

Abstract
Insertion sequences (ISs) are the simplest transposable elements and are widely distributed in bacteria; however, they also play important roles in genome evolution. We recently identified a protein called IS excision enhancer (IEE) in enterohemorrhagic Escherichia coli (EHEC) O157. IEE promotes the excision of IS elements belonging to the IS3 family, such as IS629, as well as several other families. IEE-mediated IS excision generates various genomic deletions that lead to the diversification of the bacterial genome. IEE has been found in a broad range of bacterial species; however, among sequenced E. coli strains, IEE is primarily found in EHEC isolates. In this study, we investigated non-EHEC pathogenic E. coli strains isolated from domestic animals and found that IEE is distributed in specific lineages of enterotoxigenic E. coli (ETEC) strains of serotypes O139 or O149 isolated from swine. The iee gene is located within integrative elements that are similar to SpLE1 of EHEC O157. All iee-positive ETEC lineages also contained multiple copies of IS629, a preferred substrate of IEE, and their genomic locations varied significantly between strains, as observed in O157. These data suggest that IEE may have been transferred among EHEC and ETEC in swine via SpLE1 or SpLE1-like integrative elements. In addition, IS629 is actively moving in the ETEC O139 and O149 genomes and, as in EHEC O157, is promoting the diversification of these genomes in combination with IEE.
INTRODUCTION
Insertion sequence (IS) elements are the simplest transposable elements and are considered selfish (or parasitic) genetic elements. However, they also play important roles in genome evolution (1). The transposition and proliferation of IS elements induces not only insertional gene inactivation and the modification of gene expression (1) but also a variety of genomic rearrangements, such as deletions, inversions, and duplications (2, 3). In bacteria, several thousand types of IS elements have been identified from various species and strains (4) and classified into approximately 20 families (5).
IS-mediated bacterial genome diversification has been extensively studied in enterohemorrhagic Escherichia coli (EHEC) O157. EHEC O157 strains produce highly potent cytotoxins (Shiga toxins Stx1 and/or Stx2) and causes diarrhea, hemorrhagic colitis, and hemolytic-uremic syndrome; thus, it is one of the most serious food-borne infections worldwide (6). O157 strains contain many IS elements, and these elements play important roles in the diversification of the O157 genome. For example, the O157 strain RIMD0509952 (referred to as O157 Sakai) contains 25 types of IS elements (116 copies in total), and the most abundant is an IS3 family member, IS629 (23 copies) (7, 8). Our comparative genomic analysis of O157 clinical isolates revealed that many small structural polymorphisms associated with gene inactivation and/or deletion have been generated by IS629 (9). More recently, we identified a novel protein called IS excision enhancer (IEE), which promotes IS629 excision from the O157 genome in a transposase (TPase)-dependent manner. We demonstrated that various types of genomic deletions were generated upon IEE-mediated IS excision in IS-flanking regions (10). IEE also promotes the excision of other members of the IS3, IS1, and IS30 families.
In the O157 genome, the gene encoding IEE (iee) is located in a large integrative element (IE) called SpLE1 (7). In non-O157 EHEC strains, iee is located on SpLE1-like IEs (11, 12). IEE homologs have been identified in a broad range of bacterial species and are encoded in genomic regions exhibiting low GC content and/or containing genes related to mobile genetic elements (MGEs) (10). These results suggest that IEE and its homologs have spread to a variety of bacterial strains by horizontal gene transfer. Although many E. coli strains have been sequenced, IEE is found primarily in EHEC isolates (10).
Pathogenic E. coli strains other than EHEC are also important etiological agents of zoonotic or food-borne disease in humans and of colibacillosis in domestic animals (13, 14). Enterotoxigenic E. coli (ETEC) is an important cause of diarrhea in children, which is associated with high morbidity and mortality in nonindustrialized countries. ETEC is the main cause of diarrhea in travelers to these countries (13). In swine, ETEC infections immediately after birth (neonatal diarrhea) and ETEC or Shiga toxin-producing E. coli (STEC) infections after weaning (postweaning diarrhea or edema disease) are responsible for significant economic losses due to diarrhea, growth retardation, and mortality (15, 16).
In this study, we examined the prevalence of iee in non-EHEC pathogenic E. coli strains isolated from domestic animals. Because the result indicated that the iee gene is distributed in specific lineages of ETEC isolated from swine, we further investigated the genomic structures of iee-containing IEs and the prevalence of IS elements that could be substrates for IEE in these ETEC strains.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
We investigated 256 E. coli strains, all of which were isolated from diseased domestic animals in Japan between 1991 and 2010 (Table 1). The serotypes of the strains were determined using antisera obtained from Denka Seiken Co., Ltd. (Tokyo, Japan), or Statens Serum Institut (Copenhagen, Denmark). O157 Sakai and three sequenced non-O157 EHEC strains (O26:H11 strain 11368, O111:H− strain 11128, and O103:H2 strain 12009; all three were isolated from patients in Japan [11]) were also used. All strains were grown in Luria-Bertani (LB) broth (17) at 37°C.
TABLE 1.
Prevalence of the iee gene in E. coli isolated from domestic animals
| Serotype | No. of isolates (ETEC strains) |
|||
|---|---|---|---|---|
| Carrier animal |
iee gene positive | |||
| Swine | Chickena | Other(s) | ||
| O2 | 11 (0) | 0 | 0 | 0 |
| O7 | 0 | 1 (0) | 0 | 0 |
| O8 | 5 (4) | 2 (0) | 0 | 0 |
| O10 | 0 | 4 (0) | 0 | 0 |
| O15 | 0 | 1 (0) | 0 | 0 |
| O16 | 1 (1) | 0 | 0 | 0 |
| O17 | 0 | 1 (0) | 0 | 0 |
| O18 | 0 | 1 (0) | 0 | 0 |
| O19 | 0 | 7 (0) | 0 | 0 |
| O25 | 0 | 1 (0) | 0 | 0 |
| O35 | 1 (1) | 0 | 0 | 0 |
| O39 | 0 | 3 (0) | 0 | 0 |
| O45 | 2 (1) | 2 (0) | 0 | 0 |
| O56 | 5 (4) | 0 | 0 | 0 |
| O68 | 0 | 1 (0) | 0 | 0 |
| O76 | 0 | 0 | 3b (0) | 0 |
| O84 | 0 | 1 (0) | 0 | 0 |
| O98 | 5 (5) | 0 | 0 | 0 |
| O103 | 1 (0) | 0 | 0 | 0 |
| O115 | 1 (0) | 0 | 0 | 0 |
| O116 | 15 (15) | 0 | 0 | 0 |
| O119 | 0 | 5 (0) | 0 | 0 |
| O121 | 2 (1) | 0 | 0 | 0 |
| O123 | 0 | 1 (0) | 0 | 0 |
| O132 | 1 (0) | 0 | 0 | 0 |
| O135 | 0 | 2 (0) | 0 | 0 |
| O138 | 6 (6) | 0 | 0 | 0 |
| O139 | 56 (23) | 0 | 0 | 23 (23) |
| O141 | 4 (2) | 0 | 0 | 0 |
| O149 | 17 (17) | 0 | 0 | 16 (16) |
| O157 | 2 (2) | 0 | 0 | 0 |
| O161 | 0 | 1 (0) | 0 | 0 |
| O164 | 3 (3) | 0 | 0 | 0 |
| O165 | 0 | 0 | 1c (0) | 0 |
| O167 | 0 | 1 (0) | 0 | 0 |
| O169 | 1 (0) | 0 | 0 | 0 |
| Untypeable or not tested | 19 (2) | 59 (0) | 0 | 0 |
| Total | 158 (87) | 94 (0) | 4 (0) | 39 (39) |
These strains were all isolated from internal organs of chickens with extraintestinal diseases.
These three strains were isolated from goats.
This strain was isolated from cattle.
PCR-based genotyping of E. coli strains.
The presence of iee and a panel of major IS elements (IS629, IS911, IS3, IS2, IS1, IS4, IS5, IS26, IS30, and IS621) in the E. coli strains was examined by PCR using the primers listed in Table S1 in the supplemental material. The template DNA for PCR was prepared by the alkaline-boiling method, as previously described (18). PCR was performed in a 50-μl reaction mixture containing template DNA, 0.2 μM concentrations of each primer, 0.2 mM concentrations of each deoxynucleoside triphosphate (dNTP), PCR buffer, and 1.25 U of ExTaq DNA polymerase (TaKaRa Bio, Inc., Shiga, Japan) using 30 amplification cycles of 30 s at 95°C, 30 s at 60°C, and 1 min at 72°C. PCR amplification of genes encoding various virulence factors (VFs; LT, STa, STb, EAST1, Stx1, Stx2, F4, F5, and F18) was performed as described by Vu-Khac et al. (19).
PFGE.
Pulsed-field gel electrophoresis (PFGE) was performed by clamped homogeneous electric field electrophoresis using a CHEF-DR III apparatus (Bio-Rad Laboratories, Inc., Hercules, CA). The genomic DNA of each strain was prepared as described by Akiba et al. (20). Genomic DNA in sliced plugs was digested at 37°C with 40 U of XbaI for 6 h or 30 U of SspI for 16 h (both enzymes were obtained from TaKaRa Bio, Inc.). Electrophoresis was performed in a 1% agarose gel in 0.5× Tris-borate-EDTA (TBE) buffer at 14°C at 6 V/cm for 22 h with a pulse time of 5 to 50 s (XbaI PFGE) or for 10 h with a pulse time of 4 s (SspI PFGE). For PFGE of XbaI-digested DNA, 100 μM thiourea was added to the TBE buffer to obtain clear banding patterns (21). Southern blot hybridization analysis of SspI-digested DNA was performed using an IS629-specific probe, as previously described (10).
The banding patterns obtained by PFGE of XbaI-digested DNA were analyzed using BioNumerics software version 5.1 (Applied Maths, Sint-Martens-Latem, Belgium), followed by manual band assignment. Dendrograms were then generated using the unweighted pair group method with arithmetic mean (UPGMA) based on the Dice similarity index and with an optimization parameter of 1% band position tolerance.
MLST and phylogenetic tree construction.
Multilocus sequence typing (MLST) was performed using the nucleotide sequences of the seven housekeeping genes adk, fumC, gyrB, icd, mdh, purA, and recA according to the protocols available in the E. coli MLST database (http://mlst.ucc.ie/mlst/mlst/dbs/Ecoli) (22). To determine the phylogenetic relationships of the E. coli strains, we concatenated the nucleotide sequences of the seven genes used for the MLST to generate pseudosequences and aligned them using CLUSTAL W in the software MEGA5 (23). A neighbor-joining tree was generated with a 1,000 bootstrap replicates.
Analysis of the genomic structures of iee-containing elements.
The genomic structures of the SpLE1-like elements of the O139 and O149 strains were analyzed by PCR scanning, which is a long-range PCR-based genome comparison system (24). The primers and PCR conditions were identical to those previously described by Ohnishi et al. for whole-genome PCR scanning (WGPS) of O157 strains (24), except that the primer ieeIE-f was used instead of primer 113.3-f. To analyze the region of interest in strain E0231, we used the newly designed primer 113.9-r2 instead of 113.9-r because no amplicon was produced when the primer pair 113.8-f/113.9-r was used with this strain. In addition, to amplify both SpLE1-like IE/chromosome junctions in E0231, we designed primers IE0231-f and IE0231-r based on the results of random extension-based two-step PCR (RETS-PCR, a newly developed walking method described below). The sequences for the primers used in this analysis are shown in Table S2 in the supplemental material.
RETS-PCR.
We developed a rapid walking method designated RETS-PCR to determine the sequences of the SpLE1-like IE/chromosome junctions in strain E0231. As outlined in Fig. 1, the RETS-PCR-based method comprises three steps: random extension, PCR using a single primer, and sequence determination (Fig. 1 presents the process used to analyze the left junction). In the first step, primer E0231J-R1, which comprises a known sequence in the SpLE1-like IE (denoted as “X” in Fig. 1) and a random 9-base sequence in the 3′ region, was used to generate various single-stranded DNA (ssDNA) molecules by random extension using DNA polymerase (Fig. 1A). This step was performed in a 25-μl reaction mixture containing template DNA, 0.2 μM primer E0231J-R1, 0.2 mM concentrations of each dNTP, PCR buffer, and 0.625 U ExTaq DNA polymerase for 5 min at 95°C, 30 s at 30°C, and 1 min at 72°C. In the second step (Fig. 1B), the ssDNA molecules act as templates for single-primer PCR using primer E0231J-R2, and thus only molecules containing sequence X permit the amplification of DNA segments encompassing the junction. This step was performed in a 100-μl reaction mixture containing the total product of the first step, 0.4 μM primer E0231J-R2, 0.2 mM concentrations of each dNTP, PCR buffer, and 2.5 U of ExTaq DNA polymerase with 35 amplification cycles of 30 s at 95°C, 30 s at 60°C, and 1 min at 72°C. In the third step (Fig. 1C), the sequences of the amplicons were determined using the primer E0231J-R3, the sequence of which corresponds to an upstream region of X (denoted “Y” in Fig. 1C). The nucleotide sequences were identified by the dideoxy chain termination method (25) using a BigDye terminator cycle sequencing kit and an 3130xl sequencer (Applied Biosystems, Inc., Foster City, CA) according to the manufacturer's instructions. Similarly, the right junction was determined using the primers E0231J-F1, -F2, and -F3. The sequences for the primers used in this analysis are shown in Table S3 in the supplemental material.
FIG 1.

RETS-PCR and the process used to determine unknown SpLE1-like IE/chromosome junctions. As an example, the process to determine the left SpLE1-like IE/chromosome junction in strain E0231 is presented. (A) In the first step, various ssDNA molecules are generated by random extension using primer E0231J-R1, which comprises a known sequence in the SpLE1-like IE (denoted as “X”) and a random 9-base sequence. (B) In the second step, the ssDNA molecules act as templates for single-primer PCR (using primer E0231J-R2, which anneals only to sequence X) to amplify the DNA segments encompassing the junction between the known and unknown sequences. (C) In the third step, the sequences of the amplicons are determined using primer E0231J-R3, which aligns to a region located between the junction and X (denoted Y).
Sequence determination and genomic comparison of SpLE1-like IEs.
Amplicons covering the entire SpLE1-like IE of each strain, which were generated by PCR scanning as described above, were combined and sequenced using the Illumina MiSeq platform (Illumina, Inc., San Diego, CA). Libraries for each mixture were prepared using the Nextera XT DNA Sample Prep kit (Illumina, Inc.), and pooled libraries were subjected to multiplexed paired-end sequencing (251 cycles × 2) according to the manufacturer's protocol. The sequence reads were assembled using Velvet version 1.2.05 (26). The obtained sequences were annotated with the Microbial Genome Annotation Pipeline (http://www.migap.org/) (27) and were manually curated using IMC-GE software (In Silico Biology, Inc., Kanagawa, Japan). The sequence comparison of SpLE1-like IEs was performed using GenomeMatcher software (28).
Nucleotide sequence accession numbers.
The sequences of the SpLE1-like IEs from strains E0046, E0092, E0124, E0217, E0223, and E0231 have been deposited in DDBJ/EMBL/GenBank under accession numbers AB786874 to AB786879.
RESULTS AND DISCUSSION
Screening, genotyping, and phylogenetic analysis of iee-positive E. coli strains.
We first determined the serotypes of 256 E. coli strains isolated from diseased domestic animals and identified iee-positive strains by PCR; iee was found only in strains of serotypes O139 (23 of 56 strains) and O149 (16 of 17 strains) isolated from swine (Table 1 and Fig. 2). O139 and O149 are major serotypes of ETEC that are associated with diarrhea in swine, and O139 is also one of the major serotypes of STEC that causes edema disease (15).
FIG 2.

Genotyping and phylogenetic analysis of E. coli strains. A dendrogram obtained by PFGE of XbaI-digested DNA from 73 E. coli O139 and O149 strains is shown on the left side. Information about each strain, the results of the PCR screening for genes encoding IEEs, IS elements, and VFs, and their STs as determined by MLST are aligned with the dendrogram. IS629, IS911, IS3, and IS2 are all members of the IS3 family. The presence or absence of each gene (or IS element) is indicated by an open or black square, respectively.
The 73 O139 and O149 strains were further examined by PCR for the presence of major IS elements of “pathogenic” E. coli and for genes encoding known VFs of ETEC and STEC (Fig. 2). PFGE analysis was also performed after digesting the genomic DNA with XbaI, and a dendrogram was generated to analyze the relatedness of these strains (Fig. 2). In addition, the sequence types (STs) of these strains were determined using the E. coli MLST database (22). The results of the clustering analysis of the 73 strains based on their XbaI digestion patterns correlated very well with the clustering based on the MLST analysis, and the strains were divided into three groups on the basis of their serotypes and VF profiles: ETEC O149 (ST100 or ST2273), ETEC O139 (ST42), and STEC O139 (ST1 or ST2290). ST2290 is closely related to ST1 and contains only one single-nucleotide polymorphism.
Among the 17 ETEC O149 strains, only strain E0129 lacked iee. However, E0129 belongs to ST100 (a member of the ST165 clonal complex), like most of the iee-positive ETEC O149 strains, and its genotype was also very similar to those of the iee-positive strains (Fig. 2). All ETEC O139 strains possessed iee, but this gene was not found in any of the STEC O139 strains. Interestingly, the distribution of iee among the O149 and O139 strains correlated very well with the distribution of two IS3 family members, IS629 and IS911 (Fig. 2). Although we identified ETEC strains with various serotypes isolated from swine (Table 1), the iee-positive strains were limited to strains of the O139 and O149 serotypes.
To determine the phylogenetic relationships of the ETEC O139 and O149 strains, we constructed a phylogenetic tree using the concatenated nucleotide sequences of the seven housekeeping genes used for MLST. In total, 50 genome-sequenced E. coli strains were included in this analysis (Fig. 3). Two of the 50 strains were the recently sequenced ETEC strains UMNK88 and UMNF18 (serotypes O149 and O147, respectively) (29). UMNK88 belongs to ST100, but UMNF18 belongs to ST10, which includes E. coli K-12. As shown in Fig. 3, the ETEC O139 (ST42) and STEC O139 (ST1 and ST2290) strains are relatively closely related to each other but distantly related to the two ETEC O149 lineages (ST100 or ST2273), which are located in distinct phylogenetic clusters. ST100 belongs to a cluster that also contains ST10. ST2273 belongs to a cluster that contains EHEC O26, O111, and O103 but is more closely related to O26 and O111 than to O103. These results indicate that IEE has spread to specific lineages of ETEC and EHEC strains by horizontal gene transfer.
FIG 3.
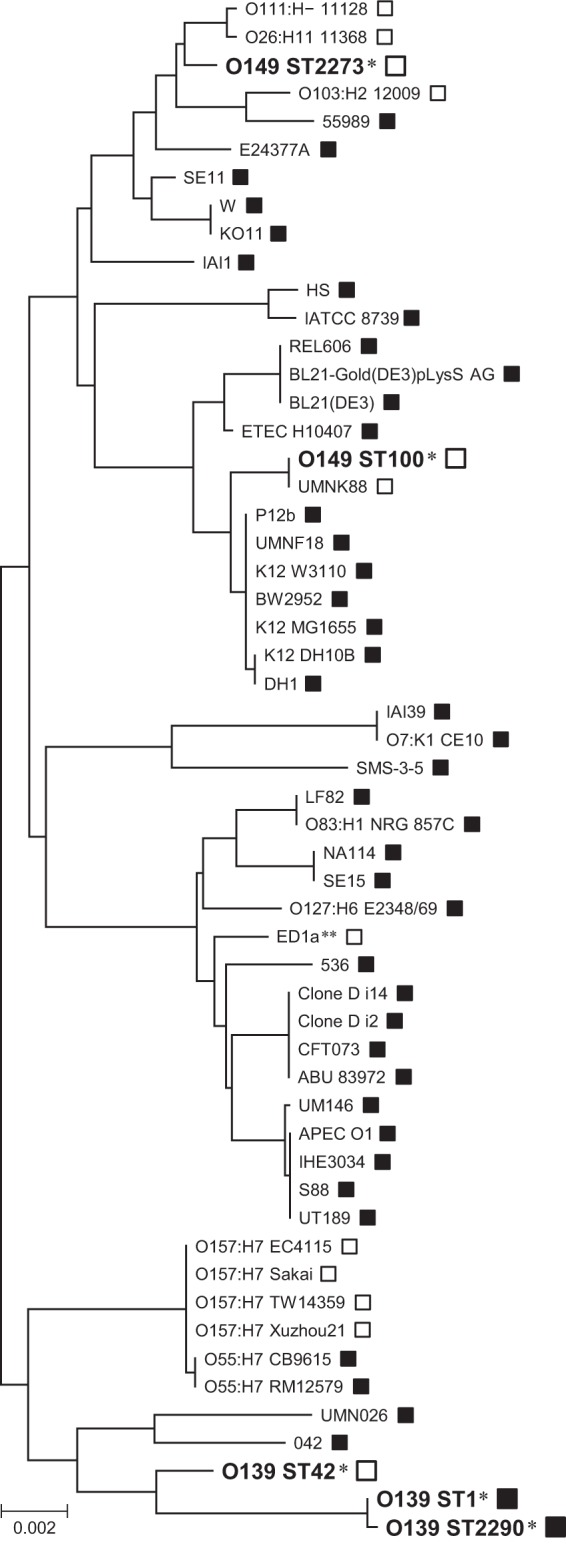
Phylogenetic tree of E. coli strains based on the sequences of seven housekeeping genes. The sequences of the seven housekeeping genes obtained in the MLST analysis were concatenated and aligned using CLUSTAL W in MEGA5 software (23), and a neighbor-joining tree was generated with 1,000 bootstrap replicates. All genome-sequenced E. coli strains are included in the phylogenetic representation, and the O139 and O149 lineages analyzed in the present study are indicated by a single asterisk (*). The scale bar represents the number of base substitutions. Open and black boxes indicate iee-positive and -negative strains, respectively. The ED1a strain marked with double asterisks (**) possesses iee on an IE that is not similar to SpLE1 (10).
Analysis of iee-containing integrated elements.
The iee gene was found on the IE “UMNK88 island 8” in the O149 strain UMNK88 (29); this IE is similar to SpLE1 and the SpLE1-like IEs of EHEC, suggesting that iee may be encoded by SpLE1-like IEs in the iee-positive ETEC strains identified in the present study. To investigate the presence of SpLE1-like IEs and analyze their genomic structures in the O139 and O149 ETEC strains, we performed PCR scanning analysis as illustrated in Fig. S1A in the supplemental material. The results indicated that these strains contain SpLE1-like IEs, with the exception of strain E0231 (see Fig. S1B in the supplemental material; see also Fig. S2 in the supplemental material for the raw data). In E0231, no amplicons were obtained from the two segments containing the left and right SpLE1-like IE/chromosome junctions (ieeIE-f/113.4-r and 113.9-f/114-r). In addition, no amplicon was generated using the primers 113.8-f/113.9-r in E0231, but this region was amplified using a newly designed primer, 113.9-r2, as a substitute for 113.9-r (see Fig. S2B in the supplemental material), suggesting that some sequence polymorphism exists in the 113.9-r site of the E0231 genome. By sequencing the ieeIE-f/113.4-r amplicons, we confirmed that the amplified regions all contained the iee genes having an identical sequence. Importantly, no amplicon was obtained from any segments of any of the iee-negative O139 STEC strains examined. These results indicate that the O139 ETEC and O149 ETEC lineages acquired iee by SpLE1-like IEs.
The sequencing analysis of the ieeIE-f/113.4-r and 113.9-f/114-r amplicons revealed that SpLE1-like IEs are integrated in the serX tRNA gene in all O139 and O149 ETEC strains examined, with the exception of strain E0231 (see Fig. S2B in the supplemental material). The integration site of the SpLE1-like IE of E0231 (the serW tRNA gene) was identified using a newly developed rapid walking method designated RETS-PCR, as outlined in Fig. 1. Similar positional variations of SpLE1 were observed in EHEC O157; strain EDL933 contains two copies of SpLE1: one at serX and the other at serW (30). In the eight O157 strains that were analyzed by Ohnishi et al. using WGPS, SpLE1 was also found at the serX and/or serW loci (24). However, this variation in the integration site is not surprising because the serX and serW genes have identical nucleotide sequences and because a highly conserved phage-type integrase is shared by SpLE1 and SpLE1-like IEs.
In strain E0129 (the only iee-negative O149 ETEC strain), the serW and serX loci were both intact and showed no sign of the insertion of MGEs, as in the iee-negative O139 STEC strains. Because E0129 also belongs to ST100, as do most of the O149 ETEC strains, it is likely that this element has been deleted in this strain.
Comparative analysis of SpLE1-like IEs.
The nucleotide sequences of SpLE1-like IEs from three ETEC O139 strains (E0046, E0124, and E0217) and three O149 strains (E0092, E0223, and E0231) were determined and compared to those of SpLE1 in O157 Sakai; SpLE1-like IEs in O26, O111, and O103 EHECs (strains 11368, 11128, and 12009, respectively); and island 8 in O149 UMNK88. Overall, the structures of the SpLE1-like IEs in the seven O139 and O149 ETEC strains were very similar to each other, whereas the deletion of an 8.7-kb segment was observed in the element of strain E0231 (Fig. 4). Although the elements of O139/O149 ETEC are also similar to SpLE1 and the SpLE1-like IEs of the O26 and O111 EHEC strains, their right end regions, particularly the far-right region have significantly diverged in sequence from those of the elements of O157, O26, and O111 (Fig. 4; see also Fig. S3 in the supplemental material). All other structural variations observed between the elements of EHEC strains and those of O139/O149 ETEC were small, and most appeared to have been generated by the insertion (or deletion) of IS elements. Considering that the three iee-positive ETEC O139 and O149 lineages are phylogenetically distant from each other and that the ST2273 lineage (O149 strain E0231) is closely related to O26 and O111 (Fig. 3), these findings suggest that the SpLE1-like IEs of these O139/O149 strains have been recently derived from an ancestor common to the EHEC elements and jumped into the three ETEC O139 and O149 lineages.
FIG 4.

Genomic comparison of the SpLE1-like IEs of the ETEC strains with the SpLE1 of EHEC O157 and the SpLE1-like IEs of non-O157 EHEC. The genes in each element are indicated by arrows; orange, blue, red, dark green, light green, and open arrows indicate the genes for IEE, integrase, IS TPase, Iha, the urease operon, and other functions, respectively. The locations of two copies of IS629 in SpLE1 (one is intact and the other is truncated) are also indicated. The nucleotide sequence identities between the elements (cutoff ≥ 70% identity) are indicated by color shading according to the scale shown at the bottom of the figure.
Yin et al. reported that the iha gene (which encodes a putative adhesin, Iha) and the ure operon (which encodes urease), both of which are encoded within SpLE1, are required for the efficient colonization of EHEC O157 strain EDL933 (the IE corresponding to SpLE1 is named “O island 48” in EDL933 [30]) in swine intestines (31). Because the iha gene and the ure operon are conserved in the SpLE1-like IEs of ETEC O139 and O149 (Fig. 4), the acquisition of SpLE1-like IEs may confer an advantage to these ETEC strains in colonizing the swine intestine. Although healthy cattle are considered the major reservoir for human infection with EHEC O157 (6), this microorganism has also been isolated from swine (32–35). Thus, although further studies are required, EHEC O157 and ETEC O139 and O149 may share an ecological niche (the swine intestine), thus allowing the transfer of SpLE1 or SpLE1-like IEs among these strains. Because genes required for conjugal transfer are not found in these IEs, a molecular mechanism underlying their transmission is another important issue to be elucidated.
IS629 in ETEC O139 and O149 strains and variation in its genomic copy number and insertion sites.
IEE promotes the excision of IS629 and other IS3 family members in a TPase-dependent manner. It also induces various genomic deletions upon IS excision and is thus implicated in the diversification of bacterial genomes (10). The genomic locations of IS629 in the O157 genomes are highly variable and show complex patterns among the O157 strains (9). Because all iee-positive ETEC O139 and O149 strains also contain IS629, we investigated the genomic locations of IS629 in these strains by PFGE analysis of SspI-digested genomic DNA, which was followed by Southern blot hybridization analysis using an IS629-specific probe (Fig. 5). This IS629 fingerprinting analysis revealed that all iee-positive O139 and O149 strains possess multiple copies of IS629, a preferred substrate of IEE (10).
FIG 5.
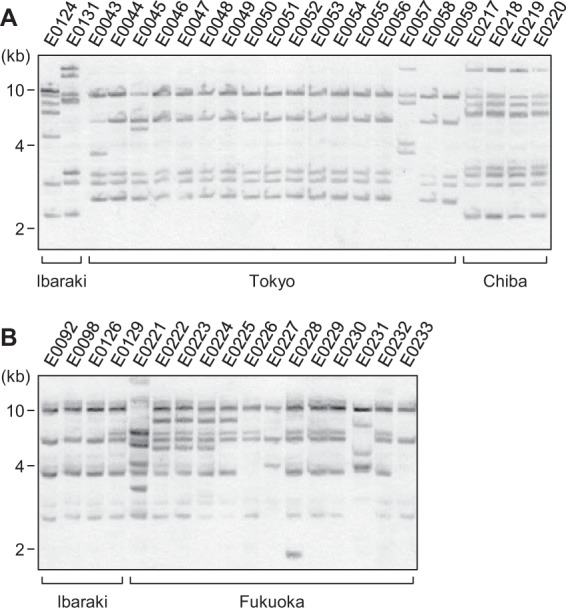
Southern blot hybridization analysis of the ETEC O139 (A) and O149 (B) strains using an IS629-specific probe. SspI-digested genomic DNA was separated by PFGE and subjected to Southern blot hybridization analysis. The prefectures where the strains were isolated are indicated. The DNA probe was derived from the central part of the IS629 sequence (nucleotide positions 308 to 607).
Intriguingly, SpLE1 of EHEC O157 carries two copies of IS629 (one is intact, and the other is truncated), and all sequenced SpLE1-like IEs of the ETEC O139 and O149 strains also carry one truncated IS629 copy. This result raises the possibility that these ETEC strains acquired at least one IS629 copy, together with iee, via the transfer of an SpLE1-like IE, followed by the transposition and proliferation of IS629 in these strains. We cannot exclude the possibility that these ETEC strains acquired IS629 independently from SpLE1-like IEs, but the potential cotransfer of IEE and IS629 merits further investigation.
The IS629 fingerprinting patterns of the ETEC O139 and O149 strains also revealed considerable variation in copy numbers between these strains, as observed in EHEC O157. Although 17 O139 strains isolated in the Tokyo prefecture (E0043 to E0059) exhibited very similar XbaI digestion patterns in PFGE (Fig. 2), suggesting that they are closely related, some of the strains displayed IS629 fingerprinting patterns that were distinct from the major pattern in this group (Fig. 5B). Similarly, O149 strains E0226 and E0227, which exhibited similar XbaI digestion patterns in PFGE, exhibited remarkable variation in their IS629 fingerprinting patterns. Thus, it appears that IS629 has actively transposed in these ETEC O139 and O149 strains, but it is also possible that some of the variation in the copy number of IS629 is attributable to IEE-mediated IS excision, which could also have generated deletions in IS-flanking regions (10).
IEE promotes the excision of other IS3 family members, such as IS911, IS3, and IS2, at the same efficiency as that for IS629 (10). All of the IS elements are present in almost all iee-positive ETEC O139 and O149 lineages (Fig. 1). Although the excision frequencies of IS1 and IS30 is lower than that for the IS3 family, these IS elements could also be substrates of IEE (10). IS1 is distributed among all iee-positive ETEC O139 and O149 strains. IS30 is found in one ETEC O149 lineage (ST100). Thus, although further research is required, the acquisition of these IS elements other than IS629 may have accelerated the genome diversification of ETEC O139 and O149, which in turn could have introduced important phenotypic variations in each lineage.
Conclusions.
This study demonstrated that IEE is distributed specifically among three distinct ETEC lineages isolated from swine and is encoded by IEs similar to SpLE1 of EHEC O157. The SpLE1-like IEs are highly conserved in genomic structure among these ETEC lineages, and similar to SpLE1, they carry the iha gene and the ure operon, which are shown to be required for the efficient colonization of O157 in the swine intestine. These data suggest that IEE may have been transferred among EHEC and ETEC in swine via the acquisition of SpLE1-like IEs. Furthermore, because the IEE-positive ETEC lineages all contained multiple copies of IS629, a preferred substrate of IEE, and their genomic locations vary significantly between strains, IS629 is likely actively moving on the ETEC genomes. As in O157, in combination with IEE, IS629 is likely promoting the diversification of the ETEC genome.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by a Grant-in-Aid for Scientific Research (C), KAKENHI grant 24590543 to M.K., from the Japan Society for the Promotion of Science.
We thank Noriko Ido, Hiroko Matsukawa, Atsuko Matsumoto, Hiroto Nishino, Torata Ogawa, Horoshi Yoshizaki, and all of the prefectural Livestock Hygiene Service Centers for providing E. coli isolates and their strain information.
Footnotes
Published ahead of print 13 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03696-13.
REFERENCES
- 1.Sinzelle L, Izsvak Z, Ivics Z. 2009. Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell. Mol. Life Sci. 66:1073–1093. 10.1007/s00018-009-8376-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kothapalli S, Nair S, Alokam S, Pang T, Khakhria R, Woodward D, Johnson W, Stocker BA, Sanderson KE, Liu SL. 2005. Diversity of genome structure in Salmonella enterica serovar Typhi populations. J. Bacteriol. 187:2638–2650. 10.1128/JB.187.8.2638-2650.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G, III, Rose DJ, Darling A, Mau B, Perna NT, Payne SM, Runyen-Janecky LJ, Zhou S, Schwartz DC, Blattner FR. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 71:2775–2786. 10.1128/IAI.71.5.2775-2786.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34:D32–D36. 10.1093/nar/gkj014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandler M, Mahillon J. 2002. Insertion sequences revisited, p 305–366 In Craig NL, Craigie R, Gellert M, Lambowitz AM. (ed), Mobile DNA II. ASM Press, Washington, DC [Google Scholar]
- 6.Mead PS, Griffin PM. 1998. Escherichia coli O157:H7. Lancet 352:1207–1212. 10.1016/S0140-6736(98)01267-7 [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Han CG, Ohtsubo E, Nakayama K, Murata T, Tanaka M, Tobe T, Iida T, Takami H, Honda T, Sasakawa C, Ogasawara N, Yasunaga T, Kuhara S, Shiba T, Hattori M, Shinagawa H. 2001. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8:11–22. 10.1093/dnares/8.1.11 [DOI] [PubMed] [Google Scholar]
- 8.Makino K, Ishii K, Yasunaga T, Hattori M, Yokoyama K, Yutsudo CH, Kubota Y, Yamaichi Y, Iida T, Yamamoto K, Honda T, Han CG, Ohtsubo E, Kasamatsu M, Hayashi T, Kuhara S, Shinagawa H. 1998. Complete nucleotide sequences of 93-kb and 3.3-kb plasmids of an enterohemorrhagic Escherichia coli O157:H7 derived from Sakai outbreak. DNA Res. 5:1–9. 10.1093/dnares/5.1.1 [DOI] [PubMed] [Google Scholar]
- 9.Ooka T, Ogura Y, Asadulghani M, Ohnishi M, Nakayama K, Terajima J, Watanabe H, Hayashi T. 2009. Inference of the impact of insertion sequence (IS) elements on bacterial genome diversification through analysis of small-size structural polymorphisms in Escherichia coli O157 genomes. Genome Res. 19:1809–1816. 10.1101/gr.089615.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kusumoto M, Ooka T, Nishiya Y, Ogura Y, Saito T, Sekine Y, Iwata T, Akiba M, Hayashi T. 2011. Insertion sequence-excision enhancer removes transposable elements from bacterial genomes and induces various genomic deletions. Nat. Commun. 2:152. 10.1038/ncomms1152 [DOI] [PubMed] [Google Scholar]
- 11.Ogura Y, Ooka T, Iguchi A, Toh H, Asadulghani M, Oshima K, Kodama T, Abe H, Nakayama K, Kurokawa K, Tobe T, Hattori M, Hayashi T. 2009. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 106:17939–17944. 10.1073/pnas.0903585106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogura Y, Ooka T, Asadulghani Terajima J, Nougayrede JP, Kurokawa K, Tashiro K, Tobe T, Nakayama K, Kuhara S, Oswald E, Watanabe H, Hayashi T. 2007. Extensive genomic diversity and selective conservation of virulence-determinants in enterohemorrhagic Escherichia coli strains of O157 and non-O157 serotypes. Genome Biol. 8:R138. 10.1186/gb-2007-8-7-r138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140. 10.1038/nrmicro818 [DOI] [PubMed] [Google Scholar]
- 14.Nataro JP, Kaper JB. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frydendahl K. 2002. Prevalence of serogroups and virulence genes in Escherichia coli associated with postweaning diarrhoea and edema disease in pigs and a comparison of diagnostic approaches. Veterinary microbiology. 85:169–182. 10.1016/S0378-1135(01)00504-1 [DOI] [PubMed] [Google Scholar]
- 16.Holland RE. 1990. Some infectious causes of diarrhea in young farm animals. Clin. Microbiol. Rev. 3:345–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J, Russel DW. 2001. Molecular cloning, 3rd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 18.Ooka T, Terajima J, Kusumoto M, Iguchi A, Kurokawa K, Ogura Y, Asadulghani M, Nakayama K, Murase K, Ohnishi M, Iyoda S, Watanabe H, Hayashi T. 2009. Development of a multiplex PCR-based rapid typing method for enterohemorrhagic Escherichia coli O157 strains. J. Clin. Microbiol. 47:2888–2894. 10.1128/JCM.00792-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vu-Khac H, Holoda E, Pilipcinec E, Blanco M, Blanco JE, Dahbi G, Mora A, Lopez C, Gonzalez EA, Blanco J. 2007. Serotypes, virulence genes, intimin types and PFGE profiles of Escherichia coli isolated from piglets with diarrhoea in Slovakia. Vet. J. 174:176–187. 10.1016/j.tvjl.2006.05.019 [DOI] [PubMed] [Google Scholar]
- 20.Akiba M, Uchida I, Nishimori K, Tanaka K, Anzai T, Kuwamoto Y, Wada R, Ohya T, Ito H. 2003. Comparison of Salmonella enterica serovar Abortusequi isolates of equine origin by pulsed-field gel electrophoresis and fluorescent amplified-fragment length polymorphism fingerprinting. Veterinary microbiology. 92:379–388. 10.1016/S0378-1135(02)00422-4 [DOI] [PubMed] [Google Scholar]
- 21.Liesegang A, Tschape H. 2002. Modified pulsed-field gel electrophoresis method for DNA degradation-sensitive Salmonella enterica and Escherichia coli strains. Int. J. Med. Microbiol: 291:645–648. 10.1078/1438-4221-00180 [DOI] [PubMed] [Google Scholar]
- 22.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60:1136–1151. 10.1111/j.1365-2958.2006.05172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum-parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohnishi M, Terajima J, Kurokawa K, Nakayama K, Murata T, Tamura K, Ogura Y, Watanabe H, Hayashi T. 2002. Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. Proc. Natl. Acad. Sci. U. S. A. 99:17043–17048. 10.1073/pnas.262441699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74:5463–5467. 10.1073/pnas.74.12.5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugawara H, Ohyama A, Mori H, Kurokawa K. 2009. Microbial genome annotation pipeline (MiGAP) for diverse users. Poster and software demonstrations, S001-1-2; 20th International Conference on Genome Informatics (GIW2009), Yokohama, Japan [Google Scholar]
- 28.Ohtsubo Y, Ikeda-Ohtsubo W, Nagata Y, Tsuda M. 2008. GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics 9:376. 10.1186/1471-2105-9-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shepard SM, Danzeisen JL, Isaacson RE, Seemann T, Achtman M, Johnson TJ. 2012. Genome sequences and phylogenetic analysis of K88- and F18-positive porcine enterotoxigenic Escherichia coli. J. Bacteriol. 194:395–405. 10.1128/JB.06225-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perna NT, Plunkett G, III, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529–533. 10.1038/35054089 [DOI] [PubMed] [Google Scholar]
- 31.Yin X, Wheatcroft R, Chambers JR, Liu B, Zhu J, Gyles CL. 2009. Contributions of O island 48 to adherence of enterohemorrhagic Escherichia coli O157:H7 to epithelial cells in vitro and in ligated pig ileal loops. Appl. Environ. Microbiol. 75:5779–5786. 10.1128/AEM.00507-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakazawa M, Akiba M, Sameshima T. 1999. Swine as a potential reservoir of Shiga toxin-producing Escherichia coli O157:H7 in Japan. Emerg. Infect. Dis. 5:833–834. 10.3201/eid0506.990618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keen JE, Wittum TE, Dunn JR, Bono JL, Durso LM. 2006. Shiga-toxigenic Escherichia coli O157 in agricultural fair livestock, United States. Emerg. Infect. Dis. 12:780–786. 10.3201/eid1205.050984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milnes AS, Stewart I, Clifton-Hadley FA, Davies RH, Newell DG, Sayers AR, Cheasty T, Cassar C, Ridley A, Cook AJ, Evans SJ, Teale CJ, Smith RP, McNally A, Toszeghy M, Futter R, Kay A, Paiba GA. 2008. Intestinal carriage of verocytotoxigenic Escherichia coli O157, Salmonella, thermophilic Campylobacter and Yersinia enterocolitica, in cattle, sheep and pigs at slaughter in Great Britain during 2003. Epidemiol. Infect. 136:739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feder I, Wallace FM, Gray JT, Fratamico P, Fedorka-Cray PJ, Pearce RA, Call JE, Perrine R, Luchansky JB. 2003. Isolation of Escherichia coli O157:H7 from intact colon fecal samples of swine. Emerg. Infect. Dis. 9:380–383. 10.3201/eid0903.020350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.