

Abstract
Three cytochrome P450 monooxygenase CYP52 gene family members were isolated from the sophorolipid-producing yeast Starmerella bombicola (former Candida bombicola), namely, CYP52E3, CYP52M1, and CYP52N1, and their open reading frames were cloned into the pYES2 vector for expression in Saccharomyces cerevisiae. The functions of the recombinant proteins were analyzed with a variety of alkane and fatty acid substrates using microsome proteins or a whole-cell system. CYP52M1 was found to oxidize C16 to C20 fatty acids preferentially. It converted oleic acid (C18:1) more efficiently than stearic acid (C18:0) and linoleic acid (C18:2) and much more effectively than α-linolenic acid (C18:3). No products were detected when C10 to C12 fatty acids were used as the substrates. Moreover, CYP52M1 hydroxylated fatty acids at their ω- and ω-1 positions. CYP52N1 oxidized C14 to C20 saturated and unsaturated fatty acids and preferentially oxidized palmitic acid, oleic acid, and linoleic acid. It only catalyzed ω-hydroxylation of fatty acids. Minor ω-hydroxylation activity against myristic acid, palmitic acid, palmitoleic acid, and oleic acid was shown for CYP52E3. Furthermore, the three P450s were coassayed with glucosyltransferase UGTA1. UGTA1 glycosylated all hydroxyl fatty acids generated by CYP52E3, CYP52M1, and CYP52N1. The transformation efficiency of fatty acids into glucolipids by CYP52M1/UGTA1 was much higher than those by CYP52N1/UGTA1 and CYP52E3/UGTA1. Taken together, CYP52M1 is demonstrated to be involved in the biosynthesis of sophorolipid, whereas CYP52E3 and CYP52N1 might be involved in alkane metabolism in S. bombicola but downstream of the initial oxidation steps.
INTRODUCTION
Cytochrome P450 proteins are ubiquitously distributed in nature and display a broad range of enzymatic activities. They are heme proteins encoded by a superfamily of genes (1). The prokaryotic P450s constitute a large class of soluble proteins, whereas the eukaryotic enzymes are almost all membrane associated with either the mitochondrion or the endoplasmic reticulum. This enzyme family is involved, among others, in the biotransformation of drugs, xenobiotics, alkanes, terpenes, and aromatic compounds and the biosynthesis of steroids, fatty acids, and bile acids, as well as the degradation of herbicides and insecticides (2).
A subfamily of P450s, CYP52, has been described in alkane-assimilating yeasts, such as Yarrowia lipolytica, Candida tropicalis, and Candida maltosa (3, 4). These P450s and their corresponding reductase, NADPH cytochrome P450 reductase, form the hydroxylase complex responsible for the first and rate-limiting step of ω-oxidation of n-alkanes and fatty acids (5, 6). Candida tropicalis metabolizes fatty acids through the ω-oxidation pathway, in which the terminal methyl carbon is oxidized by three consecutive steps to a carboxylic group, yielding a dicarboxylic acid. The first step is catalyzed by a CYP52 enzyme and its corresponding reductase (7, 8). The alcohol produced is further oxidized to an aldehyde by a fatty alcohol oxidase and finally to the dicarboxylic acid by a fatty aldehyde dehydrogenase (8, 9).
The CYP52 genes are also believed to play a key role in sophorolipid synthesis. Starmerella bombicola (former Candida bombicola) is known to synthesize this biosurfactant of commercial interest, consisting of the disaccharide sophorose, to which one ω- or (ω-1)-hydroxyl fatty acid is linked (10–12). This hydroxyl fatty acid is formed by the yeast via hydroxylation of a common saturated or unsaturated fatty acid of 16 to 18 carbon atoms by CYP52 monooxygenase activity (13). Three P450s (CYP52E3, CYP52M1, and CYP52N1) were isolated from the genome of S. bombicola, and gene expression profiles revealed that CYP52E3 and CYP52N1 transcript levels did not correlate with sophorolipid production but showed clear upregulation when the yeast cells were grown on alkanes as the sole carbon source (14). In contrast, CYP52M1 was clearly upregulated during sophorolipid synthesis and very likely takes part in sophorolipid formation (14). Recently, it has been demonstrated that CYP52M1 is responsible for the synthesis of hydroxylated fatty acids, which are essential for sophorolipid production as the disabling of the CYP52M1 function resulted in complete abolishment of sophorolipid production (15). Hence, the CYP system plays a crucial role in both alkane degradation and sophorolipid synthesis in S. bombicola cells. In addition, a glucosyltransferase gene, UGTA1, was cloned from the same organism and was shown to catalyze the first glucosylation step in the biosynthetic pathway of sophorolipids (16).
To gain a better understanding of the molecular mechanism and the biochemical roles of CYP52E3, CYP52M1, and CYP52N1 in S. bombicola and to explore potential biotechnological applications, CYP52E3, CYP52M1, and CYP52N1 were cloned into the pYES2 vector and heterologously expressed in Saccharomyces cerevisiae. Their functions were analyzed with a variety of alkane and fatty acid substrates by using microsome proteins or a whole-cell system. In addition, the kinetic parameters of CYP52M1 with fatty acids were determined. Moreover, the three CYP52 gene-transformed cells were coincubated with UGTA1-transformed cells to get more insight into the sophorolipid synthetic pathway.
MATERIALS AND METHODS
Chemicals and microorganism.
Commercial chemicals were purchased in analytical grade from the following companies: linoleic acid and α-linolenic acid, Roth, Karlsruhe, Germany; oleic acid, cis-11-eicosenoic acid, arachidonic acid, palmitic acid, palmitoleic acid, lauric acid, capric acid, myristic acid, hexadecane, octadecane, and 16-hydroxy palmitic acid, Sigma-Aldrich, Steinheim, Germany; stearic acid, Merck, Hohenbrunn, Germany; racemic cis-9,10-epoxystearic acid and racemic trans-9,10-epoxystearic acid, Biozol Diagnostica, Eching, Germany; and cis-9-octadecene-1,18-dioic acid, AnalytiCon Discovery GmbH, Potsdam, Germany.
S. bombicola (ATCC 22214) was used for the isolation of the CYP52E3, CYP52M1, CYP52N1, and UGTA1 genes. The S. cerevisiae WAT11 and INVSc1 (Invitrogen, Karlsruhe, Germany) strains were used for heterologous expression of CYP52 genes and UGTA1.
Isolation of total RNA from S. bombicola.
S. bombicola was grown on 10 ml YGU medium (10 g/liter yeast extract, 1 g/liter urea, 100 g/liter glucose) at 30°C for 24 h with shaking (150 rpm). After removal of the culture medium by centrifugation (1 min, 13,200 rpm, room temperature), the cells were frozen in liquid nitrogen and ground into powder using a mortar and a pestle. The cell powder was resuspended with RNA extraction buffer for isolation of RNA. RNA isolation was performed using a Presto SpinR universal RNA purification kit (Molzym, Bremen, Germany).
Heterologous expression of CYP52E3, CYP52M1, CYP52N1, and UGTA1 in yeast cells.
The first-strand cDNAs were synthesized from 1 μg of total RNA isolated from S. bombicola (prepared as described above) using Moloney murine leukemia virus (MMLV) reverse transcriptase (RTase) (Promega, Mannheim, Germany) and a random hexamer primer. The coding regions of CYP52E3 (GenBank accession no. EU552420), CYP52M1 (GenBank accession no. EU552419), CYP52N1 (GenBank accession no. EU552421), and UGTA1 (GenBank accession no. HM440973) were amplified by RT-PCR with the cDNA template prepared as described above. The PCR primers used for each gene are listed in Table S1 in the supplemental material. The temperature program used was 5 min at 95°C for 1 cycle, followed by 45 s at 95°C, 45 s at 55°C, and 2 min at 72°C for 35 cycles, with a final extension at 72°C for 10 min. The PCR products were double digested with restriction enzymes, the recognition sequences (underlined) of which are contained in the primers shown in Table S1, and then ligated with the pYES2 vector (Invitrogen, Karlsruhe, Germany). The recombinant genes were subjected to sequencing to confirm the sequence of the inserts. Furthermore, these four constructs and pYES2 empty vector (negative control) were transformed into the S. cerevisiae WAT11 and INVSc1 strains for heterologous expression. The transformed yeast cells were maintained on SC-U plates (0.67% yeast nitrogen base, 0.01% each adenine, arginine, cysteine, leucine, lysine, threonine, tryptophan, and uracil; 0.005% each aspartic acid, histidine, isoleucine, methionine, phenylalanine, proline, serine, tyrosine, and valine, 2% glucose, 2% agar).
Isolation of microsome protein.
Isolation of microsome protein was performed as described previously (17), with modification. Yeast colonies were inoculated into 25 ml liquid SC-U medium (as described above but without agar) containing 2% glucose and grown at 30°C for 24 h under shaking (150 rpm) to reach an optical density at 600 nm (OD600) of 2 to 4. The overnight culture was diluted into 100 ml of induction medium (SC-U medium containing 2% galactose) to obtain an OD600 of 0.4, and cells were grown at 30°C for 24 h with shaking (150 rpm). Yeast cells were harvested by centrifugation (5 min, 4°C, 1,500 × g) and resuspended in 10 ml 100 mM Tris-HCl (pH 7.5) containing 1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were incubated at room temperature for 5 min, centrifuged at 1,500 × g for 5 min at 4°C, and resuspended in 4 ml cold Tris-HCl–PMSF buffer. The same volume of glass beads (250 to 500 μM diameter; Roth, Karlsruhe, Germany) then was added. Yeast cell walls were disrupted mechanically by vigorous vortexing for 30 s, followed by 30 s on ice. The vortexing procedure was repeated 15 times for a total of 15 min. The crude extract was recovered, and the beads were washed once with 4 ml of Tris-HCl–PMSF buffer. All extracts were pooled and centrifuged for 10 min at 4°C at 21,000 × g to recover the supernatant. Polyethylene glycol 4000 (final concentration, 0.1 g/ml) and NaCl (final concentration, 0.15 M) were added to the supernatant, and the mixture was incubated for 15 min on ice and centrifuged for 10 min at 10,300 × g. Pellets were resuspended in 2 ml 100 mM Tris-HCl containing 1 mM PMSF and 10% (vol/vol) glycerol and were homogenized in a glass homogenizer. Proteins were analyzed by SDS-PAGE with 10% polyacrylamide gels and stained with Coomassie brilliant blue R-250. The protein concentration was determined by the Bradford assay (18).
Enzyme assay.
Three different systems were applied for functional analysis of CYP52E3, CYP52M1, and CYP52N1. (i) In the in vitro system, the standard assay mixture (200 μl) contained 0.4 mg of microsomal protein, 50 mM sodium phosphate buffer (pH 7), 500 μM NADPH, and 300 μM substrate. The reaction was initiated by the addition of NADPH, incubated at 30°C with constant shaking for 20 min, and stopped by the addition of 200 μl of ethyl acetate. The products were extracted with ethyl acetate, evaporated to dryness, resuspended in methanol, and analyzed by liquid chromatography-mass spectrometry (LC-MS). For further confirmation, the product extracts were methylated and trimethylsilylated, and then subjected to GC-MS. (ii) In the biotransformation system, yeast colonies were inoculated into 15 ml liquid SC-U medium containing 2% glucose and grown at 30°C for 24 h with shaking (150 rpm) to an OD600 of 2 to 4. The overnight culture was diluted into 45 ml of YPGE medium (1% yeast extract, 1% peptone, 3% ethanol, 0.5% glucose) to obtain an OD600 of 0.4, and cells were grown at 30°C with shaking (150 rpm) until the cell density reached OD600 of 0.8 to 1.2. Next, 5 ml of 20% galactose and 50 mg of fatty acid substrate were added. The cells were grown at 30°C with shaking (150 rpm) for 2 days. (iii) In the resting cell system, the overnight culture was diluted into 50 ml of induction medium (SC-U medium containing 2% galactose) to obtain an OD600 of 0.4, and the cells were grown at 30°C for 24 h with shaking (150 rpm). The yeast cells were harvested by centrifugation (5 min, 1,500 × g, 4°C), washed once with 20 ml of 100 mM Tris-HCl buffer, and then resuspended in 25 ml of 100 mM Tris-HCl (pH 7.5). After addition of 10 mg of fatty acid substrate, the cells were incubated at 30°C for additional 1 or 20 h with shaking (150 rpm). For coassay of CYP52 proteins and UGTA1, the ratio of CYP52- and UGTA1-transformed cells was 1:1. The products were extracted with the same volume of ethyl acetate from growth medium or buffer and analyzed by LC-MS (method I for detection of hydroxyl fatty acids and diacids, method II for detection of glucolipids [see below]) and gas chromatography (GC)-MS.
Product identification und quantification. (i) LC-MS.
The high-performance liquid chromatography (HPLC) system consisted of a quaternary pump and a variable-wavelength detector, both from Agilent 1100 (Bruker Daltonics, Bremen, Germany). The column was a LUNA C18 100A column that was 150 by 2 mm (Phenomenex, Aschaffenburg, Germany). HPLC was performed with the following binary gradient system: solvent A, water with 0.1% formic acid; solvent B, 100% methanol with 0.1% formic acid. Two different gradient programs were used. In method I, the protocol was 0 to 15 min 70% A–30% B to 100% B, hold for 7 min, followed by 100% B to 70% A–30% B in 3 min and then hold for 10 min. The flow rate was 0.2 ml min−1. In method II, the protocol was 0 to 30 min 70% A–30% B to 100% B, hold for 14 min, followed by 100% B to 70% A–30% B in 6 min and then hold for 20 min. The flow rate was 0.1 ml min−1. Attached to the high-performance liquid chromatograph (HPLC) was a Bruker esquire 3000plus mass spectrometer with an electrospray ionization (ESI) interface that was used to record the mass spectra. The ionization voltage of the capillary was 4,000 V, and the end plate was set to −500 V.
(ii) GC-MS.
The metabolites generated were methylated with (trimethylsilyl)diazomethane and trimethylsilylated with N,O-bis(trimethylsilyl)trifluoroacetamide (19) and then subjected to GC-MS analysis. The products were analyzed on a Thermo Finnigan Trace DSQ mass spectrometer coupled to a 0.25 μm BPX5 20 M fused silica capillary column with a 30-m by 0.25-mm inner diameter. He (1.1 ml min−1) was used as the carrier gas. The temperature program was 100°C for 5 min, followed by 100 to 190°C at a rate of 30°C min−1, 190 to 250°C at a rate of 8°C min−1, and 250 to 280°C at a rate of 20°C min−1. Finally, the temperature was held at 280°C for 10 min. The injector temperature was 220°C, set for split injection, and the ion source temperature was 250°C. The mass range was recorded from 50 to 600 m/z, and spectra were evaluated with the Xcalibur software version 1.4.
Amounts of hydroxyl fatty acid products were determined using a standard curve calculated from various known concentrations of corresponding substrates against the mass peak areas that were recorded by LC-MS. The amounts of dicarboxylic acid products were determined using a standard curve calculated from various known concentrations of cis-9-octadecene-1,18-dioic acid (AnalytiCon Discovery GmbH, Potsdam, Germany) against the mass peak areas that were recorded by LC-MS.
Determination of kinetic parameters.
The pH optimum of CYP52M1 toward oleic, arachidonic, and linoleic acids was determined using different buffers for various pH ranges in 0.5-U intervals: 50 mM citric acid at pH 4.0 to 6.0, 50 mM sodium phosphate at pH 6.0 to 8.0, and 50 mM Tris-HCl at pH 8.0 to 11.0. The optimal temperature was evaluated from 5°C to 40°C in 5°C intervals at pH 7.5 (see Fig. S3 in the supplemental material). The kinetic parameters were determined under optimum conditions and were calculated by triplicate determinations using Microsoft Excel Solver (see Fig. S4 in the supplemental material).
RESULTS
Heterologous expression of CYP52E3, CYP52M1, and CYP52N1 and functional characterization of their enzymatic activities.
To functionally characterize S. bombicola cytochrome P450s, the open reading frames of CYP52E3, CYP52M1, and CYP5N1 were cloned into the pYES2 vector for expression in yeast strains WAT11 and INVSc1. The WAT11 strain also overexpresses a cytochrome P450 reductase (20). Three different systems were used to determine the functional activities of CYP52E3, CYP52M1, and CYP52N1, namely, a biotransformation system, a resting cell system, and an in vitro system, as described in Materials and Methods.
For the in vitro system, microsomes prepared from CYP52E3-, CYP52M1-, and CYP52N1-transformed yeast cells, as well as those from the control cells (empty vector), were investigated for their ability to oxidize a series of alkanes and fatty acids. After incubation, the products were isolated and analyzed by LC-MS. When oleic acid was used as the substrate, a product with a molecular size of 298 g mol−1 was detected in the reaction mixture containing CYP52M1 microsome protein (Fig. 1A). The product was methylated and trimethylsilylated and finally analyzed by GC-MS. GC-MS analysis revealed that the new product is actually a mixture of two compounds. Compound 1 (Fig. 2A) showed a mass spectrum with prominent ions at m/z 369 (M-15, loss of CH3), 353 (M-31, loss of OCH3), 337 (M-47, loss of CH3 and CH3OH), and 117 [(CH3)3SiO+CHCH3] (Fig. 2B) and was identified as 17-hydroxy oleic acid (21). Compound 2 (Fig. 2A), displayed a mass spectrum with prominent ions at m/z 369 (M-15, loss of CH3), 337 (M-47, loss of CH3 and CH3OH), and 103 [CH2 = O+Si(CH3)3] (Fig. 2C), which is identical to those of 18-hydroxy oleic acid (22). When linoleic acid was used as the substrate, 17-hydroxy and 18-hydroxy linoleic acids were detected (data not shown). Thus, CYP52M1 hydroxylated fatty acids at their ω- and ω-1 positions. A low level of dicarboxylic acid was also detected when oleic acid was used as a substrate (data not shown). The amount of this minor product is about 1/10 of that of the hydroxyl fatty acids.
FIG 1.
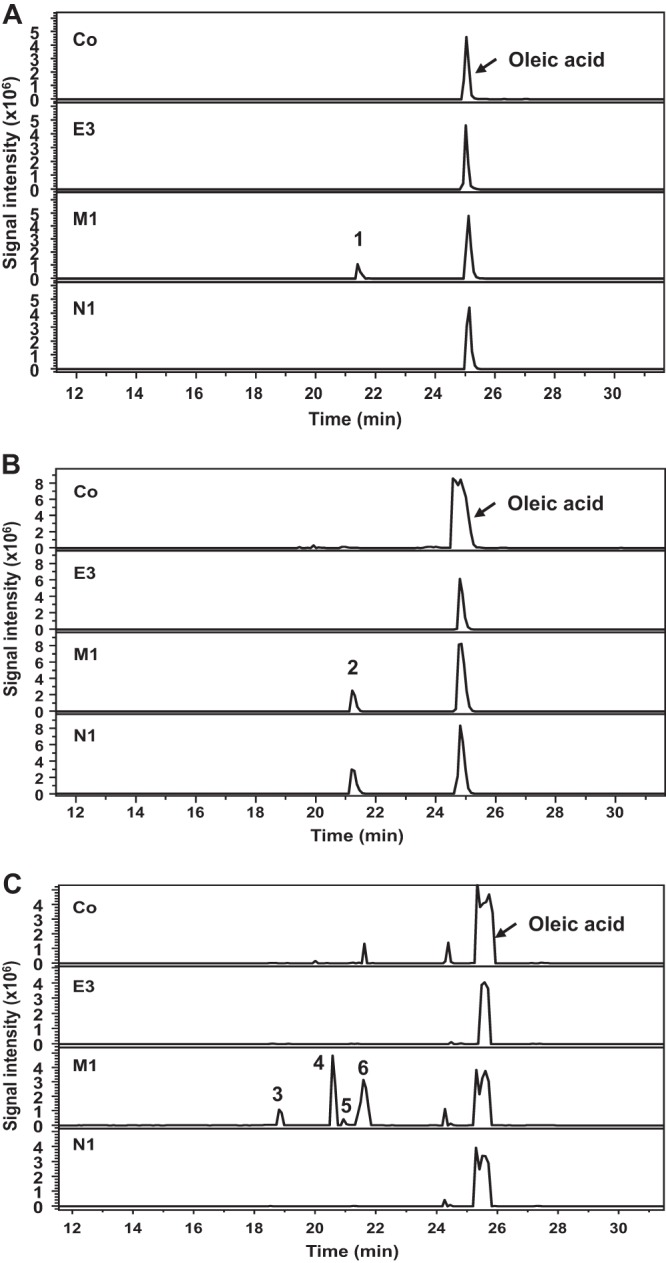
LC-MS analysis of products formed by incubation of oleic acid with three CYP52 proteins and control proteins using different systems. (A) In vitro system, the product hydroxyl oleic acid (peak 1) and the substrate oleic acid were monitored at m/z of 297 and 281, respectively. (B) The resting cell system, the product 18-carboxy oleic acid (peak 2), and the substrate oleic acid were monitored at m/z of 311 and 281, respectively. (C) The biotransformation system, the products 17,18-dihydroxy oleic acid (peak 3), 17-hydroxy-18-carboxy oleic acid (peak 4), 18-carboxy oleic acid (peak 5), and 17-hydroxy and 18-hydroxy oleic acids (peak 6) were monitored at m/z of 313, 327, 311, and 297, respectively. Co, negative control; E3, CYP52E3; M1, CYP52M1; N1, CYP52N1.
FIG 2.

GC-MS analysis of products formed by incubation of oleic acid with control (Co) and CYP52M1 (M1) microsome protein (in vitro system). (A) Chromatogram for ion trace m/z 337. The mass spectra of peaks 1 and 2 yield fragmentation patterns identical to those of 17-hydroxy oleic acid (B) (21) and 18-hydroxy oleic acid (C) (22).
When oleic acid was incubated with the resting cells, a product with a molecular size of 312 g mol−1 appeared in the reaction mixtures containing CYP52E3-, CYP52M1-, and CYP52N1-transformed cells, as shown by LC-MS analysis (Fig. 1B). However, only a very low level of product was formed by CYP52E3. The metabolite was further methylated, trimethylsilylated, and then analyzed by GC-MS. The product showed a mass spectrum with prominent ions at m/z 308 (M-32, loss of CH3OH) and 276 (M-64, loss of 2 × CH3OH) (see Fig. S2A in the supplemental material), which are identical to those of the Me3Si derivative of authentic 18-carboxy oleic acid (cis-9-octadecen-1,18-dioic acid). Hence, oleic acid was oxidized into its dicarboxylic acid in the resting cell system. Interestingly, CYP52N1 displayed activity in the resting cell system but not in the in vitro system. Moreover, 17-hydroxy linoleic acid (peak 1 in Fig. 3) was detected in addition to 18-carboxy linoleic acid (cis-9, cis-12-octadecadien-1,18-dioic acid) (peak 2 in Fig. 3) in the reaction mixture of linoleic acid with CYP52M1-transformed cells. However, this compound was not found after incubation of linoleic acid with CYP52N1-transformed cells (Fig. 3A). The same outcome was also obtained when oleic acid was used as a substrate (data not shown). This result indicated that CYP52N1 only catalyzed ω-hydroxylation. In order to find out whether CYP52M1 and CYP52N1 can oxidize ω-hydroxyl fatty acids to generate the α,ω-dicarboxylic acids, 16-hydroxy palmitic acid was incubated with control cells or CYP52E3-, CYP52M1-, and CYP52N1-transformed cells. LC-MS analysis showed that 16-hydroxy palmitic acid was converted into the dicarboxylic acid by all four cell types with the same efficiency (see Fig. S1 in the supplemental material). This implies that ω-hydroxyl fatty acids that are formed by CYP52-E3, CYP52M1, and CYP52N1 are further oxidized by endogenous enzymes of WAT11 yeast cells to the corresponding dicarboxylic acid. This observation might explain why only insignificant amounts of dicarboxylic acids were detected in the in vitro assay system.
FIG 3.

GC-MS analysis of products formed by incubation of linoleic acid with control (Co) or CYP52E3 (E3)-, CYP52M1 (M1)-, and CYP52N1 (N1)-transformed cells using the resting cell system. (A) Total-ion chromatogram. The mass spectra of peaks 1 and 2 yield fragmentation patterns identical to those of 17-hydroxy linoleic acid (B) (21) and 18-carboxy linoleic acid (C) (22).
For biotransformation studies, the fatty acid substrates were added to culture media containing the yeast cells after addition of galactose. After incubation for 2 days at 30°C with shaking, the products were extracted from the media with ethyl acetate and analyzed by LC-MS and GC-MS. Products were only formed by the CYP52M1-transformed yeast culture (Fig. 1C) whereas the negative-control and CYP52E3- and CYP52N1-transformed cell cultures lacked these compounds. Besides the primary products, 17-hydroxy oleic acid and 18-hydroxy oleic acid (peak 6 in Fig. 1C), the conversion of oleic acid yielded up to three additional metabolites, which were identified by GC-MS as putative 17,18-dihydroxy oleic acid (peak 3 in Fig. 1C), 17-hydroxy-18-carboxy oleic acid (peak 4 in Fig. 1C; see Fig. S2B in the supplemental material) (22), and 18-carboxy oleic acid (peak 5 in Fig. 1C). 17-Hydroxy oleic acid and 17-hydroxy-18-carboxy oleic acid were the main products.
Taken together, CYP52E3, CYP52M1, and CYP52N1 showed enzymatic activities toward fatty acids. CYP52M1 oxidized fatty acids into ω- and ω-1-hydroxy fatty acids, whereas CYP52E3 and CYP52N1 probably produced ω-hydroxyl fatty acids. ω-Hydroxyl acids were further converted into α,ω-dicarboxylic acids in resting cells. CYP52M1 produced two additional products—17,18-dihydroxy oleic acid, and 17-hydroxy-18-carboxy oleic acid—in the biotransformation system. CYP52M1 displayed its activity in all three systems studied; however, CYP52E3 and CYP52N1 showed activity only in the resting cells.
Substrate specificities of CYP52E3, CYP52M1, and CYP52N1.
To determine the substrate specificities of CYP52E3, CYP52M1, and CYP52N1, microsome proteins and resting cells of the respective expression strains were used for enzyme activity assays toward a variety of substrates. Product formation was measured by LC-MS. Substrates, including n-alkanes (hexadecane and octadecane), various fatty acids (capric, lauric, myristic, palmitic, palmitoleic, stearic, oleic, linoleic, α-linolenic, cis-11-eicosenoic, and arachidonic acids), and epoxy fatty acids (cis-9,10-epoxystearic acid and trans-9,10-epoxystearic acid) were used to detect P450 preferences regarding the substrate class and chain length. All substrates were also incubated with the negative (empty vector) control. No products were formed in the control experiments. When microsome proteins were used, only CYP52M1 showed enzyme activity (Table 1). CYP52M1 was found to have efficiently oxidized C16, C18, and C20 saturated and unsaturated fatty acids, namely, palmitic, palmitoleic, stearic, oleic, linoleic, cis-9,10-epoxystearic, trans-9,10-epoxystearic, and arachidonic acid to ω- and ω-1 hydroxy fatty acids. Oleic acid was the preferred substrate (Table 1). No activity was detected toward short-chain fatty acids (C10 to C14), cis-11-eicosenoic acid (C20:1), and C16 to C18 alkanes. Microsomal proteins prepared from the yeast strains expressing CYP52E3 and CYP52N1 did not oxidize any of the substrates examined, even after an incubation period of 20 h. Interestingly, when resting cells were used, CYP52N1 and CYP52M1 showed similar substrate specificities toward palmitic, palmitoleic, stearic, oleic, linoleicd, α-linolenic, and arachidonic acids (Table 1). Besides, CYP52N1 showed enzyme activity toward myristic acid. Both cell types converted these fatty acids into their corresponding ω-carboxylic fatty acids after 20 h of incubation. Moreover, ω-1-hydroxyl fatty acids were also detected in the assays containing CYP52M1. No product was detected when α-linolenic acid was incubated with CYP52M1 microsome proteins; however, a low level of 18-carboxy α-linolenic acid was formed by CYP52M1-expressing resting cells incubated with α-linolenic acid. In the resting cell system, CYP52M1 showed the highest activity toward oleic acid and CYP52N1 toward linoleic acid. CYP52E3 showed enzyme activity toward myristic acid, palmitic acid, palmitoleic acid, and oleic acid, but its activity is much lower than those of CYP52M1 and CYP52N1.
TABLE 1.
The catalytic activities of recombinant CYP52E3, CYP52M1, and CYP52N1 toward alkane and fatty acid substrates
| Substrate | Catalytic activity ofa: |
|||||
|---|---|---|---|---|---|---|
| CYP52E3 |
CYP52M1 |
CYP52N1 |
||||
| Microsomes (pmol min−1 mg−1) | Resting cells (pmol min−1 ml−1) | Microsomes (pmol min−1 mg−1) | Resting cells (pmol min−1 ml−1) | Microsomes (pmol min−1 mg−1) | Resting cells (pmol min−1 ml−1) | |
| Hexadecane | − | − | − | − | − | − |
| Octadecane | − | − | − | − | − | − |
| Capric acid | − | − | − | − | − | − |
| Lauric acid | − | − | − | − | − | − |
| Myristic acid | − | 11 ± 1 | − | 9 ± 0.2 | − | 97 ± 21 |
| Palmitic acid | − | 4 ± 0.4 | 220 ± 30 | 231 ± 26 | − | 642 ± 79 |
| Palmitoleic acid | − | 1 ± 0.2 | 195 ± 27 | 119 ± 25 | − | 177 ± 45 |
| Stearic acid | − | − | 311 ± 36 | 26 ± 3 | − | 22 ± 4 |
| Oleic acid | − | 6 ± 0.7 | 501 ± 22 | 508 ± 54 | − | 621 ± 11 |
| Linoleic acid | − | − | 343 ± 44 | 396 ± 19 | − | 740 ± 2 |
| α-Linolenic acid | − | − | − | 47 ± 1 | − | 99 ± 7 |
| cis-9,10-Epoxystearic acid | − | NA | 309 ± 63 | NA | − | NA |
| trans-9,10-Epoxystearic acid | − | NA | 301 ± 48 | NA | − | NA |
| cis-11-Eicosenoic acid | − | − | − | − | − | − |
| Arachidonic acid | − | − | 254 ± 13 | 85 ± 3 | − | 27 ± 2 |
Activities were determined by LC-MS. Values are the means of two or three separate determinations. Measurements were performed with microsome proteins (products are ω- and ω-1-hydroxyl fatty acids) or resting cells (products are hydroxyl fatty acids and α,ω-diacid). −, no activity; NA, not analyzed.
To gain more details about the catalytic properties of CYP52M1, the kinetic characteristics toward oleic, linoleic, and arachidonic acids were determined for CYP52M1 (Table 2). The microsome protein was used for kinetic study. At first, the enzymatic activity of CYP52M1 was analyzed at different pH values (pH 4 to 11) and temperatures (5 to 40°C) to find out the optimal catalytic conditions (see Fig. S3 in the supplemental material). The recombinant CYP52M1 enzyme works the best at 25 to 30°C and pH 7. The Km values for oleic, linoleic, and arachidonic acids were 40 μM, 68 μM, and 46 μM, respectively (Table 2; see Fig. S4 in the supplemental material). CYP52M1 oxidized oleic acid most efficiently, exhibiting about a 2-fold higher Vmax/Km value for this acid than for linoleic and arachidonic acids, which confirms the data in Table 1.
TABLE 2.
Kinetic parameters of CYP52M1 toward oleic acid, linoleic acid, and arachidonic acid
| Acid | Km (μM) | Vmax (pmol min−1 mg−1) | Vmax/Km (min−1 μM−1) |
|---|---|---|---|
| Oleic | 40 ± 2 | 535 ± 60 | 13.5 ± 1 |
| Linoleic | 68 ± 10 | 451 ± 50 | 7 ± 0.4 |
| Arachidonic | 54 ± 10 | 306 ± 31 | 5.7 ± 0.6 |
Components required for the oxidation of fatty acids by CYP52M1.
The activity of cytochrome P450s relies on associated proteins, such as an NADPH-cytochrome P450 reductase, that catalyzes the transfer of electrons from NADPH to the prosthetic heme group of the CYP protein. Based on this information, the components required for CYP52M1 activity were studied in vitro with CYP52M1 microsomal proteins. The omission of NADPH reduced the activity to 23%. The replacement of NADPH by NADH (26% activity) or NAD (25% activity) had almost no effect on the activity. The expression of CYP52M1 in the WAT11 strain, which overexpresses a cytochrome P450 reductase (20), did not improve the enzyme activity of CYP52M1. Its activity was the same as that expressed in the INVSc1 strain.
Heterologous expression and functional characterization of UGTA1.
The glucosyltransferase gene UGTA1 has been identified as being involved in the first glucosylation step in the biosynthetic pathway leading to sophorolipids (16). To verify its function, UGTA1 was cloned into the pYES2 vector and heterologously expressed in the S. cerevisiae WAT11 strain. Its enzymatic activity was tested with a mixture of 17- and 18-hydroxy oleic acids (prepared by incubating CYP52M1 microsome protein and oleic acid) and cis-9-octadecene-1,18-dioic acid (18-carboxy oleic acid). The products were analyzed by LC-MS. Glycosides, presumably, 17-O- and 18-O-(β-d-glucopyranosyl)-octadecenoic acids, were detected when UGTA1 was incubated with 17- and 18-hydroxy oleic acids. Surprisingly, a glucolipid was also detected when cis-9-octadecene-1,18-dioic acid was used a substrate (Fig. 4).
FIG 4.

LC-MS analysis of products generated by incubation of crude protein extract prepared from UGTA1-transformed cells with 17-hydroxy and 18-hydroxy oleic acid (top panel) and cis-9-octadecene-1,18-dioic acid (bottom panel). Peaks 1, 2, and 3 are glucolipids derived from 17-hydroxy oleic acid, 18-hydroxy oleic acid, and cis-9-octadecene-1,18-dioic acid, respectively.
Coassay of CYP52 proteins and UGTA1.
To get more insight into the sophorolipid synthetic pathway in S. bombicola, various fatty acids (palmitic, stearic, oleic, linoleic, and arachidonic acids) were incubated with UGTA1-transformed yeast cells in combination with control cells or CYP52E3-, CYP52M1-, or CYP52N1-transformed cells by using the resting cell system (for details see Materials and Methods). Glucolipids were detected in the reaction mixtures containing oleic acid and UGTA1-transformed cells in combination with CYP52E3-, CYP52M1-, or CYP52N1-transformed cells (Fig. 5A). However, only a minor amount of glucolipid was produced by CYP52E3/UGTA1, probably due to the low activity of CYP52E3 toward oleic acid (Table 1). In addition, the efficiency of glucolipid formation by CYP52M1/UGTA1 was higher than that by CYP52N1/UGTA1. LC-MS analysis also revealed that (ω-1)-O- and ω-O-glucolipids were produced from CYP52M1/UGTA1 (peak with shoulder), whereas ω-O-glucolipid was produced by CYP52E3/UGTA1 and CYP52N1/UGTA1 (Fig. 5A). Palmitic, stearic, oleic, linoleic, and arachidonic acids were converted into their corresponding glucolipids by CYP52M1/UGTA1 (Fig. 5B) and CYP52N1/UGTA1 (data not shown). However, CYP52E3/UGTA1 could only transform oleic acid and palmitic acid into glucolipids. Taken together, UGTA1 glucosylated all hydroxyl fatty acids generated by CYP52E3, CYP52M1, and CYP52N1 into their corresponding glucolipids (Fig. 5A).
FIG 5.

LC-MS analysis of products formed by the coassay of UGTA1 and CYP52 proteins. (A) Products formed by incubation of oleic acid and UGTA1-transformed cells in combination with control cells (Co) or CYP52E3 (E3)-, CYP52M1 (M1)-, and CYP52N1 (N1)-transformed cells using the resting cell system. (B) Products formed by incubation of various fatty acids and UGTA1 in combination with CYP52M1-transformed cells.
DISCUSSION
Three cytochrome P450 monooxygenase genes belonging to the CYP52 gene family were isolated from the genome of the sophorolipid-producing yeast S. bombicola, and their products were biochemically characterized. One gene (CYP52E3) displayed high identity with known CYP52E members, whereas the other genes (CYP52M1 and CYP52N1) are the first members of two new subgroups: CYP52M and CYP52N (14). Recently, CYP52M1 has been implicated in the synthesis of hydroxylated fatty acids, which are essential for sophorolipid production (15). However, the experimental confirmation of the biochemical activity of the CYP52 proteins from S. bombicola is still missing.
Several CYP52 members have been isolated from diverse sources and heterologously expressed in S. cerevisiae or insect cells, and their substrate specificities have been investigated (Table 3). Some CYP52 members display multiple functions, as, for example, CYP52A3 is able to efficiently catalyze a cascade of sequential mono- and diterminal monooxygenation reactions from n-alkanes to α,ω-dicarboxylic acid (23, 24). CYP52A3 displayed a significant preference for hexadecane and turned out to be the most important C. maltosa enzyme for the primary hydroxylation of n-alkanes. CYP52A4 and CYP52A5 exhibited broad substrate specificity toward both n-alkanes and fatty acids (24). CYP52A9 was found to catalyze preferentially the hydroxylation of fatty acids, with highest activity toward oleic acid (24). CYP52A13 preferentially oxidized oleic acid and other unsaturated acids to ω-hydroxyl fatty acids. CYP52A17 also transformed oleic acid but converted shorter, saturated fatty acids, such as myristic acid, much more effectively. Both enzymes also oxidized ω-hydroxyl fatty acids to generate the α,ω-diacid (25). In this study, we showed that CYP52M1 metabolized a number of saturated and unsaturated acids to ω-1 and ω-hydroxyl fatty acids (Fig. 6A). So far, CYP52M1 is the only CYP52 member that efficiently hydroxylates fatty acids at their ω-1 position. CYP52N1 displayed similar substrate specificity to CYP52M1, but it only catalyzed ω-hydroxylation of fatty acids, and CYP52E3 has minor ω-hydroxylation activity toward myristic acid, palmitic acid, palmitoleic acid, and oleic acid. Finally, CYP52X1 showed highest activity against medium-chain fatty acids (C12:0 and C14:0) and resembles subgroups A3 to A5 with regard to chain length preference (26).
TABLE 3.
Substrate specificities of CYP52 members
| Gene product | Favorite substrate(s) | Function | Source | Reference |
|---|---|---|---|---|
| CYP52A3 | n-Alkanes (C12, C16) | Alkane hydroxylation | C. maltosa | 24 |
| CYP52A4 | n-Alkanes (C12, C16) | Alkane hydroxylation | C. maltosa | 24 |
| Fatty acids (C14:0) | ω-Hydroxylation | |||
| CYP52A5 | n-Alkanes (C12, C16) | Alkane hydroxylation | C. maltosa | 24 |
| Fatty acids (C18:1) | ω-Hydroxylation | |||
| CYP52A9 | Fatty acids (C18:1) | ω-Hydroxylation | C. maltosa | 24 |
| CYP52A13 | C18:1, C18:2 | ω-Hydroxylation | C. tropicalis | 25 |
| CYP52A17 | C12:0, C14:0 | ω-Hydroxylation | C. tropicalis | 25 |
| CYP52E3 | C14:0 | ω-Hydroxylation | S. bombicola | This work |
| CYP52M1 | C18:1 | ω-Hydroxylation | S. bombicola | This work |
| ω-1-Hydroxylation | ||||
| CYP52N1 | C18:2 | ω-Hydroxylation | S. bombicola | This work |
| CYP52X1 | C12:0 | ω-Hydroxylation | Beauveria bassiana | 26 |
FIG 6.

(A) Products formed by CYP52M1 in different assay systems. (B) Glucolipids generated by CYP52M1/UGTA1, CYP52E3/UGTA1, and CYP52N1/UGTA1.
Recombinant CYP52M1 even converted long-chain fatty acids, such as arachidonic acid, to their corresponding ω- and ω-1-hydroxyl fatty acids in microsomal preparations and α,ω-diacid in resting cells systems (Table 1), whereas coassays of CYP52M1 and UGTA1 also produced the respective ω-1-O- and ω-O-glucolipids (Fig. 5). This observation confirms previous studies showing that S. bombicola, when grown on glucose and arachidonic acid, synthesized sophorolipids consisting of sophorose linked glycosidically to the terminal (ω) or subterminal (ω-1) hydroxyl group of a hydroxyl fatty acid (27). On acid hydrolysis, these sophorolipids yielded 19-hydroxyeicosatetraenoic acid and 20-hydroxyeicosatetraenoic acid (12).
In contrast, the use of short-chain lipid precursors (C10 to C14) in S. bombicola fermentations resulted in low sophorolipid production (28). C18 chain-length fatty acids were most suitable for sophorolipid biosynthesis, and C18:1 (oleic acid) was preferred over its saturated counterpart. Increase in the desaturation of C18 above 1 was unfavorable for sophorolipid production (29). This substrate preference of S. bombicola nicely coincides with the fatty acid specificity of CYP52M1 (Tables 1 and 2) and confirms its significance in sophoroside biosynthesis.
Interestingly different products were produced by CYP52M1 in different experimental systems (Fig. 1 and 6). It is assumed that in the whole-cell systems (resting cells and biotransformation) ω-hydroxyl fatty acids formed by CYP52M1 are further metabolized by endogenous enzymes of the cells to the corresponding dicarboxylic acids. It has been reported that ω-hydroxyl fatty acids were further oxidized to dicarboxylic acid via the action of fatty alcohol oxidase and aldehyde dehydrogenase (8, 9). Our results also showed that S. cerevisiae cells were able to transform 16-hydroxy palmitic acid into its respective dicarboxylic acid, which nicely confirms our hypothesis.
A phylogenetic tree of CYP52 proteins showed that CYP52M1 and CYP52N1 were clustered together, although the sequence identity among them is not extremely high (43%) (14). However, CYP52N1 displayed similar substrate specificity to CYP52M1 (Table 1). CYP52E3 shows a 91% match with CYP52E2 and an 82% match to CYP52E1. Nevertheless, the function or biochemical activity of the CYP52E1 and CYP52E2 gene products has not been clarified until now. CYP52M1 is clearly upregulated during sophorolipid synthesis (14), and CYP52M1 knockout mutants lack sophorolipids (15). Our results confirm the importance of CYP52M1 and UGTA1 for sophorolipid biosynthesis in S. bombicola. Besides, we achieved the biocatalytic production of (ω-1)-O- and ω-O-glucolipids by incubation of S. cerevisiae resting cells expressing CYP52M1 and UGTA1 with various fatty acids.
CYP52E3 and CYP52N1 seem not to contribute to sophorolipid synthesis but showed clear upregulation when yeast cells were grown on alkanes as the sole carbon source, indicating that they could participate in the metabolism of alkanes (14). In our experiments CYP52E3 did not convert hexadecane and octadecane but oxidized C14:0, C16:0, C16:1, and C18:1 fatty acids, albeit with low activity. In addition, CYP52N1-transformed cells were found to convert C14 to C20 fatty acids to their corresponding dicarboxylic acids, although its expression was not induced by fatty acids (14). Moreover, coincubation of CYP52E3/UGTA1 or CYP52N1/UGTA1 with oleic acid yielded glucolipids. This indicates that CYP52E2 and CYP52N1 could have a function in alkane metabolism, probably downstream of the initial oxidation step of the hydrocarbons, because hexadecane and octadecane were not accepted as the substrates by both proteins.
Expression of various microsomal P450s involved in the biotransformation of pharmaceuticals and secondary plant metabolites or the degradation of xenobiotics in S. cerevisiae, Pichia pastoris, or Yarrowia lipolytica has been reported repeatedly. These strains can be used to produce fine chemicals and to develop new potential drugs (30–32). Now, genetically engineered yeasts have also been developed to convert fatty acids and alkanes into long-chain α,ω-dicarboxylic acids (33, 34). These compounds are potentially useful in a variety of applications, including adhesives, fragrances, lubricants, coatings, and specialty polymers. Furthermore, the whole-cell biotransformation system has been found to be an effective way to synthesize ω-carboxyl fatty acids with internal functional groups from renewable feedstocks. In this study, the expression of CYP52M1 and CYP52N1 in S. cerevisiae allowed the conversion of various fatty acids into their corresponding dicarboxylic acids. As S. cerevisiae offers a low-cost and efficient way to express heterologous P450s, this system can be further developed for producing dicarboxylic acids on a large scale.
Supplementary Material
ACKNOWLEDGMENTS
Financial support by the German Federal Ministry of Nutrition, Agriculture and Consumer Protection (BMELV) and FNR (SynRg) is gratefully acknowledged.
Footnotes
Published ahead of print 15 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02886-13.
REFERENCES
- 1.Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, Gonzales FJ, Coon MJ, Gunsalus IC, Gotoh O, Okuda K, Nebert DW. 1993. The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes, and nomenclature. DNA Cell Biol. 12:1–51. 10.1089/dna.1993.12.1 [DOI] [PubMed] [Google Scholar]
- 2.Bernhardt R. 2006. Cytochrome P450 as versatile biocatalysts. J. Biotechnol. 124:128–145. 10.1016/j.jbiotec.2006.01.026 [DOI] [PubMed] [Google Scholar]
- 3.Ohkuma M, Muraoka S, Tanimoto T, Fujii M, Ohta A, Takagi M. 1995. CYP52 (cytochrome P450alk) multigene family in Candida maltosa: identification and characterization of eight members. DNA Cell Biol. 14:163–173. 10.1089/dna.1995.14.163 [DOI] [PubMed] [Google Scholar]
- 4.Seghezzi W, Melli C, Ruffiner R, Kuenzi R, Sanglard D, Fiechter A. 1992. Identification and characterization of additional members of the cytochrome P450 multigene family CYP52 of Candida tropicalis. DNA Cell Biol. 11:767–780. 10.1089/dna.1992.11.767 [DOI] [PubMed] [Google Scholar]
- 5.Gilewicz M, Zacek M, Bertrand JC, Azoulay E. 1979. Hydroxylase regulation in Candida tropicalis grown on alkanes. Can. J. Microbiol. 25:201–206. 10.1139/m79-031 [DOI] [PubMed] [Google Scholar]
- 6.Gmunder K, Kappeli O, Fiechter A. 1981. Chemostat studies on the hexadecane assimilation by the yeast Candida tropicalis: regulation of cytochromes and enzymes. Eur. J. Appl. Microbiol. Biotechnol. 12:135–142. 10.1007/BF01008333 [DOI] [Google Scholar]
- 7.Mauersberger S, Drechsler H, Oehme G, Müller HG. 1992. Substrate specificity and stereoselectivity of fatty alcohol oxidase from the yeast Candida maltosa. Appl. Microbiol. Biotechnol. 37:66–73 [Google Scholar]
- 8.Eirich LD, Craft D, Steinberg L, Wilson CR. 2004. Cloning and characterization of three fatty alcohol oxidase genes from strain Candida tropicalis ATCC 20336. Appl. Environ. Microbiol. 70:4872–4879. 10.1128/AEM.70.8.4872-4879.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemp GD, Dickinson FM, Ratledge C. 1988. Inducible long chain alcohol oxidase from alkane-grown Candida tropicalis. Appl. Microbiol. Biotechnol. 29:370–374. 10.1007/BF00265821 [DOI] [Google Scholar]
- 10.Asmer HJ, Lang S, Wagner F, Wray V. 1988. Microbial-production, structure elucidation and bioconversion of sophorose lipids. J. Am. Oil Chem. Soc. 65:1460–1466. 10.1007/BF02898308 [DOI] [Google Scholar]
- 11.Van Bogaert INA, Saerens K, De Muynck C, Develter D, Soetaert W, Vandamme EJ. 2007. Microbial production and application of sophorolipids. Appl. Microbiol. Biotechnol. 76:23–34. 10.1007/s00253-007-0988-7 [DOI] [PubMed] [Google Scholar]
- 12.Shah S, Prabhune A. 2007. Purification by silica gel chromatography using dialysis tubing and characterization of sophorolipids produced from Candida bombicola grown on glucose and arachidonic acid. Biotechnol. Lett. 29:267–272. 10.1007/s10529-006-9221-5 [DOI] [PubMed] [Google Scholar]
- 13.Lottermoser K, Schunck WH, Asperger O. 1996. Cytochromes p450 of the sophorose lipid-producing yeast Candida apicola: heterogeneity and polymerase chain reaction-mediated cloning of two genes. Yeast 12:565–575 [DOI] [PubMed] [Google Scholar]
- 14.Van Bogaert INA, Demey M, Develter D, Soetaert W, Vandamme EJ. 2009. Importance of the cytochrome P450 monooxygenase CYP52 family for the sophorolipid-producing yeast Candida bombicola. FEMS Yeast Res. 9:87–94. 10.1111/j.1567-1364.2008.00454.x [DOI] [PubMed] [Google Scholar]
- 15.Van Bogaert INA, Holvoet K, Roelants SLKW, Li B, Lin Y-C, Van der Peer Y, Soetaert W. 2013. The biosynthetic gene cluster for sophorolipids: a biotechnological interesting biosurfactant produced by Starmerella bombicola. Mol. Microbiol. 88:501–509. 10.1111/mmi.12200 [DOI] [PubMed] [Google Scholar]
- 16.Saerens KMJ, Roelants SLKW, Van Bogaert INA, Soetaert W. 2011. Identification of the UDP-glucosyltransferase gene UGTA1, responsible for the first glucosylation step in the sophorolipid biosynthetic pathway of Candida bombicola ATCC 22214. FEMS Yeast Res. 11:123–132. 10.1111/j.1567-1364.2010.00695.x [DOI] [PubMed] [Google Scholar]
- 17.Eberle D, Ullmann P, Werck-Reichhart D, Petersen M. 2009. cDNA cloning and functional characterization of CYP98A14 and NADPH:cytochrome P450 reductase from Coleus blumei involved in rosmarinic acid biosynthesis. Plant Mol. Biol. 69:239–253. 10.1007/s11103-008-9420-7 [DOI] [PubMed] [Google Scholar]
- 18.Bradford MM. 1976. A rapid and sensitive for the quantitation of microgram quantitites of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 19.Aghofack-Nguemezi J, Fuchs C, Yeh S-Y, Huang F-C, Hoffmann T, Schwab W. 2011. An oxygenase inhibitor study in Solanum lycopersicum combined with metabolite profiling analysis revealed a potent peroxygenase inactivator. J. Exp. Bot. 62:1313–1323. 10.1093/jxb/erq368 [DOI] [PubMed] [Google Scholar]
- 20.Urban P, Mignotte C, Kazmaier M, Delorme F, Pompon D. 1997. Cloning, yeast expression, and characterization of the coupling of two distantly related Arabidopsis thaliana NADPH-cytochrome P450 reductases with P450 CYP73A5. J. Biol. Chem. 272:19176–19186. 10.1074/jbc.272.31.19176 [DOI] [PubMed] [Google Scholar]
- 21.Lanser AC, Plattner RD, Bagby MO. 1992. Production of 15-, 16- and 17-hydroxy-9-octadecenoic acids by bioconversion of oleic acid with Bacillus pumilus. J. Am. Oil Chem. Soc. 69:363–366. 10.1007/BF02636069 [DOI] [Google Scholar]
- 22.Eglinton G, Hunneman DH, McCormick A. 1968. Gas chromatographic-mass spectrometric studies of long chain hydroxyl acids. III. The mass spectra of the methyl esters trimethyllsilyl ethers of aliphatic hydroxyl acids. A facile method of double bond location. Org. Mass Spectrom. 1:593–611 [Google Scholar]
- 23.Scheller U, Zimmer T, Becher D, Schauer F, Schunck W-H. 1998. Oxygenation cascade in conversion of n-alkanes to α,ω-dioic acids catalyzed by cytochrome P450 52A3. J. Biol. Chem. 273:32528–32534. 10.1074/jbc.273.49.32528 [DOI] [PubMed] [Google Scholar]
- 24.Zimmer T, Ohkuma M, Ohta A, Takagi M, Schunck W-H. 1996. The CYP52 multigene family of Candida maltosa encodes functionally diverse n-alkane-inducible cytochrome P450. Biochem. Biophys. Res. Commun. 224:784–789. 10.1006/bbrc.1996.1100 [DOI] [PubMed] [Google Scholar]
- 25.Eschenfeldt WH, Zhang Y, Samaha H, Stols L, Eirich D, Wilson CR, Donnelly MI. 2003. Transformation of fatty acids catalyzed by cytochrome P450 monooxygenase enzymes of Candida tropicalis. Appl. Environ. Microbiol. 69:5992–5999. 10.1128/AEM.69.10.5992-5999.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S, Widemann E, Bernard G, Lesot A, Pinot F, Pedrini N, Keyhani NO. 2012. CYP52X1, representing new cytochrome P450 subfamily, displays fatty acid hydroxylase activity and contributes to virulence and growth on insect cuticular substrates in entomopathogenic fungus Beauveria bassiana. J. Biol. Chem. 287:13477–13486. 10.1074/jbc.M111.338947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prabhune A, Fox SR, Ratledge C. 2002. Transformation of arachidonic acid to 19-hydroxy and 20-hydroxy-eicosatetraenoic acid using Candida bombicola. Biotechnol. Lett. 24:1041–1044. 10.1023/A:1015662013985 [DOI] [Google Scholar]
- 28.Cavalero DA, Cooper DG. 2003. The effect of medium composition on the structure and physical state of sophorolipids produced by Candida bombicola ATCC 22214. J. Biotechnol. 103:31–41. 10.1016/S0168-1656(03)00067-1 [DOI] [PubMed] [Google Scholar]
- 29.Felse PA, Shah V, Chan J, Rao KJ, Gross RA. 2007. Sophorolipid biosynthesis by Candida bombicola from industrial fatty acid residues. Enzyme Microb. Technol. 40:316–323. 10.1016/j.enzmictec.2006.04.013 [DOI] [Google Scholar]
- 30.Andersen MD, Busk PK, Svendsen I, Moller BL. 2000. Cytochromes P450 from cassava (Manihot esculenta Crantz) catalyzing the first steps in the biosynthesis of the cyanogenic glucosides linnamarin and lotaustralin. Cloning, functional expression in Pichia pastoris and substrate specificity of the isolated recombinant enzymes. J. Biol. Chem. 275:1966–1975. 10.1074/jbc.275.3.1966 [DOI] [PubMed] [Google Scholar]
- 31.Nthangeni MB, Urban P, Pompon D, Smit MS, Nicaud JM. 2004. The use of Yarrowia lipolytica for the expression of human cytochrome P450 CYP1A1. Yeast 21:583–592. 10.1002/yea.1127 [DOI] [PubMed] [Google Scholar]
- 32.Fickers P, Benetti PH, Wache Y, Marty A, Mauersberger S, Smit MS, Nicaud JM. 2005. Hydrophobic substrate utilisation by the yeast Yarrowia lipolytica, and its potential applications. FEMS Yeast Res. 5:527–543. 10.1016/j.femsyr.2004.09.004 [DOI] [PubMed] [Google Scholar]
- 33.Picataggio S, Rohrer T, Deanda K, Lanning D, Reynolds R, Mielenz J, Eirich LD. 1992. Metabolic engineering of Candida tropicalis for the production of long-chain dicarboxylic acids. Biotechnology (NY) 10:894–898. 10.1038/nbt0892-894 [DOI] [PubMed] [Google Scholar]
- 34.Picataggio S, Rohrer T, Eirich LD. July 1997. Method for increasing the omega-hydroxylase activity in Candida tropicalis. US patent 5,648,247
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.