

Abstract
Rice wine has been one of the most popular traditional alcoholic drinks in China. However, the presence of potentially carcinogenic ethyl carbamate (EC) in rice wine has raised a series of food safety issues. During rice wine production, the key reason for EC formation is urea accumulation, which occurs because of nitrogen catabolite repression (NCR) in Saccharomyces cerevisiae. NCR represses urea utilization by retaining Gln3p in the cytoplasm when preferred nitrogen sources are present. In order to increase the nuclear localization of Gln3p, some possible phosphorylation sites on the nuclear localization signal were mutated and the nuclear localization regulation signal was truncated, and the disruption of URE2 provided an additional method of reducing urea accumulation. By combining these strategies, the genes involved in urea utilization (DUR1,2 and DUR3) could be significantly activated in the presence of glutamine. During shake flask fermentations of the genetically modified strains, very little urea accumulated in the medium. Furthermore, the concentrations of urea and EC were reduced by 63% and 72%, respectively, in a model rice wine system. Examination of the normal nutrients in rice wine indicated that there were few differences in fermentation characteristics between the wild-type strain and the genetically modified strain. These results show that metabolic engineering of the NCR regulators has great potential as a method for eliminating EC during rice wine production.
INTRODUCTION
Rice wine is one of the oldest drinks in the world. It has been consumed continuously for 4,000 years in China (1). During the industrial fermentation process, Saccharomyces cerevisiae plays the leading role in the formation of ethanol and the characteristic flavor compounds (2). However, there are a few potentially harmful by-products produced by S. cerevisiae (3, 4). Among these compounds, ethyl carbamate (EC, also called urethane) is a potentially genotoxic carcinogen that can be widely found in alcoholic beverages and fermented foods (5). EC has been shown to induce dose-dependent increases in lung tumorigenesis in mice (6). There is also evidence that EC can cause liver disease, in particular, hepatic angiosarcoma, in humans (7). Therefore, the International Agency for Research on Cancer (IARC) classified EC in group 2A, which contains substances that are probably carcinogenic to humans (8).
Several methods have been applied to eliminate EC in alcoholic beverages. As urea is a precursor of EC, inhibition of the CAR1 gene, which encodes arginase, was found to decrease EC formation by 60% (9). Another solution is to express the DUR1,2 gene (encoding urea amidolyase, which can degrade urea into ammonia) or the DUR3 gene (encoding urea permease) constitutively in S. cerevisiae. By overexpressing DUR1,2 and DUR3, urea-degrading strains produced 87% and 15% less EC, respectively, than the original strain (10). However, this method produces excess ammonium that could affect the wine taste. Alternatively, the addition of acid urease can efficiently eliminate urea in wine and repress EC formation indirectly (11), but the utilization of urease is restricted because of its special requirement of nickel ions, which are harmful to humans (12). Therefore, it is necessary to find a safe and effective method to minimize the formation of EC.
In rice wine production, EC mainly forms because of urea accumulation during fermentation (13). Urea is a nonpreferred nitrogen source for S. cerevisiae that can be degraded into carbon dioxide and ammonium under suitable conditions (14). The key reason for urea accumulation is nitrogen catabolite repression (NCR) in S. cerevisiae (15). NCR is an important mode of nitrogen regulation that represses the metabolism of nonpreferred nitrogen sources when preferred nitrogen sources exist in the environment. There are many regulators involved in NCR; Gln3p is the most important activator for nonpreferred nitrogen metabolism. Its expression remains constant for different nitrogen sources, but its intracellular distribution changes. When preferred nitrogen sources exist in the environment, the activated upstream regulator (Tor1p) can interact directly with Gln3p, resulting in some serines in Gln3p being phosphorylated (16). Subsequently, phosphorylated Gln3p can combine with another negative regulator, Ure2p, so that the Gln3p-Ure2p complex cannot enter the nucleus to activate the expression of DUR1,2 and DUR3. In this way, the utilization of urea and other nonpreferred nitrogen sources is repressed.
Previous studies showed that the transportation of Gln3p into the nucleus requires a nuclear localization signal (NLS) in Gln3p. In addition, the possible phosphorylation sites of Gln3p are also in the NLS, at serines 344, 347, and 355 (17). Furthermore, one of the Tor1p-binding domains in Gln3p, which are also called nuclear localization regulatory signals (NLRS), has been identified recently. This region is located in the C terminus at residues 654 to 667 (18). Although this information about NLS and NLRS is very important for enhancing our understanding of the regulatory pattern of Gln3p in NCR, there have been few studies investigating modifications of Gln3p to reduce urea accumulation. Moreover, as Ure2p can combine with phosphorylated Gln3p so that it is retained in the cytoplasm (19), knocking out the gene encoding Ure2p (URE2) could be used as an alternative method to improve urea utilization.
In this study, by combining several metabolic engineering strategies to modify the regulators in NCR, the expression of the genes involved in urea utilization was derepressed. Then, shake flask experiments and the model rice wine system proved that the concentrations of urea and EC were significantly reduced in the genetically modified strains. These results indicated that similar methods could be applied in the commercial production of rice wine.
MATERIALS AND METHODS
Yeast strain, plasmids, and media.
Saccharomyces cerevisiae strain CEN.PK2-1C (MATa ura3-52 trp1-289 leu2-3,112 his3Δ1 MAL2-8c SUC2) was bought from the European Saccharomyces cerevisiae Archive for Functional Analysis (EUROSCARF). The multiple-copy plasmid pYX212 was obtained from R&D systems (Minneapolis, MN). The low-copy-number plasmid pY16TEF and integrative plasmid pRS306TEF were purchased from Turbobiotech in China. The yeast strains were precultured on YPD plates (10 g/liter yeast extract, 20 g/liter peptone, 20 g/liter glucose, 2% agar) at 30°C for 24 h. Based on previous research, glutamine is one of the most preferred nitrogen sources for yeast (15), and its repressive effect on the formation of enzymes and permeases for the utilization of nonpreferred nitrogen sources is stronger than the repressive effects of other preferred nitrogen sources (20). Therefore, YNB medium (1.6 g/liter yeast nitrogen base with no ammonium sulfate and amino acids, 20 g/liter glucose) with glutamine (10 mM) was used to study the derepressive effect of metabolic engineering on regulators in nitrogen catabolite repression. In addition, YNB medium agar plates (1.6%, wt/vol agar) with appropriate supplements (40 mg/liter histidine, 40 mg/liter tryptophan, 40 mg/liter leucine, 40 mg/liter uracil) were used to select the positive transformants.
Construction of recombinant vectors to express Gln3p and modified Gln3p.
The primers used in vector construction are listed in Table 1. The genomic DNA of S. cerevisiae CEN.PK2-1C was extracted using a Gentra Puregene yeast kit (Qiagen, Valencia, CA) and used as the DNA template for amplifying the GLN3 gene. The PCR-amplified GLN3 fragments were digested and inserted into the SalI/SacI site of pYX212, the BamHI/HindIII site of pRS306TEF, or the BamHI/HindIII site of pY16TEF. For truncating the GLN3 gene, one pair of special primers was designed to amplify the nucleotide sequence encoding amino acids 1 to 653 of Gln3p. The truncated GLN3 fragments were also inserted into pYX212, pRS306TEF, and pY16TEF in the same restriction enzyme sites mentioned above. All correct insertions of the target genes were confirmed by DNA sequencing. The pYX212-Gln3p and pYX212-Gln3p1-653 plasmids were transformed into S. cerevisiae CEN.PK2-1C strains using the lithium acetate method (21). Furthermore, the pYX212-Gln3p, pYX212-Gln3p1-653, pRS306TEF-Gln3p1-653, and pY16TEF-Gln3p1-653 plasmids were used to perform mutations of Gln3p.
TABLE 1.
Oligonucleotides used for construction, mutation, truncation, and disruption
| Purpose of primer pair | Directiona | Sequence (5′–3′)b |
|---|---|---|
| Gln3p and truncated Gln3p fragments for plasmid: | ||
| pYX212-Gln3p | F | ACGCGTCGACATGCAAGACGACCCCGAAAA |
| R | CGAGCTCTCATATACCAAATTTTAACCAATCCAA | |
| pYX212-Gln3p1–653 | F | ACGCGTCGACATGCAAGACGACCCCGAA |
| R | CGAGCTCTCATGAAGAAGGTCTTGAGACACTC | |
| pRS306TEF/pY16TEF-Gln3p | F | CGCGGATCCATGCAAGACGACCCCGAAAA |
| R | CCCAAGCTTTCATATACCAAATTTTAACCAATCCAA | |
| pRS306TEF/pY16TEF-Gln3p1–653 | F | CGCGGATCCATGCAAGACGACCCCGAA |
| R | CCCAAGCTTTCATGAAGAAGGTCTTGAGACACTC | |
| Mutation of possible phosphorylation sites in Gln3p NLS as follows: | ||
| S344A | F | GCCATTAGCTTTAAAATCGGAC |
| R | CTCATGGTACCATGTAATTTCTGG | |
| S347A | F | CCTTAAAAGCTGACGTTAT |
| R | ATAATGGCCTCATGGTAC | |
| S355A | F | AAGAGGATTGCTAAGAAGAGAG |
| R | TTTGATAACGTCCGATTTTAA | |
| Disruption of URE2 in: | ||
| Left arm of gene | F | CGCGGATCCGGAGACAAGGGCTTACAAACG |
| R | GAGTGTACTAGAGGAGGCCAAGAGTTTGGTGTACAACTTAATTTGCAGC | |
| HIS3 marker | F | GCTGCAAATTAAGTTGTACACCAAACTCTTGGCCTCCTCTAGTACACTC |
| R | TTCTTGTTTTTAAAGCAGCCTACGCATCTGTGCGGTATTTCAC | |
| Right arm of gene | F | GTGAAATACCGCACAGATGCGTAGGCTGCTTTAAAAACAAGAA |
| R | CGAGCTCTACATTAGTCGGGAAATCTTGC |
F, forward; R, reverse.
Restriction enzyme sites are underlined.
Site-directed mutations in the phosphorylation sites of Gln3p were carried out using the MutanBEST kit (TaKaRa, Dalian, China). The one-step PCR employed PrimeSTAR HS DNA polymerase and used the plasmids whose construction is described above as the template DNA. The primers used for the mutations are given in Table 1. The PCR products were treated with blunting kination enzyme and ligated into circular plasmids. Escherichia coli JM109 cells were transformed with these plasmids, and the transformants were confirmed by DNA sequencing. The correct plasmids were transformed into S. cerevisiae CEN.PK2-1C strains for expression.
Disruption of URE2 in S. cerevisiae CEN.PK2-1C.
The primers used for disruption of the URE2 gene are given in Table 1. The Δure2 strains were obtained from S. cerevisiae CEN.PK2-1C by HIS3 gene replacement (22). The HIS3 gene, with its promoter and terminator, was amplified from pRS303 (23) and used as the marker cassette. The 500-bp sequences upstream and downstream from the URE2 gene were amplified from the genomic DNA of S. cerevisiae CEN.PK2-1C as the left arm and right arm of the disruption cassette, respectively. The entire disruption cassette was generated by fusion PCR based on three previous PCR products (24) and was confirmed by DNA sequencing. The correct cassette was transformed into S. cerevisiae CEN.PK2-1C with selection on YNB agar plates (40 mg/liter tryptophan, 40 mg/liter leucine, and 40 mg/liter uracil).
RNA preparation and cDNA synthesis.
The yeast cells were incubated in YPD medium at 30°C for 20 h with constant shaking at 200 rpm. YNB medium with glutamine (10 mM) was then inoculated with the strains and incubated at 30°C for 10 h with continuous shaking at 200 rpm. The cells were ground in liquid nitrogen, and total RNA was prepared using TRIzol reagent (Invitrogen, Carlsbad, CA). DNA contamination was eliminated by an extra DNase digestion step. The RNA was quantified and checked in a Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE) at 260 nm and 280 nm. The integrity of the isolated RNA was verified with an automated electrophoresis system (Bio-Rad, Hercules, CA). cDNA was synthesized from 5 μg of total RNA using the PrimeScript RT reagent kit (perfect real time) (TaKaRa, Dalian, China).
qRT-PCR.
All primer sequences were designed for ACT1, DUR1,2, and DUR3 by using Beacon Designer 7.0 (Table 2). The efficiencies and specificities of the primers were tested in dilution experiments and by constructing melting curves, respectively. The real-time quantitative PCR (qRT-PCR) experiments were performed with the SYBR premix Ex Taq kit (TaKaRa) with the synthesized cDNA as the template. The cycling parameters for the PCR were as follows: preincubation at 95°C for 30 s, 40 cycles of amplification at 95°C for 5 s and 60°C for 20 s, and a final cooling step at 50°C for 30 s. The reactions were conducted in a LightCycler 480 II real-time PCR instrument (Roche Applied Science, Mannheim, Germany). All experiments were performed in biological replicates with an independent measurement of each sample, and mean values were used for further calculations. The fold changes were determined via the cycle threshold (2−ΔΔCT) method and normalized to the results for the ACT1 gene.
TABLE 2.
Oligonucleotides for RT-PCR
| Targeted gene | Directiona | Sequence (5′–3′) | Product size (bp) | Mean PCR efficiency |
|---|---|---|---|---|
| ACT1 | F | TTATTGATAACGGTTCTGGTATG | 100 | 1.904 |
| R | CCTTGGTGTCTTGGTCTAC | |||
| DUR1,2 | F | GGTGTCCCTATTGCTGTTAAG | 186 | 1.927 |
| R | CCGTGTGCCGACTAATCC | |||
| DUR3 | F | ACTGCCTGTGGGTGTTGTTG | 200 | 1.941 |
| R | CGTCTACTGGATGCCTCTTGG |
F, forward; R, reverse.
Measurement of urea, EC, and the main components in the model rice wine system.
All standards used for detection were obtained from Sigma-Aldrich (Saint Louis, MO). Other chemicals and solvents were of analytical grade. Urea was determined after the addition of 4-dimethylaminobenzaldehyde by measuring the optical absorbance at 420 nm (25). The analysis of EC was carried out on a GC-2010 gas chromatograph (GC) (Shimadzu, Kyoto, Japan) equipped with a GCMS-QP2010 plus mass spectrometer (MS) (Shimadzu, Kyoto, Japan). A polar-phase chromatography capillary column (Rtx-wax; Restek) was used according to a previous method (26). The concentrations of glucose, ethanol, and lactic acid were determined using an Agilent high-performance liquid chromatography (HPLC) system 1200 (Palo Alto, CA) with an iron-exchange Waters Sugar-Pak I column (6.5 by 300 mm) and a refractive index detector (27). Analyses of amino acids were performed using the Agilent HPLC system 1200 (Palo Alto, CA) and a Zorbax Eclipse AAA (4.6 by 150 mm) column according to the previously described method (28).
Shake flask fermentation.
The yeast cells were incubated in YPD medium at 30°C for 20 h with constant shaking at 200 rpm. YNB medium with urea and glutamine (10 mM) was then inoculated with the strains and incubated at 30°C for 48 h with continuous shaking at 200 rpm. Samples were taken every 8 h to measure the concentration of urea in the fermentation broth. The yeast growth was monitored by detecting the optical density at 600 nm (OD600). All experiments were performed in biological replicates with an independent measurement of each sample, and the mean values were used for further calculations.
Rice wine brewing in the model system.
A laboratory-scale rice wine fermentation was performed in a model system that was in strict accordance with the manufacturing process of Shaoxing rice wine in China. The yeast cells were incubated in YPD medium at 30°C for 20 h with constant shaking at 200 rpm. Steamed rice, wheat Qu (prepared by natural inoculation of molds, bacteria, and yeasts on wheat), water, and yeast were mixed in a fermentation flask, and fermentation was conducted at a constant temperature of 30°C for 5 days and then switched to 15°C for 20 days when the main fermentation was finished. Samples were taken when the fermentation was complete to measure the concentrations of urea, EC, and the main components in the model rice wine system. All experiments were performed in biological replicates with an independent measurement of each sample, and mean values were used for further calculations.
RESULTS
Influence of mutations in the NLS of Gln3p.
In order to derepress the cytoplasmic sequestration of Gln3p, three possible phosphorylation sites in the NLS were mutated to alanine by single-point mutation (Gln3pS344A, Gln3pS347A, and Gln3pS355A) or multipoint mutation (Gln3pS344A S347A S355A). All mutated Gln3p and unmodified Gln3p moieties were overexpressed with the multiple-copy plasmid pYX212 in S. cerevisiae strain CEN.PK2-1C. The expression levels of DUR1,2 and DUR3 were measured to determine the derepressive effects of mutations of Gln3p when the cells were grown in glutamine. The wild-type (WT) S. cerevisiae CEN PK2 strain was used as the control. The qRT-PCR results showed that the derepressing effects of the mutations were not obvious (Fig. 1). Overexpression of unmodified Gln3p and Gln3p with single-point mutations in the phosphorylation sites did not enhance the expression of DUR1,2 and DUR3 (all changes were <2.0-fold). Even the multipoint mutations at all three serine sites had little effect (2.9- and 2.1-fold, respectively). This meant that the effect of the existing GLN3 gene in the wild-type S. cerevisiae CEN PK2 strain was limited.
FIG 1.
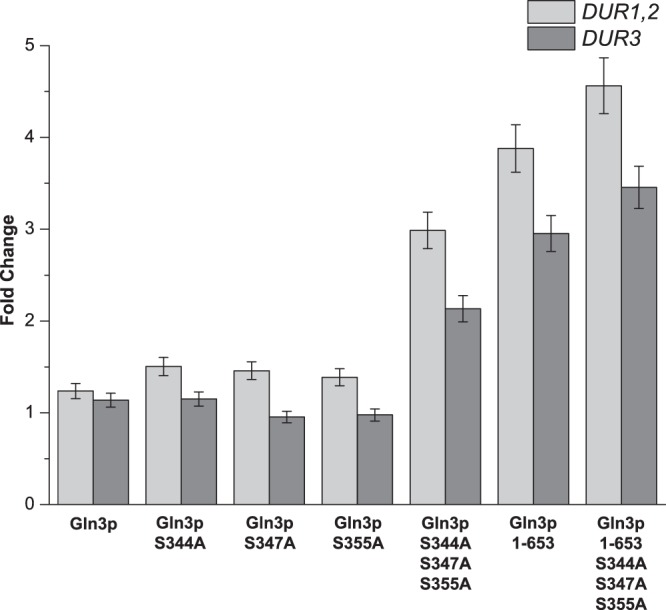
Effects of the modifications of Gln3p on the expression of DUR1,2 and DUR3. The cells were cultured in YNB medium supplemented with glutamine (10 mmol/liter). The wild-type S. cerevisiae CEN PK2 strain was used as the control. Data are normalized to the expression of the ACT1 gene. Error bars show standard deviations.
Influence of Gln3p with NLRS truncation.
In addition to mutations in the NLS, truncation of the exact C-terminal NLRS (Gln3p1–653) was employed to increase the distribution of Gln3p in the nucleus. The wild-type S. cerevisiae CEN PK2 strain was still used as the control. As there was no substantial derepressive effect of unmodified Gln3p, its effect was not examined in successive experiments. The effect of truncating the C-terminal NLRS was also evaluated by detecting the expression levels of DUR1,2 and DUR3. The qRT-PCR results showed that the derepressive effect of NLRS truncation was greater than the effects of mutations in the NLS. Further improvements in the expression of DUR1,2 and DUR3 can be achieved by truncating the NLRS (3.8- and 2.9-fold, respectively) (Fig. 1). Based on the above-described results, the effects of both the alanine point mutations and the truncation were investigated in a single experiment. The combination of these two strategies (Gln3p1–653 S344A S347A S355A) significantly enhanced the expression of DUR1,2 and DUR3 (4.5- and 3.4-fold, respectively) (Fig. 1). Therefore, Gln3p1–653 S344A S347A S355A was the optimum choice for the following metabolic engineering work.
Influence of the disruption of URE2.
The disruption of URE2 provided a supplementary means of reducing urea accumulation after a systematic analysis of the NCR mechanism. The URE2 gene was knocked out in S. cerevisiae CEN PK2. To determine the effect of this disruption, the expression levels of DUR1,2 and DUR3 were determined using the same method as described above. The results showed that disruption of URE2 significantly enhanced the expression of DUR1,2 and DUR3 (10.9- and 5.3-fold, respectively) (Fig. 2). The promising derepressive effects of disruption prompted us to express Gln3p1–653 S344A S347A S355A in the Δure2 strain. It was not surprising to find that the expression levels of DUR1,2 and DUR3 were further increased (14.8- and 9.1-fold, respectively) by combining the strategies of disruption, alanine point mutations, and truncation (Fig. 2).
FIG 2.
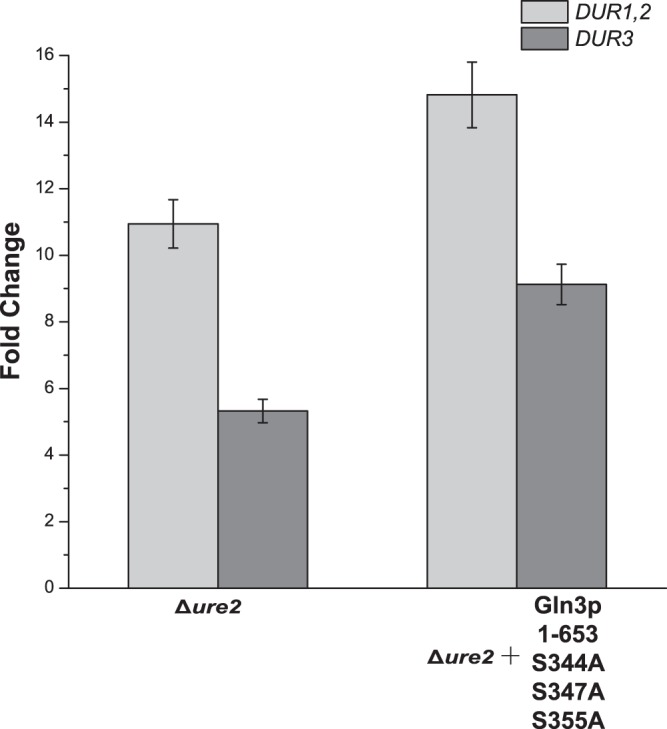
Effects of the disruption of URE2 on the expression of DUR1,2 and DUR3. The cells were cultured in YNB medium supplemented with glutamine (10 mmol/liter). Data are normalized to the expression of the ACT1 gene. Error bars show standard deviations.
Urea utilization during shake flask fermentation.
In order to determine whether or not urea utilization is enhanced during the process of yeast cultivation, the WT strain, the Δure2 (URE) strain, the strain with Gln3p1–653 S344A S347A S355A (GLN strain), and the Δure2 strain with Gln3p1–653 S344A S347A S355A (UG strain) were cultivated in YNB medium with a moderate amount of urea in shake flasks for 48 h. The concentration of urea and the OD600 were measured every 8 h. Compared with the results for the WT strain (the increase in the concentration of urea from 8 h), the consumption of urea was faster in the initial stage of fermentation and urea did not accumulate during the 48-h fermentation for all genetically modified strains (Fig. 3A). The urea utilization for the URE strain, the GLN strain, and the UG strain reached 59.1%, 55.4%, and 70.2%, respectively. Among these strains, the UG strain utilized urea best, which is consistent with previous qRT-PCR results. However, based on the comparison of the final OD600 values at 48 h between the wild-type strain and the UG strain, the growth of the strain was severely repressed, by 42.5%, because of the modification (Fig. 3B). As the growth of the URE strain was marginally affected by the disruption of URE2, the expression level of modified Gln3p could be reduced to alleviate the energy burden in the UG strain.
FIG 3.
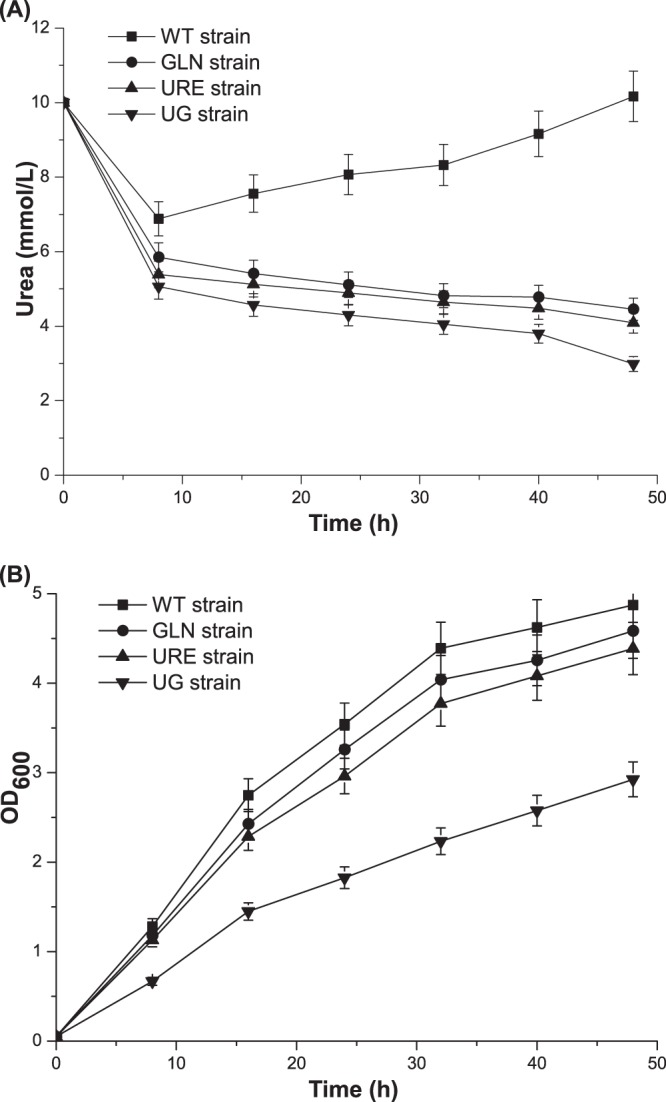
Urea utilization during shake flask fermentation. The cells were cultured in YNB medium supplemented with urea (10 mmol/liter) and were harvested every 8 h. (A) Concentrations of urea for each strain during 48-h fermentations. (B) OD600 values for each strain during 48-h fermentations. Error bars show standard deviations.
The low-copy-number expression of modified Gln3p in Δure2 strains.
To recover the growth of the UG strain, two other kinds of plasmids were used, including low-copy-number plasmid pY16TEF and integrative plasmid pRS306TEF. The effects of the new methods of expressing modified Gln3p were compared with the results for the UG strain, which was generated by using multiple-copy plasmid pYX212. The qRT-PCR results showed that the expression of DUR1,2 and DUR3 was still derepressed by pY16TEF (12.7- and 7.1-fold, respectively) and pRS306TEF (14.2- and 8.7-fold, respectively) (Fig. 4A). In addition, determinations of urea utilization further validated the qRT-PCR results. The strains with Gln3p modified by pY16TEF and pRS306TEF also did not accumulate urea during the 48-h fermentation (Fig. 4B). Furthermore, the final OD600 values reached 3.63 (pY16TEF) and 4.03 (pRS306TEF), which is close to the values for the WT strain (Fig. 4C). This meant that the growth of the UG strain had been partially recovered.
FIG 4.
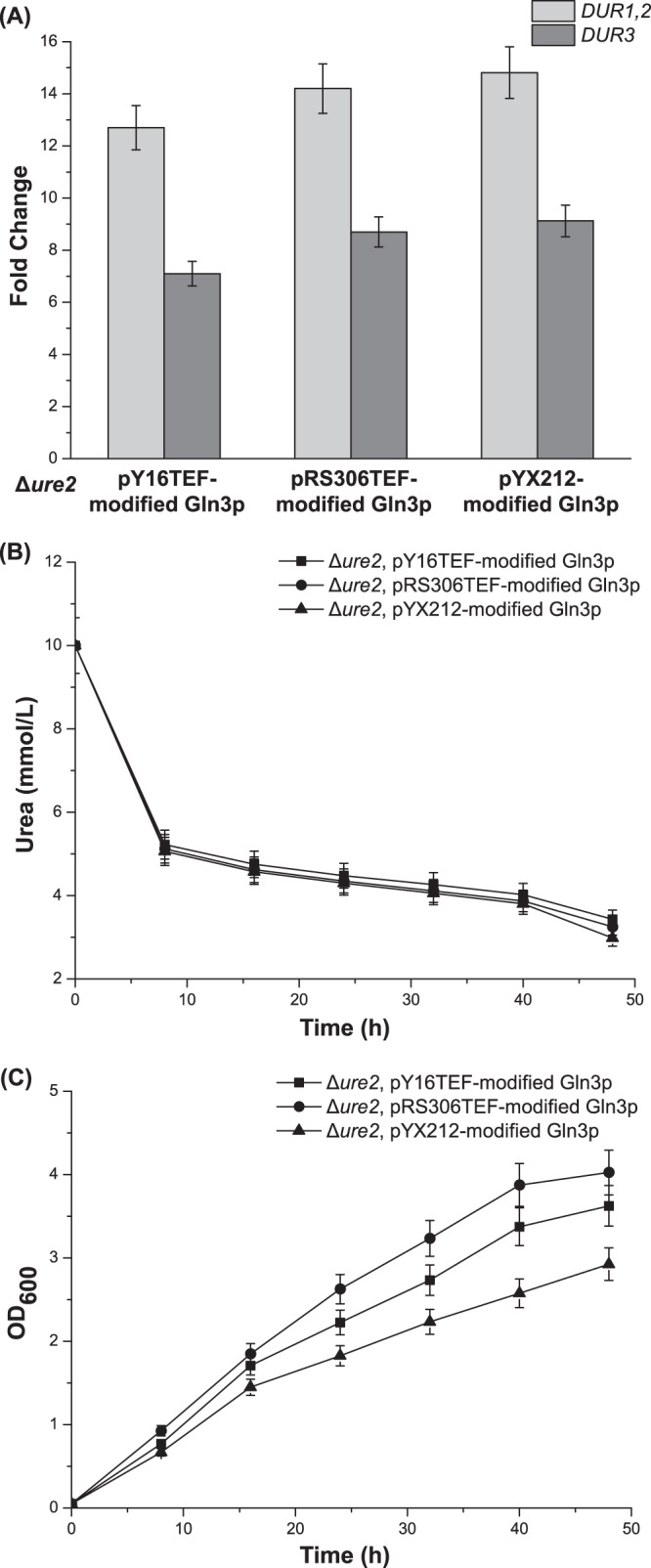
Selecting the low-copy-number expression vector for the expression of modified Gln3p in Δure2 strains. (A) Effects on the expression of DUR1,2 and DUR3 of expressing modified Gln3p via different vectors. (B) Concentrations of urea in the medium for strains containing different vectors expressing modified Gln3p during 48-h shake flask fermentations. (C) OD600 values in the same experiments whose results are shown in panel B. Error bars show standard deviations.
Rice wine brewing using the model system with genetically modified strains.
Model system rice wine brewing was carried out with the WT S. cerevisiae CEN PK2 strain and the UG strain. After 25 days of fermentation, the fermentation broth was collected to examine the concentrations of urea, EC, and the main components in the model rice wine system (Table 3). Compared with the concentrations in the WT strain, the concentrations of urea and EC were reduced by 63% and 72%, respectively. However, the concentrations of glucose, ethanol, lactic acid, and amino acids differed only slightly. These data showed that metabolically engineering the WT strain could reduce the concentrations of urea and EC without significantly changing those common fermentation characteristics.
TABLE 3.
Measurement of urea, EC, and main components in the model system of rice wine
| Compound | Value (mean ± SD) in: |
|
|---|---|---|
| WT strain | UG strain | |
| EC (μg/liter) | 198.32 ± 2.47 | 55.53 ± 0.79 |
| Urea (mg/liter) | 38.46 ± 0.42 | 14.23 ± 0.25 |
| Glucose (g/liter) | 6.84 ± 0.12 | 6.98 ± 0.08 |
| Ethanol (% vol) | 14.73 ± 0.18 | 14.56 ± 0.27 |
| Lactic acid (g/liter) | 4.32 ± 0.13 | 4.23 ± 0.06 |
| Amino acids (mg/liter): | ||
| Alanine | 690.14 ± 10.35 | 695.16 ± 18.01 |
| Arginine | 753.57 ± 9.78 | 741.24 ± 8.59 |
| Asparagine | 41.53 ± 0.54 | 42.15 ± 0.61 |
| Aspartate | 417.29 ± 8.34 | 427.71 ± 9.35 |
| Cysteine | 136.78 ± 1.91 | 133.88 ± 2.43 |
| Glutamine | 74.91 ± 0.54 | 75.62 ± 0.61 |
| Glutamate | 725.52 ± 12.33 | 758.62 ± 16.62 |
| Glycine | 275.63 ± 4.67 | 289.34 ± 5.12 |
| Histidine | 69.65 ± 0.69 | 62.19 ± 1.28 |
| Isoleucine | 244.36 ± 5.62 | 233.37 ± 6.08 |
| Leucine | 641.04 ± 13.46 | 659.06 ± 11.86 |
| Lysine | 463.28 ± 4.63 | 450.88 ± 11.29 |
| Methionine | 174.92 ± 2.09 | 167.61 ± 3.71 |
| Phenylalanine | 571.13 ± 7.42 | 554.92 ± 10.41 |
| Proline | 632.61 ± 13.91 | 619.15 ± 10.54 |
| Serine | 22.06 ± 0.38 | 24.62 ± 0.42 |
| Threonine | 166.87 ± 3.16 | 173.64 ± 2.28 |
| Tryptophan | 9.76 ± 0.18 | 9.59 ± 0.21 |
| Tyrosine | 460.52 ± 5.53 | 475.81 ± 7.72 |
| Valine | 412.34 ± 9.88 | 421.07 ± 8.86 |
DISCUSSION
Although many methods have been developed to reduce the concentration of EC in different alcoholic beverages, they all have some limitations (13). Therefore, new metabolic engineering strategies for NCR regulators were attempted. Based on previous studies, several domains in Gln3p were identified, including the NLS and the NLRS. However, this information about Gln3p had never before been applied to the regulation of the nitrogen mechanism in S. cerevisiae. According to the results for the modifications of Gln3p, multipoint mutations in the NLS and truncation of the NLRS both derepressed the expression of genes involved in urea metabolism, but the effect of truncation was stronger. In addition, the combination of these two strategies produced an enhanced effect.
The effects of the point mutations were surprising, because Kulkarni et al. mutated the possible phosphorylation sites in the NLS of Gln3p and found that the localization of Gln3p changed only a little (17). This means that, although the mutations have little effect on the localization, this strategy can still derepress the expression of DUR1,2 and DUR3. Similar to point mutations in NLS, NLRS truncation has been attempted in previous research. Rai et al. proved that truncation at the C terminus of the NLRS partially disrupted the interaction between Gln3p and Tor1p (18). However, their work did not investigate the effect of truncation on urea utilization. In this study, the relationship between NLRS truncation and the expression of the genes involved in urea metabolism was identified. Furthermore, shake flask fermentation indicated that the derepression of urea metabolic genes improved urea utilization in S. cerevisiae. Therefore, it was important to determine whether such modifications of Gln3p could be used to reduce urea accumulation.
In addition to the modifications of Gln3p, disruption of URE2 was used to derepress the expression of DUR1,2 and DUR3. In previous studies, this strategy was adopted to enhance the expression of some NCR-sensitive genes (GAP1, GDH1, and GLN1) (29, 30). However, it was unknown whether this method would have the same effect on the urea metabolic pathway. The qRT-PCR and shake flask fermentation results showed that the effect of deleting URE2 was even stronger than the effects of modifying Gln3p. It should be pointed out that deletion of URE2 could also be used to reduce urea accumulation. As both phosphorylation of Gln3p and interaction with Ure2p are required for the sequestration of Gln3p, a better derepression effect was achieved when modified Gln3p was overexpressed in the Δure2 strain.
Although the Δure2 strain with pYX212-modified Gln3p exhibited the strongest urea utilization in the presence of glutamine, the growth of this strain was strongly repressed. The inhibition of growth in a genetically modified strain has been reported in previous research (31). As the influence of overexpression of Gln3p or disruption of URE2 alone was not serious, the possible reason for the stronger inhibition of growth in the UG strain is that the combination of the two strategies could significantly enhance the expression levels of NCR-regulated genes, resulting in increasing the burden of energy. Therefore, the vector for expression of the modified Gln3p was changed. Considering the derepressing effects and stabilities of different vectors, integrative expression of modified Gln3p was selected to recover the growth of the genetically modified strain. Finally, the Δure2 strain with pRS306TEF-modified Gln3p (UG strain) was the optimal selection used in the model rice wine system.
There were two aspects to the experiments using the model rice wine system. On one hand, the effect of the UG strain on EC elimination was far superior to the effects of the other methods (9, 10, 32, 33). On the other hand, the concentrations of the main components in rice wine changed very little (all of the changes were <5%). These compounds are very important for the flavor and nutritional value of rice wine (2). The stability of the concentrations of these compounds means that the changes in fermentation characteristics were small. The stability of the fermentation characteristics is a basic requirement in metabolically engineering strains by other methods (10, 34). Therefore, the new strategies have the potential to be used in the industrial production of rice wine.
In summary, the strategies utilizing Gln3p and Ure2p provided effective methods to eliminate EC in the model rice wine system. The current work lays a foundation for further research on the control of urea accumulation to eliminate EC production in rice wine and other, similar fermented foods through metabolic engineering of regulators in NCR. Similar methods could be applied to eliminate other harmful nitrogenous compounds in alcoholic beverages. In addition, based on further global and in-depth investigations of the mechanisms of nitrogen regulation, there may be more regulators in NCR that could be new metabolic engineering targets for minimizing urea and EC production in rice wine.
ACKNOWLEDGMENTS
This work was supported by the Major State Basic Research Development Program of China (973 Program, grant 2012CB720802), the National Natural Science Foundation of China (grants 31130043, 31000807, and 21276109), the Natural Science Foundation of Jiangsu Province (grants BK2010150 and BK2011004), the Author of National Excellent Doctoral Dissertation of People's Republic of China (FANEDD, grant 2011046), the Program for New Century Excellent Talents in University (NCET-12-0876), and the 111 Project (grant 111-2-06).
Footnotes
Published ahead of print 1 November 2013
REFERENCES
- 1.Shen F, Ying YB, Li BB, Zheng YF, Hu JG. 2011. Prediction of sugars and acids in Chinese rice wine by mid-infrared spectroscopy. Food Res. Int. 44:1521–1527. 10.1016/j.foodres.2011.03.058 [DOI] [Google Scholar]
- 2.Chen SA, Xu Y. 2010. The influence of yeast strains on the volatile flavour compounds of Chinese rice wine. J. I. Brewing 116:190–196. 10.1002/j.2050-0416.2010.tb00417.x [DOI] [Google Scholar]
- 3.Fu ML, Liu J, Chen QH, Liu XJ, He GQ, Chen JC. 2010. Determination of ethyl carbamate in Chinese yellow rice wine using high-performance liquid chromatography with fluorescence detection. Int. J. Food Sci. Tech. 45:1297–1302. 10.1111/j.1365-2621.2010.02279.x [DOI] [Google Scholar]
- 4.Lu YM, Lu X, Chen XH, Jiang M, Li C, Dong MS. 2007. A survey of biogenic amines in Chinese rice wines. Food Chem. 100:1424–1428. 10.1016/j.foodchem.2005.11.035 [DOI] [Google Scholar]
- 5.Weber JV, Sharypov VI. 2009. Ethyl carbamate in foods and beverages—a review, p 429–452 In Lichtfouse E. (ed), Climate change, intercropping, pest control and beneficial microorganisms, vol 2 Springer, Dordrecht, Netherlands [Google Scholar]
- 6.Forkert PG. 2010. Mechanisms of lung tumorigenesis by ethyl carbamate and vinyl carbamate. Drug Metab. Rev. 42:355–378. 10.3109/03602531003611915 [DOI] [PubMed] [Google Scholar]
- 7.Cadranel JF, Legendre C, Desaint B, Delamarre N, Florent C, Levy VG. 1993. Liver disease from surreptitious administration of urethane. J. Clin. Gastroenterol. 17:52–56. 10.1097/00004836-199307000-00015 [DOI] [PubMed] [Google Scholar]
- 8.Lachenmeier DW. 2007. Consequences of IARC re-evaluation of alcoholic beverage consumption and ethyl carbamate on food control. Dtsch. Lebensmitt. Rundsch. 103:307–311 [Google Scholar]
- 9.Schehl B, Senn T, Lachenmeier DW, Rodicio R, Heinisch JJ. 2007. Contribution of the fermenting yeast strain to ethyl carbamate generation in stone fruit spirits. Appl. Microbiol. Biotechnol. 74:843–850. 10.1007/s00253-006-0736-4 [DOI] [PubMed] [Google Scholar]
- 10.Coulon J, Husnik JI, Inglis DL, van der Merwe GK, Lonvaud A, Erasmus DJ, van Vuuren HJJ. 2006. Metabolic engineering of Saccharomyces cerevisiae to minimize the production of ethyl carbamate in wine. Am. J. Enol. Viticult. 57:113–124 [Google Scholar]
- 11.Andrich L, Esti M, Moresi M. 2009. Urea degradation in model wine solutions by free or immobilized acid urease in a stirred bioreactor. J. Agric. Food Chem. 57:3533–3542. 10.1021/jf803962b [DOI] [PubMed] [Google Scholar]
- 12.Miyagawa K, Sumida M, Nakao M, Harada M, Yamamoto H, Kusumi T, Yoshizawa K, Amachi T, Nakayama T. 1999. Purification, characterization, and application of an acid urease from Arthrobacter mobilis. J. Biotechnol. 68:227–236. 10.1016/S0168-1656(98)00210-7 [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, Du G, Zou H, Fu J, Zhou J, Chen J. 2013. Progress in preventing the accumulation of ethyl carbamate in alcoholic beverages. Trends Food Sci. Technol. 32:97–107. 10.1016/j.tifs.2013.05.009 [DOI] [Google Scholar]
- 14.Monteiro FF, Bisson LF. 1991. Amino acid utilization and urea formation during vinification fermentations. Am. J. Enol. Viticult. 42:199–208 [Google Scholar]
- 15.Magasanik B, Kaiser CA. 2002. Nitrogen regulation in Saccharomyces cerevisiae. Gene 290:1–18. 10.1016/S0378-1119(02)00558-9 [DOI] [PubMed] [Google Scholar]
- 16.Bertram PG, Choi JH, Carvalho J, Ai W, Zeng C, Chan TF, Zheng XF. 2000. Tripartite regulation of Gln3p by TOR, Ure2p, and phosphatases. J. Biol. Chem. 275:35727–35733. 10.1074/jbc.M004235200 [DOI] [PubMed] [Google Scholar]
- 17.Kulkarni AA, Abul-Hamd AT, Rai R, El Berry H, Cooper TG. 2001. Gln3p nuclear localization and interaction with Ure2p in Saccharomyces cerevisiae. J. Biol. Chem. 276:32136–32144. 10.1074/jbc.M104580200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rai R, Tate JJ, Nelson DR, Cooper TG. 2013. gln3 mutations dissociate responses to nitrogen limitation (nitrogen catabolite repression) and rapamycin inhibition of TorC1. J. Biol. Chem. 288:2789–2804. 10.1074/jbc.M112.421826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magasanik B. 2005. The transduction of the nitrogen regulation signal in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 102:16537–16538. 10.1073/pnas.0507116102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godard P, Urrestarazu A, Vissers S, Kontos K, Bontempi G, van Helden J, Andre B. 2007. Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 27:3065–3086. 10.1128/MCB.01084-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gietz RD, Woods RA. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87–96. 10.1016/S0076-6879(02)50957-5 [DOI] [PubMed] [Google Scholar]
- 22.Wach A, Brachat A, Alberti-Segui C, Rebischung C, Philippsen P. 1997. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast 13:1065–1075 [DOI] [PubMed] [Google Scholar]
- 23.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwayama H, Obara S, Morio T, Katoh M, Urushihara H, Tanaka Y. 2002. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res. 30:e2. 10.1093/nar/30.2.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knorst MT, Neubert R, Wohlrab W. 1997. Analytical methods for measuring urea in pharmaceutical formulations. J. Pharm. Biomed. Anal. 15:1627–1632. 10.1016/S0731-7085(96)01978-4 [DOI] [PubMed] [Google Scholar]
- 26.Alcarde AR, de Souza LM, Bortoletto AM. 2012. Ethyl carbamate kinetics in double distillation of sugar cane spirit. J. Inst. Brewing 118:27–31. 10.1002/jib.14 [DOI] [Google Scholar]
- 27.Calull M, Marcé RM, Borrull F. 1992. Determination of carboxylic acids, sugars, glycerol and ethanol in wine and grape must by ion-exchange high-performance liquid chromatography with refractive index detection. J. Chromatogr. 590:215–222. 10.1016/0021-9673(92)85384-6 [DOI] [PubMed] [Google Scholar]
- 28.Cigic IK, Vodosek TV, Kosmerl T, Strlic M. 2008. Amino acid quantification in the presence of sugars using HPLC and pre-column derivatization with 3-MPA/OPA and FMOC-Cl. Acta Chim. Slov. 55:660–664 [Google Scholar]
- 29.Chen EJ, Kaiser CA. 2002. Amino acids regulate the intracellular trafficking of the general amino acid permease of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 99:14837–14842. 10.1073/pnas.232591899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tate JJ, Cooper TG. 2007. Stress-responsive Gln3 localization in Saccharomyces cerevisiae is separable from and can overwhelm nitrogen source regulation. J. Biol. Chem. 282:18467–18480. 10.1074/jbc.M609550200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin YS, Ni H, Laplaza JM, Jeffries TW. 2003. Optimal growth and ethanol production from xylose by recombinant Saccharomyces cerevisiae require moderate D-xylulokinase activity. Appl. Environ. Microbiol. 69:495–503. 10.1128/AEM.69.1.495-503.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dahabieh MS, Husnik JI, Van Vuuren HJ. 2010. Functional enhancement of sake yeast strains to minimize the production of ethyl carbamate in sake wine. J. Appl. Microbiol. 109:963–973. 10.1111/j.1365-2672.2010.04723.x [DOI] [PubMed] [Google Scholar]
- 33.Park H, Shin M, Woo I. 2001. Antisense-mediated inhibition of arginase (CAR1) gene expression in Saccharomyces cerevisiae. J. Biosci. Bioeng. 92:481–484. 10.1263/jbb.92.481 [DOI] [PubMed] [Google Scholar]
- 34.Dahabieh MS, Husnik JI, van Vuuren HJJ. 2009. Functional expression of the DUR3 gene in a wine yeast strain to minimize ethyl carbamate in chardonnay wine. Am. J. Enol. Viticult. 60:537–541 [Google Scholar]