

Abstract
Fungi can serve as highly tractable models for understanding genetic basis of sexual development in multicellular organisms. Applying a reverse-genetic approach to advance such a model, we used random and multitargeted primers to assay gene expression across perithecial development in Neurospora crassa. We found that functionally unclassified proteins accounted for most upregulated genes, whereas downregulated genes were enriched for diverse functions. Moreover, genes associated with developmental traits exhibited stage-specific peaks of expression. Expression increased significantly across sexual development for mating type gene mat a-1 and for mat A-1 specific pheromone precursor ccg-4. In addition, expression of a gene encoding a protein similar to zinc finger, stc1, was highly upregulated early in perithecial development, and a strain with a knockout of this gene exhibited arrest at the same developmental stage. A similar expression pattern was observed for genes in RNA silencing and signaling pathways, and strains with knockouts of these genes were also arrested at stages of perithecial development that paralleled their peak in expression. The observed stage specificity allowed us to correlate expression upregulation and developmental progression and to identify regulators of sexual development. Bayesian networks inferred from our expression data revealed previously known and new putative interactions between RNA silencing genes and pathways. Overall, our analysis provides a fine-scale transcriptomic landscape and novel inferences regarding the control of the multistage development process of sexual crossing and fruiting body development in N. crassa.
INTRODUCTION
Shifts in gene expression over the course of development have been attributed a primary role in the unfolding of the developmental program of animals and plants (1–4). However, the genomic basis of development in a third multicellular clade, the fungi, is arguably very different and the least well understood. Estimated as comprising 1.5 to 7.1 million species, fungi have deep evolutionary origins (5–7) and diverse body plans, ranging from highly reduced unicellular species such as microsporidia and yeasts to notoriously large hyphal mats that produce multicellular fruiting bodies such as mushrooms, which feature specialized cell types (8). To address multicellular fruiting body development from a reverse-genetic approach, model fungi can provide ideal systems, as they are easily manipulated, develop fruiting structures with a few well-characterized tissue types, and have relatively small genome sizes, so numerous fungal genomes have been sequenced.
Neurospora crassa is a multicellular ascomycete fungus in the family Sordariomycetes, which has been used as a genetic model organism due to its simple filamentous asexual stage (9, 10), and which exhibits promise for revealing the molecular basis of the more complex sexual development of fungi. N. crassa has 28 morphologically distinct cell types, including 14 that are finely differentiated during the development of its sexual reproductive structure (11, 12). Sexual development can be induced by crossing conidia (asexual spores) from one mating type with protoperithecia (presexual reproductive structures) from the opposite mating type, producing a large number of perithecia (sexual reproductive structures) that develop at sufficient synchronicity. The fertilized perithecium undergoes morphogenic processes characteristic of other complex multicellular organisms, sequentially differentiating into different tissue types: a perithecium wall, ascogenous cells, paraphyses and periphyses that are sterile hyphae emergent from ascogenous cells and perithecium wall, then asci with ascospores differentiated from ascogenous cells. These tissues and their fates within the developing perithecium represent an ideal model for studying multicellular development. From this model, shifts in gene expression related to morphological development can be revealed. However, the sexual growth of N. crassa arises as a consequence of a communion of cells of different nuclear types; the heterokaryotic reproductive cells develop into sterile paraphyses within the perithecium or undergo karyogamy and a short diploid phase prior to the production of haploid ascospores. This heterokaryosis has made it challenging to study sexual differentiation using traditional methods based on genetic screens for mutants (13, 14), and genome-wide assays so far have yielded only limited information about the genetics underlying the production of multicellular sexual reproduction structures such as perithecia (13–23).
Previous research on N. crassa has yielded knowledge and tools that facilitate the study of the genetic basis of sexual crossing and fungal multicellular development. Successive stages of morphological changes can be characterized, such as the onset of the development of the ascus and the ascospore (sexual spore). Some Neurospora mutants that affect sexual development, such as female and male fertility mutants (fmf-1), giant ascospore (prf), abnormal ascus shape (peak), and mutants that affect meiosis, such as mei-1, mei-2, and a postmeiotic mitosis mutant (mus-8), have been analyzed genetically and cytologically (10). In addition, the sexual biology of N. crassa also presents a broadly informative model for many biological processes, such as signaling pathways, genomic incompatibility, extensive chromosome rearrangements, heterochromatin silencing and DNA methylation, meiotic silencing, and repeat-induced point mutation (RIP) (24). Investigation of these processes in N. crassa has been galvanized by a combination of early breakthroughs and the sequencing and annotation of the N. crassa genome (25). In addition to their roles in genome defense, all known components of meiotic silencing pathways and some genes of the quelling pathway also affect sexual development. Phenotypes of these genes in sexual development have been observed; however, the failure of development in gene knockout strains has made it challenging to study interactions among these genes in relation to their roles in the regulatory networks across sexual development without assaying gene expression.
In animals and plants, analyses of gene expression and genetic regulatory networks in model organisms have demonstrated that genes work together in response to regulatory factors to shape metabolic processes and morphological development (2, 4, 23, 26, 27). N. crassa has evolved various mechanisms to ensure proper development during premating, mating, and postmating stages based on heterokaryotic and protoplasmic incompatibility, signaling, silencing, and secondary metabolism pathways. All of these pathways have been intensively studied for specific components of the N. crassa life cycle (24). However, studies of these components have not addressed the morphological complexity of sexual reproduction, nor have they provided an integrated understanding that can come from a genome-wide genetic and morphological characterization of sexual development.
In this study, we performed the first comprehensive genome-wide analysis of gene expression across sexual development in N. crassa. We performed transcriptomic sequencing primed by random and multitargeted primers for eight time points, which encompassed the morphological changes and tissue/cellular development of maturing perithecia. Examining expression patterns for genetic markers known to play key roles in morphological development, we performed microscopic characterization of perithecia across the same time course. Our findings provide important knowledge about genes and pathways that are known for their regulatory roles in vegetative growth and the mating process as well as about genes in the mitogen-activated protein (MAP) kinase signaling pathways and genes involved in RNA silencing and DNA methylation. Examination of both the phenotypes of strains in which differentially expressed genes were knocked out and the gene expression patterns facilitated inference of Bayesian networks (28, 29) of potential interactions between genes and pathways in controlling sexual development. Our results indicate that stage-specific gene expression correlates with developmental function during sexual development in Neurospora crassa and provide key insights into the control of the multistage development process of sexual crossing and fruiting body development in this fungus.
MATERIALS AND METHODS
Two strains of N. crassa of complementary mating types, MAT-a (FGSC4200) and MAT-A (FGSC2489), were kindly provided by the Fungal Genetics Stock Center (FGSC) (30). The strains were grown on carrot agar medium (CA) (31). Unlike traditionally used synthetic crossing medium, which often yields patches of protoperithecia, CA provided more evenly distributed protoperithecia in greater developmental synchrony. On CA, a large number of conidia are produced during the first week of growth, especially along the edge of the plate. These conidia were used for crossing to avoid different nutrient backgrounds between conidia and protoperithecia. CA was covered by a cellophane membrane to facilitate later extraction of perithecia, and cultures were maintained at a constant temperature of 26°C, exposed to constant light provided by five Ecolux bulbs (F17T8.SP41-ECO; General Electric Company). The net intensity of light exposure was 14 μMol/m2 S at the medium surface, measured at wavelengths between 400 nm and 700 nm. Conidia from the MAT-a strain were collected and suspended in 2.5% Tween 60. Formation of MAT-A protoperithecia was examined using a stereomicroscope after 7 days, and areas of evenly distributed protoperithecia of similar size were carefully selected and marked for subsequent sampling. Crosses were performed by applying 1 ml of Mat-a conidial suspension (105 to 106 conidia/ml with 2.5% Tween 60) to the surface of the MAT-A protoperithecium plates. Subsequent sexual development occurred under the same culture conditions as before the crossing and was monitored using a stereomicroscope. Representative portions of cultures were carefully excised from the plates to ensure that nearly all perithecia in each sample were at the same developmental stage. Tissues were collected from the plate surface using a razor blade just before the crossing and at 2 h after crossing. All tissues and perithecia collected from the same plate were counted as one biological replicate, and three to five biological replicates were prepared for each sampled time point. The color of protoperithecia changed from pale to dark after crossing, and perithecia could be identified with the naked eye by 24 h.
Perithecia at similar development stages (based on size and color under the stereomicroscope) were collected from the cellophane-covered agar surface using a razor blade at 24, 48, 72, 96, 120, and 144 h after crossing. To minimize physical disturbance, a minimal fraction of adjacent vegetative hyphae around sampled perithecia were not removed, especially at spots where multiple perithecia of the same development stage had formed gregariously. Morphological development, as indicated by size and color of perithecia as well as development of asci and ascospores, was observed for perithecia at all sampled time points (Fig. 1). Differentiated asci and ascospores were observed in perithecia from 144 h onward. Pieces of cellophane membrane (about 4 mm by 2 mm) carrying ∼5 to 20 perithecia of similar size were cut from cultures and fixed in 1.5 formaldehyde and 0.025 M phosphate buffer for at least 48 h. The samples were embedded in resin and prepared for transmission electron microscopy (TEM) and light microscopy as previously described (32). Blocks were sectioned at 0.7 to 1 μm using a glass knife and stained with 1% toluidine blue. A Lecia DM LB microscope (Leica Microsystem Gmbh, Wetzlar, Germany) with differential interference contrast (DIC) capabilities was used to observe samples, and images were captured using a Zeiss AxioCam MRc color camera and AxioVision 4.8.2 (Göttingen, Germany). Image processing and annotation were performed using Adobe Photoshop CS3 (San Jose, CA). All tissues and perithecia were rapidly frozen in liquid nitrogen as they were sampled and stored at −80°C.
FIG 1.
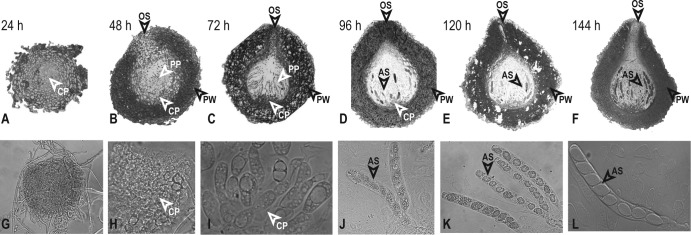
Analysis of perithecial development in N. crassa. (A to F) Sections of perithecia demonstrate the expansion of the perithecium wall (PW), composed of thick-walled cells, and the development of thin-walled cells in the centrum parenchyma (CP), except as noted. (G to L) Light microscopic analysis of perithecial squashes show internal contents of the fruiting body, including centrum parenchyma, asci (AS), and ascospores. (A) By 24 h, the perithecia show an initial expansion of the centrum parenchyma, which is surrounded by multiple layers of thick-walled cells composing the perithecium wall. Magnification, ×400. (B) At 48 h, early paraphyses (PP) appear as elongated cells within the expanded centrum parenchyma (×200). The initial ostiole (OS) forms at the apex of the fruiting body. (C) At 72 h, abundant paraphyses arise from thin-walled cells at the bottom of the centrum. The ostiole has formed and is lined with periphyses, and layers of the perithecial wall are distinct (×200). (D) At 96 h, early asci are beginning to appear (×200). (E) At 120 h, early ascospores start developing in some asci (×200). (F) At 144 h, melanized ascospores are visible in many asci. The beak and ostiole appear fully developed at the apex of the fruiting body (×200). (G) By 24 h, the perithecium has grown, but a squash does not reveal differentiation of hyphae within the perithecium (×200). (H) At 48 h, thin-walled cells of the centrum parenchyma have clearly differentiated (×400). (I) At 72 h, early paraphyses appear (×400). (J) At 96 h, early asci extend from thin-walled cells at the base of the centrum (×400). (K) At 120 h, ascospores begin to form in asci (×400). (L) At 144 h, eight ascospores can be distinguished in a well-developed ascus (×400).
Tissue samples from biological replicates were pooled for RNA extraction at each sampling time point. Tissues were homogenized with a Dounce glass grinder, and debris was filtered through Qiagen Qiashredder columns (Qiagen, Chatsworth, CA). Total RNA was extracted from homogenized tissue using TRI Reagent (Molecular Research Center) (33), and mRNA was purified using Dynabeads oligo(dT) magnetic separation (Invitrogen). About 100 ng of purified mRNA was mixed with 10× fragmentation buffer (Ambion) and incubated at 70°C for 5 min before addition of 1 μl of stop buffer (Ambion). Fragmented mRNA was precipitated using 100% ethanol with glycogen (Ambion) at −80°C. Preparation of cDNA for sequencing followed the Illumina mRNA sequencing sample preparation guide. A multitargeted primer (MTP; VWNVNNBDKGGC) (34) primed reverse transcription of the first cDNA strand, and random hexamers (N6; Invitrogen) primed the second cDNA strand. To provide technical replicates across stages of sexual development, cDNA was prepared using only N6 primers for samples at all time points. After ligation of standard adaptors for Illumina sequencing, each sample was purified using a 2% low-melting-point agarose gel. A gel band corresponding to processed cDNA fragments of 200 to 400 bp was cut and purified using a QIAquick gel extraction kit (Qiagen). Selected cDNA samples were enriched by PCR using Pfx DNA polymerase (Invitrogen) and 15 cycles of amplification at 98°C for 10 s, 65°C for 30 s, and 68°C for 30 s. The quality of purified PCR products of all samples was determined using a bioanalyzer (Agilent Technologies). Single-end 35-bp reads of MTP- and N6-primed preparations were separately sequenced, each on eight lanes of an Illumina genome analyzer, at the Yale Center for Genomic Analysis.
We used Tophat, version 1.2.0 (35), to perform spliced alignments of the reads against the N. crassa reference genome (25). As the N. crassa genome was based on the same mat A strain used in this study, a sequence of mat a-1 was added to the genome for estimating expression of this mat a-specific gene. Only reads that mapped to a single unique location within the genome with a maximum of two mismatches in the anchor region of the spliced alignment are reported. We used the default settings for all other Tophat options. We tallied the number of the reads that overlapped the exons of a gene using HTSeq, version 0.4.5p6 (unpublished data; http://www-huber.embl.de/users/anders/HTSeq/doc/), and the gene structure annotation file for the reference genome. The tally for each sample was then processed using LOX, version 1.6 (36), to estimate gene expression levels at all assayed time points during sexual development. Reads per kilobase of exon model per million mapped (RPKM) reads were estimated as in the work of Mortazavi et al. (37) (see Table S3 in the supplemental material).
The Functional Catalogue (FunCat) (38) annotation scheme was used to group genes according to their cellular or molecular functions. The statistical significance of overrepresentation of gene groups in functional categories relative to the whole genome was determined using the hypergeometric distribution, facilitated by the Munich Information Center for Protein Sequences (MIPS) FunCat online web application. Further functional annotation of genes showing statistically significant patterns of differential expression in metabolic pathways was based on the biochemical pathway and the annotation database from the Kyoto Encyclopedia of Genes and Genomes (39). Enrichment analysis was performed at the N. crassa genome web page (http://www.broadinstitute.org/annotation/genome/neurospora/) as well, where significant enrichment was reported for genes responding to light stimulus and genes involved in gene silencing.
Bayesian networks for RNA silencing genes and pathways were inferred from expression data using the Bayesian Network Webserver for Biological Network Modeling (40). Input files containing fold changes between adajcent time points across perithecium development were calculated from LOX data based on MTP-based experiments. Global structure learning settings followed the defaults and 100 high-scoring networks with a selection threshold set to 0.5 were included in model averaging without any structural constraint. Bayesian networks of different sets of genes were used to predict interactions between genes in controlling development.
Knockouts for nearly all of the 9,733 N. crassa genes, including deletion cassettes for genes in either mating type of N. crassa, were acquired from the Fungal Genetic Stock Center. All knockout strains used in this study were produced and verified, typically using Southern blot analysis, within the NIH Neurospora Genome Knock-Out Project (41), and PCR analysis was used to verify genotypes by following the recently optimized approach of Lichius et al. (42). For functional studies of genes, including het-6, het-14, het-15, mkr-5, mkr-6, qde-1, sad-1, sad-3, sms-2, sms-3, and the stc1-like gene, knockouts of both mat a and mat A were available for crossing and phenotyping across sexual development (see Table 2). Knockouts of opposite mating types were crossed on synthetic complete medium (SCM) (33), traditionally used for inducing sexual reproduction in N. crassa, and on CA (31). All cultures were grown on SCM and CA with three replicates at 26°C under constant white light. Opposite mating types were inoculated on different sides of the same 9-cm-wide plate. Three to 4 days after inoculation, crossing occurred where mycelia from the two mating types met, predominantly across the midline of the plates. Development of conidia, protoperithecia, and perithecia was examined to reveal differences in sexual development attributable to culture conditions.
TABLE 2.
Comparative expression (in RPKM) of mating types, pheromones, and pheromone receptors across N. crassa perithecial development
| Gene | 0 h | 2 h | 24 h | 48 h | 72 h | 96 h | 120 h | 144 h |
|---|---|---|---|---|---|---|---|---|
| NCU01958 mat A-1 | 5 | 13 | 11 | 4 | 6 | 6 | 12 | 9 |
| NCU01959 mat A3 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| NCU01960 mat A2 | 1 | 1 | 1 | 1 | 1 | 3 | 2 | 4 |
| mat a-1 | 0 | 0 | 1 | 0 | 3 | 9 | 25 | 42 |
| NCU05758 pre-2 | 4 | 2 | 4 | 3 | 5 | 6 | 9 | 5 |
| NCU00138 pre-1 | 6 | 6 | 6 | 4 | 4 | 5 | 9 | 8 |
| NCU01257a mfa-1 | 23 | 18 | 18 | 16 | 21 | 19 | 20 | 25 |
| NCU02500 ccg-4 | 71 | 129 | 294 | 345 | 2,208 | 1,980 | 2,897 | 3,184 |
mfa-1 is reannotated as NCU16992.7 in the latest annotation for N. crassa.
The complete raw data set generated in this study as well as the inferred expression levels are available in the supplemental material, in the filamentous fungal gene expression database (FFGED) (43), and on the Townsend Lab web site.
Gene expression data accession number.
Raw sequencing data and estimated expression levels are available at the Gene Expression Omnibus of the National Center for Biotechnology Information (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE41484.
RESULTS
Assaying our wild-type Neurospora crosses by microscopy, development of protoperithecia before crossing and of perithecia after crossing proceeded as expected (44). Except for a slight increase in size and a slight darkening in color, no obvious tissue differentiation was observed during the interval when protoperithecia became fertilized perithecia, within 24 h after crossing. Centrum parenchyma of thin-walled cells expanded with increasing perithecial size. Differentiated hyphal structures and croziers became detectable after 48 h to 72 h. Asci filled with vesicles emerged in samples after 96 h, along with narrow, hyphal paraphyses. Between 120 h and 144 h, the top of the perithecium protruded to form a beak. Inside the perithecium, mature asci developed: ascospores became clearly delimited. Perithecia darkened to nearly black with maturation.
Throughout the 144 h of perithecial development, the majority of perithecia in a culture exhibited no noticeable developmental divergence. Perithecia were often aggregated in patches that were especially tightly synchronized. Throughout our experiments, incorporation of small quantities of vegetative tissue into our samples was unavoidable, especially in samples collected during the early stages 48 h after crossing, when protoperithecia and perithecia were very small. Past 144 h, synchronicity became difficult to ensure because developmental landmarks became less clear. Generally 160 h after crossing, ascospores started being forcibly ejected and accumulated gradually onto the lower surface of petri lids. However, the intensity and duration of this long process were increasingly impacted by stochastic environmental conditions, including small fluctuations in light intensity, humidity, airflow, and position of perithecia on culture plates. Therefore, perithecial development beyond 144 h after crossing was not investigated in this study.
Multitargeted primer (MTP) and conventional random hexamer (N6) priming yielded a few differences that relate to their design. A robust estimation of the genome-wide transcriptome during the eight time points of sexual development was generated by use of these two types of primers on the Illumina platform. DNA concentrations of samples prepared with N6 primers for the time points 24 h, 96 h, and 144 h were much higher than other samples. These samples were diluted to prescribed levels, but they produced low-quality reads and were excluded from further analysis.
As in previous microarray and transcriptomic sequencing applications of MTP priming to Saccharomyces cerevisiae and N. crassa, in which MTP priming was compared to oligo(dT) priming (34), MTP priming worked well. There were no significant differences between the total number of short reads obtained with each method (Table 1). The proportion of the total reads mapping to coding genes was significantly higher for MTP than for N6 (chi-square test, P < 0.0001). The average number of genes significantly upregulated, or downregulated, was not different between MTP and N6 priming (paired t test, P > 0.05 in all four cases). MTP-primed samples yielded maximum likelihood (LOX [36]) estimates of gene expression level for 9,717 genes for all eight time points. Our analyses were based on the MTP-primed data (see Table S1 in the supplemental material), which were validated as highly consistent with the N6 samples (see Table S2 in the supplemental material).
TABLE 1.
Comparative performance of MTP and N6 priming on samples from five development stages in N. crassa sexual reproduction
| Primer and parameter | 0 h | 2 h | 48 h | 72 h | 120 h | Total | Mean | SD |
|---|---|---|---|---|---|---|---|---|
| MTP | ||||||||
| Total reads obtained | 25,344,501 | 28,901,469 | 24,546,749 | 26,620,329 | 28,459,085 | 133,872,133 | 26,774,426.6 | 1,896,929.4 |
| Mapping to the genome | 19,928,814 | 22,686,873 | 19,744,776 | 22,007,918 | 22,641,520 | 107,009,901 | 21,401,980.2 | 1,455,258.9 |
| Mapping multiple places | 2,371,545 | 3,274,722 | 1,750,275 | 2,471,673 | 1,600,175 | 11,468,390 | 2,293,678.0 | 666,413.2 |
| Mapping ambiguous places | 66,261 | 137,707 | 58,289 | 68,038 | 68,013 | 398,308 | 79,661.6 | 32,697.2 |
| Mapping genes | 17,085,018 | 19,614,422 | 17,510,480 | 19,610,346 | 20,252,575 | 94,072,841 | 18,814,568.2 | 1,417,117.0 |
| N6 | ||||||||
| Total reads obtained | 24,356,818 | 31,576,781 | 27,550,897 | 30,029,099 | 32,760,914 | 146,274,509 | 29,254,901.8 | 3,360,709.9 |
| Mapping to the genome | 21,255,501 | 28,240,311 | 22,079,771 | 25,602,909 | 29,028,130 | 126,206,622 | 25,241,324.4 | 3,512,362.8 |
| Mapping multiple places | 1,043,153 | 938,159 | 1,280,859 | 1,751,283 | 1,095,907 | 6,109,361 | 1,221,872.2 | 320,958.0 |
| Mapping ambiguous places | 90,894 | 175,281 | 85,084 | 89,449 | 101,099 | 541,807 | 108,361.4 | 37,866.9 |
| Mapping genes | 17,441,060 | 24,592,393 | 17,694,474 | 20,588,254 | 24,579,478 | 104,895,659 | 2,097,9131.8 | 3,517,025.2 |
LOX estimates and RPKMs (see Table S3 in the supplemental material) (29) from the current transcriptomic experiment were compared to a previous investigation of perithecial development using microarray analysis (22), in which more than 900 genes were measured across a completely independent experiment using the same time points of development. Observed expression patterns were generally conserved between the two experiments, especially for genes detected with high fold changes in expression as measured by microarray analysis (45). Despite a typically lower power associated with microarray methods (see Table S4 in the supplemental material), 13 genes known to regulate N. crassa sexual development were measured using both methods. Expression of these genes showed no significant discrepancies between sequencing and microarray analyses at any time point across development, including for four genes (ccg-4, 4hnr, mkr5, and sad-3) that exhibited changes of more than 20-fold based on transcriptome sequencing that showed identical expression patterns across development as measured by our microarray analysis.
Upregulation across perithecial development was the most common pattern of gene expression. More than 30% of the genes exhibited continuous upregulation of expression during late perithecial development (after 48 h). Another 30% of genes were downregulated after expression peaked at an earlier stage of development. Hierarchical clustering revealed 12 main subsets correlated by expression of individual genes up- or downregulated during late perithecial development (Fig. 2; see also Table S5 in the supplemental material). Functional enrichment analysis for each gene cluster using the FunCat database indicated dramatic shifts of expression during sexual development, with extensive upregulation of genes not annotated for function (see Table S5 in the supplemental material; hereafter, P < 0.01 unless otherwise stated).
FIG 2.
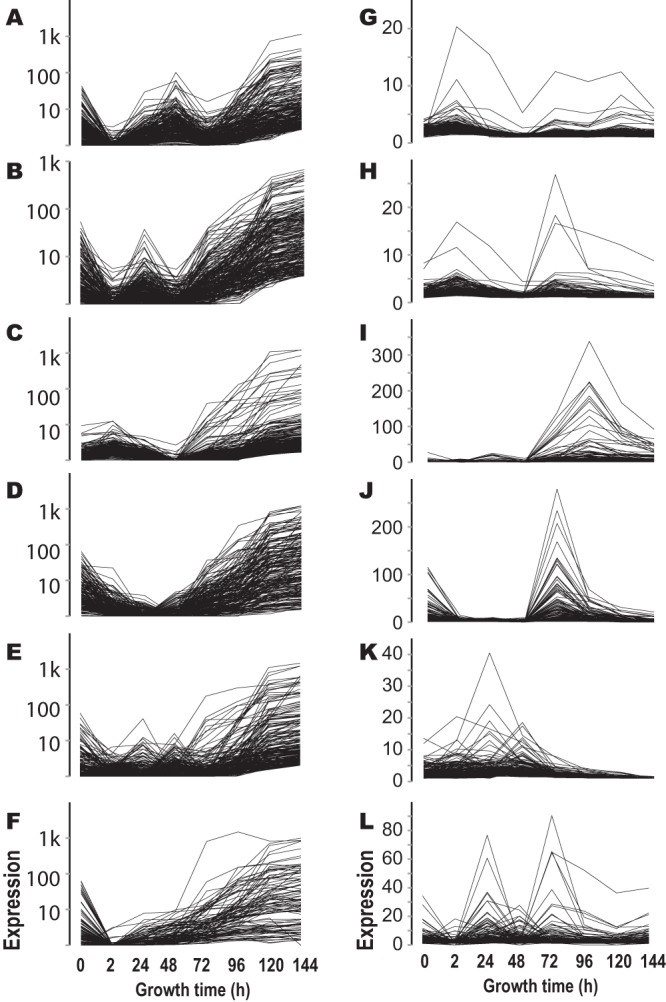
Gene clusters based on relative expression across sexual development. About 60% of the genes in the N. crassa genome are continuously upregulated or downregulated during late perithecium development. Based on their expression profiles across earlier stages, these genes can be further classified into six upregulated clusters (A to F; y axis logarithmically scaled) and six downregulated clusters (G to L; y axis linearly scaled).
Many downregulated genes were functionally annotated, and accordingly, many functional categories were identified as being significantly enriched among downregulated genes during perithecial development. A cluster of genes was upregulated between the 2-h and 48-h stages, before differentiated paraphyses and asci were detected, and then continually upregulated after 72 h (Fig. 2A). This cluster was enriched with genes that respond to light stimulus. Another major cluster of genes peaked at 24 h and was continually upregulated after 48 h, concurrent with ascus and ascospore development (Fig. 2B). Light-responsive genes were also significantly enriched in this cluster, as was extracellular protein degradation.
A third major cluster of genes exhibited a slight increase at 2 h and then was upregulated after 48 h (Fig. 2C). Functional enrichment of this cluster was significant for protein metabolism, energy, cell cycle, nucleic acid synthesis and processing, and signal transduction mechanisms. Smaller clusters of genes were upregulated at the last two sampled stages in perithecial development (Fig. 2D to F), and most of these genes have no assigned functions. A cluster of 349 genes showed a continual down-up-down-up-down expression (Fig. 2G), and these genes were functionally enriched in cellular communication and signal transduction mechanisms, interaction with the environment, including pheromone response and sex-specific proteins, and cell type differentiation, including development of ascospores. Another cluster with a similar enrichment of functional categories exhibited an upregulated expression pattern during early perithecial development (Fig. 2H).
A cluster of genes was downregulated during ascospore maturation after 96 h, exhibiting functional enrichment in nucleotide and protein metabolism, rRNA processing, cellular transport, and biogenesis of cellular components, including the cell wall (Fig. 2I). In addition, a cluster of genes was downregulated at 72 h. This cluster exhibited enrichment in extracellular metabolism, degradation and modification of exogenous components, cellular sensing and response to external stimulus, including light, pheromone response, and mating-type determination (Fig. 2J). Even earlier downregulation manifested after 24 h and 48 h in a cluster exhibiting enrichment of genes associated with protein synthesis and the heat shock response (Fig. 2K). A further cluster was enriched for many categories of metabolism and also in response to light stimulus (Fig. 2L). This cluster exhibited complex expression patterns during early perithecial development before full maturation of asci and ascospores.
More than 2,000 N. crassa genes lacking homologs to other fungal genomes outside the Neurospora genus were identified based on sequence similarity. These genes were labeled Neurospora orphan genes (46). From the recently published Sordaria macrospora genome (47), orthologs for 819 so-called Neurospora orphan genes have been identified by sequence similarity. Most of these genes have no assigned function in sexual development of Sordaria. Of the N. crassa orphan genes, 1,573 (nearly 16% of the genome) exhibited differential expression patterns during perithecium development (see Fig. S1 in the supplemental material). About 20% of these orphan genes exhibit expression patterns similar to the clusters (Fig. 2C and H) that are significantly enriched in genes that function in the cell cycle, DNA processing, mRNA and RNA synthesis, mitosis, meiosis, cellular communication/signal transduction, and cell type differentiation.
Upregulation of transcription factors and genes involved in perithecial development correlated with their stages of function. A number of genes involved in perithecial development and meiosis and some transcription factors (TFs) exhibited diverse expression patterns across perithecial development (Fig. 3). These genes include three ascus development genes, asd-1, asd-3, and asd-4, and three genes of asl-1 (ascospore lethal-1), asm-1 (ascospore maturation-1), and r (round spore) with phenotypes in ascospore development (10). Of the development-related TFs identified by Colot et al. (48), expression of TFs related to sexual development (NCU00097, NCU04561, NCU09739, NCU07392, and NCU044731) showed 5- to 20-fold changes across all time points, exhibiting patterns in which upregulated expression is correlated with gene phenotypes observed during perithecial development (Fig. 4). For transcription factors that are not well characterized for their function in metabolism and development, we often observed increasing expression during late perithecial development (see Table S6 in the supplemental material).
FIG 3.
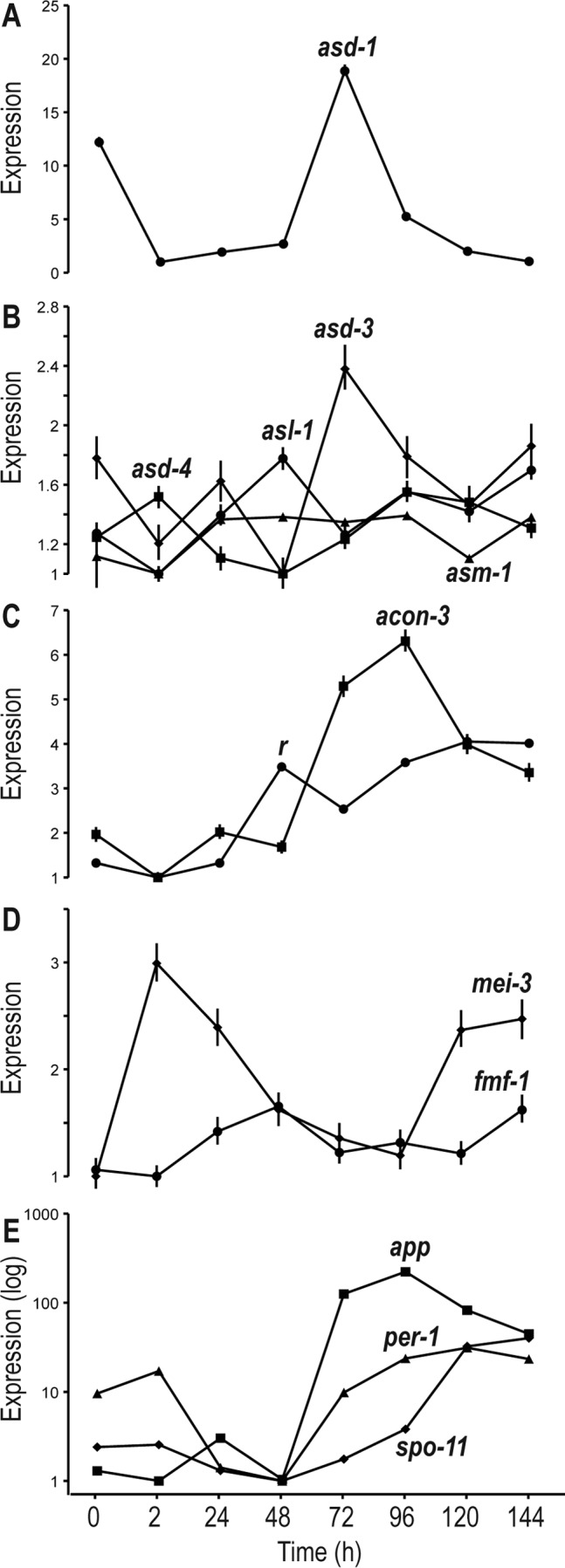
Relative expression across sexual development of genes annotated as functioning in perithecial development. (A) Expression of asd-1 (ascus development) at 72 h, when asci first form. (B) Peaks of expression for the genes asd-3, asd-4, asl-1 (ascospore lethal), and asm-1 (ascospore maturation) during intermediate stages of sexual development. (C) Expression of r (round spore) and acon-3 during perithecial development. (D) Expression of meiosis gene mei-3 and female and male fertility gene fmf-1. (F) Correlated expression of app (abundant perithecial protein), per-1 (perithecial-1), and meiosis-specific gene Spo11 during sexual development.
FIG 4.

Expression of transciption factors and arrested development in transcription factor knockouts. Solid lines indicate expression of known transcription factors, identified by their NCU number. Dashed lines associate peaks of expression with timing of appearance of knockout phenotype, as determined by Colot et al. (48). Illustrations depict perithecia at sequential developmental stages.
Two metabolic pathways, the melanin synthesis pathway and the carotenoid pathway, are responsible for the production of a dark pigment that increases across sexual development (melanin) and an orange pigment associated with conidiation that dissipates (carotene). Genes involved in these two pathways exhibited expression patterns that were highly correspondent to the observed color changes in the cultures (Fig. 5). Generally, expression of genes with known function in sexual development agreed with expectation.
FIG 5.

Relative expression across sexual development of genes whose products contribute to pigmentation in N. crassa. (A) Expression of three genes, 4hnr, 3hnr, and sd, that function in synthesis of melanin. (B) Expression patterns of genes involved in carotenoid synthesis, particularly al-1, al-2, and cao-1.
Expression of mating type loci and pheromone precursor genes was more complex than expected but in accordance with mating type specificity. Expression of mating type loci exhibited dramatic differences between the A-1, A-2, and A-3 genes at the mat A locus and the mat a-1 gene at the mat a locus (Fig. 6; Table 2). Expression of mat a-1 was not detectable in mat A protoperithecia and was almost undetectable before 48 h, but it increased continually across sexual development past 48 h, to a high of 107-fold in 144-h samples.
FIG 6.
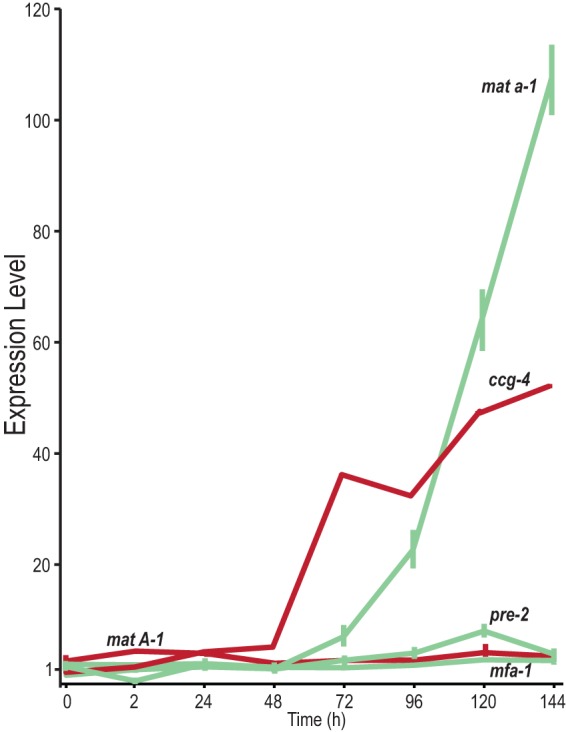
Relative expression across sexual development of mating type loci and pheromone precursors and receptors in the mat A female background. Expression profile are color-coded by their nominal mating type specificity: mat A is in red (dark) and mat a is in green (light). Expression of mating type gene mat a-1 increased 48 h and later after crossing between mat A protoperithecia and mat a conidia, where expression of mat A-1 was generally unchanged across perithecial development. Among pheromone genes, expression of the mat A-specific pheromone precursor gene ccg-4 increased during late perithecial development, and expression of the receptor PRE-2, specific for the peptide pheromone encoded by ccg-4, increased from 48 h. Expression of the receptor PRE-1, specific for the peptide pheromone encoded by mfa-1, was present and steady during perithecial development (Table 2).
Differential expression was observed for four genes encoding the mat a-specific pheromone precursor MFA-1, the pheromone receptors PRE-1 (responding to pheromone encoded by mfa-1; see Table 2) and PRE-2 (responding to pheromone encoded by ccg-4), and the mat A-specific pheromone precursor CCG-4 (Fig. 6). Whereas expression of mfa-1, pre-1, and pre-2 did not change much during perithecial development, expression of ccg-4 underwent a continuous and dramatic increase up to 45-fold. Genes mfa-1 and ccg-4 encode prepropheromones that require a posttranscription process to become mature pheromones (49). The expression of genes orthologous to the S. cerevisiae prepropheromone MFα1 and MFa1 processing genes, including kex1 (NCU04316), kex2 (NCU03219), ste13 (NCU02515), ram2 (NCU03632), ste6 (NCU07546), ste14 (NCU00034), ste24 (NCU03637), and axl1 (NCU00481), showed moderate correlation with each other (see Fig. S2 in the supplemental material). ste13 showed a continually increasing pattern after 24 h, similar to ccg-4. In addition to mating loci and pheromone genes, het (heterokaryon incompatibility) genes were also dynamically regulated during perithecium development (see Fig. S3 in the supplemental material).
Upregulated expression was observed for genes in RNA silencing pathways and genes associated with DNA methylation. We detected upregulated expression of genes that function in the vegetative silencing (quelling), repeat-induced point mutation (RIP), and meiotic silencing pathways as well as of genes involved in facultative heterochromatin formation and DNA methylation (Fig. 7A to D; see also Fig. S4 in the supplemental material). Upregulation across perithecial development was observed for sad-1 (suppressor of ascus dominance-1) and the cytosine methyltransferase homolog rid (RIP Defective), whose expression levels were increased 130-fold and 20-fold at 144 h, respectively (Fig. 7A). Similar to gene stc1 (siRNA to chromatin) in fission yeast, NCU01496 encodes a protein with a highly conserved Stc1 domain (blastp E value, 1.71e−30) that contains eight conserved cysteines that may bind to zinc. Expression of gene NCU01496 increased almost 1,000-fold by 144 h after crossing (Fig. 7A).
FIG 7.

Relative expression levels across sexual development and phenotypes of RNA silencing genes. Phenotypes of knockouts of RNA silencing genes were assayed on synthetic complete medium and carrot agar medium. Perithecia were crushed 6 days after a cross between mat A protoperithecia and mat a conidia. Expression for all genes is reported on a log scale. (A) Expression of the stc1-like gene, sad-1, and rid (RIP defective). (B) Expression of sms-2 (suppressor of meiotic silencing) and sad-2 (suppressor of ascus dominance). (C) Expression of sad-3, sad-4, sad-5, and sms-3. (D) Expression of Quelling-defective genes, including qde-1, qde-2, and qde-3 and qip (NCU00076, qde-2 interacting protein). (E) The Δsms-3 strain produced tiny hairy perithecia without differentiation of the centrum parenchyma (magnification, ×400). (F) The Δstc1 strain produced nomal-size perithecia with no apparent development of the centrum parenchyma (×400). (G and H) The Δqde-1 and Δsms-2 strains produced normal-size perithecia with undifferentiated thin-walled cells hard to detect in the centrum parenchyma area (×200). (I and J) The Δsad-1 and Δsad-3 strains produced normal-size perithecia with beaks and young asci without ascospores (×400). Scale bar = 10 mm for perithecia overview in KO phenotyping assays. Detailed images of the wild-type crushed perithecia are presented in Fig. 1G to L.
Two meiotic silencing genes, sms-2 and sad-2, exhibited similar regulatory dynamics, with a first peak at 24 h and a second increase after 48 h (Fig. 7B). Increases of 680-fold and 390-fold for sms-2 and sad-2 occurred from 2 h to 144 h (Fig. 7B). Expression of the sms-3 (suppressor of meiotic silencing-3) and sad-3 genes exhibited dynamics similar to the expression of sms-2, and sad-2, but with a 24-h delay (Fig. 7C). Expression levels of two recently identified MSUD (meiotic silencing by unpaired DNA) genes, sad-4 and sad-5 (50), dropped at 2 h after crossing but increased significantly starting at 48 h after crossing (11-fold for sad-4 and 135-fold for sad-5 in the 144-h samples [Fig. 7C]).
Bayesian networks constructed based on expression of genes recognized in RNA-silencing pathways (24), including qde-1, qde-2, qde-3, qip, sad-1, sad-2, sad-3, sms-2, sms-3, rrp-3, dcl-2, and rqh-2, always positioned genes encoding the downstream proteins in meiotic silencing, QIP, SMS-2, and the Dicer-like RNase III enzyme SMS-3, as independent parents in the top tiers (Fig. 8). This top-tier positioning suggests that the roles of the RNA silencing genes in controlling sexual development are played in an order that is inverted in comparison to their genetic dependence with regard to silencing. Removing any subset of the genes involved in the Quelling silencing pathway that functions only during asexual growth did not change the dependence orders among other genes in the network. The Bayesian network, including sad-4 and sad-5, suggested similar associations among RNA silencing genes, except that between sad-2 and sms-2, and was not consistent with phenotypes of sad-4 and sad-5. Additional genes that may interact with these genes during perithecial development are needed to be identified and included for reconstruct the network. Adding rid and stc1 to the RNA silencing genes in the Bayesian network inference demonstrated that both rid and stc1 were associated with RNA silencing pathways; they were inferred to be upstream in the network.
FIG 8.

Bayesian network inference for RNA silencing genes. Arrows indicate the dependence direction between two linked nodes (genes). Edges of posterior probability higher than 50% were present, and posterior probabilities >90% were in solid line. Illustrations of phenotypes were based on our observations, except for qip, which was drawn based on those of Hammond et al. (50), and sad-2, which was drawn based on those of Xiao et al. (76).
Phenotypes of knockouts were identifed for selected RNA silencing genes. Knockouts of many genes that were specifically upregulated in expression during crossing and perithecial development exhibited numerous phenotypes in sexual reproduction of N. crassa (Table 3; Fig. 7E to J). Knockouts of five RNA silencing genes and the stc1-like genes all yielded visible perithecia after crossing, but in all cases perithecium development failed at various stages before ascospore differentiation and production (Fig. 1C and I). Knockouts of sms-2 and the stc1-like gene produced dark, normal-size perithecia without asci. Knockouts of sms-3 produced perithecia that arrested earlier, after slight increases in size and darkening of the perithecial wall prior compared to normal perithecia 24 h to 48 h after crossing.
TABLE 3.
Phenotyping of knockouts for genes that exhibited upregulated expression during perithecial developmenta
| Function | Gene (NCU no.) | FGSC | Asexual development |
Sexual development |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Mycelium | Conidia | Protoperithecia | Perithecia | Ascus | Ascospores | Paraphyses | |||
| Heterokaryotic imcompatibility | het-14 (07511) | 11980a 11981A | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| het-6 (04453) | 11890a 11891A | Normal | Normal | Dispersalb | Normal | Normal | Normal | Normal | |
| het-15 (04694) | 16715a 16714A | Normal | Normal | Normal | Normal | Normal | Normal | Normal | |
| Mrk signaling | mkr-5 (07449) | 13456a 13457A | Normal | Normal | Normal | Youngc | No | No | Normal |
| mkr-6 (02919) | 15559a 15560A | Normal | Normal | Normal | Normal | Normal | Normal | Normal | |
| RNA silencing | sad-1 (02178) | 11151a 11152A | Normal | Normal | Normal | Normal | Normal | No | Normal |
| sad-3 (09211) | 19729a 19730A | Normal | Normal | Normal | Normal | Normal | No | Normal | |
| sms-2 (09434) | 11160a 11161A | Normal | Normal | Normal | Young | No | No | Normal | |
| sms-3 (08270) | 15892a 15891A | Normal | Normal | Normal | No | No | No | Normal | |
| qde-1 (07534) | 11156a 11157A | Normal | Normal | Normal | Normal | No | No | Normal | |
| RNAib | stc1d (01496) | 17464a 17465A | Normal | Normal | Normal | Normal | No | No | Normal |
Crossings were made between KO strains for each genes from both mating types, and descriptions were based on observation on both sides of the crossing zone.
Instead of along the crossing zone, protoperithecia were formed randomly over the plate on sides of both mating types.
Apparent changes in size and color from protoperithecia to perithecia were observed after crossing, but no development of centrum parachyma.
NCU01496 is homologous to stc1 (siRNA to chromatin-1) in fission yeast.
Neurospora crassa senses and responses to environmental signals, including blue light, via singaling pathways and light-responsive genes. Genes of the heterotrimeric G protein signaling system showed upregulation during different stages of perithecium development. Heterotrimeric G-protein signaling and cyclic AMP (cAMP) metabolism genes exhibited diverse expression patterns during perithecium development (see Fig. S5 in the supplemental material). Blue light-responsive genes exhibited distinct expression patterns across perithecial development under constant white light. Genes that putatively respond to blue light appear to be regulated in a similar pattern (see Fig. S6 in the supplemental material). A circadian pattern was not observed for the expression of the blue light sensor genes wc-1, wc-2, or related genes. Two genes encoding blue light-responsive proteins, vvd and velvet, exhibited general downregulation during perithecial development, especially after 48 h.
DISCUSSION
Here, we provide an in-depth characterization of the N. crassa transcriptome during sexual development. We observed that over 30% of the genome was expressed during early development, followed by increasing or decreasing expression of particular genes during late perithecium development. Of the genes that were upregulated late in the developmental process, many exhibited dramatic upregulation. A significant portion of these genes have not been classified or annotated for a function to date, including many Neurospora orphan genes. The lack of homologs in other fungal models, especially in well-annotated unicellular yeasts, makes it challenging to predict and test functions of these genes. Furthermore, genes whose mutants or knockouts lead to no sexual development, including meiotic recombination, are generally hard to study using traditional genetic approaches. Our approach has yielded important knowledge about such genes.
Our results show that the expression of genes involved in morphological characteristics during N. crassa perithecial development and, in particular, genes associated with the development of ascogenous hyphae and young asci, such as asd-1, asd-3, asl-1, asm-1, and round spore, peaked from 48 h to 72 h after fertilization, when those tissues developed within perithecia. Furthermore, expression of genes associated with carotenoid and melanin pigmentation contrasted across sexual development. The carotenoid biosynthetic pathway that is upregulated during asexual development in N. crassa (45) was generally downregulated during perithecium development. The expression of genes involved in the melanin synthesis pathway that produces the dark pigmentation of perithecia increased when the color of perithecia significantly darkened.
Our observations of sudden and significant shifts in the genome-wide transcriptomic landscape imply the presence of key regulators or regulatory cascades that are invoked at specific development stages. For instance, many genes exhibited low expression at 48 h, just before a dramatic increase. This set includes all genes encoding histones, many meiosis-specific genes, DNA methylation genes, the Cop9 signalosome (which plays major roles in hyphal growth, conidial development, and circadian function [51]), and some transcription factors that are involved in the sexual development of N. crassa (48). Two genes expected to have high expression in perithecia, app (abundant perithecial protein, homologous to app in Sordaria macrospora [52]) and per-1 (perithecial color [53]), increased over 100-fold after 48 h during late perithecium development. When asci and ascospores became visible under a microscope at 96 h, many genes, including a large number of functionally unannotated genes as well as mating type and pheromone genes and genes involved in signal pathways, RNA silencing pathways, and heterokaryon incompatibility, were highly expressed. Full development of beak structures and first ascospore ejection usually occur from 8 to 10 days after crossing and lasts for more than a week (44), beyond the last synchronized sampling time point used in this study.
Of all developmental genes, those with perhaps the most surprising expression patterns were the mating and pheromone genes. The mating and pheromone genes are generally thought to function in the conidia and in hyphae involved in mating but were also highly expressed after fertilization (54, 55). In our study, fertilization of mat A protoperithecia with mat a conidia revealed differences in the regulation of the mating type locus genes over the time course of sexual development. Expression of mat A-2 and A-3 was very low, and differences over time were not statistically significant. Expression of mat A-1 was meager and exhibited a net change of less than 3-fold. In contrast, expression of mat a-1 increased by more than 100-fold after 48 h. Although novel haploid hyphae from mat a conidia are rarely observed on cultures densely covered with mat A mycelia, mat a conidia could not be entirely avoided in our samples. Increased expression of mat a-1 could be due to the increase of mat a nuclei during conjugate division before karyogamy and meiotic and mitotic division after karyogamy. However, no corresponding increase in expression of the mat a-specific pheromone precursor mfa-1 was observed.
A significant increase was observed for the mat A-specific pheromone precursor ccg-4 after crossing, especially after 48 h. This finding is consistent with microarray measurements across perithecial development of N. crassa on both SCM and carrot medium (45). Expression of pre-2, which encodes a receptor that recognizes the pheromone encoded by ccg-4, has been reported to be high in male and female reproductive structures of mat a cells (56). Its expression increased significantly at 120 h after crossing but remained low in comparison to that of ccg-4. These patterns of mating type gene expression promise to help illuminate the biological function of mating type and pheromone genes after fertilization and hint at roles of mating type and pheromone genes in postcrossing sexual development. Although mating loci in both mat a and mat A strains have been known for a number of years for N. crassa and closely related species (57, 58, 82), the functions of mating loci were unknown aside from regulating homogenic incompatibility through highly regulated expression of pheromone genes and despite the distinct expression patterns of these genes at different stages in the life cycle of N. crassa and the closely related Fusarium (20).
In Neurospora and other fungal species, mating type idiomorphs impact diverse genes that are not directly involved in the mating process (21, 23, 45, 59, 60). The increased expression of mat a-1 and ccg-4 that we observed during sexual development of N. crassa calls for further investigation using strain-specific expression assays conducted with varied crosses. Although increased transcription of the pheromone precursor gene ccg-4 may not reflect a commensurate change in level of pheromone, we also observed increased expression of a homolog of ste-13 (NCU02515), which encodes the aminopeptidase required for processing prepropheromone MFα1 in S. cerevisiae (49). It has been shown that as long as the mating type idiomorphs appear in two different nuclei, the presence of one receptor and its compatible pheromone is necessary and sufficient to initiate perithecium development and ascospore production (56). Consistent with these results, we found low expression of pre-2 and high expression of ccg-4 in the mat A protoperithecia across perithecium development. Our data also demonstrate the correlated upregulation of pre-2 and ccg-4 expression on very different dynamic scales.
Mating type loci may control expression of pheromones and their receptors during presexual development before crossing in a mating type-specific manner (56). However, the mechanisms that regulate expression of pheromones and receptors after crossing when opposite mating types coexist in perithecia are unclear. Although meiosis and perithecium development can proceed in heterokaryon-containing nuclei of the opposite mating type when GNA-1 and one compatible pheromone-receptor pair are expressed, even at low levels, expression of mating types was not detected or reported in the previous study (56). Here we show the increased coexpression of mat a-1, pre-2, and ccg-4 at serial time points across perithecium development after crossing between mating types. Thus, the possibility that pre-2 specifically interacts with genes in mat a to upregulate ccg-4 requires further investigation.
A network of HMG-box transcription factors regulates mating and sexual development in Podospora anserina, a fungus that produces perithecia for fruiting bodies, and the factor PaHMG5 plays a central role in regulating expression of mating types (61). In N. crassa, NCU09387 (fmf-1) and NCU02326 are the ortholog and inparalog of the gene encoding PaHMG5. Our data support the hypothesis advanced by Ait Benkhali et al. (61) that these two genes can regulate expression of mating type genes at different stages of sexual development. Expression of fmf-1 was significantly upregulated after crossing and reached its peak at 48 h after crossing. This upregulation is consistent with the observed knockout phenotype as well: arrested development before ascospore formation. In contrast, expression of the inparalog NUC02326 significantly decreased after crossing and was maintained at a low level during late perithecial development. Its regulatory role on mating type gene expression before crossing demands further investigation.
As a heterothallic fungus, N. crassa employs a self-incompatibility mechanism termed heterokaryon incompatibility (HI) to restrict hyphal fusion to genetically identical strains and to stop growth of heterokaryotic hyphae during nonsexual development (62, 63). After crossing, cells in the developing N. crassa perithecium can be haploid, diploid, dikaryotic, or a single nucleus in some cells of the crozier structure. Dynamic expression of het and tol genes is perhaps associated with the toggling on and off of HI during sexual development, and regulation of these genes may be responsible for proper phase changes from dikaryotic cells to haploid sexual spores. Further investigation of tissue-specific expression of HI genes would be warranted.
Dynamic expression of genes within signaling pathways suggests their roles in triggering and executing sexual development. Heterotrimeric G-protein signaling, cAMP metabolism, Ras proteins, and G protein-coupled receptors (GPCRs) are responsible for transmitting extracellular signals to intracellular responses and are involved in sexual development in N. crassa (64–67). Expression of some GPCRs and G-protein subunits, especially gpr-1 and gna-3, increased after 48 h. A perithecial defect was observed for both Δbek-1 and Δgpr-1 strains (66), and abnormal perithecia with a large propotion of nonviable ascospores were found in Δgna-3 strains (67). Genetic tests suggest that RIC8 activates GNA-1 and GNA-3, but not GNA-2, and positively regulates the cAMP pathway in vegetative growth of N. crassa (68). In our study, expression of ric8, cr-1, gna-2, and gpr-6 was highly correlated. Previous studies show no apparent phenotype of Δgna-2 in asexual and sexual development of N. crassa and suggest that gna-2 plays a minor and compensatory role for gna-1 and gna-3 in the regulation of conidiation in N. crassa (65, 69). Gene coexpression data, especially finely sampled across time points, can provide numerous clues as to the relevant components and patterns of association that underlie complex gene networks.
Recently, NCU01496 was recognized as a homolog of stc1 in fission yeast, in which the gene is suggested to play important roles in RNA silencing and chromatin modification (70). It is possible that it fulfills a similar role in Neurospora, in which evidence suggests that RNA-mediated silencing and silencing by DNA methylation are independent genome defense systems (71–74). In N. crassa, diverse silencing pathways are active, including vegetative silencing (Quelling), repeat-induced point mutation (RIP), and meiotic silencing by unpaired DNA (MSUD). The last two pathways function only during sexual development (24). Quelling and MSUD detect and inactivate repeated or unpaired sequences during vegetative or sexual development, whereas RIP plays a role in deactivating transposable elements in the genome. Aside from their roles in genome defense, genes functioning in RNA silencing and quelling also play critical roles in controlling development. Details about the silencing pathways have been emerging steadily with genetic studies, but how these genes and pathways regulate sexual development remains unexplored. While we may argue that these genes are likely associated in a similar pattern between the very different functions, the structures of the networks supporting the functions can be different in terms of their components and dependencies between genes (direction of regulation). The functional dependencies of the genes in ordinary development can accumulate in the opposite direction from the functional dependencies in generalized error correction or surveillance (which each often involves retracing steps to rectify the genomic or cellular state).
Our data support previously proposed models that RNA silencing pathways and genes involved in other RNA interference pathways are closely involved with meiosis and sexual development in N. crassa. According to the latest model, an MSUD complex includes SAD-1/SAD-3, proteins that use an aberrant RNA template to synthesize a double-stranded RNA (dsRNA) molecule, DCL-1 (SMS-3), that processes the dsRNA into small interfering RNA (siRNA); QIP (QDE-2 interacting protein)/SMS-2, which destroys complementary mRNA transcribed from the aberrant RNA template generated from siRNA; and SAD-2, which recruits SAD-1 and possibly other proteins from the perinuclear region (75, 76). Our findings of same expression pattern between SAD-3 and SMS-3 support their associated functions predicted in the initial steps of producing dsRNA and siRNA molecules. In addition, we observed similar upregulation of genes coding for SMS-2 and QIP that is consistent with their same function in destroying erroneously transcribed mRNAs. The gene pair sms-2 and sad-2 and the gene pair sms-3 and sad-3 exhibited similar expression patterns, with the latter pair lagging by 24 h. However, there is not an obvious explanation for the peak expression of sms-2 and sad-2 prior to the upregulation of sms-3 and sad-3. It is possible that MSUD may be involved in RNA interference (RNAi)-induced assembly of heterochromatin in N. crassa (73), as reported in the homolog of sad-3 in Schizosaccharomyces pombe, hrr1, which is required in fission yeast for RNAi-mediated formation of heterochromatin. Our findings support this hypothesis, as we observed coexpression of the sad-1 gene involved in MSUD, the rid gene involved in RIP, and the stc1-like gene, whose homolog is also involved in RNAi and chromatin modification in fission yeast. Consistent with previous studies of sad-1 and sad-2 mutants (77, 78), we observed abortive perithecia at different development stages for knockouts of some genes involved RNA silencing pathways. These knockout phenotypes suggest that there is an interplay between silencing pathways and ordinary developmental functions associated with sexual development and meiosis. Further study on regulatory functions of different silencing pathways is needed to determine how these diverse pathways work together to regulate gene expression during sexual development of N. crassa. Consistent with these knockout phenotypes (and independent of them), model-averaged Bayesian network prediction based on expression data suggest that sms-2, qip, and sms-3 are independent parents in the top tiers of the network, and expression of sad-2 and sad-3 is dependent on those parent genes. Intriguingly, their dependency in our sexual development time course was inferred to be causally opposite in sequence order to their dependency as a genome defense response for removal of unpaired DNAs. Including rid or stc1 did not change the network structures among RNA silencing pathways.
Transcript abundance of genes involved in DNA methylation of N. crassa increased in synchrony with the RNA silencing genes in our experiment. Expression of the four DNA methylation genes, dim-2, dim-5, dim-7, and hp1 (79, 80), increased after 48 h, and the expression pattern of methyltransferase-coding gene dim-2 was similar to that of the RNA silencing genes sms-2 and sad-2. The hp1, cdp-2, hda-1, and chap genes encode the HP1 complex (HCHC), which directs histone deacetylation and DNA methylation and is required for gene silencing in centromeric regions independent of DNA methylation (74). A complicated silencing network, the sex-induced silencing of genes by RNAi before meiosis, has recently been discovered in the yeast-like basidiomyceteous fungus Cryptococcus neoformans (81). Further assessment of methylation levels during sexual development, when asci and ascospores are differentiated, would enable stronger inference from upregulated expression of these genes.
In summary, we observed stage-specific expression for some known genetic markers and transcription factors that are associated with morphological development of perithecia. Examining knockout strains for genes involved in RNA silencing and signaling pathways that were differentially expressed across development, we identified increased gene expression associated with functional stages of development, including an stc1-like gene whose homolog was recently identified as a critical link between RNAi and chromatin modification in fission yeast. Analysis of gene expression patterns during different stages of sexual development allowed us to identify conditions and developmental stages that facilitate phenotyping genes and to discern potentially novel functions of genes during sexual development of N. crassa. Although stage-specific expression was observed for genes involved in diverse functions during sexual development, the functions of numerous genes that were markedly upregulated during late perithecium development, when asci and ascospores develop, have not been reported. Our findings provide key insights into the functions of genes involved in key pathways such as expression of mating type genes and pheromones, pigmentation, heterokaryotic incompatibility, signaling pathways, and RNA silencing during sexual development of N. crassa, as well as the transcription factors and other regulatory components that are responsible for the action of these pathways in sexual development.
Supplementary Material
ACKNOWLEDGMENTS
Strains and plasmids were obtained from the Fungal Genetics Stock Center (Kansas City, MO). We thank Angelika Hofmann for helpful comments on the manuscript.
This work was supported by funding from NSF MCB 0923797 to J.P.T. and F.T. and NIH PO1 grant GM068067 to J.P.T.
Footnotes
Published ahead of print 15 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00248-13.
REFERENCES
- 1.Domazet-Lošo T, Tautz D. 2010. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468:815–818. 10.1038/nature09632 [DOI] [PubMed] [Google Scholar]
- 2.Freeman TC, Ivens A, Baillie JK, Beraldi D, Barnett MW, Dorward D, Downing A, Fairbairn L, Kapetanovic R, Raza S, Tomoiu A, Alberio R, Wu C, Su AI, Summers KM, Tuggle CK, Archibald AL, Hume DA. 2012. A gene expression atlas of the domestic pig. BMC Biol. 10:90. 10.1186/1741-7007-10-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quint M, Drost HG, Gabel A, Ullrich KK, Bonn M, Grosse I. 2012. A transcriptomic hourglass in plant embryogenesis. Nature 490:98–101. 10.1038/nature11394 [DOI] [PubMed] [Google Scholar]
- 4.Huang T, Lopez-Giraldez F, Townsend JP, Irish VF. 2012. RBE controls microRNA164 expression to effect floral organogenesis. Development 139:2161–2169. 10.1242/dev.075069 [DOI] [PubMed] [Google Scholar]
- 5.Taylor J, Berbee M. 2006. Dating divergences in the fungal tree of life: review and new analyses. Mycologia 98:838–849. 10.3852/mycologia.98.6.838 [DOI] [PubMed] [Google Scholar]
- 6.Bass D, Richards TA. 2011. Three reasons to re-evaluate fungal diversity ‘on Earth in the ocean.' Fungal Biol. Rev. 25:159–164. 10.1016/j.fbr.2011.10.003 [DOI] [Google Scholar]
- 7.Blackwell M. 2011. The fungi: 1, 2, 3. 5.1 million species? Am. J. Bot. 98:426–438. 10.3732/ajb.1000298 [DOI] [PubMed] [Google Scholar]
- 8.Stajich J, Berbee M, Blackwell M, Hibbett D, James T, Spatafora J, Taylor J. 2009. The Fungi. Curr. Biol. 19:R840–R845. 10.1016/j.cub.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beadle G, Tatum E. 1941. Genetic control of biochemical reactions in Neurospora. Proc. Natl. Acad. Sci. U. S. A. 27:499–506. 10.1073/pnas.27.11.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raju N. 2009. Neurospora as a model fungus for studies in cytogenetics and sexual biology at Stanford. J. Biosci. 34:139–159. 10.1007/s12038-009-0015-5 [DOI] [PubMed] [Google Scholar]
- 11.Lord KM, Read ND. 2011. Perithecium morphogenesis in Sordaria macrospora. Fungal Genet. Biol. 48:388–399. 10.1016/j.fgb.2010.11.009 [DOI] [PubMed] [Google Scholar]
- 12.Bistis GN, Perkins DD, Read ND. 2003. Different cell types in Neurospora crassa. Fungal Genet. Newsl. 50:17–19 [Google Scholar]
- 13.Nelson MA, Kang S, Braun EL, Crawford ME, Dolan PL, Leonard PM, Mitchell J, Armijo AM, Bean L, Blueyes E, Cushing T, Errett A, Fleharty M, Gorman M, Judson K, Miller R, Ortega J, Pavlova I, Perea J, Todisco S, Trujillo R, Valentine J, Wells A, Werner-Washburne M, Yazzie S, Natvig DO. 1997. Expressed sequences from conidial, mycelial, and sexual stages of Neurospora crassa. Fungal Genet. Biol. 21:348–363. 10.1006/fgbi.1997.0986 [DOI] [PubMed] [Google Scholar]
- 14.Teichert I, Wolff G, Kuck U, Nowrousian M. 2012. Combining laser microdissection and RNA-seq to chart the transcriptional landscape of fungal development. BMC Genomics 13:511. 10.1186/1471-2164-13-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi W, Kwon C, Trail F. 2006. Microarray analysis of transcript accumulation during perithecium development in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Mol. Genet. Genomics 276:87–100. 10.1007/s00438-006-0125-9 [DOI] [PubMed] [Google Scholar]
- 16.Nowrousian M, Frank S, Koers S, Strauch P, Weitner T, Ringelberg C, Dunlap JC, Loros JJ, Kuck U. 2007. The novel ER membrane protein PRO41 is essential for sexual development in the filamentous fungus Sordaria macrospora. Mol. Microbiol. 64:923–937. 10.1111/j.1365-2958.2007.05694.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nowrousian M, Ringelberg C, Dunlap JC, Loros JJ, Kuck U. 2005. Cross-species microarray hybridization to identify developmentally regulated genes in the filamentous fungus Sordaria macrospora. Mol. Genet. Genomics 273:137–149. 10.1007/s00438-005-1118-9 [DOI] [PubMed] [Google Scholar]
- 18.Hallen HE, Trail F. 2008. The L-type calcium ion channel cch1 affects ascospore discharge and mycelial growth in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot. Cell 7:415–424. 10.1128/EC.00248-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallen HE, Huebner M, Shiu SH, Guldener U, Trail F. 2007. Gene expression shifts during perithecium development in Gibberella zeae (anamorph Fusarium graminearum), with particular emphasis on ion transport proteins. Fungal Genet. Biol. 44:1146–1156. 10.1016/j.fgb.2007.04.007 [DOI] [PubMed] [Google Scholar]
- 20.Sikhakolli UR, Lopez-Giraldez F, Li N, Common R, Townsend JP, Trail F. 2012. Transcriptome analyses during fruiting body formation in Fusarium graminearum and Fusarium verticillioides reflect species life history and ecology. Fungal Genet. Biol. 49:663–673. 10.1016/j.fgb.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 21.Bidard F, Benkhali J, Coppin E, Imbeaud S, Grognet P, Delacroix H, Debuchy R. 2011. Genome-wide gene expression profiling of fertilization competent mycelium in opposite mating types in the heterothallic fungus Podospora anserina. PLoS One 6:321476. 10.1371/journal.pone.0021476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Lehr N, Trail F, Townsend J. 2012. Differential impact of nutrition on developmental and metabolic gene expression during fruiting body development in Neurospora crassa. Fungal Genet. Biol. 49:405–413. 10.1016/j.fgb.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Böhm J, Hoff B, O'Gorman CM, Wolfers S, Klix V, Binger D, Zadra I, Kürnsteiner H, Pöggeler S, Dyer PS, Kück U. 2013. Sexual reproduction and mating-type-mediated strain development in the penicillin-producing fungus Penicillium chrysogenum. Proc. Natl. Acad. Sci. U. S. A. 110:1476–1481. 10.1073/pnas.1217943110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borkovich K, Alex L, Yarden O, Freitag M, Turner G, Read N, Seiler S, Bell-Pedersen D, Paietta J, Plesoksky N, Plamann M, Goodrich-Tanrikulu M, Schulte U, Mannhaupt G, Nargang FE, Radford A, Selitrennikoff C, Galagan JE, Dunlap JC, Loros JJ, Catcheside D, Inoue H, Aramayo R, Polymenis M, Selker EU, Sachs MS, Marzluf GA, Paulsen I, Davis R, Ebbole DJ, Zelter A, Kalkman ER, O'Rourke R, Bowring F, Yeadon J, Ishii C, Suzuki K, Sakai W, Pratt R. 2004. Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol. Mol. Biol. Rev. 68:1–108. 10.1128/MMBR.68.1.1-108.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galagan J, Calvo S, Borkovich K, Selker E, Read N, Jaffe D, FitzHugh W, Ma L, Smirnov S, Purcell S, Rehman B, Elkins T, Engels R, Wang S, Nielsen C, Butler J, Endrizzi M, Qui D, Ianakiev P, Pedersen D, Nelson M, Werner-Washburne M, Selitrennikoff C, Kinsey J, Braun E, Zelter A, Schulte U, Kothe G, Jedd G, Mewes W, Staben C, Marcotte E, Greenberg D, Roy A, Foley K, Naylor J, Stabge-Thomann N, Barrett R, Gnerre S, Kamal M, Kamvysselis M, Mauceli E, Bielke C, Rudd S, Frishman D, Krystofova S, Rasmussen C, Metzenberg R, Perkins D, Kroken S, Cogoni C, Macino G, Catcheside D, Li W, Pratt R, Osmani S, DeSouza C, Glass L, Orbach M, Berglund J, Voelker R, Yarden O, Plamann M, Seiler S, Dunlap J, Radford A, Aramayo R, Natvig D, Alex L, Mannhaupt G, Ebbole D, Freitag M, Paulsen I, Sachs M, Lander E, Nusbaum C, Birren B. 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422:859–868. 10.1038/nature01554 [DOI] [PubMed] [Google Scholar]
- 26.Do JH, Yamaguchi R, Miyano S. 2009. Exploring temporal transcription regulation structure of Aspergillus fumigatus in heat shock by state space model. BMC Genomics 10:306. 10.1186/1471-2164-10-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bushel PR, Heard NA, Gutman R, Liu L, Peddada SD, Pyne S. 2009. Dissecting the fission yeast regulatory network reveals phase-specific control elements of its cell cycle. BMC Syst. Biol. 3:93. 10.1186/1752-0509-3-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. 2006. Inference in Bayesian networks. Nat. Biotechnol. 24:51–53. 10.1038/nbt0106-51 [DOI] [PubMed] [Google Scholar]
- 29.Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. 2007. A primer on learning in Bayesian networks for computational biology. PLoS Comput. Biol. 3:e129. 10.1371/journal.pcbi.0030129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCluskey K, Wiest A, Plamann M. 2010. The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J. Biosci. 35:119–126. 10.1007/s12038-010-0014-6 [DOI] [PubMed] [Google Scholar]
- 31.Klittich C, Leslie JF. 1988. Nitrate reduction mutants of Fusarium moniliforme (Gibberella fujikuroi). Genetics 118:417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trail F, Common R. 2000. Perithecial development by Gibberella zeae: a light microscopy study. Mycologia 92:130–138. 10.2307/3761457 [DOI] [Google Scholar]
- 33.Clark TA, Guilmette JM, Renstrom D, Townsend JP. 2008. RNA extraction, probe preparation, and competitve hybridization for transcriptional profiling using Neurospora crassa long-oligomer DNA microarrays. Fungal Genet. Rep. 55:18–28 [Google Scholar]
- 34.Adomas AB, Lopez-Giraldez F, Clark TA, Wang Z, Townsend JP. 2010. Multi-targeted priming for genome-wide gene expression assays. BMC Genomics 11:477. 10.1186/1471-2164-11-477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Lopez-Giraldez F, Townsend JP. 2010. LOX: inferring Level Of eXpression from diverse methods of census sequencing. Bioinformatics 26:1918–1919. 10.1093/bioinformatics/btq303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 38.Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Guldener U, Mannhaupt G, Munsterkotter M, Mewes HW. 2004. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 32:5539–5545. 10.1093/nar/gkh894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28:27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziebarth JD, Bhattacharya A, Cui Y. 2013. Bayesian network webserver: a comprehensive tool for biological network modeling. Bioinformatics 10.1093/bioinformatics/btt472 [DOI] [PubMed] [Google Scholar]
- 41.Dunlap J, Borkovich K, Henn M, Turner G, Sachs M, Glass N, McCluskey K, Plamann M, Galagan J, Birren B, Weiss R, Townsend J, Loros J, Nelson M, Lambreghts R, Colot H, Park G, Collopy P, Ringelberg C, Crew C, Litvinkova L, DeCaprio D, Hood H, Curilla S, Shi M, Crawford M, Koerhsen M, Montgomery P, Larson L, Pearson M, Kasuga T, Tian C, Bastuerkmen M, Altamirano L, Xu J. 2007. Enabling a community to dissect an organism: overview of the Neurospora functional genomics project. Fungal Genomics 57:49–96. 10.1016/S0065-2660(06)57002-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lichius A, Lord KM, Jeffree CE, Oborny R, Boonyarungsrit P, Read ND. 2012. Importance of MAP kinases during protoperithecial morphogenesis in Neurospora crassa. PLoS One 7:e42565. 10.1371/journal.pone.0042565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z, Townsend JP. 2010. The filamentous fungal gene expression database (FFGED). Fungal Genet. Biol. 47:199–204. 10.1016/j.fgb.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris JL, Howe HB, Jr, Roth IL. 1975. Scanning electron microscopy of surface and internal features of developing perithecia of Neurospora crassa. J. Bacteriol. 122:1239–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Kin K, Lopez-Giraldez F, Johannesson H, Townsend J. 2012. Sex-specific gene expression during asexual development of Neurospora crassa. Fungal Genet. Biol. 49:533–543. 10.1016/j.fgb.2012.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasuga T, Mannhaupt G, Glass N. 2009. Relationship between phylogenetic distribution and genomic features in Neurospora crassa. PLoS One 4:e5286. 10.1371/journal.pone.0005286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nowrousian M, Stajich JE, Chu M, Engh I, Espagne E, Halliday K, Kamerewerd J, Kempken F, Knab B, Kuo HC, Osiewacz HD, Pöggeler S, Read ND, Seiler S, Smith KM, Zickler D, Kück U, Freitag M. 2010. De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet. 6:e1000891. 10.1371/journal.pgen.1000891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colot H, Park G, Turner G, Ringelberg C, Crew C, Litvinkova L, Weiss R, Borkovich K, Dunlap J. 2006. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. U. S. A. 103:10352–10357. 10.1073/pnas.0601456103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pöggeler S. 2011. Function and evolution of pheromones and pheromone receptors in filamentous ascomycetes, p 73–95 In Pöggeler S, Wöstemeyer J. (ed), The Mycota XIV. Springer-Verlag, Berlin, Germany [Google Scholar]
- 50.Hammond TM, Xiao H, Boone EC, Decker LM, Lee SA, Perdue TD, Pukkila PJ, Shiu PK. 2013. 2013. Novel proteins required for meiotic silencing by unpaired DNA and siRNA generation in Neurospora crassa. Genetics 194:91–100. 10.1534/genetics.112.148999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Z, Wang Y, Cai G, He Q. 2012. Neurospora COP9 signalosome integrity plays major roles for hyphal growth, conidial development, and circadian function. PLoS Genet. 8:e1002712. 10.1371/journal.pgen.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nowrousian M, Piotrowski M, Kuck U. 2007. Multiple layers of temporal and spatial control regulate accumulation of the fruiting body-specific protein APP in Sordaria macrospora and Neurospora crassa. Fungal Genet. Biol. 44:602–614. 10.1016/j.fgb.2006.09.009 [DOI] [PubMed] [Google Scholar]
- 53.Howe HB, Jr, Benson EW. 1974. A perithecial color mutant of Neurospora crassa. Mol. Gen. Genet. 131:79–83. 10.1007/BF00269389 [DOI] [PubMed] [Google Scholar]
- 54.Kim H, Borkovich KA. 2004. A pheromone receptor gene, pre-1, is essential for mating type-specific directional growth and fusion of trichogynes and female fertility in Neurospora crassa. Mol. Microbiol. 52:1781–1798. 10.1111/j.1365-2958.2004.04096.x [DOI] [PubMed] [Google Scholar]
- 55.Kim H, Borkovich KA. 2006. Pheromones are essential for male fertility and sufficient to direct chemotropic polarized growth of trichogynes during mating in Neurospora crassa. Eukaryot. Cell 5:544–554. 10.1128/EC.5.3.544-554.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H, Wright SJ, Park G, Ouyang S, Krystofova S, Borkovich KA. 2012. Roles for receptors, pheromones, G proteins, and mating type genes during sexual reproduction in Neurospora crassa. Genetics 190:1389–1404. 10.1534/genetics.111.136358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nygren K, Wallberg A, Samils N, Stajich J, Townsend J, Karlsson M, Johannesson H. 2012. Analyses of expressed sequence tags in Neurospora reveal rapid evolution of genes associated with the early stages of sexual reproduction in fungi. BMC Evol. Biol. 12:229. 10.1186/1471-2148-12-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karlsson M, Nygren K, Johannesson H. 2008. The evolution of the pheromonal signal system and its potential role for reproductive isolation in heterothallic Neurospora. Mol. Biol. Evol. 25:168–178. 10.1093/molbev/msm253 [DOI] [PubMed] [Google Scholar]
- 59.Lee J, Lee T, Lee YW, Yun SH, Turgeon BG. 2003. Shifting fungal reproductive mode by manipulation of mating type genes: obligatory heterothallism of Gibberella zeae. Mol. Microbiol. 50:145–152. 10.1046/j.1365-2958.2003.03694.x [DOI] [PubMed] [Google Scholar]
- 60.Klix V, Nowrousian M, Ringelberg C, Loros J, Dunlap J, Pöggeler S. 2010. Functional characterization of MAT1-1-specific mating-type genes in the homothallic ascomycete Sordaria macrospora provides new insights into essential and nonessential sexual regulators. Eukaryot. Cell 9:894–905. 10.1128/EC.00019-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ait Benkhali J, Coppin E, Brun S, Peraza-Reyes L, Martin T, Dixelius C, Lazar N, van Tilbeurgh H, Debuchy R. 2013. A network of HMG-box transcription factors regulates sexual cycle in the fungus Podospora anserina. PLoS Genet. 9:e1003642. 10.1371/journal.pgen.1003642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glass N, Dementhon K. 2006. Non-self recognition and programmed cell death in filamentous fungi. Curr. Opin. Microbiol. 9:553–558. 10.1016/j.mib.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 63.Saupe S. 2000. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol. Mol. Biol. Rev. 64:489–502. 10.1128/MMBR.64.3.489-502.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maller J. 2003. Fishing at the cell surface. Science 300:594–595. 10.1126/science.1083725 [DOI] [PubMed] [Google Scholar]
- 65.Krystofova S, Borkovich K. 2005. The heterotrimeric G-protein subunits GNG-1 and GNB-1 form a G-beta gamma dimer required for normal female fertility, asexual development, and G alpha protein levels in Neurospora crassa. Eukaryot. Cell 4:365–378. 10.1128/EC.4.2.365-378.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krystofova S, Borkovich K. 2006. The predicted G-protein-coupled receptor GPR-1 is required for female sexual development in the multicellular fungus Neurospora crassa. Eukaryot. Cell 5:1503–1516. 10.1128/EC.00124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kays A, Rowley P, Baasiri R, Borkovich K. 2000. Regulation of conidiation and adenylyl cyclase levels by the G alpha protein GNA-3 in Neurospora crassa. Mol. Cell. Biol. 20:7693–7705. 10.1128/MCB.20.20.7693-7705.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wright S, Inchausti R, Eaton C, Krystofova S, Borkovich K. 2011. RIC8 is a guanine-nucleotide exchange factor for G alpha subunits that regulates growth and development in Neurospora crassa. Genetics 189:165–175. 10.1534/genetics.111.129270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Won S, Michkov AV, Krystofova S, Garud AV, Borkovich KA. 2012. Genetic and physical interactions between Gα subunits and components of the Gβγ dimer of heterotrimeric G proteins in Neurospora crassa. Eukaryot. Cell 11:1239–1248. 10.1128/EC.00151-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bayne E, White S, Kagansky A, Bijos D, Sanchez-Pulido L, Hoe K, Kim D, Park H, Ponting C, Rappsilber J, Allshire R. 2010. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 140:666–677. 10.1016/j.cell.2010.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freitag M, Lee D, Kothe G, Pratt R, Aramayo R, Selker E. 2004. DNA methylation is independent of RNA interference in Neurospora. Science 304:1939. 10.1126/science.1099709 [DOI] [PubMed] [Google Scholar]
- 72.Rountree M, Selker E. 2010. DNA methylation and the formation of heterochromatin in Neurospora crassa. Heredity 105:38–44. 10.1038/hdy.2010.44 [DOI] [PubMed] [Google Scholar]
- 73.Chang S, Zhang Z, Liu Y, Gottesman S, Harwood C, Schneewind O. 2012. RNA interference pathways in fungi: mechanisms and functions. Annu. Rev. Microbiol. 66:305–323. 10.1146/annurev-micro-092611-150138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Honda S, Lewis Z, Shimada K, Fischle W, Sack R, Selker E. 2012. Heterochromatin protein 1 forms distinct complexes to direct histone deacetylation and DNA methylation. Nat. Struct. Mol. Biol. 19:471–475. 10.1038/nsmb.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hammond TM, Xiao H, Boone EC, Perdue TD, Pukkila PJ, Shiu PK. 2011. SAD-3, a putative helicase required for meiotic silencing by unpaired DNA, interacts with other components of the silencing machinery. G3 (Bethesda) 1:369–376. 10.1534/g3.111.000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiao H, Alexander WG, Hammond TM, Boone EC, Perdue TD, Pukkila PJ, Shiu PK. 2010. QIP, a protein that converts duplex siRNA into single strands, is required for meiotic silencing by unpaired DNA. Genetics 186:119–126. 10.1534/genetics.110.118273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shiu P, Raju N, Zickler D, Metzenberg R. 2001. Meiotic silencing by unpaired DNA. Cell 107:905–916. 10.1016/S0092-8674(01)00609-2 [DOI] [PubMed] [Google Scholar]
- 78.Shiu P, Zickler D, Raju N, Ruprich-Robert G, Metzenberg R. 2006. SAD-2 is required for meiotic silencing by unpaired DNA and perinuclear localization of SAD-1 RNA-directed RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 103:2243–2248. 10.1073/pnas.0508896103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Honda S, Selker E. 2008. Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurospora crassa. Mol. Cell. Biol. 28:6044–6055. 10.1128/MCB.00823-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lewis ZA, Adhvaryu KK, Honda S, Shiver AL, Knip M, Sack R, Selker EU. 2010. DNA methylation and normal chromosome behavior in Neurospora depend on five components of a histone methyltransferase complex, DCDC. PLoS Genet. 6:e1001196. 10.1371/journal.pgen.1001196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, Hsueh Y, Li W, Floyd A, Skalsky R, Heitman J. 2010. Sex-induced silencing defends the genome of Cryptococcus neoformans via RNAi. Genes Dev. 24:2566–2582. 10.1101/gad.1970910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin T, Lu S, van Tilbeurgh H, Ripoll D, Dixelius C, Turgeon B, Debuchy R. 2010. Tracing the origin of the fungal alpha 1 domain places its ancestor in the HMG-box superfamily: implication for fungal mating-type evolution. PLoS One 5:e15199. 10.1371/journal.pone.0015199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.