

Abstract
The strains of Xanthomonas axonopodis pv. citri, the causative agent of citrus canker, are historically classified based on bacteriophage (phage) sensitivity. Nearly all X. axonopodis pv. citri strains isolated from different regions in Japan are lysed by either phage Cp1 or Cp2; Cp1-sensitive (Cp1s) strains have been observed to be resistant to Cp2 (Cp2r) and vice versa. In this study, genomic and molecular characterization was performed for the typing agents Cp1 and Cp2. Morphologically, Cp1 belongs to the Siphoviridae. Genomic analysis revealed that its genome comprises 43,870-bp double-stranded DNA (dsDNA), with 10-bp 3′-extruding cohesive ends, and contains 48 open reading frames. The genomic organization was similar to that of Xanthomonas phage phiL7, but it lacked a group I intron in the DNA polymerase gene. Cp2 resembles morphologically Escherichia coli T7-like phages of Podoviridae. The 42,963-bp linear dsDNA genome of Cp2 contained terminal repeats. The Cp2 genomic sequence has 40 open reading frames, many of which did not show detectable homologs in the current databases. By proteomic analysis, a gene cluster encoding structural proteins corresponding to the class III module of T7-like phages was identified on the Cp2 genome. Therefore, Cp1 and Cp2 were found to belong to completely different virus groups. In addition, we found that Cp1 and Cp2 use different molecules on the host cell surface as phage receptors and that host selection of X. axonopodis pv. citri strains by Cp1 and Cp2 is not determined at the initial stage by binding to receptors.
INTRODUCTION
Citrus canker, caused by Xanthomonas axonopodis pv. citri (syn., Xanthomonas campestris pv. citri or Xanthomonas citri), is a widespread disease in citrus-producing areas of the tropical and subtropical world (1, 2). Different types of citrus canker, corresponding to different pathotypes of X. axonopodis pv. citri, have been reported (3). The Asiatic type, caused by X. axonopodis pv. citri pathotype A, is the most widespread and the most economically important citrus canker. The host range of pathotype A strains is wider than that of the other pathotypes, including most citrus varieties (3). X. axonopodis pv. citri pathotype A strains are separated into two groups based on their sensitivity to phages Cp1 and Cp2 (4, 5). Phage typing with Cp1 and Cp2 was first demonstrated by Wakimoto (4). Wakimoto found that nearly all strains isolated from different regions in Japan were lysed by either Cp1 or Cp2 and that Cp1-sensitive (Cp1s) strains were resistant to Cp2 (Cp2r) and vice versa. Cp1 and Cp2 phages were morphologically different: Cp1 showed a head-tail structure, whereas Cp2 consisted of a polyhedral head without tail (4). In a larger survey, Obata found that Cp1r/Cp2s strains predominated in major citrus-producing regions in Japan, with the exception of Hiroshima Prefecture, where Cp1s/Cp2r strains predominated (5). Two different strain types can occur in a mixture on the same single leaf of a tree, but one single lesion usually consists of a single strain.
Notably, the sensitivity of X. axonopodis pv. citri strains to Cp1 and Cp2 is associated with differences in their physiological features and canker aggressiveness. X. axonopodis pv. citri strains that are Cp1s/Cp2r can assimilate mannitol, while Cp1r/Cp2s strains cannot (6). All strains that are Cp1r/Cp2s are canker aggressive to the citrus variety “Otachibana,” whereas all the strains with Cp1s/Cp2r are weakly aggressive (7). The Cp1r/Cp2s strains generate a 1.8-kbp-specific fragment by repetitive sequence-based PCR (rep-PCR) (8) using enterobacterial repetitive intergenic consensus (ERIC) primers. The 1.8-kbp band corresponds to a region encompassing XAC1661 (Isxac3 transposase) and XAC1662 (repA) within an insertion element in the genomic sequence of X. axonopodis pv. citri strain 306 (9). Most strains with Cp1s/Cp2r contain hssB3.0, a member of the avrBs3 and pthA (avirulence and pathogenicity) gene family (10, 11), which is responsible for the suppression of virulence on a Citrus grandis cultivar; however, Cp1r/Cp2s strains lack this gene (12). These results suggest some relatedness between Cp1/Cp2 sensitivity and the virulence and pathogenic features of X. axonopodis pv. citri strains.
In contrast to the large contribution toward characterization of host strains, very little information is available about the nature or identity of phages Cp1 and Cp2. Concerning Cp1 and Cp2, the following issues are of particular interest: (i) the virological identification and phylogenetic relationships of these phages, (ii) the origin of the above-mentioned 1.8-kbp sequence on the host genome and its possible association with a phage sequence, (iii) hssB3.0 and its possible association with a phage sequence, and (iv) the molecular mechanism of host selection by these phages. As a first step toward exploring these issues, the present study performed genomic and molecular characterization of Cp1 and Cp2.
MATERIALS AND METHODS
Bacterial strains and phages.
Ministry of Agriculture, Forestry and Fisheries (MAFF) strains of X. axonopodis pv. citri were obtained from the National Institute of Agrobiological Sciences, Japan. Strain KC33 (7) was obtained from the National Institute of Fruit Tree Science, National Agriculture and Food Research Organization (NAFRO), Japan. Their origins and sensitivity to Cp1 and Cp2 are listed in Table 1. Bacteriophages Cp1 and Cp2 (4, 5) were obtained from the Yokohama Plant Protection Station, Japan. Strains MAFF 301080 and MAFF 673010 were used as hosts for routine propagation of Cp1 and Cp2, respectively. Bacterial cells were cultured in nutrient broth (NB) medium (BBL, Becton, Dickinson and Co., Cockeysville, MD, USA) at 28°C with shaking at 200 to 300 rpm. An overnight culture of bacterial cells grown in NB was diluted 100-fold with 100 ml fresh NB in a 500-ml flask. To collect sufficient phage particles, 1 liter of bacterial culture (10 × 100-ml cultures) was grown. When the cultures reached 0.2 units of optical density at 600 nm (OD600), the phages were added at a multiplicity of infection (MOI) of 0.1. After further growth for 9 to 18 h, the cells were removed by centrifugation with an R12A2 rotor in a Hitachi heavy ion medical accelerator (HIMAC) CR21E centrifuge (Hitachi Koki Co. Ltd., Tokyo, Japan), at 8,000 × g for 15 min at 4°C. The supernatant was passed through a 0.45-μm membrane filter, and phage particles were precipitated by centrifugation with a P28S rotor in a Hitachi XII100β centrifuge at 40,000 × g for 1 h at 4°C and dissolved in SM buffer (50 mM Tris-HCl at pH 7.5, 100 mM NaCl, 10 mM MgSO4, and 0.01% gelatin). Purified phages were stored at 4°C until use. Bacteriophage particles purified by CsCl gradient ultracentrifugation (with a P28S rotor in a Hitachi XII100β ultracentrifuge) (13) were stained with Na-phosphotungstate before observation in a Hitachi H600A electron microscope, according to the method of Dykstra (14). λ phage particles were used as an internal standard marker for size determination. For host range determination, standard plaque-forming assays (15) or lysis zone formation spot tests (16) were performed.
TABLE 1.
Bacterial strains and bacteriophages used in this studya
| Strain | Host (Citrus species) | Phage typeb | Source |
|---|---|---|---|
| X. axonopodis pv. citri | |||
| MAFF 301077 | C. limon | Cp1s/Cp2r | NIASc |
| MAFF 301080 | C. sinensis | Cp1s/Cp2r | NIAS |
| 301080 R1 | Cp1r/Cp2r | This study | |
| MAFF 311130 | C. iyo | Cp1r/Cp2r | NIAS |
| MAFF 302102 | Citrus sp. | Cp1r/Cp2s | NIAS |
| MAFF 673001 | C. natsudaidai | Cp1r/Cp2s | NIAS |
| MAFF 673010 | Citrus sp. | Cp1r/Cp2s | NIAS |
| 673010 R2 | Cp1r/Cp2r | This study | |
| MAFF 673011 | C. limon | Cp1s/Cp2r | NIAS |
| MAFF 673013 | Citrus sp. | CP1s/CP2r | NIAS |
| MAFF 673018 | Citrus sp. | Cp1r/Cp2s | NIAS |
| MAFF 673021 | C. limon | Cp1r/Cp2s | NIAS |
| KC33 | C. iyo | Cp1s/Cp2s | Shiotani et al. (12) |
| Phages | |||
| Cp1 | Wakimoto (4) | ||
| Cp2 | Wakimoto (4) | ||
All strains originated in Japan.
Sensitivity to phages CP1 and CP2; s, sensitive; r, resistant.
NIAS, National Institute of Agrobiological Sciences, Japan.
Single-step growth experiment.
Single-step growth experiments were performed as previously described (17, 18), with some modifications. Strains MAFF 301080 and MAFF 673010 were used as hosts for Cp1 and Cp2, respectively. Cells (0.1 U of OD600) were harvested by centrifugation and resuspended in fresh NB (ca. 1 × 108 CFU/ml) to a final culture volume of 10 ml. Phage was added at an MOI of 1.0 and allowed to adsorb for 10 min at 28°C. After centrifugation and resuspending in the initial volume of NB with decimal dilution to a final volume of 10 ml, the cells were incubated at 28°C. Samples were taken at intervals (every 10 min up to 3.5 h for Cp1 and every 30 min up to 5 h for Cp2), and the titers were determined by the double-layered agar plate method.
Phage adsorption test.
Phage adsorption was assayed as follows: when fresh bacterial cultures (10 ml) reached 0.1 unit of OD600, the phage (10 μl) was added at an MOI of 0.1. After incubation for 10 min at 28°C, the cells were removed by centrifugation with an R12A2 rotor in a Hitachi HIMAC CR21E centrifuge at 8,000 × g for 10 min at 4°C. The supernatant was subjected to a plaque assay, where strains MAFF 301080 and MAFF 673010 were used as the hosts for Cp1 and Cp2, respectively. Escherichia coli JM109 was used as a negative control in phage adsorption.
DNA manipulation and sequencing.
Standard molecular biological techniques for DNA isolation, digestion with restriction enzymes and other nucleases, and construction of recombinant DNAs were followed, according to Sambrook and Russell (13). Phage DNA was isolated from the purified phage particles by phenol extraction. For genome size determination, the purified phage particles were embedded in 0.7% low-melting-point agarose (InCert agarose; FMC Corp., Philadelphia, PA, USA) and, after treatment with proteinase K (1 mg/ml; Merck Ltd., Tokyo, Japan) and 1% Sarkosyl, subjected to pulsed-field gel electrophoresis (PFGE) in a Chef Mapper electrophoresis apparatus (Bio-Rad Lab., Hercules, CA, USA) according to the method of Higashiyama and Yamada (19). Shotgun sequencing was performed at Hokkaido System Science Co., Ltd. (Sapporo, Japan), using the Roche GS Junior Sequence system. The draft assembly of the obtained sequences was assembled using GS De novo Assembler v2.6. The analyzed sequences corresponded to 94 and 40 times the final genome sizes of Cp1 (43,860 bp) and Cp2 (42,963 bp), respectively. Potential open reading frames (ORFs) larger than 150 bp (50 codons) were identified using Glimmer (20) and GeneMark. Homology searches were performed using BLAST/RPS-BLAST (21) against the UniProt sequence database (22) and the NCBI/CDD database (23), using an E value lower than e−4 as a cutoff for notable similarity. Multiple-sequence alignments were generated using the DNASIS program (version 3.6; Hitachi Software Engineering, Co., Ltd., Tokyo, Japan). For phylogenetic analysis of RNA polymerase (RNAP) proteins, the unrooted dendrogram was constructed with the Treeview tool using the maximum likelihood method based on a complete protein sequence alignment of RNAP proteins from other phages using ClustalX. The Cp1 cohesive ends (cos) sequence was determined as follows. A 6.8-kbp PstI fragment of Cp1 DNA was dissociated into two fragments (4.3 and 2.5 kbp) after heating at 70°C for 15 min. The dissociated bands were recovered from the agarose gel and treated with T4 DNA polymerase to form blunt ends. The nucleotide sequences of these bands were determined. By comparing the nucleotide sequences with each other and with the Cp1 genomic sequence, the cos sequence was determined according to the method of Fujiwara et al. (24).
Southern and dot blot hybridization.
Genomic DNA from bacterial cells was prepared by the minipreparation method according to Ausubel et al. (25). After digestion with various restriction enzymes (EcoRI, EcoRV, HindIII, and HincII), DNA fragments were separated by agarose gel electrophoresis, blotted onto a nylon membrane (Biodyne; Pall Gelman Laboratory, Closter, NJ, USA), hybridized with probes (the entire Cp1 DNA by combining all the HincII fragments and the entire Cp2 DNA with all the HincII fragments), labeled with fluorescein (Gene Images Random Prime labeling kit; Amersham Biosciences, Uppsala, Sweden), and detected with a Gene Images CDP-Star detection module (Amersham Biosciences). Hybridization was performed in buffer containing 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% SDS, 5% liquid block, and 5% dextran sulfate for 16 h at 65°C. The filter was washed at 60°C in 1× SSC and 0.1% SDS for 15 min and then in 0.5% SSC and 0.1% SDS for 15 min with agitation, according to the manufacturer's protocol. The hybridization signals were detected by exposing the filter onto an X-ray film (RX-U; Fuji Film, Tokyo, Japan).
SDS-PAGE and LC-MS/MS analysis.
Purified phage particles were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (10 to 12% [wt/vol] polyacrylamide) according to Laemmli (26). Protein bands were visualized by staining the gel with Coomassie brilliant blue, excised from the gel, digested with trypsin, and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) (LTQ Orbitrap XL; Thermo Fisher Scientific, Osaka, Japan) analysis at the Natural Science Center for Basic Research and Development, Hiroshima University.
Staining of bacterial cells by SYBR gold-labeled phages.
Phage labeling and observation of phage-treated bacterial cells were performed according to Mosier-Boss et al. (27). To 100 ml of the phage lysate, 10 μl of 104× SYBR gold (Molecular Probes, Inc., Eugene, OR, USA) in dimethyl sulfoxide (DMSO) was added. After 10 min, the labeled phage particles were precipitated by centrifugation with a P28S rotor in a Hitachi XII100β centrifuge at 40,000 × g for 1 h at 4°C. Three washes using 1× PBS were done to ensure the removal of excess SYBR gold. A 1-ml sample of an overnight culture of X. axonopodis pv. citri was mixed with 4 ml NB and allowed to grow until an OD600 of 0.5 was reached. To a 10-μl sample of the bacterial culture was added 10 μl SYBR gold-labeled phage, and the mixture was incubated for 10 to 60 min. As a control, a culture of E. coli JM109 was treated in the same way. After fixation of the mixture with 30 μl 4% paraformaldehyde in PBS, 5 ml double-distilled water (ddH2O) was added, and the mixture was filtered through a membrane filter (0.2-μm pore size, Steradisc; Krabo, Osaka, Japan). The bacterial cells were observed under a fluorescence microscope system with filter sets (Olympus BH2 fluorescence microscope; Olympus, Tokyo, Japan). Microscopic images were recorded with a charge-coupled-device (CCD) camera (Kyence VB-6010; Osaka, Japan).
Nucleotide sequence accession numbers.
The sequence data for the Cp1 and Cp2 genomes have been deposited in the DDBJ database under accession no. AB720063 and AB720064, respectively.
RESULTS AND DISCUSSION
Cp1 and Cp2 belong to different virus families.
Morphology indicates that phages Cp1 and Cp2 belong to different virus families: Cp1 as a member of Siphoviridae and Cp2 as a member of Podoviridae. The purified particles of Cp1 and Cp2 were negatively stained and examined by transmission electron microscopy. Cp1 particles had an icosahedral capsid of 60 ± 5 nm in diameter with a long noncontractile tail of 135 ± 10 nm long by 12 ± 2 nm wide (see Fig. S1 in the supplemental material). This morphology was almost the same (though in smaller dimensions) as that reported preliminarily for CP1 particles replicated in strain N6101 as a host (28), indicating that, morphologically, Cp1 belongs to the family Siphoviridae. In contrast, Cp2 particles showed an icosahedral capsid of 60 ± 5 nm in diameter with a short tail of 15 ± 5 nm long (see Fig. S1 in the supplemental material), indicating that Cp2 has a structure typical of members of the family Podoviridae. In a preliminary work, this phage was reported to have larger polyhedral particles without a tail (28). These results raised the question of whether Cp1 and Cp2 are related to each other in infection and replication in host strains.
Comparison of infection cycles of Cp1 and Cp2.
Both Cp1 and Cp2 form clear plaques with various X. axonopodis pv. citri strains, including those shown in Table 1, as hosts (4, 5). The infection cycle of each phage was characterized by a single-step growth experiment with appropriate hosts. The growth curves are shown in Fig. 1. In the case of Cp1 replication, the latent period was ∼60 min, followed by a rise period of 20 to 30 min, giving an entire cycle of 80 to 90 min. The average burst size was 20 PFU per infected cell. For Cp2 replication, the latent period was ∼90 min, with a 60-min rise period, taking 150 to 180 min for a single growth cycle. The burst size was approximately 100 PFU/cell. These results showed that Cp1 infected and replicated more rapidly than Cp2, but the burst size was much smaller than that of Cp2. These results contrasted with the observation that more than 600 progeny phages were formed in an infected bacterial cell for both Cp1 and Cp2, as detected by electron microscopy (28). The burst size of a phage may depend on the host strain, culture medium, culture conditions, cell age, and MOI (29). The net ratio of infective to noninfective phage particles in the progeny population and the frequent readsorption of phage particles onto cell debris may partly explain this discrepancy.
FIG 1.
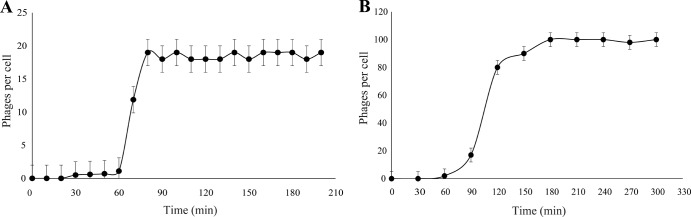
Single-step growth curves for Cp1 (A) and Cp2 (B). The PFU per infected cell in cultures of MAFF 301080 for Cp1 and MAFF 673010 for Cp2 at different times postinfection are shown. Samples were taken at intervals (every 10 min up to 3.5 h for Cp1 and every 30 min up to 5 h for Cp2). Error bars indicate standard deviations (n = 3).
Genomic analyses of Cp1: gene organization and homology to other phages.
The Cp1 genome was a linear double-stranded (ds) DNA of approximately 44 kbp, as determined by PFGE (data not shown). The nucleotide sequence of the Cp1 genome was determined using shotgun sequencing of DNA purified from phage particles. Sequences were assembled into a circular contig of 43,870 bp, suggesting the presence of terminal repeats. The exact termini of the Cp1 genome were determined to have a 10-nucleotide (nt) 3′-protruding cos site (5′-CCAGTTGTCT, corresponding to positions 43,861 to 43,870) at either end.
The Cp1 genome had a G+C content of 53.3%; this value was significantly lower than that of the host genome (e.g., 64.7% for strain 306, accession no. NC_003919). When the databases were searched using BLAST and BLASTX programs for sequences homologous to the Cp1 DNA sequence, extensive homologies were detected in the genome sequence of X. campestris phage phiL7 (accession no. EU717894), X. oryzae phage OP1 (accession no. AP008979), and Xanthomonas phage Xp10 (accession no. AY299121). All these phages are siphoviruses infecting species of Xanthomonas, which is consistent with the Cp1 morphological features. An extended colinearity of the nucleotide sequence homology, with several short interspersed divergent islands, was detected throughout almost the entire genomic region between Cp1 and phiL7, which gave the highest similarity score (see Fig. S2 in the supplemental material).
Forty-eight potential ORFs comprising 50 or more codons, starting with ATG or GTG as the initiation codon, and with a Shine-Dalgarno ribosome-binding sequence preceding the initiation codon, were detected in the Cp1 genome. The genome was divided into left and right arms by the ORF29 and ORF30 intergenic region, with genes on the two arms transcribed convergently (Fig. 2). The 48 deduced proteins included (i) 27 proteins that had database homologs of known function, including phage structural proteins, DNA packaging proteins, and proteins involved in DNA replication, transcription, and lysis; (ii) 19 hypothetical proteins in the databases, including many phiL7 proteins; and (iii) 2 proteins with no similarities in the databases (see Table S1 in the supplemental material). The gene organization of Cp1 was compared with that of phiL7 (Fig. 2). This comparison showed that p21, p25, p26, p30-p34, p36-p38, p42, p45, p49-p52, p55, and p56 of phiL7, most of which were without similarity in the databases, were missing from Cp1, and instead orf3, orf5, orf30-orf32, orf34, orf44, and orf45 were inserted or replaced in the Cp1 genome. Some of these genes, such as p45, p49, and p56 of phiL7 and orf3, orf5, and orf30 of Cp1, encoded HNH endonuclease, GIY-YIG endonuclease, or group I intron endonuclease, suggesting their involvement in gene rearrangements, especially horizontal gene movements. However, the group I intron inserted in the DNA polymerase gene (p44-p46) in phiL7 (30) was missing from the corresponding region of the Cp1 gene (orf40).
FIG 2.
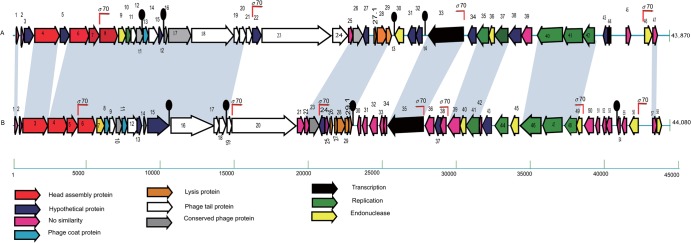
Genomic organization of phages Cp1 (A) and phiL7 (B). Colored arrows indicate the directions and categories of the genes. Broken arrows and black knobs indicate the σ70-type promoters and predicted terminators for transcription, respectively. “No similarity” was determined using an E value lower than e−4 as a cutoff for notable similarity.
Interestingly, Cp1 encodes a viral RNA polymerase (RNAP; ORF33) (a single-subunit RNAP similar to the T7-type enzymes). Sequence analysis showed that Cp1 ORF33 was 71.4% identical to phiL7 p35 (ACE75775.1) and 31.5% identical to T7 RNAP (NP_041960) (Fig. 3A). Cp1 ORF33 also showed 40 to 50% amino acid sequence identity with RNAPs of other Xanthomonas phages, such as Xp10 (AAP58699.1), OP1 (BAE72738.1), and Xop411 (ABK00180.1) (Fig. 3A). All important amino acid residues identified in T7 RNAP for structure and function (31, 32) were conserved among these RNAPs. Their phylogenetic relationship is shown in Fig. 3B. The siphovirus Xp10 was shown to rely on both host and phage RNAPs, and the shift from host to phage RNAP is regulated by phage protein p7 (33). Xp10 p7 (73 amino acids [aa]) is encoded by gene p45L, located after the replication module of the Xp10 genome. A similar regulation may work in Cp1 because ORF44, encoding 66 aa with a sequence similarity to inhibitors of transcriptional initiators and terminators (33% identical to Xp10 p45L; AAP58713.1), was located at the corresponding position on Cp1 DNA (Fig. 2). Like Xp10, the protein encoded by ORF44 may function to inhibit host RNAP and act as an antiterminator, allowing RNAP to pass through the intrinsic terminator for expression of the late genes (33).
FIG 3.

Comparison of amino acid sequences of RNA polymerases (RNAP) encoded by Xanthomonas phages. (A) The amino acid sequence of Cp1 ORF33 (AB720063) was aligned with those of phiL7 p35 (ACE75775.1)., Xop411 p32 (ABK00180.1), Xp10 32L (AAP58699.1), OP1 ORF33 (BAE72738.1), and coliphage T7 RNAP (NP_041960.1) using ClustalX. The ClustalX coloring scheme depends on both the residue type and the pattern of conservation within a column (http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/multalignviewer/cxcolor.html). Conservation scores are drawn below the alignment. (B) The unrooted dendrogram was constructed with the Treeview tool using the maximum likelihood method based on a complete protein sequence alignment of RNAP proteins from other phages.
Furthermore, we detected a homolog of the OP1 tail fiber protein (Tfb, OP1-ORF25), which was thought to be involved in host range determination, mediated by variation in the combination of repetitive motifs at the N terminus (34).
Genomic analyses of Cp2: gene organization and homology to other phages.
The Cp2 genome was also a linear dsDNA of approximately 43.0 kbp, based on PFGE analysis (data not shown). The Cp2 DNA isolated from phage particles was subjected to shotgun sequencing. Sequences were assembled into a circular contig of 42,963 bp, also suggesting the presence of terminal repeats. The precise sequence of the repeat was not determined. The Cp2 genome had a G+C content of 66.7%, comparable with that of the host genome (ca. 64.7%). To find homologous sequences, nucleotide sequences from Cp2 were used to search sequence databases. Short-ranged homologies were found in the sequences of Azospirillum brasilense Sp245 plasmid (accession no. HE577328; E value, 2e−11 [84 bit]) and Burkholderia pseudomallei bacteriophage phi1026b (accession no. AY453853; E value, 5e−09 [76 bit]). Interestingly, a sequence in the genome of X. axonopodis pv. citri strain 306 also showed some homology to the Cp2 genome (accession no. AE008923; E value, 8e−08 [72 bit]). The homologous sequence corresponded to a single gene (orf32 of Cp2, encoding a single-strand [ss] binding protein). However, the 1.8-kbp region, including XAC1661 (Isxac3 transposase) and XAC1662 (repA) of strain 306, which was specifically amplified by rep-PCR for Cp1r/Cp2s strains (9), did not show any sequence homology with the Cp2 genome. Forty potential ORFs were identified in the Cp2 genome. The 40 deduced proteins included (i) 20 proteins that had database homologs of known functions, including phage structural proteins, DNA processing proteins, and lysis proteins; (ii) 16 hypothetical proteins showing marginal similarities with unknown proteins of various origins; and (iii) four proteins with no similarities in the databases (see Table S2 in the supplemental material). The ORF map is shown in Fig. 4. Morphologically, Cp2 belongs to the family Podoviridae. The genome of coliphage T7, the representative of T7-like phages of the Podoviridae, generally consists of three functional gene clusters: one for early functions (class I), one for DNA metabolism (class II), and the other for structural proteins and virion assembly (class III) (35). For the Cp2 genome, the assignment of classes I to III was difficult because of the lack of sufficient information about each gene, especially about key genes, such as those encoding RNAP, DNA metabolism, and structural proteins. After identifying genes for structural proteins, we tentatively assigned the three functional modules according to the T7 gene arrangement (Fig. 4). In this Cp2 gene arrangement, the putative class I module contained ORF32 (with similarity to ssDNA-binding proteins), ORF33 (HNH endonuclease), ORF34 (YqaJ-like recombinase), and ORF35 (ERF family protein) (see Table S2 in the supplemental material). The Cp2 putative class II region contained ORF39 (with a marginal similarity to DNA polymerase gamma-1 subunit), ORF40 (pyocin-like), ORF3 (Holliday junction resolvase), and ORF5 (lysozyme) (see Table S2 in the supplemental material). The details for class III, consisting of ORF13 to ORF29, are described below.
FIG 4.

Genomic organization of phages of Cp2 (A) and Escherichia coli T7 (B). Arrows indicate the sizes and directions of the genes. The typical podovirus genome represented by T7 phage consists of three functional modules: class I, class II, and class III, as shown in panel B (35). Putative class I (white), class II (white), and class III (red) modules for Cp2 are shown in panel A.
We could not find a gene for RNAP in the Cp2 genome. In general, T7-like podoviruses use phage-encoded RNAP for predominant expression of phage genes (36). T7-RNAP encoded in the class I module (early expressed by host RNAP) specifically recognizes phage promoters of class II and class III genes for the shift of gene expression. However, several podoviruses that have the RNAP gene in the class II module showed a high dependency on the host RNAP for the expression of phage genes (18, 37). Marine phages VpV262 and SI01, which share extensive homology with T7, lack a phage RNAP (38), indicating absolute dependency on the host RNAP for the expression of phage genes. Therefore, the lack of an RNAP gene in the Cp2 genome is not surprising. Searching promoter sequences throughout the Cp2 genome using ORFinder revealed only typical σ70 promoters (data not shown). Notably, the siphovirus Cp1 contained an RNAP gene that is phylogenetically related to RNAP genes of T7-like phages. In the genomes of siphoviruses such as Xanthomonas phage Xp10, as well as Xop411 and OP1, a cluster of λ-like structural genes is connected to other gene clusters arranged in the reverse orientation, such as the T7-like class II and class I genes (39).
Proteomic analyses of Cp1 and Cp2 virions.
Using SDS-PAGE in conjunction with LC-MS/MS, we identified virion proteins of Cp1 and Cp2. In the case of Cp1, at least 10 proteins, ranging from 13 kDa to ca. 120 kDa, were separated by SDS-PAGE (Fig. 5A). All these proteins were recovered, in-gel digested, and subjected to mass spectrometric analysis. The results are shown on the right side of Fig. 5A. The identified proteins include p5 (unknown protein), p6 (head portal protein), p8 (head protein), p14 (major tail protein), p18 (tail length tape major protein), p23 (tail fiber protein), p28 (lysozyme), p29 (hypothetical protein), and p31 (unknown protein). We observed oligomerization of two proteins. The major head protein (p8; calculated to be 41.7 kDa) was detected at a position corresponding to ∼200 kDa by SDS-PAGE, suggesting oligomers consisting of five subunits. In addition, p5 (unknown protein) has a calculated molecular mass of 23.71 kDa but was observed at a position corresponding to 60 to 70 kDa, suggesting that this protein exists as a trimer. These subunits may be covalently linked in the phage particles. For other proteins, the observed size was close to the calculated size. In the Cp1 genomic analysis, we detected a homolog of the OP1 tail fiber protein (Tfb, OP1-ORF25), which was thought to be involved in host range determination, mediated by variation in the combination of repetitive motifs at the N terminus (33). However, the corresponding Cp1 ORF (ORF24) was much smaller (358 aa) than OP1-ORF25 (431 aa) and the similarity was limited to 42 aa at the N terminus (67% identity) without any following repetitive motifs. Indeed, this protein was not detected among the Cp1 structural proteins (Fig. 5A). It is unknown whether this protein is involved in host range determination. Instead, a large protein corresponding to ORF23 (1,574 aa residues) was detected (Fig. 5A), which showed significantly high similarity to tail fiber proteins of several phages: 22R of Xp10 (accession no. Q7Y5J5), p22 of Xop411 (accession no. YP_001285691) and p20 of phiL7 (accession no. YP_002922634). This protein might be involved in determining the host range, as suggested by Lee et al. (30, 40).
FIG 5.

Proteomic analysis of Cp1 (A) and Cp2 (B) particles. Proteins of purified phage particles were separated in 10% (wt/vol) polyacrylamide gel by SDS-PAGE and stained with Coomassie brilliant blue. The protein bands were recovered, digested in gel, and subjected to LC-MS/MS analysis. On the right are the descriptions of the genes, their deduced molecular sizes based on the ORF sequences, and their possible functions. Positions of size markers are shown on the left. For some protein bands, possible oligomerization of the phage protein was observed.
For Cp2 virions, at least 13 proteins, ranging from 16 kDa to ca. 300 kDa, were separated by SDS-PAGE (Fig. 5B). All of these proteins were recovered and subjected to mass spectrometric analysis as above. The results are shown on the right side of Fig. 5B. The proteins identified were from the cluster of ORFs 13 to 28 on the Cp2 map (Fig. 4), giving a putative structural module (class III) of T7-like phages. From this cluster, p14 (endoproteinase), p17 (unknown protein), and p21 (hypothetical protein) were not detected. The identified proteins included p13 (head-tail connecting protein), p15 (major capsid protein), p18 (tail tubular protein A), p23 (structural lysozyme), p24 (lysin-like protein), p25 (end-tail-spike protein), P26 (LysM domain protein), p27 (tail fiber protein), and p28 (tail fiber protein). Among these, p23 (structural lysozyme; calculated to be 79 kDa) gave three ladder bands around 80 to 130 kDa, which suggested processing and oligomerization of this protein.
Host selection by Cp1 and Cp2.
As described above, phages Cp1 and Cp2, used practically for phage typing of X. axonopodis pv. citri strains, were found to belong to completely different virus groups. This raises the question of how these phages discriminate host strains. The initial and essential event for a phage to succeed in infection is attachment of the phage particles to the host surface receptors. Therefore, differential adsorption of Cp1 and Cp2 to host cells was examined, according to the method described in Materials and Methods. The results shown in Table 2 indicated that both Cp1s/Cp2r and Cp1r/Cp2s strains (MAFF 301080 and MAFF 673010, respectively) adsorbed Cp1 and Cp2 efficiently. Even a Cp1r/Cp2r strain (MAFF 31130) also adsorbed Cp1 and Cp2 almost equally. To further investigate the relationship between Cp1 and Cp2 infection, we isolated resistant mutants from the host strains. When a spontaneous mutant showing Cp1r from MAFF 301080 was subjected to phage adsorption assay, it did not adsorb Cp1 but did absorb Cp2, as did its wild type (Table 2). This mutant also showed Cp2r. In the same way, a spontaneous mutant showing Cp1r/Cp2r from MAFF 673010 did not adsorb Cp2 but did absorb Cp1 (Table 2). Moreover, when cells of MAFF 301080 (Cp1s/Cp2r) were first treated with Cp2 particles at an MOI of 5 and then subjected to plaque assay with Cp1, the number of plaques that appeared were almost the same as that with untreated cells (data not shown). Similar results were obtained for MAFF 673010 (Cp1r/Cp2s) cells with pretreated with Cp1 and assayed for Cp2 infection. Taken together, these results indicated that Cp1 and Cp2 use different molecules on the host cell surface as phage receptors and that discrimination of strains by Cp1 and Cp2 is not at the initial stage by binding to receptors, but at some stage(s) afterwards. Another question arose as to what happens to the phage DNA after cell attachment in the case of nonhost strains. To answer this question, SYBR gold-labeled phages were added to cells of X. axonopodis pv. citri strains and the movement of SYBR gold-labeled phage DNA was monitored. SYBR gold-labeled phage attached to host cells (observed after 10 to 20 min postinfection), and bacterial cells were not stained at 10 min postinfection (Fig. 6A and B, left panels), but some cells became stained at 20 min postinfection, possibly by injection of phage DNA (Fig. 6AB, right panels). As shown in Fig. 6C and D, after 30 min postinfection, Cp1 DNA seemed to be injected into most cells of MAFF 673010, MAFF 31130, and MAFF 301080. Similarly, Cp2 DNA seemed to be injected into most cells of MAFF 301080, MAFF 31130, and MAFF 673010. With cells of E. coli as the control, Cp1 and Cp2 did not attach to the cells and no injection of DNA occurred (data not shown). Once the phage DNA was introduced into cells, it was retained stably for a certain period. In the case of resistant strains, cell growth continued after phage addition. Host restriction/ modification systems may contribute to this host selection, but its importance is not clear because Cp1 and Cp2 progenies produced from strain KC33 (Cp1s/Cp2s) showed exactly the same host range as their original phages (data not shown). These results suggested that host selection by Cp1 and Cp2 may occur at or after immediate early expression of phage genes.
TABLE 2.
Adsorption of Cp1 and Cp2 to bacterial strains
| Strain | Phage type | Phage adsorption (%) |
|
|---|---|---|---|
| Cp1 | Cp2 | ||
| X. axonopodis pv. citri | |||
| MAFF 301080 | Cp1s/Cp2r | 100 | >99 |
| 301080 R1 | Cp1r/Cp2r | 0 | >99 |
| MAFF 673010 | Cp1r/Cp2s | >99 | 100 |
| 673010 R2 | Cp1r/Cp2r | >99 | 0 |
| MAFF 31130 | Cp1r/Cp2r | >99 | >99 |
| E. coli JM109 | 0 | 0 | |
FIG 6.

Attachment of SYBR gold-labeled phage particles to bacterial cells and staining of the cells by possible injection of phage DNA. (A) Cp1 particles were added to cells of host strain MAFF 301080; (B) Cp2 particles were added to cells of host strain MAFF 673010. Cells from panels A and B were observed under a fluorescence microscope at 10 min (MOI = 1, left panel) and 20 min (MOI = 10, right panel) postinfection. The attachment of the phage particles to the bacterial cell surface is shown. Phage particles appear as tiny spots (arrows). At 10 min postinfection, bacteria cell were not stained (bright and dark fields), while cells became stained at 20 min postinfection (dark field). Cp1 (C) and Cp2 (D) particles were also added to cells of MAFF 301080 (Cp1s/Cp2r), MAFF 673010 (Cp1r/Cp2s), and MAFF 311130 (Cp1r/Cp2r). Cells were observed under a fluorescence microscope at 30 min postinfection. Bacterial cells were stained by SYBR gold, indicating injection of phage DNA into the Cp1s/Cp2r or Cp1r/Cp2s as well as Cp1r/Cp2r cells. Bar, 10 μm.
In the genome of hosts such as strain 306 (accession no. NC_603919), there are no sequence elements that showed significant homology with Cp1 or Cp2 sequences. The 1.8-kbp region containing a transposon that was specifically amplified by rep-PCR from genomic DNA of Cp1r/Cp2s strains (9) and the region containing avrBs3 and pthA, varying between Cp1s/Cp2r and Cp1r/Cp2s strains (9), did not show any nucleotide sequence homology with the Cp1 and Cp2 genomes. Dot blot and Southern blot hybridization of genomic DNA from 11 strains tested (Table 1) showed no significant hybridization signals with Cp1 DNA or Cp2 DNA as a probe (data not shown). Therefore, no direct connection between the host genome and Cp1/Cp2 genomes was detected.
Conclusion.
Bacteriophages Cp1 and Cp2, traditionally used as phage-typing agents for X. axonopodis pv. citri strains, were found to belong to completely different virus groups. Cp1 was characterized as a phiL7-like siphovirus, but without a group I intron in the genome, and Cp2 was classified as a new podovirus with genes lacking detectable homologs in the current databases. The host hssB3.0 and XAC1661-XAC1662 sequences were not related to either Cp1 or Cp2 sequences. Both Cp1 and Cp2 efficiently attached to host cells, even if those were resistant strains.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Shiotani of the National Institute of Fruit Tree Science, NAFRO, for X. axonopodis pv. citri strain KC33 and helpful suggestions about the strains, H. Ogata of Tokyo Institute of Technology for Cp2 gene annotation, and M. Nakano of ADSM, Hiroshima University, for her technical guidance on LC-MS/MS analysis.
Footnotes
Published ahead of print 11 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02310-13.
REFERENCES
- 1.Civerelo EL. 1984. Bacterial canker disease of citrus. J. Rio. Grande Val. Hort. Soc. 37:127–145 [Google Scholar]
- 2.Gottwald TR, Graham JH, Schubert TS. 2002. Citrus canker: the pathogen and its impact. Plant health progress. http://www.plantmanagementnetwork.org/pub/php/review/citruscanker/
- 3.Stall RE, Civerolo EL. 1991. Research relating to the recent outbreak of citrus canker in Florida. Annu. Rev. Phytopathol. 29:399–420 [DOI] [PubMed] [Google Scholar]
- 4.Wakimoto S. 1967. Some characteristics of citrus canker bacteria, Xanthomonas citri (Hasse) Dowson, and the related phages isolated from Japan. Ann. Phytopathol. Soc. Jpn. 33:301–310 [Google Scholar]
- 5.Obata T. 1974. Distribution of Xanthomonas citri strain in relation to the sensitivity to phage CP1 and CP2. Ann. Phytopathol. Soc. Jpn. 40:6–13 [Google Scholar]
- 6.Goto M, Takahashi T, Messina MA. 1980. A comparative study of the strains of Xanthomonas campestris pv. citri isolated from citrus canker in Japan and cancrosis B in Argentina. Ann. Phytopathol. Soc. Jpn. 46:329–338 [Google Scholar]
- 7.Shiotani H, Tsuyumu S, Ozaki K. 2000. Pathogenic interactions between Xanthomonas axonopodis pv. citri and cultivars of Pummelo (Citrus grandis). Phytopathology 90:1383–1389. 10.1094/PHYTO.2000.90.12.1383 [DOI] [PubMed] [Google Scholar]
- 8.Louws FJ, Fulbright DW, Stephens CT, de Bruijn FJ. 1994. Specific genomic fingerprints of phytopathogenic Xanthomonas and Pseudomonas pathovars and strains generated with repetitive sequences and PCR. Appl. Environ. Microbiol. 60:2286–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiotani H. 2007. Dissertation. The United Graduate School of Agricultural Science, Gifu University, Gifu, Japan [Google Scholar]
- 10.Swarup S, Yang Y, Kingsley MT, Gabriel DW. 1992. An Xanthomonas citri pathogenicity gene, phcA, pleiotropically encodes gratuitous avirulence on nonhosts. Mol. Plant Microbe Interact. 5:204–213 [DOI] [PubMed] [Google Scholar]
- 11.Szurek B, Rossier O, Hause G, Bonas U. 2002. Type III dependent translocation of the Xanthomonas AvrBs3 protein into the plant cell. Mol. Microbiol. 46:13–23. 10.1046/j.1365-2958.2002.03139.x [DOI] [PubMed] [Google Scholar]
- 12.Shiotani H, Fujikawa T, Ishihara H, Tsuyumu S, Ozaki K. 2007. A pthA homolog from Xanthomonas axonopodis pv. citri responsible for host-specific suppression of virulence. J. Bacteriol. 189:3271–3279. 10.1128/JB.01790-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 14.Dykstra MJ. 1993. A manual of applied technique for biological electron microscopy. Plenum Press, New York, NY [Google Scholar]
- 15.Yamada T, Kawasaki T, Nagata S, Fujiwara A, Usami S, Fujie M. 2007. New bacteriophages that infect the phytopathogen Ralstonia solanacearum. Microbiology 153:2630–2639. 10.1099/mic.0.2006/001453-0 [DOI] [PubMed] [Google Scholar]
- 16.Hung CH, Yang CF, Yang CY, Tseng YH. 2003. Involvement of tonB-exbBD1D2 operon in infection of Xanthomonas campestris phage phiL7. Biochem. Biophys. Res. Commun. 302:878–884. 10.1016/S0006-291X(03)00255-9 [DOI] [PubMed] [Google Scholar]
- 17.Carlson K. 1994. Single-step growth, p 434–437 In Karam J, Drake JW, Kreuzer KN, Mosig G, Hall DH, Eiserlig FA, Black LW, Spicer EK, Kutter E, Carlson K, Miller ES. (ed), Molecular biology of bacteriophage T4. ASM Press, Washington, DC [Google Scholar]
- 18.Kawasaki T, Shimizu M, Satsuma H, Fujiwara A, Fujie M, Usami S, Yamada T. 2009. Genomic characterization of Ralstonia solanacearum phage ϕRSB1, a T7-like wide-host-range phage. J. Bacteriol. 191:422–427. 10.1128/JB.01263-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higashiyama T, Yamada T. 1991. Electrophoretic karyotyping and chromosomal gene mapping of Chlorella. Nucleic Acids Res. 19:6191–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul SF, Madden TL, Schaffer AA, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UniProt Consortium 2007. The Universal Protein Resource (UniProt). Nucleic Acids Res. 35:D193–D197. 10.1093/nar/gkl929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvrnin V, Church DM, DiCuccio M, Edgar E, Federhen S, Geer LY, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Ostell J, Miller V, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. 2007. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 35:D5–D12. 10.1093/nar/gkl1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiwara A, Fujisawa M, Hamasaki R, Kawasaki T, Fujie M, Yamada T. 2011. Biocontrol of Ralstonia solanacearum by treatment with lytic bacteriophages. Appl. Environ. Microbiol. 77:4155–4162. 10.1128/AEM.02847-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ausubel F, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1995. Short protocols in molecular biology, 3rd ed. John Wiley and Sons, Inc., Hoboken, NJ [Google Scholar]
- 26.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 27.Mosier-Boss PA, Lieberman SH, Andrews JM, Rohwer FL, Wegley LE, Breitbart M. 2003. Use of fluorescently labeled phage in the detection and identification of bacterial species. Appl. Spectrosc. 57:1138–1144. 10.1366/00037020360696008 [DOI] [PubMed] [Google Scholar]
- 28.Arai K, Shimo H, Doi Y, Yira K. 1974. Electron microscopy of Xanthomonas citri phages CP1 and CP2 infection. Ann. Phytopathol. Soc. Jpn. 40:98–102 [Google Scholar]
- 29.Hadas H, Einav M, Fishov I, Zaritsky A. 1997. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 143:179–185 [DOI] [PubMed] [Google Scholar]
- 30.Lee CN, Lin JW, Weng SF, Tseng YH. 2009. Genomic characterization of the intron-containing T7-like phage phiL7 of Xanthomonas campestris. Appl. Environ. Microbiol. 75:7828–7837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheetham GM, Jeruzalmi D, Steiz TA. 1999. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature 399:80–83 [DOI] [PubMed] [Google Scholar]
- 32.Osumi-Davis PA, de Aguilera MC, Woody RW, Woody AY. 1992. Asp537, Asp812 are essential and Lys631, His811 are catalytically significant in bacteriophage T7 RNA polymerase activity. J. Mol. Biol. 226:37–45 [DOI] [PubMed] [Google Scholar]
- 33.Semenova E, Djordjevie M, Shraiman B, Severinov K. 2005. The tale of two RNA polymerases: transcription profiling and gene expression strategy of bacteriophage Xp10. Mol. Microbiol. 55:764–777. 10.1111/j.1365-2958.2004.04442.x [DOI] [PubMed] [Google Scholar]
- 34.Inoue Y, Matsuura T, Ohara T, Azekami K. 2006. Bacteriophage OP1, lytic for Xanthomonas oryzae pv. oryzae, changes its host range by duplication and deletion of the small domain in the deduced tail fiber gene. J. Gen. Plant Pathol. 72:111–118. 10.1007/s10327-005-0252-x [DOI] [Google Scholar]
- 35.Dunn JJ, Studier FW. 1983. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166:477–535 [DOI] [PubMed] [Google Scholar]
- 36.Molineux IJ. 1999. T7-like phages (Podoviridae), p 1722–1729 In Granoff A, Webster R. (ed), Encyclopedia of virology. Academic Press, London, United Kingdom [Google Scholar]
- 37.Ceyssens PJ, Lavigne R, Mattheus W, Chibeu A, Hertveldt K, Mast J, Robben J, Volckaert G. 2006. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: establishment of the ϕKMV subgroup within the T7 subgroup. J. Bacteriol. 188:6924–6931. 10.1128/JB.00831-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardies SC, Comeau AM, Serwer P, Suttle CA. 2003. The complete sequence of marine bacteriophage VpV262 infecting Vibrio parahaemolyticus indicates that an ancestral component of a T7 viral supergroup is widespread in the marine environment. Virology 310:359–371. 10.1016/S0042-6822(03)00172-7 [DOI] [PubMed] [Google Scholar]
- 39.Yuzenkova J, Nechaev S, Berlin J, Rogulja D, Kuznedelov K, Inman R, Mushegian A, Severinov K. 2003. Genome of Xanthomonas oryzae bacteriophage XP10: an odd T-odd phage. J. Mol. Biol. 330:735–748. 10.1016/S0022-2836(03)00634-X [DOI] [PubMed] [Google Scholar]
- 40.Lee CN, Hu RM, Chow TY, Lin JW, Chen HY, Tseng YH, Weng SF. 2007. Comparison of genomes of three Xanthomonas oryzae bacteriophages. BMC Genomics 8:442–453. 10.1186/1471-2164-8-442 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.