed] [Google Scholar]
- 6.Cid-Arregui A. 2009. Therapeutic vaccines against human papillomavirus and cervical cancer. Open Virol. J. 3:67–83. 10.2174/1874357900903010067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rozendaal L, Walboomers JM, van der Linden JC, Voorhorst FJ, Kenemans P, Helmerhorst TJ, van Ballegooijen M, Meijer CJ. 1996. PCR-based high-risk HPV test in cervical cancer screening gives objective risk assessment of women with cytomorphologically normal cervical smears. Int. J. Cancer 68:766–769. [DOI] [PubMed] [Google Scholar]
- 8.Moore RA, Santos EB, Nicholls PK, White KL, Anderson DM, Lloyd A, Topley P, Romanos M, Thomsen L, Parmar V, Walcott S, Gough GW, Stanley MA. 2002. Intraepithelial DNA immunisation with a plasmid encoding a codon optimised COPV E1 gene sequence, but not the wild-type gene sequence completely protects against mucosal challenge with infectious COPV in beagles. Virology 304:451–459. 10.1006/viro.2002.1726 [DOI] [PubMed] [Google Scholar]
- 9.Jensen ER, Selvakumar R, Shen H, Ahmed R, Wettstein FO, Miller JF. 1997. Recombinant Listeria monocytogenes vaccination eliminates papillomavirus-induced tumors and prevents papilloma formation from viral DNA. J. Virol. 71:8467–8474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egawa N, Nakahara T, Ohno SI, Narisawa-Saito M, Yugawa T, Fujita M, Yamato K, Natori Y, Kiyono T. 2012. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol. 86:3276–3283. 10.1128/JVI.06450-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acres B, Bonnefoy JY. 2008. Clinical development of MVA-based therapeutic cancer vaccines. Expert Rev. Vaccines 7:889–893. 10.1586/14760584.7.7.889 [DOI] [PubMed] [Google Scholar]
- 12.Quoix E, Ramlau R, Westeel V, Papai Z, Madroszyk A, Riviere A, Koralewski P, Breton JL, Stoelben E, Braun D, Debieuvre D, Lena H, Buyse M, Chenard MP, Acres B, Lacoste G, Bastien B, Tavernaro A, Bizouarne N, Bonnefoy JY, Limacher JM. 2011. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. 12:1125–1133. 10.1016/S1470-2045(11)70259-5 [DOI] [PubMed] [Google Scholar]
- 13.Brun JL, Dalstein V, Leveque J, Mathevet P, Raulic P, Baldauf JJ, Scholl S, Huynh B, Douvier S, Riethmuller D, Clavel C, Birembaut P, Calenda V, Baudin M, Bory JP. 2011. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am. J. Obstet. Gynecol. 204:169.e1–169.e8. 10.1016/j.ajog.2010.09.020 [DOI] [PubMed] [Google Scholar]
- 14.Harrop R, Drury N, Shingler W, Chikoti P, Redchenko I, Carroll MW, Kingsman SM, Naylor S, Melcher A, Nicholls J, Wassan H, Habib N, Anthoney A. 2007. Vaccination of colorectal cancer patients with modified vaccinia Ankara encoding the tumor antigen 5T4 (TroVax) given alongside chemotherapy induces potent immune responses. Clin. Cancer Res. 13:4487–4494. 10.1158/1078-0432.CCR-07-0704 [DOI] [PubMed] [Google Scholar]
- 15.Gomez CE, Najera JL, Krupa M, Esteban M. 2008. The poxvirus vectors MVA and NYVAC as gene delivery systems for vaccination against infectious diseases and cancer. Curr. Gene Ther. 8:97–120. 10.2174/156652308784049363 [DOI] [PubMed] [Google Scholar]
- 16.Harrop R, Ryan MG, Myers KA, Redchenko I, Kingsman SM, Carroll MW. 2006. Active treatment of murine tumors with a highly attenuated vaccinia virus expressing the tumor associated antigen 5T4 (TroVax) is CD4+ T cell dependent and antibody mediated. Cancer Immunol. Immunother. 55:1081–1090. 10.1007/s00262-005-0096-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corona Gutierrez CM, Tinoco A, Navarro T, Contreras ML, Cortes RR, Calzado P, Reyes L, Posternak R, Morosoli G, Verde ML, Rosales R. 2004. Therapeutic vaccination with MVA E2 can eliminate precancerous lesions (CIN 1, CIN 2, and CIN 3) associated with infection by oncogenic human papillomavirus. Hum. Gene Ther. 15:421–431. 10.1089/10430340460745757 [DOI] [PubMed] [Google Scholar]
- 18.Grandpre LE, Duke-Cohan JS, Ewald BA, Devoy C, Barouch DH, Letvin NL, Reinherz EL, Baden LR, Dolin R, Seaman MS. 2009. Immunogenicity of recombinant modified vaccinia Ankara following a single or multi-dose vaccine regimen in rhesus monkeys. Vaccine 27:1549–1556. 10.1016/j.vaccine.2009.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habersetzer F, Honnet G, Bain C, Maynard–Muet M, Leroy V, Zarski JP, Feray C, Baumert TF, Bronowicki JP, Doffoël M, Trépo C, Agathon D, Toh ML, Baudin M, Bonnefoy JY, Limacher JM, Inchauspé G. 2011. A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 141:890–899.e4. 10.1053/j.gastro.2011.06.009 [DOI] [PubMed] [Google Scholar]
- 20.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. 1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219. 10.1007/s002510050595 [DOI] [PubMed] [Google Scholar]
- 21.Yasugi T, Vidal M, Sakai H, Howley PM, Benson JD. 1997. Two classes of human papillomavirus type 16 E1 mutants suggest pleiotropic conformational constraints affecting E1 multimerization, E2 interaction, and interaction with cellular proteins. J. Virol. 71:5942–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun S, Thioudellet C, Rodriguez P, Ali-Hadji D, Perraud F, Accart N, Balloul JM, Halluard C, Acres B, Cavallini B, Pavirani A. 2000. Immune rejection of human dystrophin following intramuscular injections of naked DNA in mdx mice. Gene Ther. 7:1447–1457. 10.1038/sj.gt.3301261 [DOI] [PubMed] [Google Scholar]
- 23.Sutter G, Moss B. 1992. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. U. S. A. 89:10847–10851. 10.1073/pnas.89.22.10847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cochran MA, Puckett C, Moss B. 1985. In vitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J. Virol. 54:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masopust D, Ha S-J, Vezys V, Ahmed R. 2006. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J. Immunol. 177:831–839 [DOI] [PubMed] [Google Scholar]
- 26.Jabbari A, Harty JT. 2006. Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. J. Exp. Med. 203:919–932. 10.1084/jem.20052237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penna A, Pilli M, Zerbini A, Orlandini A, Mezzadri S, Sacchelli L, Missale G, Ferrari C. 2007. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology 45:588–601. 10.1002/hep.21541 [DOI] [PubMed] [Google Scholar]
- 28.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, Sekaly RP. 2006. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 12:1198–1202. 10.1038/nm1482 [DOI] [PubMed] [Google Scholar]
- 29.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. 10.1038/nature04444 [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Xu L, Jiang Y, Sun J, He X. 2007. Phenotypic and functional analysis of LCMV gp33-41-specific CD8 T cells elicited by multiple peptide immunization in mice revealed the up-regulation of PD-1 expression on antigen-specific CD8 T cells. Cell. Mol. Immunol. 4:431–437 [PubMed] [Google Scholar]
- 31.Rouse BT, Sarangi PP, Suvas S. 2006. Regulatory T cells in virus infections. Immunol. Rev. 212:272–286. 10.1111/j.0105-2896.2006.00412.x [DOI] [PubMed] [Google Scholar]
- 32.Bachmann MF, Wolint P, Schwarz K, Jager P, Oxenius A. 2005. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol. 175:4686–4696 [DOI] [PubMed] [Google Scholar]
- 33.Seder RA, Darrah PA, Roederer M. 2008. T-cell quality in memory and protection: implications for vaccine design. Nat. Rev. Immunol. 8:247–258. 10.1038/nri2274 [DOI] [PubMed] [Google Scholar]
- 34.Fournillier A, Gerossier E, Evlashev A, Schmitt D, Simon B, Chatel L, Martin P, Silvestre N, Balloul JM, Barry R, Inchauspe G. 2007. An accelerated vaccine schedule with a poly-antigenic hepatitis C virus MVA-based candidate vaccine induces potent, long lasting and in vivo cross-reactive T cell responses. Vaccine 25:7339–7353. 10.1016/j.vaccine.2007.08.020 [DOI] [PubMed] [Google Scholar]
- 35.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927. 10.1128/JVI.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kristensen NN, Christensen JP, Thomsen AR. 2002. High numbers of IL-2-producing CD8+ T cells during viral infection: correlation with stable memory development. J. Gen. Virol. 83:2123–2133 [DOI] [PubMed] [Google Scholar]
- 37.Geiben-Lynn R, Greenland JR, Frimpong-Boateng K, Letvin NL. 2008. Kinetics of recombinant adenovirus type 5, vaccinia virus, modified vaccinia Ankara virus, and DNA antigen expression in vivo and the induction of memory T-lymphocyte responses. Clin. Vaccine Immunol. 15:691–696. 10.1128/CVI.00418-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanley M, Pinto LA, Trimble C. 2012. Human papillomavirus vaccines—immune responses. Vaccine 30(Suppl 5):F83–F87. 10.1016/j.vaccine.2012.04.106 [DOI] [PubMed] [Google Scholar]
- 39.Kawana K, Adachi K, Kojima S, Kozuma S, Fujii T. 2012. Therapeutic human papillomavirus (HPV) vaccines: a novel approach. Open Virol. J. 6:264–269. 10.2174/1874357901206010264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kojima S, Kawana K, Fujii T, Yokoyama T, Miura S, Tomio K, Tomio A, Yamashita A, Adachi K, Sato H, Nagamatsu T, Schust DJ, Kozuma S, Taketani Y. 2011. Characterization of gut-derived intraepithelial lymphocyte (IEL) residing in human papillomavirus (HPV)-infected intraepithelial neoplastic lesions. Am. J. Reprod. Immunol. 66:435–443. 10.1111/j.1600-0897.2011.01041.x [DOI] [PubMed] [Google Scholar]