

Abstract
Allosteric regulation of phosphoenolpyruvate carboxylase (PEPC) controls the metabolic flux distribution of anaplerotic pathways. In this study, the feedback inhibition of Corynebacterium glutamicum PEPC was rationally deregulated, and its effect on metabolic flux redistribution was evaluated. Based on rational protein design, six PEPC mutants were designed, and all of them showed significantly reduced sensitivity toward aspartate and malate inhibition. Introducing one of the point mutations (N917G) into the ppc gene, encoding PEPC of the lysine-producing strain C. glutamicum LC298, resulted in ∼37% improved lysine production. In vitro enzyme assays and 13C-based metabolic flux analysis showed ca. 20 and 30% increases in the PEPC activity and corresponding flux, respectively, in the mutant strain. Higher demand for NADPH in the mutant strain increased the flux toward pentose phosphate pathway, which increased the supply of NADPH for enhanced lysine production. The present study highlights the importance of allosteric regulation on the flux control of central metabolism. The strategy described here can also be implemented to improve other oxaloacetate-derived products.
INTRODUCTION
The phosphoenolpyruvate (PEP)-pyruvate-oxaloacetate (OAA) node is one of the most important links between glycolysis/gluconeogenesis and the tricarboxylic acid (TCA) cycle (1). A set of reactions at this node direct the carbon flux into appropriate directions, making it a highly relevant switch point for carbon flux distribution within the central metabolism (Fig. 1). In most bacteria, the cells regenerate OAA through anaplerotic pathways either from PEP by phosphoenolpyruvate carboxylase (PEPC) or from pyruvate by pyruvate carboxylase (PC). Corynebacterium glutamicum possesses both PEPC and PC for replenishing OAA consumed for biosynthesis during cellular growth or OAA-derived amino acid production in industrial fermentations (2). The OAA supply has been considered a bottleneck for lysine production and different strategies have been applied to increase OAA availability such as overexpression of PEPC (3, 4) or PC (5, 6), deletion of pyruvate kinase (7), or phosphoenolpyruvate carboxykinase (PEPCK) (8).
FIG 1.
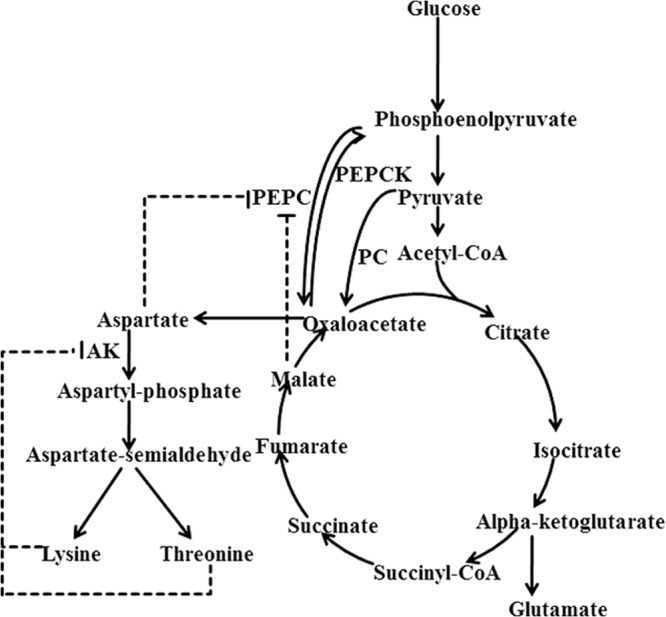
Simplified scheme of allosteric regulation of the central metabolism and lysine synthesis pathways in C. glutamicum. Feedback inhibitions of PEPC by aspartate and malate and AK by threonine and lysine strictly control the metabolic flux to lysine synthesis pathway. Dotted arrows represent pathways consisting of several reactions. PC, pyruvate carboxylase, PEPCK, phosphoenolpyruvate carboxykinase; AK, aspartokinase.
Although PEPC showed a higher in vitro activity than PC, it has been reported that it contributed only ca. 10% of the total oxaloacetate synthesis in glucose-growing cells of C. glutamicum (9). Overexpression of PC in C. glutamicum increased lysine accumulation by 50% (6), while the overexpression of PEPC showed only marginal effect on lysine production (3, 4). The contradictory phenomenon of the PEPC for its high in vitro activity with its low effect on lysine production may be due to the fact that PEPC is subjected to the rigid feedback inhibitions of aspartate and malate (4) (Fig. 1), whereas PC is only slightly inhibited by aspartate (10). Thus, deregulation of the feedback inhibition of PEPC by aspartate and malate may enhance the anaplerotic function of PEPC and improve lysine production. This conjecture, however, has not been verified thus far. In the present work, we report for the first time a rational deregulation of the feedback inhibition of PEPC and demonstrate that it can significantly improve lysine production in C. glutamicum.
MATERIALS AND METHODS
Structure modeling of PEPC from C. glutamicum.
Since the structure of PEPC from C. glutamicum has not been crystallized until now, a homology modeling of its three-dimensional structure was generated by using the software Modeller (http://salilab.org/modeller/) based on the structure of Escherichia coli PEPC (PDB code 1FIY). C. glutamicum PEPC shared 33.5% identity with the amino acid sequence of E. coli PEPC, and the residues involved in aspartate binding are conserved.
Bacterial strains and plasmids.
The strains and plasmids used in the present study are listed in Table 1. E. coli JM109 was used for the construction of plasmids, and E. coli BL21(DE3)-RIL cells (Stratagene) was used as a host for enzyme overexpression and purification. C. glutamicum LC298, a lysine-producing strain that has a deregulated aspartokinase (11), was used as the host to construct C. glutamicum LP917.
TABLE 1.
Strains and plasmids used in the present study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| JM109 | Host for plasmid construction | Stratagene |
| BL21-CodonPlus | Host for protein overexpression | Stratagene |
| DH5α MCR | Host for suicide plasmid construction | 12 |
| C. glutamicum | ||
| ATCC 13032 | Wild-type strain | Lab collection |
| LC298 | C. glutamicum ATCC 13032, with a lysC mutation (Q298G) | 11 |
| LP917 | C. glutamicum ATCC 13032, with lysC (Q298G) and PEPC (N917G) mutations | This study |
| Plasmids | ||
| pET-28a(+) | Plasmid for protein overexpression | Novagen |
| pK18mobsacB | Suicide vector | 13 |
| pET-pEppc | pET-28a(+) containing the PEPC gene | This study |
| pK18-pepc | pK18mobsacB containing the PEPC recombinant fragment with point mutation of N917G | This study |
Molecular cloning and enzyme overexpression.
The wild-type ppc gene encoding PEPC was amplified by PCR from the genomic DNA of C. glutamicum ATCC 13032 using the primers 5′-GCGCGCCCATATGACTGATTTTTTACGCGATGACAT-3′ and 5′-TATAGTCGACCTAGCCGGAGTTGCGCAGCGCAGTGG-3′ and inserted into NdeI-XhoI sites of pET-28a(+) (Novagen). The expression vector was designated pEppc. Six PEPC mutants with selected point mutations were constructed by using a QuikChange site-directed mutagenesis kit (Stratagene) according to the standard protocol. Wild-type PEPC and its mutants were overexpressed in E. coli BL21(DE3) RIPL (Stratagene). The cells were cultured in Luria-Bertani (LB) medium at 37 °C until the optical density at 600 nm (OD600) reached 0.6, and 0.1 mmol of IPTG (isopropyl-β-d-thiogalactopyranoside) was added to induce enzyme overexpression for another 12 h at 20°C. Purified enzymes were obtained by using a Ni2+-nitrilotriacetic acid (NTA) column (GE Healthcare Bio-Sciences, Piscataway, NJ). Protein content was quantified by the method of Bradford (14).
Determination of the enzyme activities of PEPC and its mutants.
The enzyme activity of the purified PEPC was assayed by a coupling reaction catalyzed by malate dehydrogenase at 25°C as previously described (15–17). The standard reaction mixture contained 100 mM Tris-HCl (pH 7.5), 10 mM MnSO4, 10 mM NaHCO3, 2 mM PEP, 0.1 mM NADH, and 1.5 IU of malate dehydrogenase. The decrease of NADH in absorbance at 340 nm was monitored by a spectrophotometer. One unit of the enzyme activity is defined as the oxidization of 1 μmol of NADH per min. Kinetic constants were obtained from nonlinear regression data analysis and are expressed as means ± the standard deviations (SD; n = 3).
Strain construction.
Homologous recombination was used to introduce a point mutation of N917G into the ppc gene of the strain C. glutamicum LC298 (11). Transformation was performed with the integrative vector pK18mobsacB (13) by using kanamycin resistance and sucrose tolerance as positive and negative selection markers in two different recombination events as described previously (11). Sequence analysis was performed after the second recombination and the strain with the correct mutation was designated C. glutamicum LP917.
Fermentations and metabolic flux analysis.
Batch fermentations were carried out in 1.5-liter well-equipped bioreactors (DASGIP parallel bioreactor system, Jülich, Germany) with an initial working volume of 370 ml. The cultivations were performed in an optimized defined minimal medium containing 30 g of glucose, 15 g of (NH4)2SO4, 15 g of NH4Cl, 0.05 g of CaCl2·2H2O, 0.5 g of MgSO4·7H2O, 2 g of NaCl, 2 g of KH2PO4, 2 g of K2HPO4, 15 mg of 3,4-dihydroxybenzoic acid, 0.5 mg of biotin, 1.6 mg of thiamine-HCl, 3 mg of nicotinic acid, and 10 ml of 100× trace elements liter−1 (18). For the labeled glucose experiments, naturally labeled glucose was replaced by 99% [1-13C]glucose (Sigma-Aldrich, Germany) or by an equimolar mixture of naturally labeled and 99% [13C6]glucose (Euriso-Top, France). The fermentations were controlled at 30°C and pH 7.2, with aeration of 1 volume per volume per minute. Dissolved oxygen was controlled at 30% saturation by automatic control of impeller rpm in the DASGIP parallel bioreactors.
Substrate and product quantification.
Quantification of glucose, trehalose, and organic acids was carried out using high-performance liquid chromatography (HPLC; Kontron Instruments, United Kingdom) with separation on an Aminex HPX-87H column at 60°C with 0.005 M H2SO4 and detection via refractive index or by UV absorption at 210 nm. Quantification of extracellular lysine concentration was performed using HPLC (Knauer, Germany) after derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AccQ-Fluor reagent kit; Waters, Milford, MA). The cell concentration was determined by measuring the OD660 and by dry-biomass measurements. The correlation factors between cell dry weight (CDW) and the ODs were determined to be CDW = 0.28 × OD (g liter−1) for C. glutamicum LC298 and CDW = 0.25 × OD (g liter−1) for C. glutamicum LP917.
Mass isotopomer fractions of protein bound amino acids from the hydrolyzed biomass harvested during the exponential phase from the tracer experiments were determined by gas chromatography-mass spectrometry (GC-MS) (19, 20). The preparations of samples and derivatization were performed as described by Zamboni et al. (20).
Metabolic flux parameter estimation.
To investigate the PEPC flux, the PEP/PYR node has to be differentiated. Two separate experiments, with 100% [1-13C]glucose and a 1:1 mixture of [13C6]glucose and naturally labeled glucose, were performed in parallel (7, 21). The metabolic model was constructed as described previously (7), with separate PEP and PYR nodes. Only one decarboxylating reaction designated to phosphoenolpyruvate carboxykinase (PEPCK) was considered in the present model since it was reported that the activities of malic enzyme and oxaloacetate decarboxylase were absent during cell growth on glucose (9). The precursor demands implemented in the model were estimated based on the biomass yield for each strain and the biomass composition previously measured for C. glutamicum (22). Parameter estimation and sensitivity analysis was performed using the software package 13CFLUX2 (23).
RESULTS AND DISCUSSION
Identification of targeted residues relating to the feedback inhibition of PEPC.
PEPC can be inhibited by both aspartate and malate. The aspartate binding sites in E. coli PEPC have been determined, while no information about the malate binding sites has been reported (24). As indicated in the homology modeling of C. glutamicum PEPC (Fig. 2A), four residues—R620, K813, R873, and N917—are directly involved in aspartate binding. The main chain of aspartate is stabilized by R620 and N917 through hydrogen bonds and salt bridges. Interestingly, R620 is the second arginine in the uniquely conserved sequence of GRGGXXGRGG in the PEPC family (24). This flexible loop is considered to be essential to the catalytic activity of PEPC. The trap of R620 by aspartate may affect the flexibility of this loop and thus inhibit the enzyme activity. Another two residues, K813 and R873, are both strongly salt bridged to the carboxyl group in the side chain of aspartate. All four residues are thus selected as targets for site-directed mutagenesis. Two additional residues, K653 and S869, which are not directly involved in aspartate binding but share high evolutionary correlation (11, 25, 26), as well as hydrogen bonds with R620 and R873, respectively, are also selected as targets for site-directed mutagenesis. The six targeted residues were mutated to glycine independently in order to investigate their effect for the feedback inhibition of PEPC by aspartate and malate.
FIG 2.

Identification of targets for the deregulation of phosphoenolpyruvate carboxylase from feedback inhibition and experimental verification. (A) Homology modeling of aspartate binding sites. Dotted lines represent hydrogen bonds. (B) Inhibition profiles of phosphoenolpyruvate carboxylase mutants by aspartate (left) and malate (right). The inhibition curves are expressed as relative activities, taking the activities in the absence of inhibitors as 100% (Table 1). The data from three independent measurements and the means of values were plotted with error bars.
Characterization of wild-type and mutant PEPC.
The kinetics of wild-type and mutant PEPC, determined by using purified enzymes with His tag at the N terminus, are summarized in Table 2. The mutations of K813, S869, R873, and N917 to glycine did not significantly alter the enzyme activity or kinetic constants of PEPC. The mutation of R620 and K653 to glycine, however, dramatically reduces the enzyme activity due to the increased Km value of PEP. Thus, R620 and K653 may be involved in PEP binding, as discussed above.
TABLE 2.
Specific activities and kinetic parameters of wild-type PEPC and its mutants
| PEPC type/ mutant |
Mean sp acta (U/mg of protein) ± SD | Mean Km (mM) ± SDb |
|
|---|---|---|---|
| KHCO3 | PEP | ||
| Wild type | 47 ± 5 | 2.8 ± 0.18 | 0.62 ± 0.13 |
| R620G | 0.48 ± 0.12 | 3.3 ± 0.22 | 4.88 ± 0.58 |
| K653G | 2.24 ± 0.23 | 3.5 ± 0.28 | 3.31 ± 0.54 |
| K813G | 38 ± 3 | 3.1 ± 0.37 | 0.78 ± 0.21 |
| S869G | 42 ± 3 | 2.9 ± 0.18 | 0.68 ± 0.14 |
| R873G | 43 ± 4 | 2.8 ± 0.25 | 0.66 ± 0.11 |
| N917G | 44 ± 4 | 2.6 ± 0.26 | 0.69 ± 0.22 |
All of the enzyme assays were carried out at 25°C. The standard assay contained 100 mM Tris-HCl (pH 8.5), 10 mM KHCO3, 2 mM PEP, 0.1 mM acetyl coenzyme A, 10 mM MgSO4, 0.1 mM NADH, 1.5 IU of malate dehydrogenase, and an appropriate amount of enzyme. All of the assays were repeated three times, and the mean values are shown.
The kinetic experiments were carried out under the same condition except for the substrate concentration.
The inhibition profiles for the wild type and mutants by aspartate and malate are shown in Fig. 2B. The wild-type PEPC was significantly inhibited by aspartate and lost ca. 95% activity when the aspartate concentration was >2 mM. This may explain the fact that the overexpression of PEPC did not significantly enhance lysine production since the intracellular concentration of aspartate was reported to be much higher than 2 mM (3, 4). All six constructed mutants could partially or completely release the feedback inhibition of PEPC by aspartate, indicating that they are significant for the allosteric inhibition of PEPC by aspartate. PEPC with mutation of K813G, R873G, or N917G could keep >80% activity even when the aspartate concentration was greater than 20 mM (data not shown). Wild-type PEPC was also inhibited by malate with a higher half-saturation concentration compared to aspartate. Interestingly, all of the constructed mutants can also release the feedback inhibition of PEPC by malate. This may be due to the fact that malate and aspartate are structural analogues and may share the same binding sites.
Introduction and evaluation of PEPC mutation N917G.
Based on the kinetic study and inhibition profiles of PEPC mutants, the point mutation N917 was introduced into the ppc gene of l-lysine-producing strain C. glutamicum LC298 (11) by allelic replacement, resulting in the creation of strain LP917. Strain LP917 thus bears both feedback insensitive aspartokinase and PEPC.
The cell growth and l-lysine production of strain LC298 and LP917 are depicted in Fig. 3. The growth of strain LP917 was lower than that of strain LC298, which may be because more PEP was used for lysine production and thus not available for cell growth. On the other hand, both the production rate and the yield of l-lysine of the strain LP917 was significantly higher than those of the strain LC298. The yield of l-lysine by the strain LP917 was 0.211 ± 0.004 mol mol of glucose−1 (Table 3), which is 37% higher than that of the strain LC298. Since the difference between the two strains involve only one point mutation in ppc gene, the increased lysine production could therefore be clearly attributed to the deregulation of feedback inhibition of PEPC.
FIG 3.

Biomass, lysine production, and glucose consumption of C. glutamicum LP917 and LC298. Strain LC298 shares a point mutation of Q298G in the lysC gene encoding a feedback-insensitive aspartokinase. Strain LP917 is derived from strain LC298 by the integration of point mutation of N917G in the ppc gene encoding the feedback insensitive phosphoenolpyruvate carboxylase. The data represent mean values and standard deviations from three parallel cultures.
TABLE 3.
Concentrations of biomass and metabolites of C. glutamicum LC298 and C. glutamicum LP917 grown on glucose in bioreactors
| Biomass or metabolite | Mean yield ± SDa |
|
|---|---|---|
| C. glutamicum LC298 | C. glutamicum LP917 | |
| Biomass | 64.6 ± 1.6 | 56.1 ± 1.3 |
| Lysine | 154.2 ± 3.3 | 211.3 ± 4.2 |
| Trehalose | 17.3 ± 1.1 | 21.4 ± 1.5 |
| Lactate | 21.1 ± 1.3 | 17.2 ± 1.1 |
All yields are expressed as mmol mol of glucose−1 except for biomass, which is expressed as g mol glucose−1, and are taken as mean values from three parallel experiments with the corresponding standard deviations. The yields were determined as the slope of the linear fit between glucose consumption and product formation.
Metabolic flux analysis.
To further characterize the strains, 13C-based metabolic flux analysis was carried out. We used 100% [1-13C]glucose to estimate the fluxes in the upper glycolytic pathways, whereas we performed experiments with a mixture of 13C6 and naturally labeled glucose for the differentiation of the PEP and PYR node (21). The obtained flux estimates from 13C6 labeling data were used as free constraints in the [1-13C]glucose-labeled simulations.
The isotopic steady states were confirmed after measuring the labeling pattern of the protein bound amino acids during the exponential phase. As shown in Fig. 4, an approximately 30% higher flux through the PEPC was observed in the mutant strain (LP917) compared to the parent strain, leading to an increased availability of oxaloacetate for lysine production. Interestingly, the anaplerotic flux through the pyruvate carboxylase was slightly reduced due to a decreased flux from PEP to pyruvate. The flux to TCA cycle is almost identical in both strains, whereas the flux to PPP pathway was ca. 9% higher in the mutant strain. The higher flux to lysine required the increased demand of NADPH, which was supplied by the PPP pathway. In both strains the NADPH balance is almost closed where the PPP pathway served as the major supplier.
FIG 4.
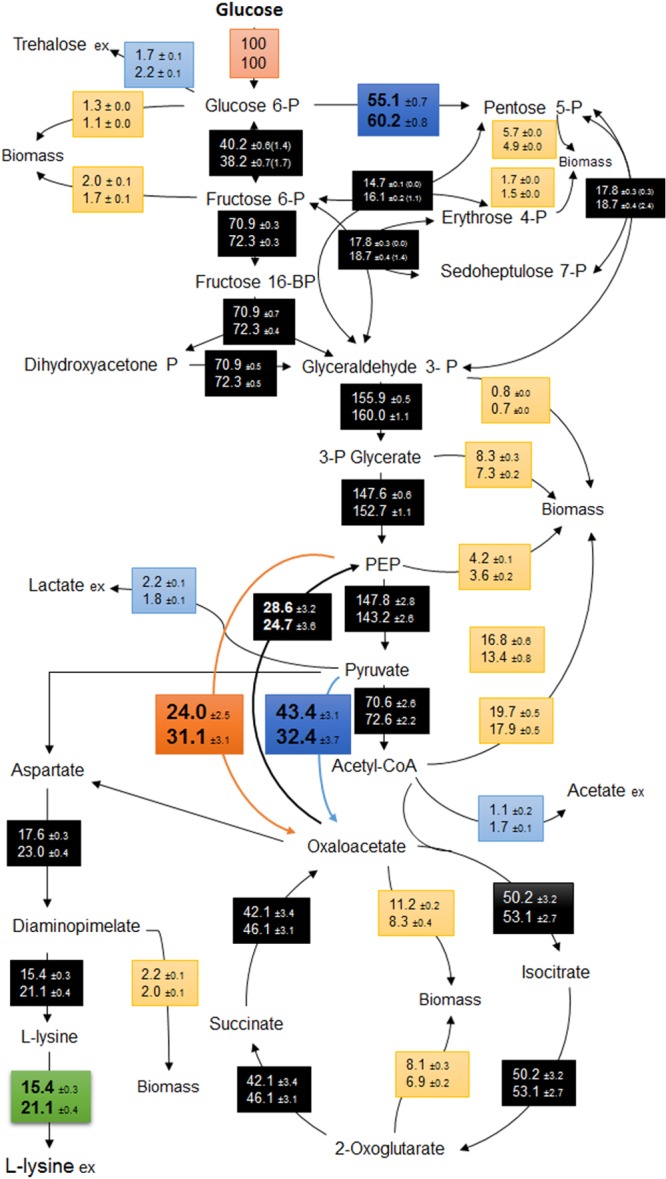
Metabolic flux distributions in C. glutamicum LC298 (top) and C. glutamicum LP917 (bottom) during the exponential growth phase. All fluxes are expressed as the molar percentages of mean specific glucose uptake rates (LC298, 4.5 mmol g−1 h−1; LP917, 4.1 mmol g−1 h−1). The deviations correspond to 90% confidence intervals obtained by Monte-Carlo analysis. The values in parentheses represent the exchange coefficients of the fluxes.
Conclusions.
The construction of a minimally mutated industrial strain with a high production rate and yield is an attractive approach for current strain development. Integration of beneficial mutations identified from comparative genomics analysis has permitted the creation of a strain demonstrating high lysine production (10, 28). We also showed that the application of structural synthetic biology could accelerate this process (11, 25, 26). In the present work, we focused on the regulation of a PEP-pyruvate-OAA node and investigated the feedback inhibition of PEPC for the overproduction of OAA-derived metabolites. Previous studies of C. glutamicum's carboxylation mechanism suggested that pyruvate carboxylase (PC) was the main enzyme responsible for the anaplerotic reactions and that PEPC is dispensable for cell growth and lysine production (6, 9, 27). It was shown here that the strict allosteric regulation of PEPC by aspartate and malate is one of the most important mechanisms to control the metabolic flux distribution at this node. Deregulation of the feedback inhibition of PEPC can significantly enhance the anaplerotic function of PEPC and improve OAA-derived metabolite production.
Footnotes
Published ahead of print 13 December 2013
REFERENCES
- 1.Sauer U, Eikmanns BJ. 2005. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol. Rev. 29:765–794. 10.1016/j.femsre.2004.11.002 [DOI] [PubMed] [Google Scholar]
- 2.Kjeldsen KR, Nielsen J. 2009. In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol. Bioeng. 102:583–597. 10.1002/bit.22067 [DOI] [PubMed] [Google Scholar]
- 3.Cremer J, Eggeling L, Sahm H. 1991. Control of the lysine biosynthetic sequence in Corynebacterium glutamicum as analyzed by overexpression of the individual corresponding genes. Appl. Environ. Microbiol. 57:1746–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Regan M, Thierbach G, Bachmann B, Villeval D, Lepage P, Viret JF. 1989. Cloning and nucleotide sequence of the phosphoenolpyruvate carboxylase-coding gene of Corynebacterium glutamicum ATCC 13032. Gene 77:237–251. 10.1016/0378-1119(89)90072-3 [DOI] [PubMed] [Google Scholar]
- 5.Eikmanns BJ. 2005. Central metabolism: tricarboxylic acid cycle and anaplerotic reactions, p 241–276 In Eggeling L, Bott M. (ed), Handbook of Corynebacterium glutamicum. CRC Press, Inc, Boca Raton, FL [Google Scholar]
- 6.Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H. 2001. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J. Mol. Microbiol. Biotechnol. 3:295–300 [PubMed] [Google Scholar]
- 7.Becker J, Klopprogge C, Wittmann C. 2008. Metabolic responses to pyruvate kinase deletion in lysine-producing Corynebacterium glutamicum. Microb. Cell Fact. 7:8. 10.1186/1475-2859-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen S, Mack C, de Graaf AA, Riedel C, Eikmanns BJ, Sahm H. 2001. Metabolic consequences of altered phosphoenolpyruvate carboxykinase activity in Corynebacterium glutamicum reveal anaplerotic regulation mechanisms in vivo. Metab. Eng. 3:344–361. 10.1006/mben.2001.0198 [DOI] [PubMed] [Google Scholar]
- 9.Petersen S, de Graaf AA, Eggeling L, Möllney M, Wiechert W, Sahm H. 2000. In vivo quantification of parallel and bidirectional fluxes in the anaplerosis of Corynebacterium glutamicum. J. Biol. Chem. 275:35932–35941. 10.1074/jbc.M908728199 [DOI] [PubMed] [Google Scholar]
- 10.Ikeda M, Ohnishi J, Hayashi M, Mitsuhashi S. 2006. A genome based approach to create a minimally mutated Corynebacterium glutamicum strain for efficient l-lysine production. J. Ind. Microbiol. Biotechnol. 33:610–615. 10.1007/s10295-006-0104-5 [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Meyer WQ, Rappert S, Sun JB, Zeng AP. 2011. Coevolutionary analysis-enabled rational deregulation of allosteric enzyme inhibition in Corynebacterium glutamicum for lysine production. Appl. Environ. Microbiol. 77:4352–4360. 10.1128/AEM.02912-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant SG, Jesse NJ, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA 87:4645–4649. 10.1073/pnas.87.12.4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schafer A, Tauch A, Jager W, Kalinowski K, Thierbach G, Puhler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. 10.1016/0378-1119(94)90324-7 [DOI] [PubMed] [Google Scholar]
- 14.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 15.Endo T, Mihara Y, Furumoto T, Matsumura H, Kai Y, Izui K. 2007. Maize C4-form phosphoenolpyruvate carboxylase engineered to be functional in C3 plants: mutations for diminished sensitivity to feedback inhibitors and for increased substrate affinity. J. Exp. Bot. 59:1811–1818. 10.1093/jxb/ern018 [DOI] [PubMed] [Google Scholar]
- 16.Mori M, Shiio I. 1985. Purification and some properties of phosphoenolpyruvate carboxylase from Brevibacterium flavum and its aspartate-overproducing mutant. J. Biochem. 97:1119–1128 [DOI] [PubMed] [Google Scholar]
- 17.Yano M, Izui K. 1997. The replacement of Lys620 by serine desensitizes Escherichia coli phosphoenolpyruvate carboxylase to the effects of the feedback inhibitors l-aspartate and l-malate. Eur. J. Biochem. 247:74–81. 10.1111/j.1432-1033.1997.t01-1-00074.x [DOI] [PubMed] [Google Scholar]
- 18.Vallino JJ, Stephanopoulos G. 1993. Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol. Bioeng. 41:633–646. 10.1002/bit.260410606 [DOI] [PubMed] [Google Scholar]
- 19.Christensen B, Nielsen J. 1999. Isotopomer analysis using GC-MS. Metab. Eng. 1:282–290. 10.1006/mben.1999.0117 [DOI] [PubMed] [Google Scholar]
- 20.Zamboni N, Fendt SM, Rühl M, Sauer U. 2009. 13C-based metabolic flux analysis. Nat. Protoc. 4:878–898. 10.1038/nprot.2009.58 [DOI] [PubMed] [Google Scholar]
- 21.Wittmann C, Heinzle E. 2001. Modeling and experimental design for metabolic flux analysis of lysine-producing corynebacteria by mass spectrometry. Metab. Eng. 3:173–191. 10.1006/mben.2000.0178 [DOI] [PubMed] [Google Scholar]
- 22.Wittmann C, de Graaf AA. 2005. Metabolic flux analysis in Corynebacterium glutamicum, p 277–304 ln Eggeling L, Bott M. (ed), Handbook of Corynebacterium glutamicum. CRC Press, Inc, Boca Raton, FL [Google Scholar]
- 23.Weitzel M, Nöh K, Dalman T, Niedenführ S, Stute B, Wiechert W. 2013. 13CFLUX2: high performance software suite for 13C metabolic flux analysis. Bioinformatics 29:143–145. 10.1093/bioinformatics/bts646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kai Y, Matsumura H, Inoue T, Terada K, Nagara Y, Yoshinaga Y. 1999. Three-dimensional structure of phosphoenolpyruvate carboxylase: a proposed mechanism for allosteric inhibition. Proc. Natl. Acad. Sci. U. S. A. 96:823–828. 10.1073/pnas.96.3.823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Rappert S, Sun JB, Zeng AP. 2011. Integrating molecular dynamics and co-evolutionary analysis for reliable target prediction and deregulation of the allosteric inhibition of aspartokinase for amino acid production. J. Biotechnol. 154:248–254. 10.1016/j.jbiotec.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Wilmanns M, Zeng AP. 2010. Structural synthetic biotechnology: from molecular structure to predictable design for industrial strain development. Trends. Biotechnol. 28:534–542. 10.1016/j.tibtech.2010.07.004 [DOI] [PubMed] [Google Scholar]
- 27.Peters-Wendisch PG, Eikmanns BJ, Thierbach G, Bachmann B, Sahm H. 1993. Phosphoenolpyruvate carboxylase is indispensable for growth and lysine production. FEMS Microbiol. Lett. 112:269–274. 10.1111/j.1574-6968.1993.tb06461.x [DOI] [Google Scholar]
- 28.Ohnishi J, Katahira R, Mitsuhashi S, Kakita S, Ikeda M. 2005. A novel gnd mutation leading to increased l-lysine production in Corynebacterium glutamicum. FEMS Microbial Lett. 242:265–274. 10.1016/j.femsle.2004.11.014 [DOI] [PubMed] [Google Scholar]