

Abstract
Accurate sequence analysis of mononucleotide repeat regions is difficult, complicating the use of short sequence repeats (SSRs) as a tool for bacterial strain discrimination. Although multiple SSR loci in the genome of Mycobacterium avium subsp. paratuberculosis allow genotyping of M. avium subsp. paratuberculosis isolates with high discriminatory power, further characterization of the most discriminatory loci is limited due to inherent difficulties in sequencing mononucleotide repeats. Here, a method was evaluated using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) as an alternative to Sanger sequencing to further differentiate the dominant mycobacterial interspersed repetitive-unit (MIRU)–variable-number tandem-repeat (VNTR) M. avium subsp. paratuberculosis type (n = 37) in Canadian dairy herds by targeting a highly discriminatory mononucleotide SSR locus. First, PCR-amplified DNA was digested with two restriction enzymes to yield a sufficiently small fragment containing the SSR locus. Second, MALDI-TOF MS was performed to identify the mass, and thus repeat length, of the target. Sufficiently intense, discriminating spectra were obtained to determine repeat lengths up to 15, an improvement over the limit of 11 using traditional sequencing techniques. Comparison to synthetic oligonucleotides and Sanger sequencing results confirmed a valid and reproducible assay that increased discrimination of the dominant M. avium subsp. paratuberculosis MIRU-VNTR type. Thus, MALDI-TOF MS was a reliable, fast, and automatable technique to accurately resolve M. avium subsp. paratuberculosis genotypes based on SSRs.
INTRODUCTION
Evaluating genetic diversity of a pathogen is critical in understanding its transmission. Historically, this has been challenging in organisms with limited genetic variation, as many common molecular techniques are not sufficiently discriminatory to differentiate strains (1). However, a limited number of genomic targets, typically repeat regions, have been identified and used for strain discrimination.
Mycobacterium avium subsp. paratuberculosis is the causative bacterium of Johne's disease, a severe gastroenteritis in domestic ruminants (2). Furthermore, there is an unproven association between Johne's disease in cattle and Crohn's disease in humans (3, 4). Although M. avium subsp. paratuberculosis is fastidious and slow growing and has limited genetic variation, numerous typing techniques have been developed to differentiate isolates (5–9). All typing techniques developed thus far, however, fail to provide sufficient discriminatory power to be used for detailed epidemiological analyses.
Variable-number tandem-repeat (VNTR) analysis has been moderately successful in differentiation of M. avium subsp. paratuberculosis strains. Two dominant VNTR types, however, account for the vast majority of M. avium subsp. paratuberculosis isolates characterized. Therefore, a more discriminatory genotyping method, such as short-sequence-repeat (SSR) typing, is required to further differentiate M. avium subsp. paratuberculosis isolates (10).
Compared to other genotyping methods, characterization of SSRs in the M. avium subsp. paratuberculosis genome is powerful in identifying epidemiologically and geographically distinct strains (8). Due to their instability, SSRs evolve more rapidly than single nucleotide polymorphisms (SNPs); therefore, SSR typing is particularly useful for differentiating monomorphic bacteria (11). SSRs are repetitive tracts of nucleotides of 1 to 10 bp in which mutations occur during replication. When DNA polymerase encounters long repeats, it can stall, sometimes leading to dissociation of the nascent DNA strand, which may subsequently realign out of register (12). This slip-strand mispairing leads to a different repeat length from the template strand. M. avium subsp. paratuberculosis, along with many other mycobacterial species, lacks mismatch repair proteins, which favors instability of SSRs and thus variability in these regions (13).
Eleven SSRs in the M. avium subsp. paratuberculosis genome were previously identified as polymorphic (8). The two most discriminatory loci are mononucleotide repeats, in which accurate sequence analysis is difficult (10, 14–16). The ambiguous sequencing chromatograms of long repeats are characterized by stutter peaks, a critical limitation of this genotyping method, as it relies entirely on accurate interpretation of repeat length. Mass spectrometry (MS) is based on measurement of the mass of a molecule of interest, inherently minimizing ambiguity and interindividual interpretation differences. Current MS technologies enable mass resolution of a single nucleotide (17), creating an ideal platform for characterization of long mononucleotide repeats that are not resolved using Sanger sequencing. The objective of this study was to develop a reliable and reproducible assay based on matrix-assisted laser desorption ionization–time of flight MS (MALDI-TOF MS) to characterize the most discriminatory SSR locus in the M. avium subsp. paratuberculosis genome.
MATERIALS AND METHODS
M. avium subsp. paratuberculosis culture and DNA extraction.
Environmental fecal samples collected from dairy farms participating in the Alberta Johne's Disease Initiative (AJDI; R. Wolf, F. Clement, H. W. Barkema, and K. Orsel, submitted for publication) were processed as described previously (18). Briefly, samples were decontaminated and incubated in the Trek ESP Culture System II (Trek Diagnostic Systems, Cleveland, OH, USA) for 8 weeks. A confirmatory IS900 PCR was performed on all cultures (19). Using a disposable loop, 10 μl of culture broth from samples positive for IS900 was plated onto Middlebrook 7H11 agar supplemented with mycobactin. Plates were incubated at 37°C until individual colonies were visible (approximately 4 to 6 weeks). A single colony was subsequently substreaked to a new 7H11 plate, which was then incubated at 37°C until enough growth was available for DNA extraction (approximately 6 to 8 weeks). Extraction of DNA was achieved using a modified protocol of the Qiagen DNeasy blood and tissue kit (Qiagen, Mississauga, ON, Canada). The M. avium subsp. paratuberculosis growth was removed from the agar plate using a disposable loop and added to tubes containing 40 mg of 0.1-mm silica beads and 180 μl ATL lysis buffer. After 2 min of bead beating, the spin-column protocol was followed according to the manufacturer's instructions.
Isolate selection.
One isolate per farm (n = 53) was characterized by mycobacterial interspersed repetitive-unit (MIRU)–VNTR typing using eight targets, as described previously (9). Isolates belonging to the most common genotype (INMV2) were chosen for further analysis.
PCR.
The whole-genome sequence of M. avium subsp. paratuberculosis strain K10 (20) was used to design primers in Geneious version 6.1.5 (Biomatters, Auckland, New Zealand) to amplify the most discriminating mononucleotide locus (locus 1) (8). A FokI restriction enzyme recognition site was incorporated into the primer sequences (forward, AAAAAAGGATGCGGCCCTGGTGGTCGACGAC; reverse, AAAAAAGGATGTCACCACCCAGTGCACGTACGG). This type IIS restriction enzyme cleaves 9 bp downstream and 13 bp upstream of the recognition site on the forward and reverse strand, respectively. A poly(A) sequence (n = 6) was added to the ends of the primers to ensure binding of the enzyme, and biotin was attached to the 5′ end of the forward primer. The SSR locus 1 was amplified in a PCR mixture containing 2.5 units of Herculase enhanced DNA polymerase, 200 μM (each) deoxynucleoside triphosphate (dNTP), 1.5 pmol forward primer, 3.0 pmol reverse primer, 5 μl 10× buffer, and 2 μl template DNA (20 to 40 ng) in a total volume of 50 μl. This DNA polymerase produces blunt-ended PCR amplicons and has proofreading capabilities with lower error rates (21, 22). The forward primer concentration was limited in order to minimize the binding of nonextended biotinylated primers to the streptavidin-coated beads in the subsequent step. The thermocycling conditions were as follows: an initial step of 98°C for 3 min, followed by a touchdown PCR of 98°C for 40 s, 71°C for 30 s (−0.5°C/cycle), and 72°C for 1 min, for 20 cycles, and 98°C for 40 s, 61°C for 30 s, and 72°C for 1 min, for 20 cycles, followed by a final elongation step at 72°C for 10 min.
Restriction enzyme digestion.
An aliquot (40 μl) of the final PCR product was mixed with an equal volume of washed streptavidin Dynabeads and rotated at room temperature for 15 min. The beads were immobilized with a magnet and washed according to the manufacturer's instructions. After washing, beads were resuspended in 7 μl NEB4 buffer containing 1 unit each of FokI and BsaHI. Samples were incubated at 37°C for 2 h, followed by 65°C for 20 min for enzyme inactivation. Then, 3 μl Clean Resin (Sequenom, USA) was added to each tube to remove salts and each tube was rotated at room temperature for 5 to 10 min. Tubes were centrifuged for 10 s, and the supernatant was saved for further analysis.
MALDI-TOF.
An aliquot (0.5 μl) of supernatant was mixed in equal volumes with a 3-hydroxypicolinic acid (HPA) matrix. This sample-matrix mixture was spotted on the MALDI plate and allowed to dry before being introduced into the instrument. Mass spectrometry was performed on a MALDI TOF/TOF 5800 spectrometer (AB Sciex, Framingham, MA, USA) operating in linear, positive-ion mode. An oligonucleotide mass standard of 6,352 Da was used for calibration.
Validation and reproducibility.
Oligonucleotides representing fragments of various alleles (7 through 15) of the G1 locus obtained after PCR and digestion were synthesized (Core DNA Services, University of Calgary). The expected masses of the digested DNA fragments were calculated using the Mongo Oligo Mass Calculator (v2.06; The RNA Institute, University at Albany, Albany, NY, USA). The oligonucleotides were reconstituted in high-pressure liquid-chromatography (HPLC)-grade water (concentration, 20 pmol) and analyzed using MALDI-TOF MS.
Sanger sequencing was used to characterize the set of INMV2 isolates. The SSR locus 1 was amplified in a PCR mixture containing 1× SsoFast EvaGreen Supermix, 1 μl dimethyl sulfoxide (DMSO), 2 pmol forward primer, 2 pmol reverse primer, and 2 μl genomic DNA. Thermocycling conditions were an initial denaturation step of 95°C for 3 min, followed by 35 cycles of 95°C for 10 s and annealing/elongation at 55°C for 10 s. Then, 20 μl of PCR product was purified using a PCR purification kit (Qiagen Inc., Venlo, The Netherlands) and submitted to the sequencing center at the University of Calgary. Reproducibility of the MALDI-TOF assay was assessed by performing the procedure beginning at PCR amplification on 3 isolates of each genotype (when available) 3 times.
RESULTS
The amplified region of the G1 locus, restriction enzyme cut sites, and the corresponding expected mass of each fragment are shown in Fig. 1. The mass of fragments C to G remained constant, whereas expected masses of both A and B fragments of various repeat lengths (7 to 15) were variable (Table 1). Analysis of synthetic oligonucleotides representing A and B in the range of previously observed alleles is shown in Fig. 2. Two distinct peaks were generated which correspond to the expected masses within 10 Da, demonstrating that a single nucleotide difference was clearly resolved.
FIG 1.

Diagram representing PCR-amplified DNA containing the mononucleotide SSR locus 1 (underlined). Restriction enzyme cut sites are presented as boxed arrowheads along with the corresponding masses of each fragment.
TABLE 1.
Expected mass of each repeat length of polymorphic fragments A and B
| Repeat length | Expected mass (Da) |
|
|---|---|---|
| A | B | |
| 7 | 6,794 | 7,341 |
| 8 | 7,123 | 7,629 |
| 9 | 7,453 | 7,919 |
| 10 | 7,782 | 8,208 |
| 11 | 8,111 | 8,498 |
| 12 | 8,440 | 8,787 |
| 13 | 8,770 | 9,076 |
| 14 | 9,099 | 9,365 |
| 15 | 9,428 | 9,654 |
| >15 | >9,700 | >9,700 |
FIG 2.
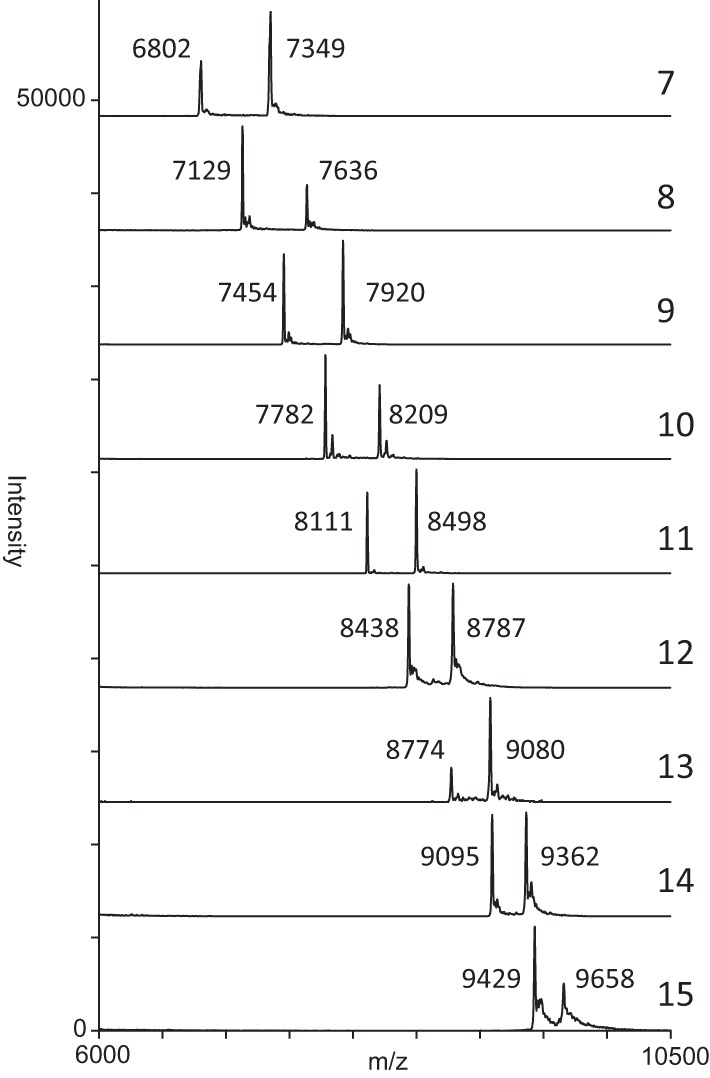
Mass spectra of synthetic oligonucleotides representing fragments A and B of different repeat lengths (7 to 15).
M. avium subsp. paratuberculosis isolates.
Of the 53 isolates, 37 isolates belonged to the MIRU-VNTR type INMV2 and the remaining isolates belonged to 6 other types. The MALDI-TOF MS analysis of M. avium subsp. paratuberculosis DNA yielded consistent peaks for conserved fragments (C through G), although observed masses were consistently higher (approximately 90 Da) than the predicted mass for both the conserved and polymorphic fragments (A and B). Small peaks, which could be attributed to PCR-induced slip-strand mispairing, were sometimes visible in the mass spectra. Since these peaks have a lower intensity, they were disregarded based on operator discretion and did not interfere with the assignment of repeat length. Polymorphic fragments under 9,800 Da were discernible; however, increasing mass of the fragments led to decreasing intensity, resolution, and stability of the peak. Therefore, fragments over 9,700 Da were designated >15 repeat units. A summary of the genotypes observed and examples of spectra for each repeat length are shown in Table 2 and Fig. 3, respectively. Overall, there were 9 SSR types identified in the 37 Alberta M. avium subsp. paratuberculosis isolates tested.
TABLE 2.
SSR (G1) genotypes of INMV2 M. avium subsp. paratuberculosis isolates analyzed in this study
| Repeat | No. of M. avium subsp. paratuberculosis isolates (n = 37) |
|---|---|
| 7 | 13 |
| 8 | 1 |
| 9 | 3 |
| 10 | 1 |
| 11 | 0 |
| 12 | 1 |
| 13 | 1 |
| 14 | 1 |
| 15 | 3 |
| >15 | 13 |
FIG 3.

(A) Examples of spectra of each repeat length with observed masses. Polymorphic peaks are illustrated with inverted triangles (A, solid; B, shaded). (B) Enlarged image of the polymorphic peaks.
Validation.
To test the reproducibility of this assay, 3 isolates per genotype (when available) were tested 3 times, yielding 17 isolates in which masses were recorded and assigned a corresponding allele. All 17 isolates were assigned the same allele each time. Additionally, all isolates included in this study were sequenced using Sanger techniques. As expected, alleles with repeats greater than 11 bp were not resolved. Regardless, alleles 7 through 11 matched our previous designation as determined by MALDI-TOF MS.
DISCUSSION
Typing of repetitive DNA markers, such as SSRs, is invaluable in genotyping organisms with limited genetic diversity (1, 23). However, inherent problems in sequencing mononucleotide repeats limit its application, as Sanger sequencing often fails to resolve poly(G) repeats exceeding 11 bp (10, 15). In the present study, using MALDI-TOF MS, we increased the discriminatory power of SSR typing in M. avium subsp. paratuberculosis isolates by resolving mononucleotide repeats up to 15 bp.
Purified biotinylated PCR amplicons were used, followed by restriction endonuclease digestion to obtain a short fragment containing the mononucleotide repeat suitable for MALDI-TOF MS analysis. Resulting spectra revealed stable peaks formed by nonpolymorphic fragments suitable for calibration purposes, whereas the mass of the polymorphic repeat fragments changed according to the expected masses. As previously observed, Sanger sequencing chromatograms of repeats greater than 11 were characterized by stutter peaks and were not resolved.
In previous studies, SSRs were used to discriminate bacterial isolates of the same VNTR type (10, 24). Furthermore, mononucleotide SSRs in the highest-resolution analysis of Bacillus anthracis strains were used to further differentiate isolates with identical SNP and VNTR profiles (25). Similarly, MIRU-VNTR analysis was useful as a first step in discriminating M. avium subsp. paratuberculosis isolates. The dominant MIRU-VNTR type in Alberta was further differentiated into at least 9 genotypes by MALDI-TOF MS analysis of SSR locus 1.
Compared to shorter repeats, long mononucleotide SSRs have been linked to higher diversity, as they are more likely to undergo slip-strand mispairing and thus greater variability in repeat length (24). Therefore, the ability to discriminate long mononucleotide repeats is particularly advantageous to characterize genetic diversity. Sanger sequencing is a critical limitation in identification of these highly discriminatory loci exceeding 11 bp.
Advancement of new technologies, such as next-generation sequencing, has enabled the identification of SSRs across the genomes of plants, animals (26), and microorganisms. This has facilitated the use of SSR variability in several applications, including epidemiological typing (27, 28), phylogenetic analysis (29), and use as molecular markers in population genetic studies (30, 31) and human forensics (32). In addition, SSR length may be associated with increased virulence or immune escape, as it has a biological role in pathogen adaptation and modulation of gene expression (33–35). Regardless, all of these applications rely on accurate and discriminatory analysis of repeat length. Pyrosequencing is increasingly used in SNP screening assays and in identifying short indels (36); however, it produces errors in sequences with long homopolymer tracts (37). The use of MALDI-TOF MS to identify mononucleotide SSR length overcomes previous limitations, allowing researchers to explore these applications with increased confidence and precision.
Sanger sequencing was less successful than MALDI-TOF MS for differentiating mononucleotide repeat locus 1. The current method of clustering isolates into a ≥11 category using sequencing, when in fact they are different, may be misleading. Therefore, care should be taken when interpreting the >15 isolates in this study as well. The MALDI-TOF MS assay may require more hands-on time than sequencing techniques; however, the entire process from PCR to spectrum interpretation can be completed for at least 37 isolates in a single workday. Dominant SSR types based on locus 1 also exist within the dominant MIRU-VNTR type. In that regard, repeat lengths of 7 and >15 bp each represent 35% of isolates in this study. Additional typing of different SSR loci, such as 2 and 8, would likely further differentiate these dominant types.
Slip-strand mispairing is the mechanism believed to induce variability in SSR length in vivo; the same mechanism occurred during PCR (21). Although a proofreading polymerase was used to minimize amplification errors in the present study, small peaks outside the expected mass were visible. Repeated analysis of different alleles consistently resulted in the same designation, which highlights the reproducibility of this technique.
This technique can be applied to other organisms and should be particularly useful for determining the number of long mononucleotide repeat units. The procedure described could be fully automated (using a robot), including spotting onto the MALDI plate. Additional SSR loci could be included to multiplex this assay, increasing the discriminatory power while concurrently decreasing time and costs. In addition, MALDI-TOF MS may be used to identify mixed infections of different M. avium subsp. paratuberculosis genotypes, as multiple peaks can be observed in a single spectrum and could be further expanded to investigate evolution or strain competition in vitro and in vivo. SSRs in M. avium subsp. paratuberculosis and other organisms are unstable, with some loci more likely to undergo mutation than others. This was recently demonstrated at mononucleotide locus 2 when M. avium subsp. paratuberculosis was passaged through a goat, apparently changing the SSR from a repeat of 9 to a repeat of 10 in two separate isolates (15). This instability must be taken into consideration when applying this genotyping technique in the absence of other genomic targets and/or on closely related isolates.
In conclusion, improved characterization of mononucleotide SSRs in the M. avium subsp. paratuberculosis genome enhanced differentiation of isolates, essential for accurate identification of epidemiologically relevant strains. When combined with other typing techniques, mononucleotide SSRs are powerful in further differentiating seemingly identical isolates. Purification of a PCR-amplified mononucleotide SSR followed by restriction digestion prior to MALDI-TOF MS analysis overcame limitations posed by Sanger sequencing, resulting in an improved and reproducible genotyping assay for use in molecular epidemiological studies.
ACKNOWLEDGMENTS
This research was supported by the Alberta Livestock and Meat Agency and the Dairy Farmers of Canada.
Footnotes
Published ahead of print 8 November 2013
REFERENCES
- 1.Comas I, Homolka S, Niemann S, Gagneux S. 2009. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS One 4:e7815. 10.1371/journal.pone.0007815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweeney RW. 1996. Transmission of paratuberculosis. Vet. Clin. North Am. Food Anim. Pract. 12:305–312 [DOI] [PubMed] [Google Scholar]
- 3.Barkema HW, Hendrick S, de Buck JM, Ghosh S, Kaplan GG, Rioux KP, Krause DO. 2010. Zoonotic pathogens in the food chain. CABI, Wallingford, United Kingdom [Google Scholar]
- 4.Behr MA. 2010. The path to Crohn's disease: is mucosal pathology a secondary event? Inflamm. Bowel Dis. 16:896–902. 10.1002/ibd.21171 [DOI] [PubMed] [Google Scholar]
- 5.Collins DM, de Lisle GW. 1986. Restriction endonuclease analysis of various strains of Mycobacterium paratuberculosis isolated from cattle. Am. J. Vet. Res. 47:2226–2229 [PubMed] [Google Scholar]
- 6.Coffin JW, Condon C, Compston CA, Potter KN, Lamontagne LR, Shafiq J, Kunimoto DY. 1992. Use of restriction fragment length polymorphisms resolved by pulsed-field gel electrophoresis for subspecies identification of mycobacteria in the Mycobacterium avium complex and for isolation of DNA probes. J. Clin. Microbiol. 30:1829–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motiwala AS, Strother M, Amonsin A, Byrum B, Naser SA, Stabel JR, Shulaw WP, Bannantine JP, Kapur V, Sreevatsan S. 2003. Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis: evidence for limited strain diversity, strain sharing, and identification of unique targets for diagnosis. J. Clin. Microbiol. 41:2015–2026. 10.1128/JCM.41.5.2015-2026.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amonsin A, Li LL, Zhang Q, Bannantine JP, Motiwala AS, Sreevatsan S, Kapur V. 2004. Multilocus short sequence repeat sequencing approach for differentiating among Mycobacterium avium subsp. paratuberculosis strains. J. Clin. Microbiol. 42:1694–1702. 10.1128/JCM.42.4.1694-1702.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thibault VC, Grayon M, Boschiroli ML, Hubbans C, Overduin P, Stevenson K, Gutierrez MC, Supply P, Biet F. 2007. New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J. Clin. Microbiol. 45:2404–2410. 10.1128/JCM.00476-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thibault VC, Grayon M, Boschiroli ML, Willery E, Allix-Béguec C, Stevenson K, Biet F, Supply P. 2008. Combined multilocus short-sequence-repeat and mycobacterial interspersed repetitive unit-variable-number tandem-repeat typing of Mycobacterium avium subsp. paratuberculosis isolates. J. Clin. Microbiol. 46:4091–4094. 10.1128/JCM.01349-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Belkum A, Scherer S, van Alphen L, Verbrugh H. 1998. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. 62:275–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levinson G, Gutman GA. 1987. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4:203–221 [DOI] [PubMed] [Google Scholar]
- 13.Springer B, Sander P, Sedlacek L, Hardt W-D, Mizrahi V, Schär P, Böttger EC. 2004. Lack of mismatch correction facilitates genome evolution in mycobacteria. Mol. Microbiol. 53:1601–1609. 10.1111/j.1365-2958.2004.04231.x [DOI] [PubMed] [Google Scholar]
- 14.Douarre PE, Cashman W, Buckley J, Coffey A, O'Mahony JM. 2010. Isolation and detection of Mycobacterium avium subsp. paratuberculosis (MAP) from cattle in Ireland using both traditional culture and molecular based methods. Gut Pathog. 2:11. 10.1186/1757-4749-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasnitz N, Köhler H, Weigoldt M, Gerlach GF, Möbius P. 13 September 2013. Stability of genotyping target sequences of Mycobacterium avium subsp. paratuberculosis upon cultivation on different media, in vitro- and in vivo passage, and natural infection.Vet. Microbiol. 10.1016/j.vetmic.2013.09.008 [DOI] [PubMed] [Google Scholar]
- 16.Cernicchiaro N, Wells SJ, Janagama H, Sreevatsan S. 2008. Influence of type of culture medium on characterization of Mycobacterium avium subsp. paratuberculosis subtypes. J. Clin. Microbiol. 46:145–149. 10.1128/JCM.01769-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haff LA, Smirnov IP. 1997. Single-nucleotide polymorphism identification assays using a thermostable DNA polymerase and delayed extraction MALDI-TOF mass spectrometry. Genome Res. 7:378–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna SLB, Keefe GP, Barkema HW, Sockett DC. 2005. Evaluation of three ELISAs for Mycobacterium avium subsp. paratuberculosis using tissue and fecal culture as comparison standards. Vet. Microbiol. 110:105–111. 10.1016/j.vetmic.2005.07.010 [DOI] [PubMed] [Google Scholar]
- 19.Vary PH, Andersen PR, Green E, Hermon-Taylor J, McFadden JJ. 1990. Use of highly specific DNA probes and the polymerase chain reaction to detect Mycobacterium paratuberculosis in Johne's disease. J. Clin. Microbiol. 28:933–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, Banerji N, Kanjilal S, Kapur V. 2005. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc. Natl. Acad. Sci. U. S. A. 102:12344–12349. 10.1073/pnas.0505662102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fazekas A, Steeves R, Newmaster S. 2010. Improving sequencing quality from PCR products containing long mononucleotide repeats. Biotechniques 48:277–285. 10.2144/000113369 [DOI] [PubMed] [Google Scholar]
- 22.Ross PL, Belgrader P. 1997. Analysis of short tandem repeat polymorphisms in human DNA by matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 69:3966–3972. 10.1021/ac970312t [DOI] [PubMed] [Google Scholar]
- 23.Le Fleche P, Hauck Y, Onteniente L, Prieur A, Denoeud F, Ramisse V, Sylvestre P, Benson G, Ramisse F, Vergnaud G. 2001. A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestis and Bacillus anthracis. BMC Microbiol. 1:2. 10.1186/1471-2180-1-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stratilo CW, Lewis CT, Bryden L, Mulvey MR, Bader D. 2006. Single-nucleotide repeat analysis for subtyping Bacillus anthracis isolates. J. Clin. Microbiol. 44:777–782. 10.1128/JCM.44.3.777-782.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keim P, Van Ert MN, Pearson T, Vogler AJ, Huynh LY, Wagner DM. 2004. Anthrax molecular epidemiology and forensics: using the appropriate marker for different evolutionary scales. Infect. Genet. Evol. 4:205–213. 10.1016/j.meegid.2004.02.005 [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Silva I, Whitney J, Wainwright B, Andrews KR, Ylitalo-Ward H, Bowen BW, Toonen RJ, Goetze E, Karl SA. 2013. Microsatellites for next-generation ecologists: a post-sequencing bioinformatics pipeline. PLoS One 8:e55990. 10.1371/journal.pone.0055990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danin-Poleg Y, Cohen LA, Gancz H, Broza YY, Goldshmidt H, Malul E, Valinsky L, Lerner L, Broza M, Kashi Y. 2007. Vibrio cholerae strain typing and phylogeny study based on simple sequence repeats. J. Clin. Microbiol. 45:736–746. 10.1128/JCM.01895-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Belkum A. 1999. The role of short sequence repeats in epidemiologic typing. Curr. Opin. Microbiol. 2:306–311. 10.1016/S1369-5274(99)80053-8 [DOI] [PubMed] [Google Scholar]
- 29.Barthe S, Gugerli F, Barkley NA, Maggia L, Cardi C, Scotti I. 2012. Always look on both sides: phylogenetic information conveyed by simple sequence repeat allele sequences. PLoS One 7:e40699. 10.1371/journal.pone.0040699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruford MW, Wayne RK. 1993. Microsatellites and their application to population genetic studies. Curr. Opin. Genet. Dev. 3:939–943. 10.1016/0959-437X(93)90017-J [DOI] [PubMed] [Google Scholar]
- 31.Gupta PK, Varshney RK. 2000. The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185. 10.1023/A:1003910819967 [DOI] [Google Scholar]
- 32.Kayser M, de Knijff P. 2011. Improving human forensics through advances in genetics, genomics and molecular biology. Nat. Rev. Genet. 12:179–192. 10.1038/nrg2952 [DOI] [PubMed] [Google Scholar]
- 33.Bayliss CD, Dixon KM, Moxon ER. 2004. Simple sequence repeats (microsatellites): mutational mechanisms and contributions to bacterial pathogenesis. A meeting review. FEMS Immunol. Med. Microbiol. 40:11–19. 10.1016/S0928-8244(03)00325-0 [DOI] [PubMed] [Google Scholar]
- 34.Coenye T, Vandamme P. 2005. Characterization of mononucleotide repeats in sequenced prokaryotic genomes. DNA Res. 12:221–233. 10.1093/dnares/dsi009 [DOI] [PubMed] [Google Scholar]
- 35.Mrázek J, Guo X, Shah A. 2007. Simple sequence repeats in prokaryotic genomes. Proc. Natl. Acad. Sci. U. S. A. 104:8472–8477. 10.1073/pnas.0702412104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King CR, Marsh S. 2013. Pyrosequencing of clinically relevant polymorphisms. Methods Mol. Biol. 1015:97–114. 10.1007/978-1-62703-435-7_6 [DOI] [PubMed] [Google Scholar]
- 37.Moore MJ, Dhingra A, Soltis PS, Shaw R, Farmerie WG, Folta KM, Soltis DE. 2006. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 6:17. 10.1186/1471-2229-6-17 [DOI] [PMC free article] [PubMed] [Google Scholar]