

Abstract
The Amazon rainforest, the largest equatorial forest in the world, is being cleared for pasture and agricultural use at alarming rates. Tropical deforestation is known to cause alterations in microbial communities at taxonomic and phylogenetic levels, but it is unclear whether microbial functional groups are altered. We asked whether free-living nitrogen-fixing microorganisms (diazotrophs) respond to deforestation in the Amazon rainforest, using analysis of the marker gene nifH. Clone libraries were generated from soil samples collected from a primary forest, a 5-year-old pasture originally converted from primary forest, and a secondary forest established after pasture abandonment. Although diazotroph richness did not significantly change among the three plots, diazotroph community composition was altered with forest-to-pasture conversion, and phylogenetic similarity was higher among pasture communities than among those in forests. There was also 10-fold increase in nifH gene abundance following conversion from primary forest to pasture. Three environmental factors were associated with the observed changes: soil acidity, total N concentration, and C/N ratio. Our results suggest a partial restoration to initial levels of abundance and community structure of diazotrophs following pasture abandonment, with primary and secondary forests sharing similar communities. We postulate that the response of diazotrophs to land use change is a direct consequence of changes in plant communities, particularly the higher N demand of pasture plant communities for supporting aboveground plant growth.
INTRODUCTION
The Amazon rainforest is the largest equatorial forest in the world and acts as one of the major carbon dioxide (CO2) sinks, absorbing 0.4 Pg C · year−1 (1 Pg = 1015 g) of this greenhouse gas annually (1, 2). To absorb this enormous amount of CO2, a significant input of nitrogen (N) is required. This can occur through the decomposition of the organic material, atmospheric deposition of N, and biological N2 fixation. In terrestrial ecosystems, 97% of the natural N input comes from biological N2 fixation (3) performed by Bacteria and Archaea (4, 5). The majority of these nitrogen-fixing microorganisms (diazotrophs) are present in a free-living state, and their contribution to the N budget in tropical ecosystems can be from 12.2 to 36.1 kg · ha−1 · year−1 (6).
The composition and abundance of the free-living diazotrophic community in soil is directly related to its N2 fixation rate (7) and can be altered by a number of factors, including land management practices (7–10), soil N content (11–14), C and P availability (15–18), soil texture (19), pH and clay content (20, 21), season (22), and presence of different plant species (23, 24). Despite the importance of free-living diazotrophic communities to ecosystem processes, our knowledge of their diversity, abundance, and community composition in tropical forests remains very limited.
The Amazon rainforest continues to be threatened by deforestation at alarming rates, with 70% of the conversion associated with pasture formation. Primary forests, which can contain up to 179 unique tree species ha−1 (25, 26), are replaced with one or two fast-growing grasses (primarily Urochloa brizantha and Panicum maximum). This shift in plant community composition is likely to impact ecosystems through microbially mediated processes. For example, the processes of mineralization, nitrification, and denitrification have been reported to decrease in response to the conversion of Amazon forest to pasture (27–29). Similarly, methane consumption has been reported to decrease in response to deforestation (30).
The impact of deforestation on the bacterial diversity of Amazon rainforest soils has been limited to a few studies, all of which have either used phylogenetic markers (i.e., the 16S rRNA gene) (31–34) or the functional gene marker pmoA (a genetic marker for methane-oxidizing bacteria) (30). These studies reported significant differences in bacterial community compositions in forest and pasture soils. However, it remains virtually unknown how other microbial groups responsible for specific functional processes respond to deforestation. In addition, it is also unclear to what extent altered functional diversity can recover after the reestablishment of forest following pasture abandonment.
In this study, we sought to address the above gap in knowledge by studying the free-living diazotrophic community, a functional group of microorganisms that makes a significant contribution to the N pool in tropical systems (6, 35). The objectives of the present study were (i) to assess the diazotrophic community composition and abundance of the Amazon rainforest and determine its response to land use change from primary forest to pasture, (ii) to evaluate the extent of recovery for diazotrophic communities following pasture abandonment (and the growth of secondary forest), (iii) to determine whether diazotrophic community composition and abundance are linked to the changes in soil physiochemical characteristics, and (iv) to identify indicator diazotrophic taxa associated with changes in land use in tropical forest ecosystems.
MATERIALS AND METHODS
Soil collection and DNA extraction.
Soil samples were collected from Fazenda Nova Vida (10°10′18.71″S, 62°47′15.67″W), situated in the state of Rondônia, Brazil, in 2009. The state of Rondônia was selected for sampling because it was one of the three states (Rondônia, Mato Grosso, and Pará) which accounted for more than 85% of all Amazon deforestation from 1996 to 2005, converting an average of 16,600 km2 year−1 of forest to pasture land (36). Samples were collected from a primary forest, a pasture established in 2004, and a secondary forest established after pasture abandonment in 1999. All plots share common boundaries within a 2-km distance. A 100-m2 transect was placed in each plot, and nested transects of 10, 1, 0.1, and 0.01 m2 were established from the same initial point as previously described by Rodrigues et al. (34). A total of five soil samples per plot were collected: three on the transect with the cardinal direction north (1, 10, and 100 m) and one each (100 m) on the transects from the directions northeast and east.
After removal of the litter layer, soils were taken with a 5-cm-diameter corer to a 10-cm depth, kept on ice, and transported to the laboratory, where they were stored at −80°C until used for DNA extraction. Soil cores were individually sieved with a 2-mm mesh and subdivided for total soil carbon and nitrogen determination with the Auto Analyzer LECO TruSpec CN at the Centro de Energia Nuclear na Agricultura, University of Sao Paulo, Brazil, and elemental concentrations were analyzed at the Laboratorio de Fertilidade do Solo, Department of Soil Sciences, University of Sao Paulo, as described previously (34). The details of the sampling sites and soil physiochemical characteristics are presented in Table S1 in the supplemental material.
DNA extraction and PCR amplification.
Total genomic DNA was extracted from soil using the Power Lyzer PowerSoil DNA isolation kit (MO BIO, Carlsbad, CA) according to the manufacturer's instructions. Soil DNA was quantified, diluted to a concentration of 25 ng/μl, and used to amplify the nifH gene with primers PolF (TGCGAYCCSAARGCBGACTC) and PolR (ATSGCCATCATYTCRCCGGA) (37). The reaction volume of 50 μl contained 0.2 mM each deoxynucleoside triphosphate, 0.4 μM each primer, 1× PCR buffer, 2 mM MgCl2, 50 ng of template DNA, and 2 U of Taq DNA polymerase (Gene Script, Piscataway, NJ). The PCR conditions were 2 min of denaturation, followed by 35 rounds of temperature cycling (95°C for 30 s, 59°C for 30 s, and 72°C for 45 s), and a final extension at 72°C for 7 min. Aliquots (5 μl) of the amplified products were visualized on ethidium bromide-stained 1% agarose gels. Along with polymerase (Pol) primers, three other primer sets—Ueda19F-Ueda470R (15), PicenoF/R (38), and Z-primer (39)—were tested for nifH amplification (40). Only the Pol primer set resulted in successful amplification for all 15 samples, and hence it was used for the subsequent analyses.
Cloning and DNA sequencing.
Amplified products were purified with the Ultra Clean 15 DNA purification kit (MO BIO, Carlsbad, CA), ligated into the TOPO TA pcr2.1 vector, and transformed into Escherichia coli One Shot TOP10 competent cells (TOPO TA cloning kit; MO BIO, Carlsbad, CA), as recommended by the manufacturer. Transformants were grown in Luria-Bertani medium supplemented with kanamycin (50 ng/μl) and analyzed for the presence of an insert of approximately 362 bp by PCR amplification with primers M13F (GTAAAACGACGGCCAG) and M13R (CAGGAAACAGCTATGAC). Amplified fragments were treated with ExoSAP-IT (USB, Cleveland, OH) and sequenced with the M13F primer. A total of 825 nifH gene clones (55 clones per sample) were sequenced with the 3130XL genetic analyzer (Applied Biosystems, Foster City, CA) at the University of Texas—Arlington Genomics Core Facility.
Sequence analyses and OTU assignment.
A total of 825 sequences of the nifH gene were first aligned using CLUSTAL_X (41) and trimmed to 362 bp using Sequencher 4.2.2 (Gene Codes Corp., Ann Arbor, MI), and a final alignment was performed with MUSCLE (42). All sequences were compared to those in NCBI databases for confirmation as nifH gene sequences and deposited in GenBank.
Aligned nifH gene sequences were used to create a distance matrix followed by clustering into operational taxonomic units (OTU) at 99, 97, 95, 90, and 80% DNA identity using the software Mothur (43). Rarefaction curves were generated using the same software.
DNA translation and OPU assignment.
In order to increase the taxonomic resolution of the nifH sequences (44) and associate their presence with a specific land use, DNA sequences were translated into deduced amino acid sequences and aligned using the functional gene pipeline (Fungene) of the Ribosomal Database Project (RDP) (http://rdp.cme.msu.edu) (45). Aligned protein sequences were used to generate the distance matrix and clustered into operational protein units (OPU) at 99, 97, 95, 90, and 80% protein similarity using Mothur as described above.
OPU representing more than five sequences were selected for construction of an amino acid-based maximum likelihood (ML) phylogenetic tree using the PHYML 3.0 software (46) with settings previously described elsewhere (47). The software MEGA (version 4) was used to collapse sequences at a 97% amino acid similarity level (48).
Phylogenetic diversity.
Phylogenetic diversity of diazotrophic communities was compared among different land use systems using the unweighted UniFrac algorithm (49). A DNA-based ML phylogenetic tree along with the branch length matrix was used as the input file and a principal coordinate analysis (PCoA) was performed using the Fast UniFrac matrix (http://bmf2.colorado.edu/fastunifrac/) (50).
Diazotrophic community structure.
To assess changes in diazotrophic community composition across different land use systems, an OTU-based DNA similarity data matrix (at 99, 97, 95, 90, and 80% identity) was square root transformed and analyzed using Primer 6 (version 6.1.11) computer software (Primer-E Ltd., Plymouth, United Kingdom) with the permutational multivariate analysis of variance (PerMANOVA) add-on package (51). Differences in the diazotrophic communities were evaluated through total species richness, Margalef's richness index, and the Chao1 estimator (52). Changes in OTU composition among communities were evaluated with the Sørensen index of similarity calculated from presence/absence data (53).
Patterns of the diazotrophic community structure were visualized by nonmetric multidimensional scaling (NMDS) of the Bray-Curtis similarity matrix. Statistical significance of differences among the data sets was analyzed by PerMANOVA (54), followed by a calculation of a pseudo-F statistic and comparison of the total variance explained by sample identities to that explained by random permutations. Treatments were compared to 9,999 permutations.
Associations between the diazotrophic species composition data matrix and the environmental data matrix (soil moisture, N, C, C/N, pH, P, S, K+, Ca2+, Mg2+, Al3+, H+, CEC, V, and Al + H [potential acidity]) were examined using the “BEST” procedure in Primer (51). All environmental data were log transformed and standardized prior to the BEST analysis.
Indicator species analysis.
Translated amino acid-based data (OPU 97% amino acid similarity) was also assessed with PerMANOVA. In order to identify the indicator species and the relative contribution of each OPU to the differences in groups observed by PerMANOVA, we performed a similarity percentage (SIMPER) analysis (51).
qPCR.
For quantitative PCR (qPCR), reactions were performed in triplicate for each biological sample and carried out in a 20-μl volume containing 10 μl of iTaq Fast SYBR green super mix with ROX (Bio-Rad, Inc., Hercules, CA), 100 nM the same primers used for cloning, PolF and PolR, and 10 ng of DNA. Amplification was performed with the Applied Biosystems 7300 real-time PCR system under the following conditions: 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 45 s, annealing at 59°C for 1 min, and extension at 72°C for 1 min. PCR-grade water (no template) was used as a negative control. The specificity of the qPCR products was confirmed by melting curve analysis and gel-based post-PCR analysis. Standard curves (100 to 107 nifH copies per reaction) were generated with plasmid DNA containing a partial fragment of the nifH gene from Herbaspirillum seropedicae ATCC 35892. The qPCR efficiency (E) was calculate according to the equation E = 10[−1/slope]. The real-time efficiency of nifH was 1.80 ± 0.01, and the R2 of the standard curve was 0.99. The detection limit of the qPCR assay was ca. 100 copies/μl. Differences in the abundance of the nifH gene were assessed by single-factor ANOVA using the software package R, version 2.9.2 (www.R-project.org).
Nucleotide sequence accession numbers.
All sequences confirmed as nifH gene sequences have been deposited in GenBank under accession no. JX865783 through JX866606.
RESULTS
OTU assignment and univariate analyses.
A total of 825 nifH gene sequences were clustered based on different percent identity values (99, 97, 95, 90, and 80%), which resulted in 632, 466, 367, 262, and 80 OTU, respectively. We present the results below based on 97% identity, as different cutoff values yielded similar community compositional patterns for the same treatment plot.
The total number of unique nifH sequences, the Chao1 estimate, and the value for the Margalef's species richness index were not significantly different between forest and pasture sites (Fig. 1). Rarefaction curve analysis indicates that the number of OTU did not reach an asymptote, which suggests that additional sequencing effort could capture more diversity (see Fig. S1 in the supplemental material).
FIG 1.
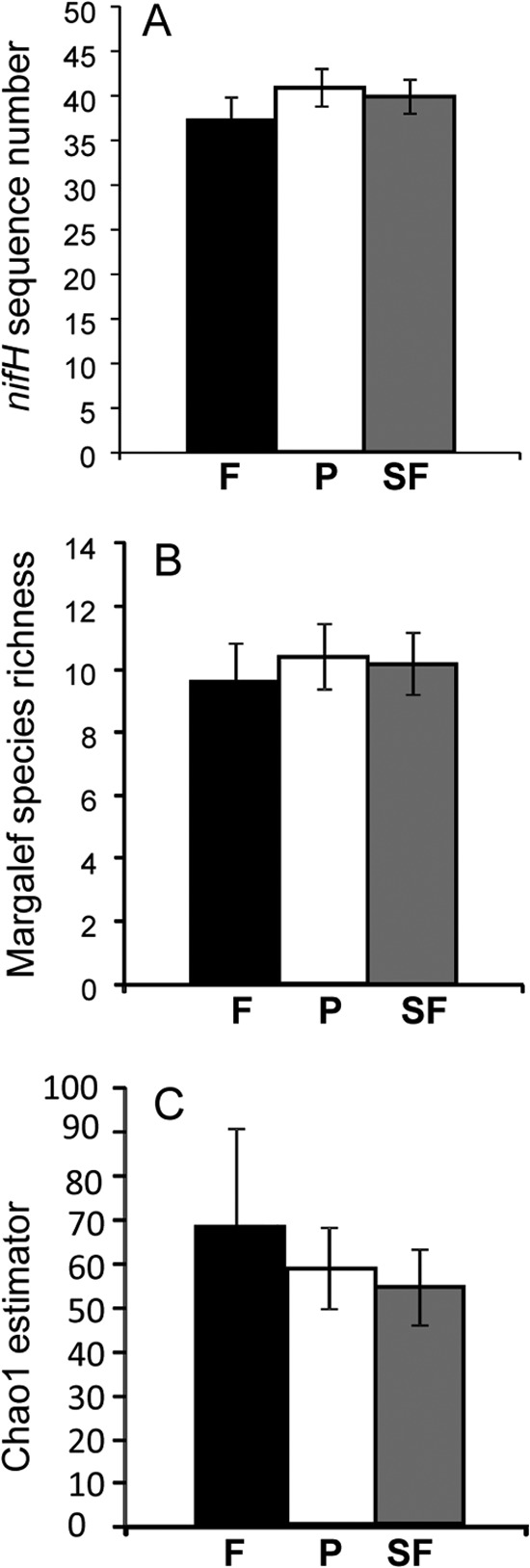
nifH gene diversity in response to land use change in the Amazon. (A) Number of unique genes; (B) Margalef species richness; (C) Chao1 estimate. Land use treatments are follows: F, primary forest (black bars); P, pasture (white bars); SF, secondary forest (gray bars). Error bars represent standard errors.
Community composition and phylogenetic diversity.
The diazotrophic community composition across different land uses was assessed by multivariate analyses of the nifH OTU. There was a distinct clustering of samples by land use with the Bray-Curtis index (Fig. 2A), and subsequent PerMANOVA analysis confirmed that the observed differences were statistically significant [F(2, 12) = 1.72, P = 0.001]. Pairwise comparisons of diazotrophic communities between land uses indicated that they were significantly different (P < 0.05). To test whether these differences were associated with changes in composition only (as opposed to changes in relative abundance and composition), we calculated the Sørensen index using an OTU data set transformed to a presence/absence matrix. The NMDS ordination of the Sørensen index also showed a distinct clustering of the diazotrophic community across the three sites (Fig. 2B). These groups were significantly different from each other [F(2, 12) = 1.81, P = 0.001], which was subsequently confirmed with pairwise t test comparisons. These results were consistent regardless of the selected percent similarity cutoff value for the OTU (i.e., OTU95, OTU90, or OTU80) (see Fig. S2 in the supplemental material), although differences in community composition became nonsignificant between primary and secondary forests at higher taxonomic levels (see Fig. S2).
FIG 2.

Nonmetric multidimensional scaling representation of the nifH gene sequence composition based on 97% DNA identity for three different land use systems in the Amazon: black triangles, primary forest samples (F); white circles, pasture samples (P); gray squares, secondary forest samples (SF). (A) Bray-Curtis similarity index; (B) Sørensen similarity index. The two-dimensional stress for each panel was 0.11.
Diazotrophic communities from pastures were different from those recovered from forests. A principal coordinate analysis of Fast UniFrac distances resulted in separation between the pasture and forest sites (Fig. 3). Furthermore, the relatively tight clustering of all five replicates from pastures suggests that the diazotrophic communities within pastures were much more closely related than those in forest (Fig. 3).
FIG 3.
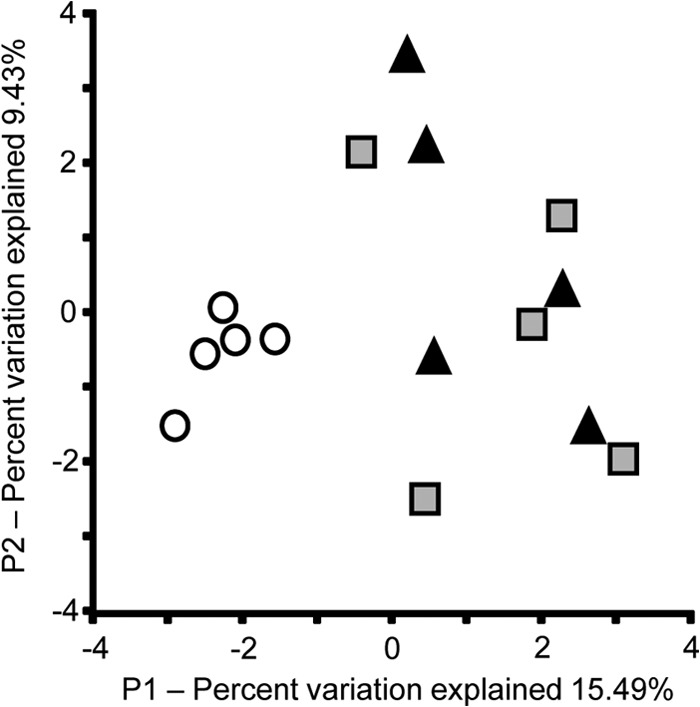
Principal coordinate analysis plot of phylogenetic similarities among nifH gene sequences from three different land use systems in the Amazon: black triangles, primary forest samples; white circles, pasture samples; gray squares, secondary forest samples. Plots were generated using unweighted UniFrac distances from a maximum likelihood-based analysis.
Environmental attributes.
Despite significant differences in the plant communities, all three different land use systems had similar values for 15 soil physicochemical characteristics (see Table S1 in the supplemental material). The total soil N concentration was slightly higher at the forest and secondary forest sites (140 mg/kg soil) than in the pasture (100 mg/kg soil), but these differences were not statistically significant. To test whether there was an association between the diazotrophic community and environmental factors, an individual regression analysis was performed between the Bray-Curtis similarity results and each environmental parameter. Only three factors—total soil N, C/N ratio, and Al + H (potential acidity)—showed a significant association with the diazotrophic community (P < 0.001, R2 = 0.367).
OPU analyses.
We tested whether the observed differences were maintained at the protein level and performed a phylogenetic analysis with translated amino acid sequences (44). We selected OPU comprising a minimum of 5 sequences, and a representative from each group was used for identification. The numbers of OPU were 19, 14, 2, and 1, for similarities of 99, 97, 95, and 90%, respectively. We used the 97% similarity cutoff value because it represented the majority of the sequences (85%; n = 691) observed in this study. The 14 different OPU groups used for constructing a maximum-likelihood phylogenetic tree (Fig. 4) yielded two distinct NifH clusters (I and III), corresponding to the two previously recognized NifH groups (55). No sequences belonging to clusters II, IV, and V were observed. The majority of the sequences (84%) belonged to NifH cluster I, and within this cluster, OPU1 and OPU5 (identified as belonging to Proteobacteria) were detected as predominant groups (90%) among the total number of sequences (see Table S2 in the supplemental material). These OPU varied between the pasture and the two forest sites. For example, OPU1 (most closely related to the Deltaproteobacteria) was more commonly found at the pasture site (53% of the total sequences in that cluster were from pasture), whereas OPU5 (most closely related to Alpha- and Betaproteobacteria) was predominantly composed of sequences from primary and secondary forest sites (38% and 42%, respectively). Four OPU were only detected at the primary and secondary forest sites (OPU56, OPU4, OPU63, and OPU51; most closely related to Paenibacillus, Nostoc, and Firmicutes). NifH cluster III was also diverse and was represented by seven OPU. Three of these (OPU2, OPU9, and OPU72) were only observed at the primary and secondary forest sites. OPU7 and OPU9 (related to archaeal species in the genera Methanocella and Methanosphaerula) also varied between the forest and pasture sites. Three OPU (OPU2, OPU16, and OPU19) were most closely related to uncultured bacteria, suggesting that these groups could be novel diazotrophic taxa (see Table S2 in the supplemental material).
FIG 4.

Maximum likelihood phylogenetic tree based on translated amino acid sequences of the nifH gene from three different land use systems in the Amazon rainforest: black, primary forest (F); white, pasture (P); gray, secondary forest (SF). Only operational protein units (OPU) representing ≥5 sequences were used. The size of the circle is proportionate to the number of sequences within an OPU. Numbers at the nodes reflect bootstrap support values above 70% with branches within clusters being collapsed. The percent contribution of sequences from different land uses and percent similarities of translated nifH gene sequences to closely related NifH protein sequences are provided.
A pairwise comparative analysis among sites using translated sequences (grouped at 97% similarity) indicated that primary and secondary forests were not significantly different (t = 0.74, P = 0.761). However, the diazotrophic communities in both secondary and primary forests were significantly different from those observed in the pasture [PerMANOVA; F(2, 12) = 3.80, P = 0.006]. We performed an indicator species analysis (SIMPER) to identify OPU associated with different land uses. Three OPU (OPU1, OPU5, and OPU16) contributed to 32.68% of the total dissimilarity detected between the primary forest and pasture. OPU1 and OPU16 were predominantly detected in pasture samples, whereas OPU5 was dominant in primary forest samples. Likewise, three OPU (OPU1, OPU5, and OPU51) were found to be important for discrimination of secondary forest and pasture sites, contributing to 42.09% of the total dissimilarity.
nifH gene abundance.
The number of copies of the nifH gene differed significantly at each site (P < 0.05), with pasture samples containing the highest values (mean of 2.2 × 105 copies g−1 dry soil), while the primary forest had the smallest number of copies of the nifH gene (2.1 × 104 copies g−1 dry soil) (Fig. 5; see Fig. S3 in the supplemental material). The secondary forest contained an intermediate number of the copies of the nifH gene (5.0 × 104 copies g−1 dry soil).
FIG 5.
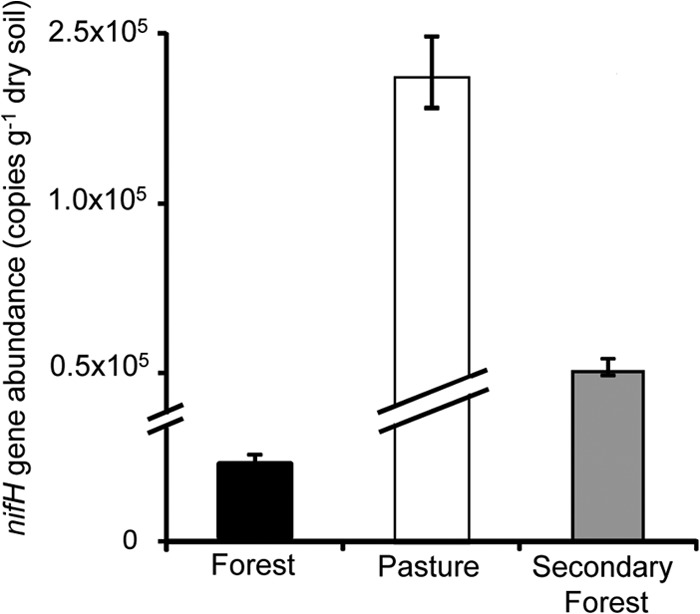
Copy numbers of the nifH gene per gram of dry soil determined for three different land use systems in the Amazon. Each bar represents the average of the 15 replicates (5 biological replicates × 3 technical replicates).
DISCUSSION
The nifH gene, encoding the reductase subunit of nitrogenase, has been widely used as a genetic marker to study the diversity and abundance of N2-fixing microorganisms (55–57). Previous studies, based on the 16S rRNA gene, reported increases in richness and α diversity following deforestation and pasture establishment in the Amazon (33, 34). We observed a different response to deforestation for diazotrophic communities than that reported for the total bacterial community (31–34). In our study, while richness and diversity of diazotrophic communities remained similar among the three land uses (Fig. 1), the community composition and the nifH gene abundance were drastically altered with land use (Fig. 2A and B). To our knowledge, this work is the first to report that microbial communities associated with a particular ecosystem function, biological nitrogen fixation, are altered with Amazon deforestation.
We postulate that the changes in composition and abundance of diazotroph communities that we observed are a direct consequence of the plant composition observed in each land use. An increase in plant diversity is often associated with higher rates of N mineralization carried out by microorganisms and a greater net N supply (58, 59). Consistent with this idea, higher rates of N mineralization were observed in forests compared to pastures at our field site (28). Consequently, the forest demand for N is mainly filled (>80%) by the recycling of above- and belowground litter (60), with N2 fixers providing for the remaining N requirements. In contrast, pastures are constantly N depleted due to grazing of aboveground biomass and rely heavily on free-living diazotrophs to fulfill the N requirements for supporting plant growth. The grass species Urochloa brizantha and Panicum maximum obtain up to 41% of their N through biological nitrogen fixation by allocating large quantities of easily degradable carbon sources as root exudates (61). A combination of C-rich and N-depleted conditions in pastures may provide a competitive advantage for free-living diazotrophs. Therefore, we sought to identify soil physicochemical variables associated with differences in nifH gene sequences. Three soil variables, namely, N, C/N ratio, and potential acidity (Al + H), showed significant association values (P < 0.001) with land use. The decrease in soil N concentration and increase in C/N ratio are suggested drivers of environmental selection for N2 fixers (19). The third variable, potential acidity, is related to pH, a well-established factor affecting the diversity of microbial communities (33, 62). In agreement with the above results, we observed a dramatic shift in nifH gene abundances with land use change. Future in situ estimates of biological nitrogen fixation (BNF) in the Amazon rainforest will help to improve nutrient flux models for this terrestrial ecosystem.
We investigated the possibility of diazotrophic community recovery after pasture abandonment and establishment of a secondary forest. Retrieved nifH sequences from both primary and secondary forests showed high levels of similarity at taxonomic and phylogenetic levels (Fig. 2 and 3). We also detected a decrease in nifH gene abundance in secondary forest samples relative to pasture samples, although gene abundance was still higher in secondary forests than in primary forests. Together, these results suggest that recovery of diazotrophic communities may be possible following pasture abandonment. This has important implications for restoration of degraded areas since 30 to 50% of cleared land in the Amazon is under secondary forest succession (63). At the phylogenetic level, the return of nifH gene sequences similar to those observed in primary forest suggests compositional restoration within 10 years or less after abandonment. At the functional level, the decrease in copy numbers of the nifH gene implies a less N-limited environment as secondary forests mature. We recognize that fewer gene copies may not translate into lower BNF rates and remain cautious about establishing a direct link; however, there is strong evidence suggesting that as plant diversity increases, the total N accumulated and recycled through the forest alleviates the need for external N input (64). A botanical survey at our research site showed that shrubs and woody plant species comprised 37% of the secondary forest plant community after 2 years of pasture abandonment (65), which may help to explain the resilience of the free-living diazotrophic community.
We performed an indicator species analysis to assess operational protein units contributing to the compositional differences we observed among land use types. The two distinct clusters observed in our study are typically found in soils: while cluster I is represented by aerobic N2 fixers, cluster III consists of anaerobic diazotrophs from the domains Bacteria and Archaea (55). Most of the sequences were related to Proteobacteria (64.2%), the most abundant group in terrestrial ecosystems (57), and sequences in this group contributed greatly to the observed differences between the pasture and the two forest sites. For example, OPU5, the largest group of nifH sequences we detected, is related to Alpha- and Betaproteobacteria, and we found it predominantly in primary (38%) and secondary (42%) forests relative to pastures (20%). On the other hand, the proportion of the community comprised of OPU1 (related to the Deltaproteobacteria) increased from 28% to 53% with forest-to-pasture conversion (Fig. 4). Many OPU were observed only in forest samples, suggesting an impact on diazotrophic diversity with ecosystem conversion. The effects of the loss of particular functional groups in tropical forests are not known, nor have they been extensively evaluated in other forest ecosystems, but they do raise concerns about the long-term stability of the biogeochemical process.
Noteworthy was the presence of sequences associated with Archaea. Our results indicate a clear shift, with OPU9 present only in forest sites, while 85% of sequences binned as OPU7 came from the pasture sample. OPU9 is related to Methanoregula and Methanosphaerula, two genera isolated from soils with pH below 5.5, while OPU7 belongs to Methanocella, a genus with representatives obtained from soils with pH 6.5 to 7.0 (Fig. 4). Despite the fact that the ability of methanogenic Archaea to fix N2 has long been known (66, 67) and interconnections between the C and N biogeochemical cycles have been well established, how the processes of methanogenesis and BNF are related in nature remains virtually unknown. Because Fazenda Nova Vida pastures have been characterized as methane sources (68), one would expect that archaeal biological N2 fixation plays an important role in the Amazon rainforest under threat of deforestation.
Conclusions.
Land use changes in the Amazon rainforest result in a series of ecosystem alterations that include an increase in belowground productivity (32) and a more open N cycle (69), two ecological controls known to influence free-living diazotrophic communities (64). Our study found that diazotrophic communities in tropical terrestrial ecosystems can indeed respond to land use change, and we documented a shift in their community composition and abundance following forest-to-pasture conversion. Understanding the causes and consequences of these alterations may lead to refined models that incorporate molecular-level dynamics into ecosystem-level processes.
Supplementary Material
ACKNOWLEDGMENTS
We thank the owners and staff of Agropecuária Nova Vida for logistical support and permission to work on their property, the ARMO team members for assistance with sampling, Siu M. Tsai for soil analyses, and Vivian H. Pellizari for assistance with export permits.
This project was supported by Agriculture and Food Research Initiative Competitive Grant 2009-35319-05186 from the U.S. Department of Agriculture National Institute of Food and Agriculture.
Footnotes
Published ahead of print 25 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02362-13.
REFERENCES
- 1.Phillips OL, Aragao LE, Lewis SL, Fisher JB, Lloyd J, Lopez-Gonzalez G, Malhi Y, Monteagudo A, Peacock J, Quesada CA, van der Heijden G, Almeida S, Amaral I, Arroyo L, Aymard G, Baker TR, Bánki O, Blanc L, Bonal D, Brando P, Chave J, de Oliveira ÁC, Cardozo ND, Czimczik CI, Feldpausch TR, Freitas MA, Gloor E, Higuchi N, Jiménez E, Lloyd G, Meir P, Mendoza C, Morel A, Neill DA, Nepstad D, Patiño S, Peñuela MC, Prieto A, Ramírez F, Schwarz M, Silva J, Silveira M, Thomas AS, ter Steege H, Stropp J, Vásquez R, Zelazowski P, Dávila EA, Andelman S, Andrade A, et al. 2009. Drought sensitivity of the Amazon rainforest. Science 323:1344–1347. 10.1126/science.1164033 [DOI] [PubMed] [Google Scholar]
- 2.Davidson EA, de Araujo AC, Artaxo P, Balch JK, Brown IF, Bustamante C MM, Coe MT, DeFries RS, Keller M, Longo M, Munger JW, Schroeder W, Soares-Filho BS, Souza CM, Jr, Wofsy SC. 2012. The Amazon basin in transition. Nature 481:321–328. 10.1038/nature10717 [DOI] [PubMed] [Google Scholar]
- 3.Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA. 2008. Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320:889–892. 10.1126/science.1136674 [DOI] [PubMed] [Google Scholar]
- 4.Eady RR. 1992. The dinitrogen-fixing bacteria, p 534–553 In Balows A, Tr̈uper HG, Dworkin M, Harder W, Schleifer KH. (ed), The prokaryotes. A handbook on the biology of bacteria: ecophysiology, isolation, identification, application. Springer-Verlag, New York, NY [Google Scholar]
- 5.Young JPW. 1992. Phylogenetic classification of nitrogen-fixing organisms, p 43–86 In Stacey G, Evans HJ, Burris RH. (ed), Biological nitrogen fixation. Chapman and Hall, New York, NY [Google Scholar]
- 6.Cleveland CC, Townsend AR, Schimel DS, Fisher H, Howarth RW, Hedin LO, Perakis SS, Latty EF, Von Fischer JC, Elseroad A, Wasson MF. 1999. Global patterns of terrestrial biological nitrogen (N2) fixation in natural ecosystems. Global Biogeochem. Cycles 13:623–645. 10.1029/1999GB900014 [DOI] [Google Scholar]
- 7.Hsu S-F, Buckley DH. 2009. Evidence for the functional significance of diazotroph community structure in soil. ISME J. 3:124–136. 10.1038/ismej.2008.82 [DOI] [PubMed] [Google Scholar]
- 8.Gros R, Monrozier LJ, Faivre P. 2006. Does disturbance and restoration of alpine grassland soils affect the genetic structure and diversity of bacterial and N2-fixing populations? Environ. Microbiol. 8:1889–1901. 10.1111/j.1462-2920.2006.01106.x [DOI] [PubMed] [Google Scholar]
- 9.Wakelin SA, Gregg AL, Simpson RJ, Li GD, Riley IT, McKay AC. 2009. Pasture management clearly affects soil microbial community structure and N-cycling bacteria. Pedobiologia 52:237–251. 10.1016/j.pedobi.2008.10.001 [DOI] [Google Scholar]
- 10.Orr CH, James A, Leifert C, Cooper JM, Cummings SP. 2011. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl. Environ. Microbiol. 77:911–919. 10.1128/AEM.01250-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalininskaya TA. 1989. The influence of different forms of combined nitrogen on nitrogen-fixing activity of Azospirilla in the rhizosphere of rice plants, p 283–286 In Vancura V, Kunc F. (ed), Proceedings of the International Symposium on Interrelationships between Microorganisms and Plants in Soil. Elsevier Science Publishing, New York, NY [Google Scholar]
- 12.DeLuca TH, Drinkwater LE, Wiefling BA, DeNicola DM. 1996. Free-living nitrogen-fixing bacteria in temperate cropping systems: influence of nitrogen source. Biol. Fertil. Soils 23:140–144. 10.1007/BF00336054 [DOI] [Google Scholar]
- 13.Rudnick P, Meletzus D, Green A, He LH, Kennedy C. 1997. Regulation of nitrogen fixation by ammonium in diazotrophic species of proteobacteria. Soil Biol. Biochem. 29:831–841. 10.1016/S0038-0717(96)00238-6 [DOI] [Google Scholar]
- 14.Limmer C, Drake HL. 1998. Effects of carbon, nitrogen, and electron acceptor availability on anaerobic N2-fixation in a beech forest soil. Soil Biol. Biochem. 30:153–158. 10.1016/S0038-0717(97)00099-0 [DOI] [Google Scholar]
- 15.Widmer F, Shaffer BT, Porteous LA, Seidler RJ. 1999. Analysis of nifH gene pool complexity in soil and litter at a Douglas fir forest site in the Oregon Cascade Mountain Range. Appl. Environ. Microbiol. 65:374–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaffer BT, Widmer F, Porteous LA, Seidler RJ. 2000. Temporal and spatial distribution of the nifH gene of N2 fixing bacteria in forests and clearcuts in western Oregon. Microb. Ecol. 39:12–21. 10.1007/s002489900183 [DOI] [PubMed] [Google Scholar]
- 17.Bürgmann H, Meier S, Bunge M, Widmer F, Zeyer J. 2005. Effects of model root exudates on structure and activity of a soil diazotroph community. Environ. Microbiol. 7:1711–1724. 10.1111/j.1462-2920.2005.00818.x [DOI] [PubMed] [Google Scholar]
- 18.Reed SC, Townsend AR, Cleveland CC, Nemergut DR. 2010. Microbial community shifts influence patterns in tropical forest nitrogen fixation. Oecologia 164:521–531. 10.1007/s00442-010-1649-6 [DOI] [PubMed] [Google Scholar]
- 19.Poly F, Ranjard L, Nazaret S, Gourbière F, Monrozier LJ. 2001. Comparison of nifH gene pools in soils and soil microenvironments with contrasting properties. Appl. Environ. Microbiol. 67:2255–2262. 10.1128/AEM.67.5.2255-2262.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roper MM, Smith NA. 1991. Straw decomposition and nitrogenase activity (C2H2 reduction) by free-living microorganisms from soil: effects of pH and clay content. Soil Biol. Biochem. 23:275–283. 10.1016/0038-0717(91)90064-Q [DOI] [Google Scholar]
- 21.Limmer C, Drake HL. 1996. Non-symbiotic N2 fixation in acidic and pH-neutral forest soils: aerobic and anaerobic differentials. Soil Biol. Biochem. 28:177–183. 10.1016/0038-0717(95)00118-2 [DOI] [Google Scholar]
- 22.Mergel A, Kloos K, Bothe H. 2001. Seasonal fluctuations in the population of denitrifying and N2-fixing bacteria in an acid soil of a Norway spruce forest. Plant Soil 230:145–160. 10.1023/A:1004826116981 [DOI] [Google Scholar]
- 23.Patra AK, Abbadie L, Clays-Josserand A, Degrange V, Grayston SJ, Guillaumaud N, Loiseau P, Louault F, Mahmood S, Nazaret S, Philippot L, Poly F, Prosser JI, Le Roux X. 2006. Effects of management regime and plant species on the enzyme activity and genetic structure of N-fixing, denitrifying and nitrifying bacterial communities in grassland soils. Environ. Microbiol. 8:1005–1016. 10.1111/j.1462-2920.2006.00992.x [DOI] [PubMed] [Google Scholar]
- 24.Hai B, Diallo NH, Sall S, Haesler F, Schauss K, Bonzi M, Assigbetse K, Chotte JL, Munch JC, Schloter M. 2009. Quantification of key genes steering the microbial nitrogen cycle in the rhizosphere of sorghum cultivars in tropical agroecosystems. Appl. Environ. Microbiol. 75:4993–5000. 10.1128/AEM.02917-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirça Pires J, Dobzhansky T, Black GA. 1953. An estimate of the number of species of trees in an Amazonian forest community. Bot. Gaz. 114:467–477. 10.1086/335790 [DOI] [Google Scholar]
- 26.Hubbell SP, He F, Condit R, Borda-de-Agua L, Kellner J, ter Steege H. 2008. How many tree species are there in the Amazon and how many will go extinct? Proc. Natl. Acad. Sci. U. S. A. 105:11498–11504. 10.1073/pnas.0801915105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neill C, Piccolo MC, Steudler PA, Melillo JM, Feigl BJ, Cerri CC. 1995. Nitrogen dynamics in soils of forests and active pastures in the western Brazilian Amazon basin. Soil Biol. Biochem. 27:1167–1175. 10.1016/0038-0717(95)00036-E [DOI] [Google Scholar]
- 28.Neill C, Piccolo MC, Cerri CC, Steudler PA, Melillo JM, Brito M. 1997. Net nitrogen mineralization and net nitrification rates in soils following deforestation for pasture across the southwestern Brazilian Amazon Basin landscape. Oecologia 110:243–252. 10.1007/s004420050157 [DOI] [PubMed] [Google Scholar]
- 29.Neill C, Piccolo MC, Melillo JM, Steudler PA, Cerri CC. 1999. Nitrogen dynamics in Amazon forest and pasture soils measured by 15N pool dilution. Soil Biol. Biochem. 31:567–572. 10.1016/S0038-0717(98)00159-X [DOI] [Google Scholar]
- 30.Dörr N, Glaser B, Kolb S. 2010. Methanotrophic communities in Brazilian ferralsols from naturally forested, afforested, and agricultural sites. Appl. Environ. Microbiol. 76:1307–1310. 10.1128/AEM.02282-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borneman J, Triplett EW. 1997. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl. Environ. Microbiol. 63:2647–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cenciani K, Lambais MR, Cerri CC, Basilio de Azevedo LC, Feigl BJ. 2009. Bacteria diversity and microbial biomass in forest, pasture and fallow soils in the southwestern Amazon Basin. Rev. Bras. Cienc. Solo 33:907–916. 10.1590/S0100-06832009000400015 [DOI] [Google Scholar]
- 33.Jesus ED, Marsh TL, Tiedje JM, Moreira FMD. 2009. Changes in land use alter the structure of bacterial communities in western Amazon soils. ISME J. 3:1004–1011. 10.1038/ismej.2009.47 [DOI] [PubMed] [Google Scholar]
- 34.Rodrigues JMR, Pellizari VH, Mueller R, Baek K, Jesus Eda C, Paula FS, Mirza B, Hamaoui GS, Jr, Tsai SM, Feigl B, Tiedje JM, Bohannan BJ, Nüsslein K. 2013. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of bacterial microbial communities. Proc. Natl. Acad. Sci. U. S. A. 110:988–993. 10.1073/pnas.1220608110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galloway JN, Dentener FJ, Capone DG, Boyer EW, Howarth RW, Seitzinger SP, Asner GP, Cleveland CC, Green PA, Holland EA, Karl DM, Michaels AF, Porter JH, Townsend AR, Vöosmarty CJ. 2004. Nitrogen cycles: past, present, and future. Biogeochemistry 70:153–226. 10.1007/s10533-004-0370-0 [DOI] [Google Scholar]
- 36.INPE 2011. Program for the Estimation of Amazon Deforestation (Projeto PRODES Digital). Brazilian National Institute for Space Research. http://www.dpi.inpe.br/prodesdigital/prodes.php
- 37.Poly F, Monrozier LJ, Bally R. 2001. Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152:95–103. 10.1016/S0923-2508(00)01172-4 [DOI] [PubMed] [Google Scholar]
- 38.Piceno YM, Lovell CR. 2000. Stability in natural bacterial communities: I. Nutrient addition effects on rhizosphere diazotroph assemblage composition. Microb. Ecol. 39:32–40. 10.1007/s002489900192 [DOI] [PubMed] [Google Scholar]
- 39.Zehr JP, McReynolds L. 1989. Use of degenerate oligonucleotides for amplifcation of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl. Environ. Microbiol. 60:2113–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirza BS, Rodrigues JLM. 2012. Development of a direct isolation procedure for free-living diazotrophs under controlled hypoxic conditions. Appl. Environ. Microbiol. 78:5542–5549. 10.1128/AEM.00714-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lema KA, Willis BL, Bourne DG. 2012. Corals form specific associations with diazotrophic bacteria. Appl. Environ. Microbiol. 78:3136–3144. 10.1128/AEM.07800-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guindon S, Lethiec F, Duroux P, Gascuel O. 2005. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33:W557–W559. 10.1093/nar/gki352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirza BS, Welsh A, Rasul G, Rieder JP, Paschke MW, Hahn D. 2009. Variation in Frankia populations of the Elaeagnus host infection group in nodules of six host plant species after inoculation with soil. Microb. Ecol. 58:384–393. 10.1007/s00248-009-9513-0 [DOI] [PubMed] [Google Scholar]
- 48.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599. 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- 49.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamady M, Lozupone C, Knight R. 2010. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4:17–27. 10.1038/ismej.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clarke KR, Gorley RN. 2006. PRIMER v6: user manual/tutorial. PRIMER-E, Plymouth, United Kingdom [Google Scholar]
- 52.Hughes JB, Hellman JJ, Ricketts TH, Bohannan BJM. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67:4399–4406. 10.1128/AEM.67.10.4399-4406.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magurran AE. 1988. Ecological diversity and its measurement. Princeton University Press, Princeton, NJ [Google Scholar]
- 54.Anderson MJ, Gorley RN, Clarke KR. 2008. PERMANOVA+ for PRIMER: guide to software and statistical methods. PRIMER-E Ltd., Plymouth Marine Laboratory, Plymouth, United Kingdom [Google Scholar]
- 55.Gaby JC, Buckley DH. 2012. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7:e42149. 10.1371/journal.pone.0042149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zehr JP, Jenkins BD, Short SM, Steward GF. 2003. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ. Microbiol. 5:539–554. 10.1046/j.1462-2920.2003.00451.x [DOI] [PubMed] [Google Scholar]
- 57.Gaby JC, Buckley DH. 2011. A global census of nitrogenase diversity. Environ. Microbiol. 13:1790–1799. 10.1111/j.1462-2920.2011.02488.x [DOI] [PubMed] [Google Scholar]
- 58.Tilman D, Reich PB, Knops J, Wedin D, Mielke T, Lehman C. 2001. Diversity and productivity in a long-term grassland experiment. Science 294:843–845. 10.1126/science.1060391 [DOI] [PubMed] [Google Scholar]
- 59.Zak DR, Holmes WE, White DC, Peacock AD, Tilman D. 2003. Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84:2042–2050. 10.1890/02-0433 [DOI] [Google Scholar]
- 60.Norby RJ, Iversen CM. 2006. Nitrogen uptake, distribution, turnover, and efficiency of use in a CO2-enriched sweetgum forest. Ecology 87:5–14. 10.1890/04-1950 [DOI] [PubMed] [Google Scholar]
- 61.Reis VM, dos Reis FB, Jr, Quesada DM, de Oliveira OCA, Alves BJR, Urquiaga S, Boddey RM. 2001. Biological nitrogen fixation associated with tropical pasture grasses. Aust. J. Plant Physiol. 28:837–844. 10.1071/PP01079 [DOI] [Google Scholar]
- 62.Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U. S. A. 103:626–631. 10.1073/pnas.0507535103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hirsh AI, Little WS, Houghton RA, Scott NA, White JD. 2004. The net carbon flux due to deforestation and forest re-growth in the Brazilian Amazon: analysis using a process-based model. Global Change Biol. 10:908–924. 10.1111/j.1529-8817.2003.00765.x [DOI] [Google Scholar]
- 64.Vitousek PM, Cassman K, Cleveland C, Crews T, Field CB, Grimm NB, Howarth RW, Marino R, Martinelli L, Rastetter EB, Sprent JI. 2002. Towards an ecological understanding of biological nitrogen fixation. Biogeochemistry 57-58:1–45. 10.1023/a:1015798428743 [DOI] [Google Scholar]
- 65.Feigl B, Cerri C, Piccolo M, Noronha N, Augusti K, Melillo J, Eschenbrenner V, Melo L. 2006. Biological survey of a low-productivity pasture in Rondonia State, Brazil. Outlook Agric. 35:199–208. 10.5367/000000006778536738 [DOI] [Google Scholar]
- 66.Belay N, Sparling R, Daniels L. 1984. Dinitrogen fixation by a thermophilic methanogenic bacterium. Nature 312:286–288. 10.1038/312286a0 [DOI] [PubMed] [Google Scholar]
- 67.Murray PA, Zinder SH. 1984. Nitrogen fixation by a methanogenic archaebacterium. Nature 312:284–286. 10.1038/312284a0 [DOI] [Google Scholar]
- 68.Fernandes SAP, Bernoux M, Cerri CC, Feigl BJ, Piccolo MC. 2002. Seasonal variation of soil chemical properties and CO2 and CH4 fluxes in unfertilized and P-fertilized pastures in an Ultisol of the Brazilian Amazon. Geoderma 107:227–241. 10.1016/S0016-7061(01)00150-1 [DOI] [Google Scholar]
- 69.Davidson EA, de Carvalho CJR, Figueira AM, Ishida FY, Ometto JP, Nardoto GB, Saba RT, Hayashi SN, Leal EC, Vieira IC, Martinelli LA. 2007. Recuperation of nitrogen cycling in Amazonian forests following agricultural abandonment. Nature 447:995–996. 10.1038/nature05900 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.