

Abstract
Developing an effective anthrax vaccine that can induce a rapid and sustained immune response is a priority for the prevention of bioterrorism-associated anthrax infection. Here, we developed a recombinant replication-deficient adenovirus serotype 5-based vaccine expressing the humanized protective antigen (Ad5-PAopt). A single intramuscular injection of Ad5-PAopt resulted in rapid and robust humoral and cellular immune responses in Fisher 344 rats. Animals intramuscularly inoculated with a single dose of 108 infectious units of Ad5-PAopt achieved 100% protection from challenge with 10 times the 50% lethal dose (LD50) of anthrax lethal toxin 7 days after vaccination. Although preexisting intranasally induced immunity to Ad5 slightly weakened the humoral and cellular immune responses to Ad5-PAopt via intramuscular inoculation, 100% protection was achieved 15 days after vaccination in Fisher 344 rats. The protective efficacy conferred by intramuscular vaccination in the presence of preexisting intranasally induced immunity was significantly better than that of intranasal delivery of Ad5-PAopt and intramuscular injection with recombinant PA and aluminum adjuvant without preexisting immunity. As natural Ad5 infection often occurs via the mucosal route, the work here largely illuminates that intramuscular inoculation with Ad5-PAopt can overcome the negative effects of immunity induced by prior adenovirus infection and represents an efficient approach for protecting against emerging anthrax.
INTRODUCTION
Bacillus anthracis, the causative agent of anthrax, is a Gram-positive spore-forming bacterium. It has two main virulence factors, the poly-gamma-d-glutamic acid (PGA) capsule, which protects the bacilli from complement fixation and phagocytes (1), and the tripartite exotoxin composed of protective antigen (PA), lethal factor (LF), and edema factor (EF) (2). Because the highly toxic strains of B. anthracis are easy to obtain, culture, and mass produce, and their highly stable spores can be stored for a long time, anthrax became one of the earliest biological warfare agents (3, 4). In view of the potential threat of anthrax from bioterrorism and biological warfare, developing rapid and efficient medical countermeasures remains a national priority.
A number of antibiotics, such as ciprofloxacin and doxycycline, are very effective at eliminating vegetative B. anthracis but have limited effects on antibiotic-resistant spores (5). Vaccination with an anthrax vaccine is the best strategy for preexposure and postexposure prophylaxis. Due to its central role in virulence, PA has been the principal target for the development of vaccines against anthrax (6–8). The first generation of such vaccines included Anthrax Vaccine Adsorbed (AVA) (BioThrax) in the United States and Anthrax Vaccine Precipitated (AVP) in the United Kingdom (6). Although they provide good protection in various animal models and are readily tolerated in humans (9), multiple doses are required over a protracted period to achieve adequate levels of protective immunity, making these vaccines less than optimal for use in response to a bioterrorism incident. A second generation of vaccines based on highly purified recombinant PA (rPA) are under development. However, the immunogenicity and potency of these well-defined and homogeneous rPA-based vaccines are similar to those of AVA and AVP (10). Therefore, developing a more efficacious vaccine that can confer rapid and robust immunity against B. anthracis in a biological emergency incident is imperative.
Viral vectors can be used to express proteins from pathogens for immunization against infectious diseases (11). Due to their safety, ease of manipulation, and capacity to elicit potent innate and adaptive immune responses by achieving high levels of transgene expression, recombinant adenoviruses (Ad) have been widely applied as gene transfer vehicles for vaccines (12). They have been extensively tested in several preclinical and clinical studies for a number of infectious diseases, including Ebola virus, Marburg virus, and human immunodeficiency virus 1 (HIV-1), (13–15). Although the high prevalence of neutralizing antibodies to Ad in the human population has the potential to limit the effectiveness of Ad-based vaccines, several strategies have been developed to circumvent the anti-vector preexisting immunity (PEI) (16). In animal models, Ad-vectored vaccines can confer rapid and more robust protection against live pathogens than other types of vaccines (12). Thus, Ad is an attractive vector candidate for an anthrax vaccine.
In this report, we describe the development and testing of an Ad vector-based vaccine candidate containing a full-length codon-optimized PA gene. The immunogenicity of the vaccine in the presence of PEI to the Ad5 vector is also discussed. The results demonstrate that a single intramuscular injection with the vaccine can elicit rapid and robust hormonal and cellular immune responses that protect rats from lethal toxin (LT) challenge, even in the presence of PEI.
MATERIALS AND METHODS
Cells and culture conditions.
293 cells (human embryonic kidney cells transformed by the Ad E1 region) and J774A.1 cells (murine macrophage cell line; ATCC TIB-67) were maintained in modified Eagle's medium (MEM) with 10% fetal bovine serum, 100 U penicillin/ml, and 100 μg streptomycin/ml at 37°C with 5% CO2.
Construction of the Ad5-PAopt vaccine.
The codon-optimized PA gene with a signal peptide derived from the tissue plasminogen activator polypeptide was synthesized and cloned into the adenoviral shuttle plasmid pDC316 (pDC316-PAopt). The shuttle plasmid and the adenoviral backbone plasmid (pBHGlox_E1, 3Cre) were cotransfected into 293 cells using Lipofectamine reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. The transfected cells were maintained until Ad-related cytopathic effects were observed. The Ads were harvested and confirmed by PCR. Positive Ads were reamplified in 293 cells and purified by ion exchange (SOURCE 15Q) and size exclusion. The virus was titrated on 293 cells using an Adeno-X rapid titer kit (Clontech, Saint-Germain-en-Laye, France). The resulting titers were measured as infectious units (IFU)/ml. As controls, the Ad5 vector expressing the enhanced green fluorescence protein (Ad5-EGFP) was also constructed.
In vitro assessment of PA expression.
The expression of the PA protein from pDC316-PAopt and Ad5-PAopt was determined by Western blot analysis. For detecting the expression level of PA from the shuttle plasmid pDC316, plasmids cloned with optimized PA gene, the wild-type PA gene, and the pDC316 backbone were transfected into 293 cells using Lipofectamine reagent. At 48 h posttransfection, the cells were collected and lysed in 1× SDS-PAGE buffer with 50 mM dithiothreitol (DTT) (Merck KGaA, Darmstadt, Germany), 1× protein inhibitor (Thermo Pierce, Rockford, IL), and nuclease (250 U/ml) (Merck KGaA). Subsequently, Western blot analysis was performed with a PA-specific monoclonal antibody and a goat anti-mouse horseradish peroxidase (HRP)-conjugated IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA). For detecting the expression level of PA from Ad5-PAopt, Ad5-PAopt and Ad-EGFP were used to infect 293 cells, which were collected and lysed for Western blot analysis 48 h postinfection.
Vaccination and collection of blood serum samples, lung lavage fluid samples, and splenocytes.
Female 4- 6-week-old BALB/c (H2d) mice were purchased from the Laboratory Animal Centre in the National Institute for the Control of Pharmaceutical and Biological Products (People's Republic of China). Male Fisher 344 rats (190 to 210 g) were purchased from Vital River Laboratories (Beijing, China). The mice and rats were immunized with Ad5-PAopt, Ad5-EGFP, or rPA (rPA protein with aluminum adjuvant) via the intramuscular or intranasal route. All animals were handled according to protocols approved by the Laboratory Animal Care and Use Committee of the Beijing Institute of Biotechnology and confirmed according to national guidelines on the ethical use of laboratory animals.
At the indicated time after immunization, each mouse and rat was bled from the tail vein (100 μl for each). The blood samples were incubated at 37°C for 1 h, followed by centrifugation (5,000 rpm, 10 min), and the obtained serum samples were stored at −20°C until assayed. Lung lavage fluid samples were collected by flushing the lung with 400 μl phosphate-buffered saline (PBS) for at least 10 times.
Splenocytes from the immunized and control mice were harvested for cytokine detection. Under aseptic conditions, spleens were pushed through a 70-μm cell strainer in complete RPMI 1640 medium to prepare a single-cell suspension. After centrifugation of the splenocytes at 500 × g for 5 min, the supernatant was discarded and red blood cells were removed with ACK lysing buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA [pH 7.2 to ∼7.4]). The cells were washed twice in complete RPMI 1640 medium, counted, and kept on ice until required for testing.
Antibodies against PA.
Anti-PA antibodies were quantified by enzyme-linked immunosorbent assay (ELISA) and toxin-neutralizing assay (TNA). For ELISA, flat-bottomed 96-well plates were coated with 100 μl of purified PA protein at a concentration of 2 μg/ml at 4°C overnight. The plates were washed, blocked with 2% bovine serum albumin (BSA) for 1 h at 37°C, and washed again two times with washing buffer (PBS plus 0.1% Tween 20). Serial dilutions of blood serum or lung lavage fluid (100 μl per well, 1:2 dilution) starting at the indicated dilution were added to each well and incubated for 1 h at 37°C. The plates were washed four times, and 100 μl/well of HRP-conjugated anti-mouse IgG, IgG1, IgG2a, IgM, or IgA antibody was added and incubated for 1 h at 37°C. After washing four times, the plates were incubated with 100 μl/well of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Merck KGaA) for 5 min to develop the color reaction. The reaction was stopped with 50 μl of 2 M H2SO4, and the absorbance at 450 nm was measured with a microplate reader (Bio-Rad, Hercules, CA). Secondary antibodies to IgG, IgM, and IgA were obtained from Santa Cruz Biotechnology, while antibodies to IgG1 and IgG2a were obtained from Abcam (Cambridge, United Kingdom). Antibody titers were calculated using a log optical density-log dilution interpolation model and a cutoff value equal to 2-fold the absorbance of the background.
The TNA was carried out using J774A.1 cells, as previously described (17). The cells were planted in flat-bottom 96-well cell culture plates at a concentration of 3.5 × 104 cells/well and incubated for 24 h at 37°C. Serum or lung lavage samples from immunized mice or rats were 2-fold serially diluted in culture medium with 50 ng/ml of rPA and 40 ng/ml of recombinant LF (rLF) and incubated for 30 min at 37°C. Medium from cells in 96-well plates was aspirated and replaced by the serum-rPA-rLF mixture at 100 μl/well. The cells in the control wells were incubated with 100 μl of medium only, rPA only, rLF only, or rPA plus rLF without serum. After 4 h of incubation at 37°C, 25 μl of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (5 mg/ml) (Merck KGaA) was added to each well. The cells were incubated for 1 h at 37°C and lysed with MTT lysing buffer (0.5% [wt/vol] SDS, 90% [vol/vol] isopropanol, 25 mM HCl) after the medium was discarded. The absorbances at 570 nm and 630 nm were measured with a microplate reader (Bio-Rad).
Flow cytometry.
Splenocytes from the immunized mice were cultured at 37°C for 6 h with 20 μg/ml of synthetic peptide, with no peptide as the background control or with 100 ng/ml of phorbol myristate acetate (PMA) (Sigma, St. Louis, MO) and 1 μg/ml of ionomycin (Sigma) as the positive control. For the last 4 h of culture, 10 μg/ml of brefeldin A (BFA) (Sigma) was added to block the secretion of gamma interferon (IFN-γ). The cells were stained with peridinin-chlorophyll protein/cyanide 5.5 (PerCP/Cy5.5)-conjugated anti-CD3 (clone 145-2c11; BioLegend, San Diego, CA) and fluorescein isothiocyanate (FITC)-conjugated anti-CD8 (clone 53-6.7; BioLegend) monoclonal antibodies and then were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences, San Jose, CA). The permeabilized cells were incubated with phycoerythrin (PE)-conjugated anti-IFN-γ (clone XMG1.2; BD Biosciences), washed, and resuspended in PBS. The samples were analyzed using a Beckman Coulter CyAn ADP flow cytometer (Beckman Coulter, Brea, CA) and Summit software.
ELISPOT assay.
The BD enzyme-linked immunosorbent spot assay (ELISPOT) mouse IFN-γ set was used to count peptide-specific T cells. The ELISPOT plates were coated overnight at 4°C with 5 μg/ml of an anti-mouse IFN-γ antibody. The antibody-coated plates were washed two times with sterile PBS and blocked with complete RPMI medium for 2 h at room temperature. After blocking, 100 μl of splenocyte suspension (2 × 106 cells/ml) containing different peptides (20 μg/ml) was added to each well. A positive control (50 ng/ml of PMA [Sigma] and 500 ng/ml of ionomycin Sigma]) and a “no peptide” negative control were included in all assays. The plates were incubated for 18 h at 37°C/5% CO2. Following incubation, the wells were washed twice with deionized water and three times with washing buffer (PBS containing 0.05% Tween 20). Biotinylated anti-mouse IFN-γ was added to each well at a concentration of 2 μg/ml, and the plates were incubated for 2 h at room temperature. Following three washes, streptavidin-HRP was added to each well, and the plates were incubated for 1 h at room temperature. After four washes with washing buffer and two washes with PBS, the colorimetric reactions were developed using 3-amino-9-ethylcarbazole as a substrate. Upon visualization of the spots, the reaction was stopped by rinsing in tap water. The membranes were allowed to dry overnight in the dark, and the spots were counted with a BioReaderW 4000 Pro-X (Bio-Sys GmbH, Karben, Germany). The results were expressed as the number of spot-forming cells (SFCs)/2 × 105 splenocytes.
Anthrax LT challenge.
Challenge with the anthrax LT was carried out on immunized male Fisher 344 (F344) rats by intravenous injection with 300 μl of mixed rPA (20 μg/rat) and rLF (5 μg/rat). Survival was monitored for 48 h after challenge.
Statistical analysis.
The data were calculated as the means ± standard errors of the means (SEM). Statistical analysis was performed with two-tailed unpaired t tests using Prism 5 (GraphPad Software). P values of ≤0.05 were considered statistically significant.
RESULTS
Humoral immune response of Ad5-PAopt-immunized BALB/c mice.
PA has been the principal target for the development of anthrax vaccines (6). Since PA is a prokaryotic antigen, codons of the PA gene sequence were optimized for human expression using the UpGene software. The pDC316 plasmid containing the wild-type PA gene (pDC316-PAwt) or codon-optimized PA gene (pDC316-PAopt) was transfected into 293 cells, and the expression level of PA with codon optimization was significantly increased at 48 h after transfection (Fig. 1A). The in vitro PA expression of Ad5-PAopt also was examined by Western blotting of transduced 293 cells (Fig. 1B).
FIG 1.
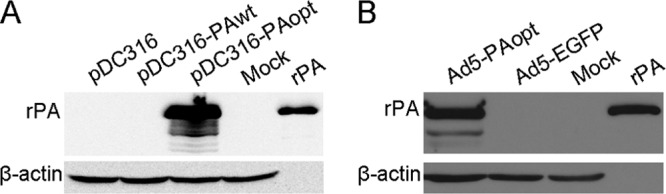
Western blot analysis of PA expression in vitro. (A) Expression of PA in plasmid-transfected 293 cells. (B) Expression of PA in Ad5-PAopt-infected 293 cells. rPA, recombinant protective antigen; pDC316, shuttle plasmid of the Ad system; pDC316-PAopt, pDC316 containing optimized PA gene; pDC316-PAwt, pDC316 containing wild-type PA gene; Ad5-PAopt, Ad5 expressing humanized PA; Ad5-EGFP, Ad5 expressing enhanced green fluorescence protein; Mock, untransfected or uninfected cells.
Blood serum antigen-specific antibody and neutralizing antibody titers against anthrax PA after vaccination are the best correlates of protection against anthrax (6–8). The humoral immune responses of the Ad5-PAopt-immunized mice were measured using a direct enzyme-linked immunosorbent assay (ELISA) and toxin-neutralizing assay (TNA) (Fig. 2). It showed that intramuscular inoculation of Ad5-PAopt resulted in rapid and robust antibody responses. At a dose of 107 infectious units (IFU), a relatively high serum immunoglobulin G (IgG) response was detected 1 week after intramuscular injection, reaching the highest level at week 4, and was maintained for at least 6 months (Fig. 2A). The intranasally immunized mice produced a low IgG response at week 1, but the antibody levels continuously increased thereafter, peaking at a slightly higher level than the peak titer of the intramuscularly immunized mice. However, a significantly lower IgG response was observed in rPA immunized mice. The relative levels of IgM responses between the groups of mice were similar to those of IgG (Fig. 2B). We next detected the subclasses of antigen-specific antibodies. The main antibody subclass of intranasally Ad5-PAopt-immunized mice and intramuscularly rPA-immunized mice was IgG1 (Fig. 2C), while intramuscularly Ad5-PAopt-immunized mice produced higher IgG2a antibody titers (Fig. 2D). These results indicate that intranasal inoculation with Ad5-PAopt stimulated a Th2-type immune response, which is similar to the response to the rPA vaccine, while the main immune response induced by intramuscular delivery was a Th1 type.
FIG 2.

Humoral immune response to Ad5-PAopt in mice. (A to E) Groups of BALB/c mice were intramuscularly or intranasally immunized with 106 or 107 IFU of Ad5-PAopt or intramuscularly injected with 5 μg of rPA, and serum samples were collected at indicated times for detection of IgG (A), IgM (B), IgG1 (C), and IgG2a (D) antibodies and toxin-neutralizing antibodies (E). (F) Dynamic changes of serum neutralizing antibody 2 weeks after intramuscular and intranasal Ad5-PAopt vaccination and intramuscular rPA inoculation. The data are presented as the means ± SEM (n = 6 per group). i.m., intramuscular; i.n., intranasal; rPA+Alum, rPA protein with aluminum adjuvant; IFU, infectious units.
To determine the anti-PA neutralizing response (Fig. 2E), mice intramuscularly inoculated with 107 IFU of Ad-PAopt achieved a high level of neutralizing antibodies at week 2, which is significantly higher than that in intranasally immunized mice. Although the peak IgG antibody level in the intranasally immunized mice was slightly higher than in those immunized intramuscularly, the latter group of mice generated higher neutralizing antibody titers. It is likely that the intramuscularly immunized mice produced different types of antibodies from the intranasally immunized mice. Furthermore, we detected dynamic changes in the neutralizing antibody levels over the course of 2 weeks (days 4, 6, 7, 8, 10, 12, and 14) after immunization (Fig. 2F). The results showed that the neutralizing antibody titers reached a high level 10 days after intramuscular inoculation. Meanwhile, neutralizing antibody titers in the intranasally vaccinated mice developed slowly, with the titer far from the peak value of the intramuscularly inoculated group even at day 14 after immunization. To our surprise, neutralizing antibodies from the mice given intramuscular injections of rPA (5 μg, which is one-tenth of the single dose used in humans) were not detected within the 2 weeks postvaccination. Taken together, the above results suggest that intramuscular inoculation with Ad5-PAopt can induce rapid and strong humoral immune responses.
Cellular immune responses to Ad5-PAopt.
Ad vectors can strongly stimulate both humoral and cellular immune responses. Here, the cellular immune response induced by Ad5-PAopt was examined by first predicting the H2d-specific cytotoxic T-lymphocyte (CTL) epitopes (for BALB/c mice) in PA using computer-assisted algorithms (18). The top-ranking peptides were selected and synthesized for further evaluation (Table 1) and of the four predicted peptides, only NA-9 specifically stimulated a gamma interferon (IFN-γ) response by flow cytometry analysis (Fig. 3A). The purified PA showed a low IFN-γ response, with no significant difference compared with that of the nonstimulated cells. Further intracellular IFN-γ staining confirmed that immunization with Ad5-PAopt was sufficient to stimulate a strong CD8+ T-cell response against NA-9, whereas control Ad5-EGFP did not elicit this response (Fig. 3B and C). Thus, NA-9 represents the first H2d-restricted CTL epitope described in anthrax PA.
TABLE 1.
Amino acid sequence of predicted H2d-specific anthrax PA CTL epitopes
| Epitope | Amino acid sequence |
|---|---|
| IF-9 | IALNAQDDF |
| HI-9 | HPLVAAYPI |
| IS-9 | IYNVLPTTS |
| NA-9 | NYYPSKNLA |
FIG 3.
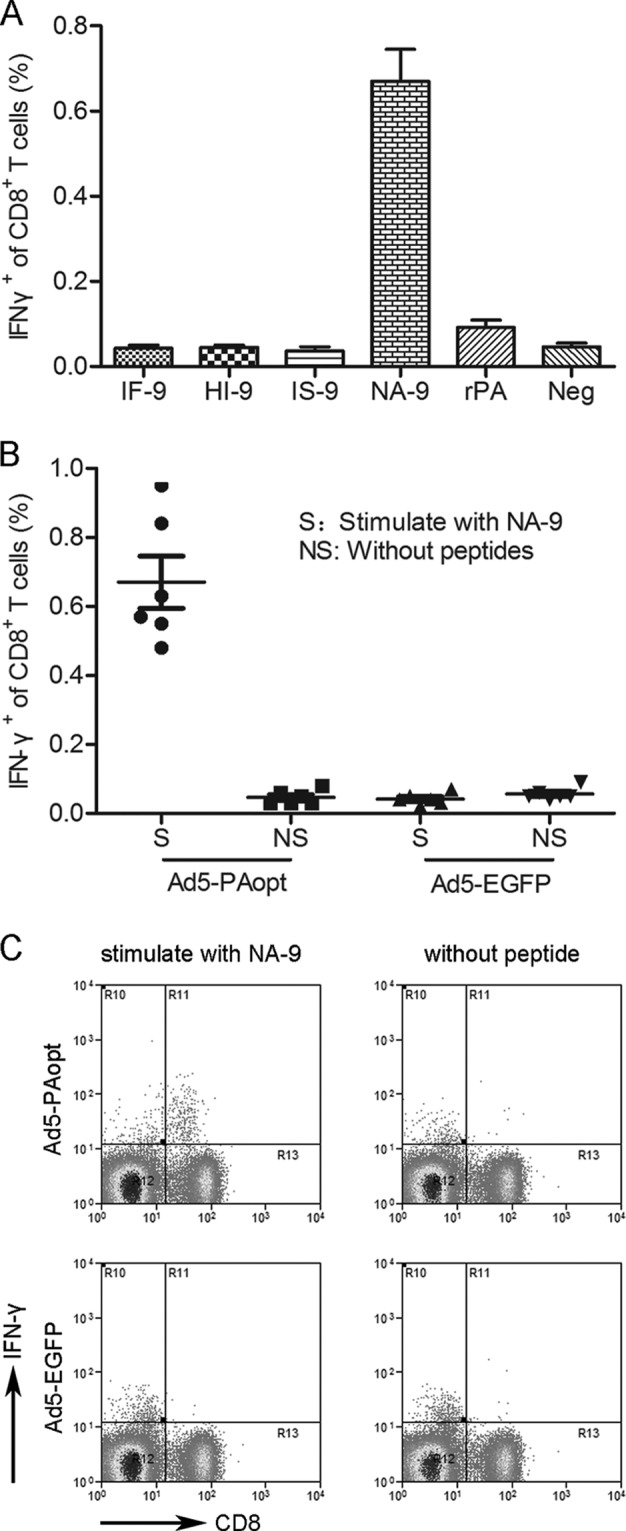
Identification of H2d-specific CTL epitopes in PA. (A) BALB/c mice were immunized with Ad5-PAopt twice, at an interval of 4 weeks. Splenocytes were harvested 10 days after the second immunization and restimulated in vitro with the predicted peptides from PA for use in a cytokine staining assay. Cells restimulated with purified rPA and a negative control without peptide were included. (B and C) To further confirm that the peptide identified in the cytokine staining assay was PA specific, splenocytes from BALB/c mice immunized with Ad5-PAopt or Ad5-EGFP (as a control) twice were restimulated in vitro with NA-9 or without peptides, and the responding CD8+ T cells were visualized by intracellular IFN-γ staining. The data are presented as the means ± SEM (n = 6 per group).
The cellular immune response in mice after intramuscular vaccination with Ad5-PAopt (107 IFU per mouse) was measured by intracellular cytokine staining and the IFN-γ enzyme-linked immunosorbent spot assay (ELISPOT) (Fig. 4). At 2 weeks after immunization, splenocytes from Ad5-PAopt-immunized mice produced high levels of IFN-γ (Fig. 4A) and tumor necrosis factor (TNF) (Fig. 4B) after restimulation with NA-9 peptide in vitro, while those from Ad5-EGFP and rPA-immunized mice did not. In fact, the secretion of IFN-γ and TNF in the splenocytes from rPA-immunized mice also was not detected after in vitro rPA restimulation (data not shown). At the same time, ELISPOT assays were performed 10 and 25 weeks after intramuscular immunization (Fig. 4C). The analysis showed that the cellular immune response slightly decreased with the passage of time, yet it was maintained at a high level even at week 25. These results suggest that intramuscular inoculation with Ad5-PAopt can induce a strong and sustained PA-specific CD8 CTL immune response.
FIG 4.
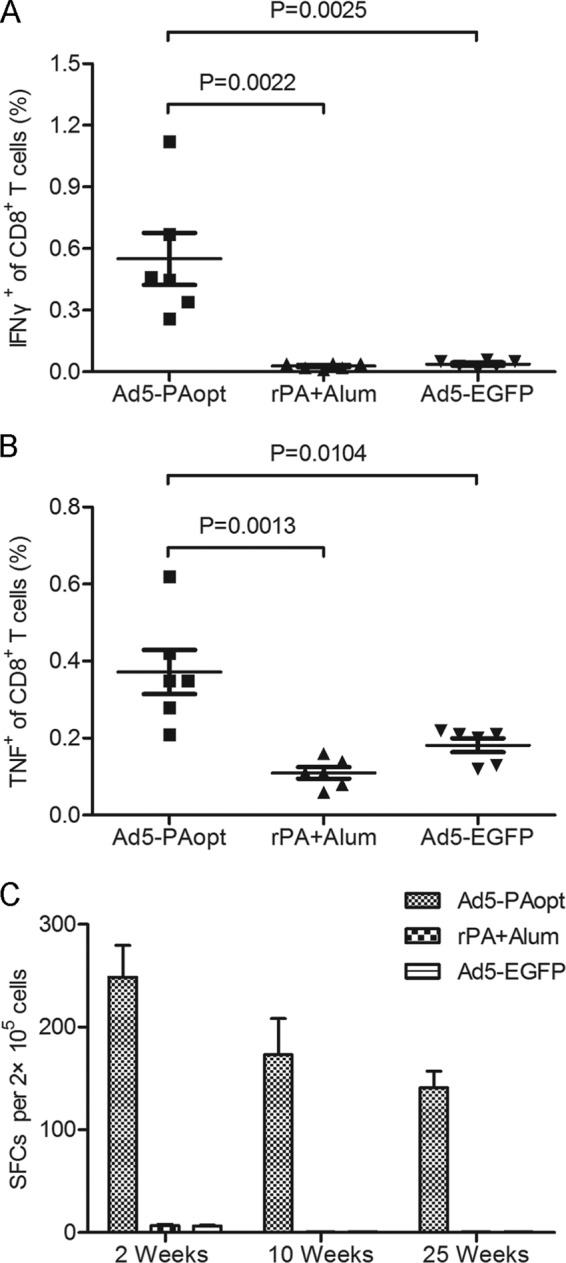
Cellular immune responses to Ad5-PAopt. Mice were intramuscularly vaccinated with Ad5-PAopt, Ad5-EGFP, or rPA, and splenocytes were harvested at indicated times and restimulated in vitro with NA-9 for intracellular cytokine staining and ELISPOT IFN-γ assay. (A) Intracellular IFN-γ assay at week 2 after vaccination. (B) Intracellular TNF assay at week 2 after vaccination. (C) ELISPOT IFN-γ assay for examining the maintenance of the cellular immune response. The data are presented as the means ± SEM (n = 6 per group). rPA+Alum, rPA protein with aluminum adjuvant; SFCs, spot-forming cells.
Immunogenicity of Ad5-PAopt in the presence of PEI to Ad5 vector.
For Ad5-based vaccines, PEI to the vector is an issue of concern (19). In order to analyze the immunogenicity of Ad5-PAopt in the presence of PEI to the Ad5 vector, BALB/c mice were intramuscularly or intranasally inoculated without (controls) or with 1010 viral particles of Ad5-EGFP and maintained for 10 weeks. Thereafter, the mice were vaccinated with 107 IFU of Ad5-PAopt via the intramuscular or intranasal route. The humoral immune response was detected at various times by ELISA and TNA (Fig. 5).
FIG 5.
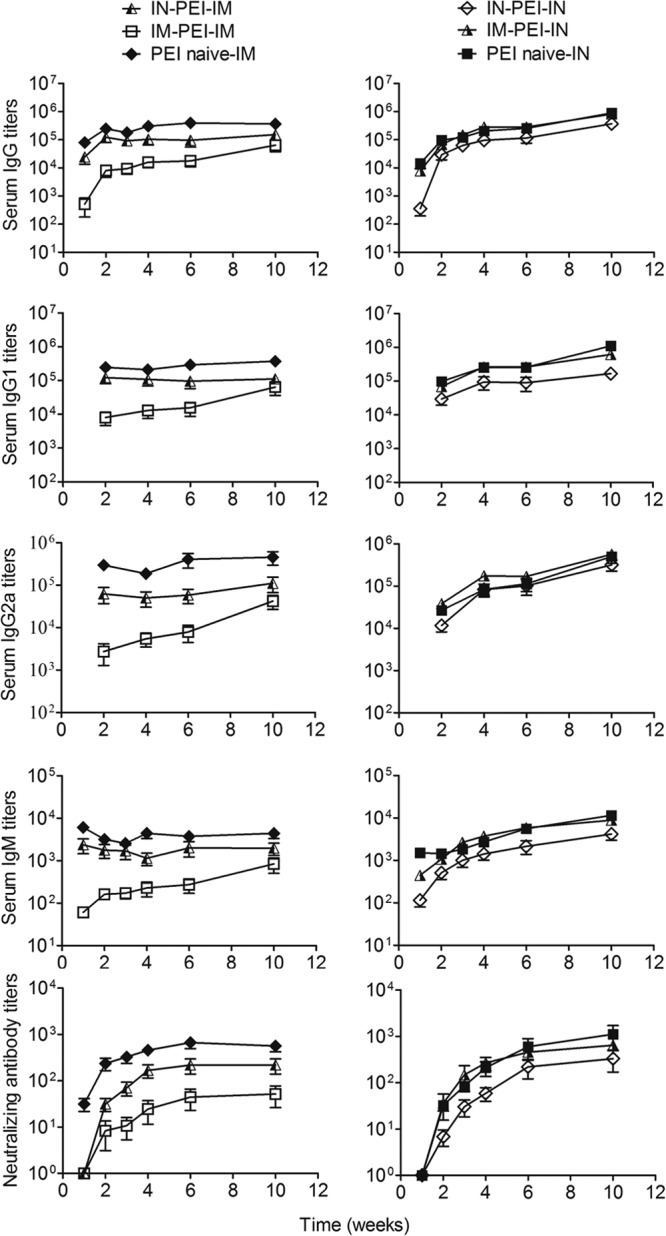
Blood serum antibody and toxin-neutralizing antibody responses to Ad5-PAopt in the presence of PEI to Ad5 vector. Groups of mice were preinoculated without (controls) or with 1010 viral particles of Ad5-EGFP intramuscularly or intranasally to induce PEI to the Ad5 vector. Ten weeks later, those mice were intramuscularly or intranasally vaccinated with Ad5-PAopt, and serum samples were collected for detection of antibody and toxin-neutralizing antibodies. Right panels, antibody and neutralizing antibody responses in PEI induced and naive mice after intranasal Ad5-PAopt vaccination. Left panels, antibody and neutralizing antibody responses in PEI induced and naive mice after intramuscular Ad5-PAopt vaccination. The data are presented as the means ± SEM (n = 10 per group). PEI, preexisting immunity; i.m., intramuscular; i.n., intranasal; i.n.-PEI, intranasal induction of PEI; i.m.-PEI, intramuscular induction of PEI.
Similar to the humoral immune response to Ad5-PAopt, the Ad5 binding antibody and neutralizing antibody titers peaked at week 10 after Ad5-EGFP immunization and had already entered the stabilization period (data not shown). For intranasal Ad5-PAopt-immunization (Fig. 5, right panels), the induction of PEI via intramuscular inoculation (i.m.-PEI) did not affect the immune response, yielding equivalent IgG, IgM, and IgG1 levels to those in naive mice. Surprisingly, the IgG2a levels of i.m.-PEI mice were slightly higher than those of naive mice. Meanwhile, intranasally induced PEI (i.n.-PEI) slightly affected the intranasal Ad5-PAopt immune response, with slightly lower IgG, IgM, and IgG1 levels but an equivalent IgG2a level to that of naive mice. For intramuscular Ad5-PAopt immunization (Fig. 5, left panels), the i.m.-PEI significantly affected the immune response, with significantly lower IgG, IgM, IgG1, IgG2a, and toxin-neutralizing antibody levels than those in naive mice. However, the i.n.-PEI only partially affected the immune responses of intramuscular Ad5-PAopt-immunized mice. Although the total antibody and toxin-neutralizing antibody titers of the i.n.-PEI and intramuscular Ad5-PAopt-immunized mice were slightly lower than those of naive mice, they were significantly higher than those from intramuscular Ad5-PAopt-immunized mice in the presence of i.m.-PEI. As Ad usually infects via the mucosal system, i.n.-PEI simulated natural infection better than i.m.-PEI. These results largely indicate that intramuscular vaccination with an Ad5-vectored vaccine will still induce a relatively high antigen-specific immune response given a prior natural infection of Ad5.
Comparing the humoral responses induced by intramuscular and intranasal vaccination with or without PEI (Fig. 5), we found that the naive mice and i.n.-PEI mice generated rapid immune responses to intramuscular vaccination, with high IgG and IgM antibody levels at 1 week after inoculation, which then peaked at week 2. However, the total antibody and toxin-neutralizing antibody levels increased slowly in intranasally inoculated mice but ultimately exceeded those of the intramuscularly immunized mice. These results indicate that intramuscular injection can induce rapid immune responses, while intranasal vaccination can obtain higher antibody titers.
We next evaluated the cellular immune responses in intramuscularly vaccinated mice and collected lung lavage fluid samples from intranasally vaccinated mice to detect IgA, IgG, and toxin-neutralizing antibodies (Fig. 6). The results showed that the cellular immune responses of mice with i.m.-PEI were significantly lower than those of mice without PEI or with i.n.-PEI (Fig. 6A); however, i.n.-PEI did not affect the cellular immune responses of the intramuscularly vaccinated mice. The IgA, IgG, and toxin-neutralizing antibody levels from lung lavage fluid samples of mice with i.n.-PEI were significantly lower than those in mice without PEI (Fig. 6B to D). This finding indicated that i.n.-PEI did affect the antibody titer in the lung lavage fluid but not in serum of those intranasally vaccinated mice. In summary, i.m.-PEI affects subsequent intramuscular immune responses to intramuscular but not intranasal vaccination. However, intramuscular vaccination with Ad5-PAopt followed by i.n.-PEI can also induce robust humoral and cellular immune responses.
FIG 6.
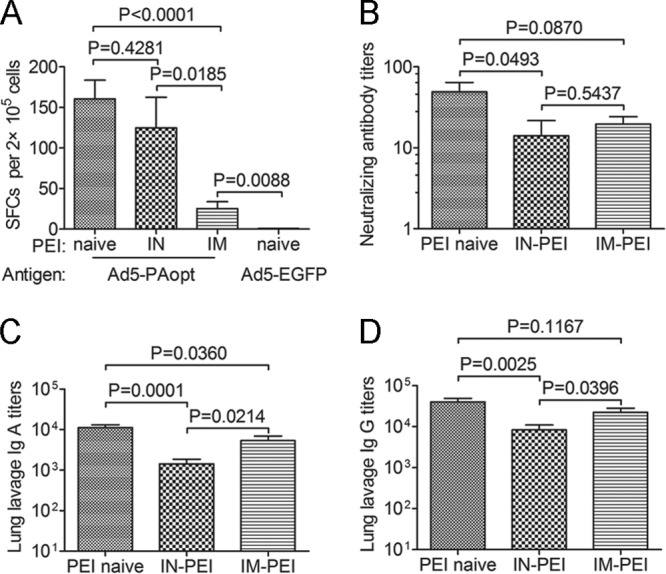
Cellular and antibody immune responses (lung lavage fluid samples) to Ad5-PAopt in the presence of PEI to Ad5 vector. Groups of mice were preinoculated without (controls) or with 1010 viral particles of Ad5-EGFP intramuscularly or intranasally to induce PEI to the Ad5 vector. Ten weeks later, those mice were intramuscularly or intranasally vaccinated with Ad5-PAopt. After 10 weeks, intramuscular Ad5-PAopt-vaccinated mice were sacrificed for the ELISPOT IFN-γ assay, and intranasal Ad5-PAopt-vaccinated mice were sacrificed to collect lung lavage fluid samples. (A) Cellular immune responses of intramuscular Ad5-PAopt-vaccinated mice. (B) Neutralizing antibody titers in lung lavage fluid samples from intranasal Ad5-PAopt-vaccinated mice. (C) IgA titer of lung lavage fluid samples from intranasal Ad5-PAopt-vaccinated mice. (D) IgG titer of lung lavage fluid samples from intranasal Ad5-PAopt-vaccinated mice. The data are presented as the means ± SEM (n = 10 per group). PEI, preexisting immunity; i.m., intramuscular; i.n., intranasal; i.n.-PEI, intranasally induced PEI; i.m.-PEI, intramuscularly induced PEI; SFCs, spot-forming cells.
Anthrax LT challenge.
To determine if the immune response generated against PA was sufficient for providing protection against lethal toxin in vivo, Fisher 344 rats were selected for anthrax LT challenge. Groups of rats were intramuscularly injected with 108 or 107 IFU of Ad5-PAopt or with 10 μg of rPA as a control. Seven days later, those rats were intravenously challenged with 20 μg of rPA and 5 μg of rLF (equivalent to 10 times the LD50) and monitored for survival over 48 h (Fig. 7A). All rats that were immunized with 108 IFU of Ad5-PAopt survived after challenge, while all rats that were vaccinated with rPA succumbed within 1 h. Additionally, of the six rats that were immunized with 107 IFU of Ad5-PAopt, only one survived after challenge, while the other five rats were observed to have a significant delay until death. Surprisingly, all of the rats immunized with 108 IFU of Ad5-PAopt died within 1 h after LT challenge on day 6 after intramuscular inoculation. By observing the dynamics of toxin-neutralizing antibodies within 14 days postvaccination (Fig. 7D), we saw that on day 5 after 108 IFU of Ad5-PAopt intramuscular injection, only a small proportion of the naive rats were detected with a very low level of anti-anthrax toxin-neutralizing antibody; however, the neutralizing antibody developed rapidly on day 6 and reached a relatively high level on day 7. Those results largely indicate that 108 IFU of Ad5-PAopt intramuscular vaccination can provide protection from LT challenge as early as day 7 postimmunization.
FIG 7.
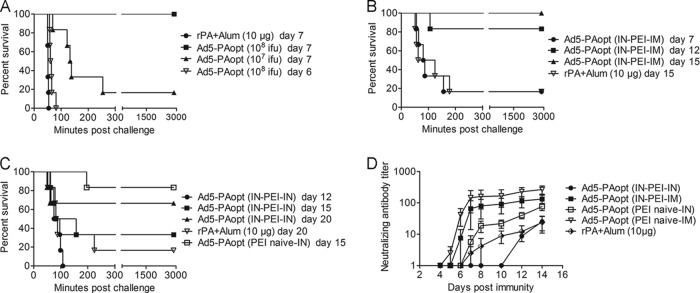
Anthrax LT challenge in Ad5-PAopt-immunized rats. Naive Fisher 344 rats or those with PEI were intramuscularly or intranasally vaccinated with Ad5-PAopt or intramuscularly injected with rPA as control. At the indicated time, rats were intravenously challenged with 10 times the LD50 of anthrax lethal toxin and monitored for survival over 48 h. (A) Protective efficacy in naive rats vaccinated intramuscularly with Ad5-PAopt. (B) Protective efficacy of intramuscular Ad5-PAopt vaccination in rats with intranasally induced PEI to Ad. (C) Protective efficiency of intranasal Ad5-PAopt vaccination in rats with intranasally induced PEI to Ad. (D) Dynamic changes of toxin-neutralizing antibodies within 14 days in naive and Ad preinoculated rats. The data are presented as the means ± SEM (n = 6 per group). PEI, preexisting immunity; i.m., intramuscular; i.n., intranasal; i.n.-PEI, intranasally induced PEI; i.m.-PEI, intramuscularly induced PEI.
In order to assess the immunogenicity of Ad5-PAopt in the presence of PEI to Ad5, groups of F344 male rats were intranasally inoculated with 108 IFU of Ad5-EGFP and screened after 4 weeks to allow for the induction of sufficient antigen-specific and neutralizing antibodies to Ad5. Those rats with positive Ad5-neutralizing antibodies were then injected with 108 IFU of Ad5-PAopt and challenged with anthrax LT on days 7, 12, and 15 (Fig. 7B). The results showed that 100% protection was achieved on day 15 (>80% on day 12), while a single injection with rPA vaccine achieved low protective efficacy on day 15, demonstrating that a single intramuscular immunization with Ad5-PAopt can still provide rapid and high-level protection in the presence of Ad5 PEI.
For the intranasal route (Fig. 7C), vaccination with 108 IFU of Ad5-PAopt did not achieve 100% protection on day 15 in Ad5-naive rats. Meanwhile, in the presence of i.n.-PEI to Ad5, the protective efficacy decreased, with all rats succumbing to infection within 2 h to 12 days after vaccination and only 33.3% of rats surviving on day 15. These results illustrate that intranasal immunization induces a much slower immune response than intramuscular vaccination. Similarly, a single injection with the rPA vaccine showed poor protective efficacy on day 15 and even on day 20 after immunization. The dynamic changes of toxin-neutralizing antibodies within 14 days in the PEI-induced rats (Fig. 7D) also supported this conclusion. These results indicate that compared to the intranasal immunity generated with Ad5-PAopt and intramuscular injection with rPA, intramuscular vaccination with Ad5-PAopt can induce rapid and robust immune responses to protect the rats from LT challenge. Even in the presence of i.n.-PEI to Ad5, this immune model may still be advantageous and able to generate rapid protection.
DISCUSSION
Developing an anthrax vaccine that can induce rapid and sustained protective immunity is critically needed, since B. anthracis continues to present a major threat as a potential biological weapon. Although the effectiveness of the protein-based first- and second-generation anthrax vaccines has been verified, they are still not ideal due to the requirement of multiple inoculations to reach an effective level of protection. Ad have been widely studied as vaccine vectors due to their ability to induce rapid and robust T-cell and antibody responses in species ranging from mice to humans. Here, we developed an Ad5-based vaccine for B. anthracis and showed that a single intramuscular injection of Ad5-PAopt protected rats from LT challenge 7 days after immunization. This protective efficacy is also achieved within 15 days in the presence of intranasally induced PEI to Ad5, which is significantly better than that with a single intramuscular injection with rPA or a single intranasal inoculation with Ad5-PAopt.
Previous studies have suggested that the PA-neutralizing humoral response is well-correlated with protective immunity (20). Here, a single inoculation of Ad5-PAopt resulted in robust and sustained anti-PA serum antibody and neutralizing antibody responses. Compared to intranasal inoculation, intramuscular vaccination with Ad5-PAopt more rapidly induced antibody production, with the neutralizing antibody level reaching a high level 10 days after immunization. However, anti-PA neutralizing antibodies were not detected in mice within 2 weeks of receiving a single intramuscular injection of 5 μg of rPA. In fact, the rPA vaccine requires at least two injections to reach high antibody levels, with the peak antibody level occurring 2 weeks after the second injection (21). Thus, the Ad5-based vaccine can induce a high level of anti-anthrax toxin antibodies in a relative short time, supporting the feasibility of its use in emergency situations.
The role of cell-mediated immunity in providing protection against anthrax is unclear. Here, we found that a single intramuscular injection of Ad5-PAopt induced a robust and sustained CD8 CTL immune response, while mice immunized with the rPA vaccine did not have this response. Those data correlate well with the induced antibody subtypes, indicating that the intramuscular injection of Ad5-PAopt stimulated a Th1-type immune response and rPA immunization stimulated the Th2-type response. The intranasal Ad5-PAopt-immunized mice generated a robust IgG1 immune response but only a moderate IgG2a immune response; furthermore, this vaccination model induced significantly slower immune responses than with intramuscular injection. It seemed that the rapidly generated humoral immune response in intramuscular Ad5-PAopt-immunized mice may partly be attributed to its robust cellular immune response.
The potential preexisting anti-vector immunity must be considered in developing Ad5-based vaccines. Several strategies have been developed to overcome the effects of preexisting Ad immunity and improve the efficacy of Ad-based vaccines (22–25). Alternative inoculation modes, such as intranasal and oral vaccinations, have shown good immune effects in the presence of PEI (26, 27). Here, we also found that the intranasal inoculation of Ad5-PAopt bypassed the anti-Ad5 PEI initiated not only from the intramuscular route but also from the intranasal route, although the intranasally induced PEI to Ad slightly affected the response to subsequent intranasal vaccination. However, intranasal vaccination with Ad5-PAopt resulted in slower immune responses, and it did not achieve full protection 15 days after a single dose, even in the naive rats. Although i.m.-PEI significantly affected the intramuscular vaccination that followed, i.n.-PEI only slightly affected this inoculation method. Furthermore, the intramuscular Ad5-PAopt vaccination induced rapid and robust humoral and cellular immune responses. In the presence of i.n.-PEI, a single intramuscular injection with Ad5-PAopt provided full protection from LT challenge 15 days after vaccination. Natural Ad5 infection occurs via the nasopharynx or gut and replicates in epithelial cells of mucosal tissues, thereby inducing mucosal immunity (28). The results here suggest that intramuscular injection of the Ad-based vaccine can still induce a good immune response in individuals with a prior natural infection of Ad5.
Compared with the rPA aluminum adjuvanted vaccine, Ad-based vaccines can achieve rapid immune responses after a single inoculation. To our surprise, the immunization of rats with the Ad5-based vaccine achieved protection from LT challenge with a relatively lower level of neutralizing antibodies than that for the rPA immunized rats (data not shown). It may be that the Ad5-PAopt- and rPA-immunized rats produced different subtypes of antibodies or that the cellular immune response in the Ad5-PAopt-immunized rats contributed to the protection. Studies from our group and others have demonstrated that intranasal vaccination with Ad5-based vaccines can bypass PEI (27, 29). There, we observed that compared to intranasal immunization, even in the presence of intranasal anti-Ad5 PEI, the intramuscular injection induced effective immune responses to protect rats from anthrax LT challenge in a relatively short time. Perhaps, antigen-specific T cells induced by intramuscular inoculation of the Ad5-based vaccine stimulated B cells and promoted those subjects to produce antibodies more rapidly. The results there show that the protective efficacy in the Ad5-primed Ad5-PAopt intramuscularly boosted rats was better than that in naive Ad5-PAopt intranasally vaccinated subjects within 15 days after vaccination, demonstrating again that intramuscular injection with Ad5-PAopt is more suitable for emergency use than the rPA vaccine.
There are several anthrax vaccine candidates based on Ad vectors that have been reported. Tan et al. (30) first developed an adenovirus-based vaccine encoding humanized PA and evaluated the efficiency of this vaccine in a mouse model; the LT challenge showed that 72% of the mice survived 27 days after vaccination and 27% survived on day 11. Next, a nonhuman primate adenovirus, AdC7, was used as the vaccine vector in order to overcome the preexisting neutralizing antibodies against Ad5 in humans (31). After that, the Ad vector expressing the B. anthracis PA domain 4 was developed for security reasons (32), and the prime-boost vaccination strategy was also studied (33). Recently, Zhang et al. (34) proved that an adenovirus-vectored nasal vaccine conferred rapid and sustained protection against anthrax in a single-dose regimen. However, intramuscular inoculation with Ad5-PAopt in our work induced a faster immune response than intranasal route did in Zhang's work, even in the presence of PEI.
In summary, the results presented here demonstrate that the Ad5-based vector expressing humanized anthrax PA can induce rapid and robust humoral and cellular immune responses after a single intramuscular inoculation. Moreover, this vaccination model can achieve good immune responses to protect intranasal Ad5-primed rats from lethal anthrax LT challenge within 15 days at a significantly better level than with a single intramuscular rPA injection or intranasal Ad5-PAopt inoculation. Of note, the H2d-specific CTL epitope in PA discovered here will facilitate the detection of the cellular immune responses to anthrax vaccines in future studies. Thus, intramuscular vaccination with Ad5-PAopt may provide a rapid and efficient method for counteracting emerging anthrax.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant no. 81025018) and the National Major Project for Infectious Disease Control and Prevention (grant no. 2013ZX10004001).
Footnotes
Published ahead of print 4 December 2013
REFERENCES
- 1.Scorpio A, Chabot DJ, Day WA, O'Brien DK, Vietri NJ, Itoh Y, Mohamadzadeh M, Friedlander AM. 2007. Poly-gamma-glutamate capsule-degrading enzyme treatment enhances phagocytosis and killing of encapsulated Bacillus anthracis. Antimicrob. Agents Chemother. 51:215–222. 10.1128/AAC.00706-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedlander AM, Welkos SL, Pitt ML, Ezzell JW, Worsham PL, Rose KJ, Ivins BE, Lowe JR, Howe GB, Mikesell P, Lawrence WB. 1993. Postexposure prophylaxis against experimental inhalation anthrax. J. Infect. Dis. 167:1239–1243 [DOI] [PubMed] [Google Scholar]
- 3.Anderson PD. 2012. Bioterrorism: toxins as weapons. J. Pharm. Pract. 25:121–129. 10.1177/0897190012442351 [DOI] [PubMed] [Google Scholar]
- 4.Thavaselvam D, Vijayaraghavan R. 2010. Biological warfare agents. J. Pharm. Bioallied Sci. 2:179–188. 10.4103/0975-7406.68499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson DW, Peacock S, Belton FC. 1956. Observations on the prophylaxis of experimental pulmonary anthrax in the monkey. J. Hyg. (Lond.) 54:28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cybulski RJ, Jr, Sanz P, O'Brien AD. 2009. Anthrax vaccination strategies. Mol. Aspects Med. 30:490–502. 10.1016/j.mam.2009.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedlander AM, Little SF. 2009. Advances in the development of next-generation anthrax vaccines. Vaccine 27(Suppl 4):D28–D32. 10.1016/j.vaccine.2009.08.102 [DOI] [PubMed] [Google Scholar]
- 8.Grabenstein JD. 2008. Vaccines: countering anthrax: vaccines and immunoglobulins. Clin. Infect. Dis. 46:129–136. 10.1086/523578 [DOI] [PubMed] [Google Scholar]
- 9.Puziss M, Wright GG. 1963. Studies on immunity in anthrax. X. Gel-adsorbed protective antigen for immunization of man. J. Bacteriol. 85:230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keitel WA. 2006. Recombinant protective antigen 102 (rPA102): profile of a second-generation anthrax vaccine. Expert Rev. Vaccines 5:417–430. 10.1586/14760584.5.4.417 [DOI] [PubMed] [Google Scholar]
- 11.Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M. 2013. Viral vectors: a look back and ahead on gene transfer technology. New Microbiol. 36:1–22 [PubMed] [Google Scholar]
- 12.Zhang J, Tarbet EB, Toro H, Tang DC. 2011. Adenovirus-vectored drug-vaccine duo as a potential driver for conferring mass protection against infectious diseases. Expert Rev. Vaccines 10:1539–1552. 10.1586/erv.11.141 [DOI] [PubMed] [Google Scholar]
- 13.Ledgerwood JE, Costner P, Desai N, Holman L, Enama ME, Yamshchikov G, Mulangu S, Hu Z, Andrews CA, Sheets RA, Koup RA, Roederer M, Bailer R, Mascola JR, Pau MG, Sullivan NJ, Goudsmit J, Nabel GJ, Graham BS, VRC 205 Study Team 2010. A replication defective recombinant Ad5 vaccine expressing Ebola virus GP is safe and immunogenic in healthy adults. Vaccine 29:304–313. 10.1016/j.vaccine.2010.10.037 [DOI] [PubMed] [Google Scholar]
- 14.Natuk RJ, Chanda PK, Lubeck MD, Davis AR, Wilhelm J, Hjorth R, Wade MS, Bhat BM, Mizutani S, Lee S. 1992. Adenovirus-human immunodeficiency virus (HIV) envelope recombinant vaccines elicit high-titered HIV-neutralizing antibodies in the dog model. Proc. Natl. Acad. Sci. U. S. A. 89:7777–7781. 10.1073/pnas.89.16.7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang D, Schmaljohn AL, Raja NU, Trubey CM, Juompan LY, Luo M, Deitz SB, Yu H, Woraratanadharm J, Holman DH, Moore KM, Swain BM, Pratt WD, Dong JY. 2006. De novo syntheses of Marburg virus antigens from adenovirus vectors induce potent humoral and cellular immune responses. Vaccine 24:2975–2986. 10.1016/j.vaccine.2005.11.046 [DOI] [PubMed] [Google Scholar]
- 16.Seregin SS, Amalfitano A. 2009. Overcoming pre-existing adenovirus immunity by genetic engineering of adenovirus-based vectors. Expert Opin. Biol. Ther. 9:1521–1531. 10.1517/14712590903307388 [DOI] [PubMed] [Google Scholar]
- 17.Li H, Soroka SD, Taylor TH, Jr, Stamey KL, Stinson KW, Freeman AE, Abramson DR, Desai R, Cronin LX, Oxford JW, Caba J, Pleatman C, Pathak S, Schmidt DS, Semenova VA, Martin SK, Wilkins PP, Quinn CP. 2008. Standardized, mathematical model-based and validated in vitro analysis of anthrax lethal toxin neutralization. J. Immunol. Methods 333:89–106. 10.1016/j.jim.2008.01.007 [DOI] [PubMed] [Google Scholar]
- 18.Wu S, Yu T, Song X, Yi S, Hou L, Chen W. 2012. Prediction and identification of mouse cytotoxic T lymphocyte epitopes in Ebola virus glycoproteins. Virol. J. 9:111. 10.1186/1743-422X-9-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun C, Zhang Y, Feng L, Pan W, Zhang M, Hong Z, Ma X, Chen X, Chen L. 2011. Epidemiology of adenovirus type 5 neutralizing antibodies in healthy people and AIDS patients in Guangzhou, southern China. Vaccine 29:3837–3841. 10.1016/j.vaccine.2011.03.042 [DOI] [PubMed] [Google Scholar]
- 20.Fay MP, Follmann DA, Lynn F, Schiffer JM, Stark GV, Kohberger R, Quinn CP, Nuzum EO. 2012. Anthrax vaccine-induced antibodies provide cross-species prediction of survival to aerosol challenge. Sci. Transl. Med. 4:151ra126. 10.1126/scitranslmed.3004073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorse GJ, Keitel W, Keyserling H, Taylor DN, Lock M, Alves K, Kenner J, Deans L, Gurwith M. 2006. Immunogenicity and tolerance of ascending doses of a recombinant protective antigen (rPA102) anthrax vaccine: a randomized, double-blinded, controlled, multicenter trial. Vaccine 24:5950–5959. 10.1016/j.vaccine.2006.05.044 [DOI] [PubMed] [Google Scholar]
- 22.Barouch DH, Pau MG, Custers JH, Koudstaal W, Kostense S, Havenga MJ, Truitt DM, Sumida SM, Kishko MG, Arthur JC, Korioth-Schmitz B, Newberg MH, Gorgone DA, Lifton MA, Panicali DL, Nabel GJ, Letvin NL, Goudsmit J. 2004. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 172:6290–6297 [DOI] [PubMed] [Google Scholar]
- 23.Krause A, Whu WZ, Xu Y, Joh J, Crystal RG, Worgall S. 2011. Protective anti-Pseudomonas aeruginosa humoral and cellular mucosal immunity by AdC7-mediated expression of the P. aeruginosa protein OprF. Vaccine 29:2131–2139. 10.1016/j.vaccine.2010.12.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts DM, Nanda A, Havenga MJ, Abbink P, Lynch DM, Ewald BA, Liu J, Thorner AR, Swanson PE, Gorgone DA, Lifton MA, Lemckert AAC, Holterman L, Chen B, Dilraj A, Carville A, Mansfield KG, Goudsmit J, Barouch DH. 2006. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 441:239–243. 10.1038/nature04721 [DOI] [PubMed] [Google Scholar]
- 25.Gabitzsch ES, Balint-Junior JP, Xu Y, Balcaitis S, Sanders-Beer B, Karl J, Weinhold KJ, Paessler S, Jones FR. 2012. Control of SIV infection and subsequent induction of pandemic H1N1 immunity in rhesus macaques using an Ad5 [E1-, E2b-] vector platform. Vaccine 30:7265–7270. 10.1016/j.vaccine.2012.09.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiang ZQ, Gao GP, Reyes-Sandoval A, Li Y, Wilson JM, Ertl HC. 2003. Oral vaccination of mice with adenoviral vectors is not impaired by preexisting immunity to the vaccine carrier. J. Virol. 77:10780–10789. 10.1128/JVI.77.20.10780-10789.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, Feldmann H, Kobinger GP. 2008. Nasal delivery of an adenovirus-based vaccine bypasses pre-existing immunity to the vaccine carrier and improves the immune response in mice. PLoS One 3:e3548. 10.1371/journal.pone.0003548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merkel TJ, Perera PY, Kelly VK, Verma A, Llewellyn ZN, Waldmann TA, Mosca JD, Perera LP. 2010. Development of a highly efficacious vaccinia-based dual vaccine against smallpox and anthrax, two important bioterror entities. Proc. Natl. Acad. Sci. U. S. A. 107:18091–18096. 10.1073/pnas.1013083107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richardson JS, Pillet S, Bello AJ, Kobinger GP. 2013. Airway delivery of an adenovirus-based Ebola virus vaccine bypasses existing immunity to homologous adenovirus in nonhuman primates. J. Virol. 87:3668–3677. 10.1128/JVI.02864-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan Y, Hackett NR, Boyer JL, Crystal RG. 2003. Protective immunity evoked against anthrax lethal toxin after a single intramuscular administration of an adenovirus-based vaccine encoding humanized protective antigen. Hum. Gene Ther. 14:1673–1682. 10.1089/104303403322542310 [DOI] [PubMed] [Google Scholar]
- 31.Hashimoto M, Boyer JL, Hackett NR, Wilson JM, Crystal RG. 2005. Induction of protective immunity to anthrax lethal toxin with a nonhuman primate adenovirus-based vaccine in the presence of preexisting anti-human adenovirus immunity. Infect. Immun. 73:6885–6891. 10.1128/IAI.73.10.6885-6891.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McConnell MJ, Hanna PC, Imperiale MJ. 2006. Cytokine response and survival of mice immunized with an adenovirus expressing Bacillus anthracis protective antigen domain 4. Infect. Immun. 74:1009–1015. 10.1128/IAI.74.2.1009-1015.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McConnell MJ, Hanna PC, Imperiale MJ. 2007. Adenovirus-based prime-boost immunization for rapid vaccination against anthrax. Mol. Ther. 15:203–210. 10.1038/sj.mt.6300034 [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Jex E, Feng T, Sivko GS, Baillie LW, Goldman S, Van Kampen KR, Tang DC. 2013. An adenovirus-vectored nasal vaccine confers rapid and sustained protection against anthrax in a single-dose regimen. Clin. Vaccine Immunol. 20:1–8. 10.1128/CVI.00280-12 [DOI] [PMC free article] [PubMed] [Google Scholar]