

Abstract
Rapid and accurate strain identification is paramount in the battle against microbial outbreaks, and several subtyping approaches have been developed. One such method uses clustered regular interspaced short palindromic repeats (CRISPRs), DNA repeat elements that are present in approximately half of all bacteria. Though their signature function is as an adaptive immune system against invading DNA such as bacteriophages and plasmids, CRISPRs also provide an excellent framework for pathogen tracking and evolutionary studies. Analysis of the spacer DNA sequences that reside between the repeats has been tremendously useful for bacterial subtyping during molecular epidemiological investigations. Subtyping, or strain identification, using CRISPRs has been employed in diverse Gram-positive and Gram-negative bacteria, including Mycobacterium tuberculosis, Salmonella enterica, and the plant pathogen Erwinia amylovora. This review discusses the several ways in which CRISPR sequences are exploited for subtyping. This includes the well-established spoligotyping methodologies that have been used for 2 decades to type Mycobacterium species, as well as in-depth consideration of newer, higher-throughput CRISPR-based protocols.
INTRODUCTION
Subtyping, the differentiation of bacteria below the species or subspecies level (i.e., to the strain level), is a vital epidemiological tool in the recognition of outbreaks and identification of infection sources. It is imperative to accurately identify isolates that are part of an outbreak in as timely manner as possible in order to mount an appropriate public health response. An ideal subtyping method is highly discriminatory in that it can differentiate between strains but is not so discriminatory that epidemiologic concordance is compromised (1). Beyond strain identification, high-resolution subtyping methods can provide opportunities to improve our understanding of bacterial population genetics, evolution, and epidemiology.
Replacing traditional methods such as phage typing and antibiograms, several higher-throughput and higher-resolution subtyping methods have been developed in the past 2 decades. These include PCR-centered approaches such as multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) and multilocus sequence typing (MLST) and protocols relying on restriction digestion such as restriction fragment length polymorphism (RFLP), pulsed-field gel electrophoresis (PFGE), and ribotyping analyses, plus, more recently, whole-genome sequence-based techniques (for a review, see reference 2). Clustered regularly interspaced short palindromic repeats, or CRISPRs, are bacterial loci whose dynamic nature has allowed them to be harnessed as ideal targets for molecular subtyping. This review details the use of CRISPRs for subtyping and highlights the diverse typing applications that use these loci.
ORGANIZATION OF CRISPR LOCI
CRISPRs were first identified over 25 years ago as ambiguous repeats in Escherichia coli (3); the repeats are now referred to as CRISPR spacer arrays (4–6). CRISPR arrays consist of tandem direct repeats (DRs) of 23 to 55 bp in length separated by similarly sized variable spacer sequences that are generally derived from bacteriophages or plasmids (7–10).
In their best-characterized capacity, CRISPR elements function as an elegant nucleic acid-based adaptive immune system in both archaea and bacteria (reviewed in references 11, 12, and 13). Approximately 85% and 48% of archaea and bacteria, respectively, that have been sequenced to date harbor CRISPR elements (14).
WHAT ARE CRISPRS?
CRISPR loci comprise of two main elements, the CRISPR spacer array and a group of CRISPR-associated (cas) genes (Fig. 1), collectively referred to as a “CRISPR-Cas system” (15). An AT-rich region, known as the leader sequence, is present directly upstream of the spacer array (5) and thought to function as a promoter (16). The length of a CRISPR array is dependent on the number of spacers and varies dramatically among different organisms and also among different bacterial serotypes or strains. The smallest spacer arrays comprise one spacer flanked by direct repeats, and to date, the largest single spacer array has been identified in Haliangium ochraceum DSM 14365, with 587 spacers (14).
FIG 1.
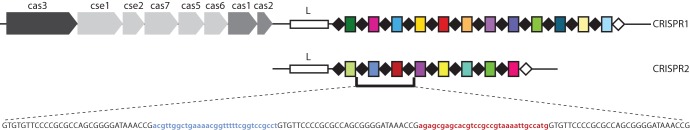
CRISPR-cas system. There are two CRISPR loci in Salmonella enterica and seven cas genes (light gray arrows). All CRISPR-Cas systems contain cas1 and cas2 (medium gray boxes). S. enterica has a type I CRISPR-Cas system of which cas3 is the signature gene (dark gray box). An AT-rich leader sequence is immediately upstream of each spacer array (white boxes; L). The direct repeats (DRs) are shown as black diamonds, and the terminal DR, which differs from the consensus DR, is shown as a white diamond. Spacers are shown as colored rectangles, and unique spacers are represented by unique colors. Below the spacer array, the sequence of two spacers and the respective three flanking DRs is shown with the DRs in black uppercase characters and the spacers color coded and in lowercase characters.
The activity of a CRISPR locus occurs in three stages: acquisition, expression, and interference. Acquisition, or “adaptation,” of the CRISPR locus involves addition of new spacers, generally to the 5′ end, or “leader proximal end,” of the spacer array and occurs as the CRISPR-Cas system adapts to a new invader (7). The CRISPR spacer array is constitutively transcribed into a precursor CRISPR RNA (pre-crRNA) that is cleaved by specific Cas proteins and further processed into mature, small interfering crRNAs. These crRNAs typically comprise the spacer flanked on either side by portions of the DRs (17–20). Subsequently, mature crRNAs guide the Cas-crRNA ribonucleoprotein complex to complementary nucleic acids, typically invading bacteriophages or plasmids, resulting in degradation of the target (21).
As new spacers are always added to one end of the CRISPR array, a polarity exists: spacers at the leader distal end are more ancient and are often shared among common ancestors (10, 22). Thus, the specific spacer composition of a CRISPR array can elegantly reflect the divergence of bacterial strains or serotypes (for an example, see reference 23). Spacer acquisition itself differs dramatically among different species, and endogenous acquisition has been observed in the laboratory in only a few bacteria (7, 24, 25). Acquisition, along with spacer loss and duplication, makes CRISPR elements among the fastest evolving loci in bacteria (26–30).
Given the temporal organization of spacers, sequencing of CRISPR arrays has been a tremendously useful tool in investigating and constructing phylogenetic relationships between different bacterial lineages, specifically, in Yersinia species, Erwinia amylovora, E. coli, and Salmonella enterica and, most recently, in the periodontal pathogen Porphyromonas gingivalis (23, 31–38). CRISPR analysis from metagenomic data can also be used to identify both the presence of and relationship between viruses and hosts within complex and diverse ecological niches (26, 30, 39–41). Beyond subtyping and these examples, more-versatile CRISPR-based applications exist, namely, their use in development of commercial phage-resistant bacterial strains (22, 42, 43) and as genome editing tools in eukaryotes (44–48).
In this review, we focus on how CRISPR spacer sequence information has been utilized for efficient bacterial subtyping in the context of molecular epidemiology. We discuss initial CRISPR-based subtyping methodologies, as well as review sequenced-based CRISPR subtyping approaches. We finish with a few examples of unique and alternative uses of CRISPRs for subtyping bacteria.
WHAT MAKES AN IDEAL CRISPR-TYPING LOCUS?
Though present in 48% of bacteria, not all CRISPRs are appropriate for molecular subtyping. Bacteria that acquire spacers at a higher rate are not likely to be tremendously useful for subtyping as, in the presence of bacteriophages or replicating plasmids, acquisition may happen within the time frame of an outbreak investigation. Spacer acquisition is not the only manner in which CRISPR loci from different strains can be modified; internal spacers can also be lost (31, 38, 42, 49–51), and single nucleotide polymorphisms (SNPs) can be introduced into the spacers or direct repeats (7). These changes can also contribute strain-to-strain differences (52). Conversely, bacterial species that contain homogenous CRISPR loci which are not adapting at all, or whose genetic integrity is diminishing (frequent mutations/deletions within cas genes and/or disruption of the spacer array), are not likely to exhibit strain-to-strain differences.
SPOLIGOTYPING
CRISPR applications existed long before their function was elucidated. The first use of spacer information for subtyping was in spacer-oligonucleotide typing, or “spoligotyping,” of Mycobacterium tuberculosis strains (53, 54). The principle of spoligotyping is PCR amplification of the CRISPR array with labeled primers that recognize the DR sequences (Fig. 2A), followed by hybridization of the PCR products to a membrane that contains probes bearing spacer DNA sequences (Fig. 2B). Due to strain-specific spacer content, differential hybridization patterns enable separation of different strains (Fig. 2C). Spoligotyping was able to separate the members of a group of closely related Mycobacterium species that comprise the M. tuberculosis complex (MTBC) (55).
FIG 2.

Spoligotyping. (A) Labeled primers (arrows; labeled primer indicated by asterisks [*]) complementary to the DRs are used to amplify the CRISPR spacer array and result in several PCR products of various lengths. (B) The mixture of labeled PCR products is hybridized to an array of probes, each of which corresponds to a unique spacer. The schematic shows unhybridized (top) and hybridized (bottom) membranes, with probes color coordinated for clarity. Positively hybridized probes are depicted with black borders. In this example, the hybridization pattern correlates to Strain a in panel C. (C) The spoligotype patterns show black boxes for a positive signal (spacer present) and white boxes for a negative signal (spacer absent). Different bacterial strains (Strain a, Strain b, and Strain c) can be identified on the basis of different hybridization profiles.
Improvements to classical spoligotyping include automation and the use of microbeads to replace the membrane, enabling a higher-throughput procedure (56). Addition of 25 new probes to the original 43 probes has also increased genotyping ability using this approach (57, 58). Spoligotyping remains the gold standard for MTBC subtyping, and data are shared in the international spoligotyping database, spo1DB4, which contains over 2,000 unique spoligotype patterns (59). The disadvantage of spoligotyping is that it determines the presence of only preestablished spacers. Fortuitously, in the case of MTBC, sequencing of several isolates has shown that these loci have ceased acquiring new spacers. The number of spacers in the CRISPR region of M. tuberculosis ranges from 6 to 47, and yet the number of different spacers in a set of more than 1,000 strains tested was limited to approximately 50, showing that strain-to-strain differences in M. tuberculosis CRISPR occur by loss of spacers (51).
The “next-generation” microbead-spoligotyping approach was recently applied to Salmonella, in an assay termed CRISPOL (for “CRISPR polymorphism”) (49). Weill and colleagues showed 100% concordance between CRISPOL types and CRISPR sequence data from 150 Salmonella enterica subsp. enterica serovar Typhimurium isolates. When the method was applied to over 2,000 isolates of Salmonella serovar Typhimurium and the corresponding monophasic variant I 4,5,12:i:- (so-called on the basis of its antigenic formula, as defined by the Kauffman and White classification scheme; see reference 60), 245 unique CRISPOL types were identified.
Traditional spoligotyping has also been applied to Corynebacterium diptheriae (61–63) and Legionella pneumophila (64). Since not all Legionella spp. contain CRISPR elements, spoligotyping lacks general utility with regard to routine genotyping but can certainly be used to further discriminate among common strains (64). Another alternative to the classical spoligotyping method is the use of a primer extension assay combined with mass spectrometry analysis. Use of 23- or 25-multiplex primers in this assay provided concordance of 96.9% with traditional membrane-based spoligotyping (65).
SEQUENCE-BASED CRISPR TYPING
DNA sequence-based molecular subtyping methods are becoming increasingly popular as cost continues to decrease while the fidelity, base-pair output, and high-throughput nature of the technology grow. As a result, sequence-based analysis of CRISPR arrays has been increasingly used for microbial subtyping.
CRISPR TYPING
Beyond spoligotyping, the first use of CRISPR spacer alleles for subtyping was in the group A Streptococcus (GAS) M1 serotype (66). Initial investigations in a limited number (14) of isolates showed that the size of PCR products corresponding to the spacer array correlated with the RFLP types of two insertion sequences (IS1548 and IS1562). Sequence analysis of these and an additional 30 isolates showed variation arising from altered numbers of spacers in different isolates and from different spacer compositions. The number of spacers present ranged from two to seven. Due to the presence of a better subtyping target (the sic gene), CRISPR typing in GAS was not extended. Nonetheless, this work showed for the first time the potential utility of sequence-based CRISPR typing in bacteria.
The next pathogen to be subtyped using CRISPR was Yersinia pestis, which is responsible for causing the plague and contains three CRISPR arrays, Ypa, Ypb, and Ypc (10). A limited study of strains representing three Y. pestis biovars showed that CRISPR typing was able to separate some isolates that had the same MLVA type (10). Subsequent investigations of a larger and more diverse number of isolates showed that different CRISPR types were associated with different geographical regions of origin (31).
Subsequently, the majority of sequenced-based CRISPR typing has been performed in Salmonella, which contains two CRISPR loci, CRISPR1 and CRISPR2 (Fig. 1). There are over 2,500 S. enterica subsp. enterica serovars (or serotypes). Sequence analysis of over 1,000 Salmonella CRISPRs showed that this pathogen is not rapidly acquiring new spacers (49, 50, 52, 67–69). Within individual serovars, CRISPR polymorphisms are due to spacer “microevolution,” specifically, multiplication or loss of internal spacers, as well as to the presence of SNP spacer or direct-repeat variants (49) (Fig. 3). Spacer multiplication and/or loss occurs much more frequently than the introduction of SNPs, and such multiplication or loss is likely the result of replicative errors promoted by the homologous direct repeats (31). That spacer microevolution appears to occur relatively frequently, specifically, in Salmonella serovar Typhimurium, may provide strain discrimination capacity comparable to that of other subtyping techniques such as PFGE (49). Whether this is true of all serovars will require further analysis.
FIG 3.

CRISPR-based typing. (A) Entire CRISPR spacer arrays are PCR amplified (the primers are indicated with red and blue arrows) and sequenced. Spacer sequence information is extracted, and unique spacers are represented as differently colored rectangles. A unique composition of spacers defines an allele, shown to the left of the array. In the example shown, where there are two CRISPR arrays, a CRISPR type is defined by a combination of two unique alleles as shown in the table. Polymorphisms between strains occur from loss of some spacers and/or duplication of others; for example, the light green spacer in CRISPR1 is duplicated in allele 5. (B) When a unique spacer (orange spacer; indicated by an arrow) is present in a pathogenic or predominant strain, these isolates can be easily identified using real-time PCR with fluorescent probes specific for that particular spacer, as shown for strain A. Since strain B does not contain that spacer, it would not be detected using real-time PCR. (C) Rapid CRISPR size typing screen based on comparative amplicon size analysis. Bacterial strains that differ in numbers of spacers can be easily visualized by analyzing the size difference after gel electrophoresis of the amplified PCR product.
Analysis of 34 S. enterica 6,7:c:1,5 isolates showed that CRISPR types correlate with MLST types (49). Furthermore, the spacer content of several Salmonella serovar Typhimurium and Salmonella serovar Enteritidis isolates from 10 documented outbreaks showed that all isolates from individual outbreaks exhibited the same CRISPR type (49), thus demonstrating superb epidemiological concordance. A new multiplex PCR assay has been developed for detection of Salmonella serovar Typhi and Salmonella serovar Paratyphi A, based on the presence of specific spacers in CRISPR2 and CRISPR1 of these serovars, respectively (F.-X. Weill, personal communication). This method shows 100% specificity when tested in several different Salmonella serovar Typhi and Salmonella serovar Paratyphi A strains from diverse genetic and geographical origins as well as when tested among a large number of different bacterial species.
In-depth spacer analysis of both CRISPR loci from over 1,500 Salmonella isolates (representing 130 different serovars) shows a high degree of CRISPR polymorphism within some serovars (49, 50, 52), enabling the use of these elements as excellent subtyping tools. Within a serovar, the order of spacers is strictly conserved and, importantly, spacer composition correlates strongly with serovar (49). These findings, also corroborated by a CRISPR-based phylogenetic analysis (23), suggest that CRISPR sequence analysis could provide a one-shot approach for both serotyping and subtyping, with the benefit of much-reduced time and expense to public health laboratories.
CRISPR sequence-based typing in Campylobacter jejuni, which is a leading bacterial cause of gastroenteritis, is also comparable to other subtyping methods. In one study, a clear association of CRISPR types (CTs) was found predominantly in certain MLST or amplified fragment length polymorphism (AFLP) clusters (70). CRISPR sequence typing of multiple isolates representing three outbreaks of C. jejuni was concordant with the epidemiological data (70). Unfortunately, ∼25% of Campylobacter species investigated either lacked an amplifiable CRISPR locus (10%) or contained a single DR (without a spacer; 15%), thus, CRISPR typing does not appear to be a versatile approach for routine Campylobacter genotyping. Given this, however, there was an association between isolates with a particular MLST sequence type and those that contained a single DR and between isolates with another sequence type and those that lacked a CRISPR locus (70).
Beyond human pathogens, CRISPR typing has also been adapted to Erwinia amylovora, the Gram-negative plant pathogen, which contains three CRISPR arrays. Different E. amylovora strains exhibit extremely low levels of genome diversity (71, 72) and are challenging to separate by traditional subtyping methods such as PFGE (73), ribotyping (74), and VNTR (75), among others. Two in-depth investigations showed that CRISPR typing was able to successfully separate strains that had previously been considered indistinguishable by PFGE and ribotyping (32, 34). Interestingly, in one of the studies, there were clear differences in spacer content, array length, and diversity that appeared to be based on geography and plant host (32).
CRISPR-MVLST
It has been shown that multi-virulence-locus sequence typing (MVLST) is a more powerful typing approach than traditional MLST (76–79). Addition of such a scheme to CRISPR sequencing (CRISPR-MVLST) in Salmonella (using fimH and sseL virulence genes) increased discriminatory power compared to either method used alone (50). Subsequent analysis of over 400 isolates, representing four of the five most prevalent clinical serovars in the United States, showed that the discrimination provided by CRISPR-MVLST is comparable to that provided by PFGE (52, 69, 80). In clonal serovars such as Salmonella serovar Enteritidis and Salmonella serovar Heidelberg, neither subtyping method provided sufficient discrimination to separate unrelated strains. A higher and more appropriate discrimination index (as defined in reference 81) was achieved by combining CRISPR-MVLST and PFGE. For example, in a screen of 141 Salmonella serovar Enteritidis isolates, CRISPR-MVLST defined 13 sequence types (discrimination index [D] = 0.71) and PFGE defined 22 pulsotypes (D = 0.79) but a combination of the two typing methods provided 45 unique types with a discrimination index of 0.92 (52). In Salmonella serovars Typhimurium (diphasic) and Newport, which collectively account for almost a quarter of salmonellosis cases (82), strain discrimination data provided by CRISPR-MVLST are sufficient (0.94 and 0.96, respectively) (69, 80).
These two studies of several Salmonella serovar Newport and Salmonella serovar Typhimurium isolates showed for the first time that there is an extremely high level of correlation between CRISPR-MVLST sequence types and PFGE patterns (69, 80). This is somewhat surprising, given that differences in PFGE typically arise from horizontal gene transfer whereas CRISPR-MVLST differences in Salmonella generally arise through vertical transmission. Interestingly, a correlation between patterns of antibiotic resistance/sensitivity and CRISPR-MVLST sequence types has also recently been observed in Salmonella serovar Typhimurium (67).
Importantly, as the first example used in the context of an outbreak, CRISPR-MVLST was able to identify outbreak-specific isolates. In a blinded study, Salmonella serovar Newport isolates, from a 2012 Pennsylvanian tomato-associated outbreak that sickened nearly 40 people, were successfully separated from sporadic case control isolates (69). Notably, in that study, epidemiological concordance determined by CRISPR-MVLST was as good as that by PFGE and better than that by MLVA. In another investigation involving two outbreaks of Salmonella serovar Typhimurium, CRISPR-MVLST was similarly able to identify and separate outbreak isolates (80). These findings clearly demonstrate that CRISPR-MVLST provides levels of strain discrimination similar to those of other subtyping approaches, notably PFGE, and that this discrimination is not detrimental to epidemiological concordance.
ALTERNATIVE CRISPR-BASED TYPING APPROACHES
There are several other ways that CRISPR arrays can be utilized for subtyping, including the application of real-time PCR protocols or exploitation of the inherent variations in the sizes of different CRISPR arrays.
REAL-TIME PCR METHODOLOGIES
E. coli strains are serogrouped based on the O antigen and serotyped based on both the O and H antigens (83). Shiga-toxin-producing E. coli (STEC) strains represent a pathotype with over 100 different serotypes that can cause clinical outcomes, running the gamut in terms of symptoms from mild or bloody diarrhea to hemolytic uremic syndrome (HUS) (84). Not all STEC strains are equally pathogenic, although a new policy from the USDA Food Safety and Inspection Service (FSIS) mandates the classification of six E. coli serogroups (the “Big Six” [O26, O45, O103, O111, O121, and O145]), along with the prototypical STEC, O157:H7, as adulterants in beef products (FSIS Notice 47-13; www.fsis.usda.gov). This new policy has resulted in efforts to develop rapid and specific typing methodologies to identify these bacteria. Two groups have recently analyzed the two E. coli CRISPR loci with an aim to develop CRISPR-based typing protocols. Delannoy et al. developed real-time PCR assays with primers designed using specific spacers present in the following Big Six serotypes: O26:H11, O45:H2, O103:H2, O111:H8, O121:H19, O145:H28, and O157:H7 (85). These serotypes represent the most clinically predominant STECs within a particular O serogroup in the United States and Europe (86). Testing these real-time PCR assays among 958 strains provided 95.7% to 100% sensitivity (ability of an individual assay to identify all strains of a given serotype) and 97.5% to 100% specificity (lack of cross-reactivity with another serotype) (85). The same group also developed a real-time PCR assay that was specific for O104:H4 (87), a rare E. coli serotype that was responsible for a large European outbreak in 2012, affecting 3,950 people and killing 53 (88). A schematic for strain-specific CRISPR-based real-time PCR is shown in Fig. 3B. An in-depth analysis by Yin et al. of several Big Six and O157:H7 STEC strains of both clinical and nonclinical origin showed that CRISPR sequences were similar but were not the same among isolates of the same O serogroup (38). In that study, the authors examined the CRISPR regions from 252 isolates and also analyzed CRISPR data from over 1,100 sequences available on GenBank. Their data seem to suggest that there are occasional correlations between H antigen and spacer composition, especially in phylogenetically related E. coli strains. The development of real-time PCR protocols to identify specific and dominant STEC strains appears promising, though the data from both groups seem to suggest that CRISPR sequence analysis in E. coli is more suited to use as a tool for identification rather than for subtyping (38, 85, 87).
As mentioned earlier, some Campylobacter strains contain a single DR in the CRISPR array (70). Price and Smith utilized an elegant high-resolution melt (HRM) approach to differentiate between some isolates that exhibited a SNP in this sole DR as well as between isolates that harbor multiple spacers (89).
SUBTYPING BASED ON CRISPR LOCUS SIZE
Variations in spacer numbers produce different lengths of CRISPR arrays that can be exploited to rapidly screen isolates by PCR and electrophoretic analysis (Fig. 3C). As mentioned previously, data from early work in GAS suggested that PCR/CRISPR array size correlated with RFLP typing results (66). Following this work, Vergnaud and colleagues used the YPa (Yp1) locus as a VNTR marker for MLVA in Y. pestis (33, 90). More recently, using both outbreak and control isolates, two groups have shown that “CRISPR size typing” can be successfully implemented in Salmonella serovar Typhimurium and Salmonella serovar Newport (49, 69). This approach can likely be extended to other Salmonella serovars, with its main utility being in developing countries where access to sequencing equipment can be limited.
CONCLUSIONS
CRISPR-based typing techniques have been well established for some bacterial species such as Mycobacterium and are currently being developed and studied extensively, especially for human pathogens but also for some agriculturally important species such as Erwinia. In most cases, CRISPR-based subtyping techniques provide discriminatory power and epidemiological concordance that are at least similar to, if not improved from, those of other methodologies. The benefits of CRISPR-based subtyping, especially in Salmonella, where this method can be used for serotyping as well as subtyping, include a dramatic decrease in the time to serovar and strain identification plus the capacity to fully automate the process. Additionally, as has been proven for spoligotyping, the great advantage is the capability of interlaboratory sharing of tractable sequencing data or numerically coded information that corresponds to the presence of specific spacers (91). CRISPR analysis again requires less time and money than subtyping by whole-genome sequencing to both perform and analyze the data; plus, data storage and transfer are much simpler.
Regarding CRISPR-based typing, there are several tools available to researchers and public health laboratories. Databases that contain CRISPR spacer sequence information are currently being populated for both Salmonella and Legionella (92). We foresee such databases growing with respect to the number of pathogens represented as well as the depth of strain diversity, providing resources similar to those of the international PFGE database, PulseNet. Additionally, there are user-friendly, free-access tools available: the CRISPRs Web Server (http://crispr.u-psud.fr/) provides several excellent tools, including CRISPRdb and CRISPRfinder (CRISPI; http://crispi.genouest.org/) (14, 93).
As seen in the genera Legionella and Campylobacter, not all strains of a particular bacterial species must contain CRISPRs. Though this limitation precludes the use of CRISPR typing as a general subtyping tool in such species, CRISPR could still be used to refine common strains. Another caveat is that some CRISPR alleles are extremely long and are thus not amenable to sequencing. In such cases, alternative approaches could involve amplifying certain strain-specific portions of a CRISPR allele using spacer-specific primers or third-generation long-read sequencing such as the PacBio platform.
FUTURE PERSPECTIVES
There is no doubt that next-generation sequencing has become cheaper and more amenable to standard laboratory practices at a tremendous rate. It is quite feasible that, in the near future, whole-genome sequencing will become the new gold standard and replace subtyping techniques such as PFGE. Would this spell the end for the short-lived use for CRISPR-based sequence typing? We argue that it would not: CRISPRs, along with other genetic targets such as virulence genes and genes with serotype-specific alleles, could be included as sequence targets that would be routinely extracted from draft genomes using automated bioinformatics tools.
Harnessing CRISPR biology, specifically, identifying the presence/absence of spacer sequences for subtyping purposes, is distinct from studying the functionality of CRISPR-Cas systems. Though 48% of bacterial genomes that have been sequenced to date contain CRISPR-Cas sytems (14), for many of these species, only a single strain or a few strains in a limited number of species have been investigated in detail with regard to the function and composition of the spacer arrays. We anticipate that projects such as the “100K Genome Project,” headed by the Food and Drug Administration and the University of California, Davis, which aims at completing whole-genome sequencing of 100,000 food-borne pathogens, will provide insights into CRISPR occurrence, diversity, and activity in many relevant pathogenic species (http://100kgenome.vetmed.ucdavis.edu/). This will be especially insightful because the majority of the strains that are being used in that study are primary clinical strains rather than “laboratory strains” such as E. coli K-12.
Additionally, the accumulation of spacer sequence information generated by extensive CRISPR typing studies will provide greater insight into microbial evolution, similar to what has been revealed for the phylogenetics of Salmonella (23). While evolutionary relationships are simpler to define when CRISPR arrays are actively acquiring spacers, it is more challenging to determine evolutionary relationships between bacterial strains when CRISPR loci differ primarily by loss of internal spacers. It has not been ascertained whether there is any global selection for loss of particular spacers, although there seems to be a bias toward the loss of ancestral spacers (22, 94). Why are some spacers maintained whereas others are lost? Is it due to relief of functional selection (the target bacteriophage no longer exists)? Is it sequence/structure dependent? Are there positional parameters within the array that determine loss? Does loss occur one spacer at a time, or are “blocks” of spacers lost simultaneously?
For most of the CRISPR arrays investigated during typing studies, acquisition does not appear to be occurring rapidly, if at all. It is therefore interesting that in these species, the genetic integrity of CRISPR arrays (with respect to spacer and direct-repeat length and organization) appear to be well maintained. This suggests that CRISPRs may have an alternative function beyond immunity to exogenous DNA (95, 96).
Given that almost half of all bacterial genomes sequenced to date harbor these remarkable elements, it would not be surprising to see adaptation of the CRISPR methodologies discussed here to other bacterial pathogens. There are three distinct CRISPR types, type I, type II, and type III, each defined by its complement of Cas proteins and processing of crRNAs (15). It is interesting, however, that in the majority of bacteria where CRISPRs have been used for subtyping, namely, Salmonella (types I to E), E. coli (types I to E), Erwinia (types I to E), Legionella (types I to F), Yersinia species (types I to F), and P. gingivalis (types I to C), type I CRISPR-Cas systems are predominant. This suggests that CRISPR typing may be ideally suited to other pathogenic bacteria harboring type I CRISPR-Cas systems such as Clostridium and Klebsiella.
ACKNOWLEDGMENTS
We thank Rodolphe Barrangou, Stephen Knabel, and Matt Crook and members of the Dudley laboratory for suggestions during the preparation of this review. We also thank Francois-Xavier Weill for sharing unpublished data.
Our CRISPR work has been funded by the United States Army Research Office (grant number W911NF-11-1-0442).
Biographies

Nikki Shariat studied molecular biology and genetics at the University of East Anglia in Norwich, United Kingdom, with a focus on microbiology. In 2008, she received her Ph.D from the Department of Biological Sciences, Vanderbilt University, Nashville, TN, where she studied the role of small interfering RNAs in mice. Her first postdoctoral position was at the University of California, San Francisco, where she examined the regulation of small RNAs. She is currently a postdoctoral scholar at Penn State, where she is investigating CRISPR elements in Salmonella enterica. Her research focus is on the evolution and function of CRISPR RNAs in gram-negative bacteria.

Edward Dudley received an M.S. in food science from the University of Wisconsin in 1994 and a Ph.D in bacteriology from the same institution in 2000, studying the genetics and physiology of lactic acid bacteria used in food fermentations. In 2001, he moved to the University of Maryland—Baltimore to examine virulence gene regulation in the human pathogen enteroaggregative Escherichia coli. He joined the Department of Food Science at Penn State in 2007 and established a program investigating the biology of E. coli O157:H7 and methods of subtyping food-borne pathogens, particularly the use of CRISPR for separating strains of S. enterica and E. coli.
Footnotes
Published ahead of print 25 October 2013
REFERENCES
- 1.Struelens MJ. 1996. Consensus guidelines for appropriate use and evaluation of microbial epidemiologic typing systems. Clin. Microbiol. Infect. 2:2–11. 10.1111/j.1469-0691.1996.tb00193.x [DOI] [PubMed] [Google Scholar]
- 2.Sabat A, Budimir A, Nashev D, Sá-Leão R, van Dijl J, Laurent F, Grundmann H, Friedrich AW; ESCMID Study Group of Epidemiological Markers (ESGEM) 2013. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 18:20380. [DOI] [PubMed] [Google Scholar]
- 3.Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. 1987. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 169:5429–5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen R, van Embden JD, Gaastra W, Schouls LM. 2002. Identification of a novel family of sequence repeats among prokaryotes. OMICS 6:23–33. 10.1089/15362310252780816 [DOI] [PubMed] [Google Scholar]
- 5.Jansen R, van Embden JDA, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43:1565–1575. 10.1046/j.1365-2958.2002.02839.x [DOI] [PubMed] [Google Scholar]
- 6.Mojica F, Díez-Villaseñor C, Soria E, Juez G. 2000. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 36:244–246. 10.1046/j.1365-2958.2000.01838.x [DOI] [PubMed] [Google Scholar]
- 7.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 8.Bolotin A, Quinquis B, Sorokin A, Ehrlich S. 2005. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561. 10.1099/mic.0.28048-0 [DOI] [PubMed] [Google Scholar]
- 9.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60:174–182. 10.1007/s00239-004-0046-3 [DOI] [PubMed] [Google Scholar]
- 10.Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663. 10.1099/mic.0.27437-0 [DOI] [PubMed] [Google Scholar]
- 11.Barrangou R. 2013. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip. Rev. RNA 4:267–278. 10.1002/wrna.1159 [DOI] [PubMed] [Google Scholar]
- 12.Bhaya D, Davison M, Barrangou R. 2011. CRISPR-Cas systems in Bacteria and Archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45:273–297. 10.1146/annurev-genet-110410-132430 [DOI] [PubMed] [Google Scholar]
- 13.Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338. 10.1038/nature10886 [DOI] [PubMed] [Google Scholar]
- 14.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. 10.1186/1471-2105-8-172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica F, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9:467–477. 10.1038/nrmicro2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R. 2010. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 75:1495–1512. 10.1111/j.1365-2958.2010.07073.x [DOI] [PubMed] [Google Scholar]
- 17.Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. 10.1126/science.1159689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hale C, Kleppe K, Terns R, Terns M. 2008. Prokaryotic silencing (psi) RNAs in Pyrococcus furiosus. RNA 4:2572–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hale C, Zhao P, Olson S, Duff M, Graveley B, Wells L, Terns R, Terns M. 2009. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956. 10.1016/j.cell.2009.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lillestøl R, Redder P, Garrett R, Brügger K. 2006. A putative viral defence mechanism in archaeal cells. Archaea 2:59–72. 10.1155/2006/542818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garneau J, Dupuis M, Villion M, Romero D, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan A, Moineau S. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71. 10.1038/nature09523 [DOI] [PubMed] [Google Scholar]
- 22.Horvath P, Romero DA, Coûté-Monvoisin A-C, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190:1401–1412. 10.1128/JB.01415-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fricke W, Mammel M, McDermott P, Tartera C, White D, Leclerc J, Ravel J, Cebula T. 2011. Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 193:3556–3568. 10.1128/JB.00297-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O'Toole GA. 2012. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 194:5728–5738. 10.1128/JB.01184-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Ploeg J. 2009. Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages. Microbiology 155(Pt 6):1966–1976. 10.1099/mic.0.027508-0 [DOI] [PubMed] [Google Scholar]
- 26.Andersson AF, Banfield JF. 2008. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 320:1047–1050. 10.1126/science.1157358 [DOI] [PubMed] [Google Scholar]
- 27.Held N, Whitaker R. 2009. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ. Microbiol. 11:457–466. 10.1111/j.1462-2920.2008.01784.x [DOI] [PubMed] [Google Scholar]
- 28.Paez-Espino D, Morovic W, Sun CL, Thomas BC, Ueda K, Stahl B, Barrangou R, Banfield JF. 2013. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat. Commun. 4:1430. 10.1038/ncomms2440 [DOI] [PubMed] [Google Scholar]
- 29.Pride DT, Sun CL, Salzman J, Rao N, Loomer P, Armitage GC, Banfield JF, Relman DA. 2011. Analysis of streptococcal CRISPRs from human saliva reveals substantial sequence diversity within and between subjects over time. Genome Res. 21:126–136. 10.1101/gr.111732.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyson GW, Banfield JF. 2008. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ. Microbiol. 10:200–207. 10.1111/j.1462-2920.2007.01444.x [DOI] [PubMed] [Google Scholar]
- 31.Cui Y, Li Y, Gorgé O, Platonov ME, Yan Y, Guo Z, Pourcel C, Dentovskaya SV, Balakhonov SV, Wang X, Song Y, Anisimov AP, Vergnaud G, Yang R. 2008. Insight into microevolution of Yersinia pestis by clustered regularly interspaced short palindromic repeats. PLoS One 3:e2652. 10.1371/journal.pone.0002652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGhee G, Sundin G. 2012. Erwinia amylovora CRISPR elements provide new tools for evaluating strain diversity and for microbial source tracking. PLoS One 7:e41706. 10.1371/journal.pone.0041706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pourcel C, Andre-Mazeaud F, Neubauer H, Ramisse F, Vergnaud G. 2004. Tandem repeats analysis for the high resolution phylogenetic analysis of Yersinia pestis. BMC Microbiol. 4:22. 10.1186/1471-2180-4-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rezzonico F, Smits T, Duffy B. 2011. Diversity, evolution and functionality of CRISPR regions in the fire blight pathogen Erwinia amylovora. Appl. Environ. Microbiol. 77:3819–3829. 10.1128/AEM.00177-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riehm JM, Vergnaud G, Kiefer D, Damdindorj T, Dashdavaa O, Khurelsukh T, Zöller L, Wölfel R, Le Flèche P, Scholz HC. 2012. Yersinia pestis lineages in Mongolia. PLoS One 7:e30624. 10.1371/journal.pone.0030624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vergnaud G, Li Y, Gorge O, Cui Y, Song Y, Zhou D, Grissa I, Dentovskaya SV, Platonov ME, Rakin A, Balakhonov SV, Neubauer H, Pourcel C, Anisimov AP, Yang R. 2007. Analysis of the three Yersinia pestis CRISPR loci provides new tools for phylogenetic studies and possibly for the investigation of ancient DNA. Adv. Exp. Med. Biol. 603:327–338. 10.1007/978-0-387-72124-8_30 [DOI] [PubMed] [Google Scholar]
- 37.Watanabe T, Nozawa T, Aikawa C, Amano A, Maruyama F, Nakagawa I. 2013. CRISPR regulation of intraspecies diversification by limiting IS transposition and intercellular recombination. Genome Biol. Evol. 5:1099–1114. 10.1093/gbe/evt075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin S, Jensen MA, Bai J, DebRoy C, Barrangou R, Dudley EG. 2013. Evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in CRISPR spacer composition. Appl. Environ. Microbiol. 79:5710–5720. 10.1128/AEM.00950-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heidelberg JF, Nelson WC, Schoenfeld T, Bhaya D. 2009. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One 4:e4169. 10.1371/journal.pone.0004169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mick E, Stern A, Sorek R. 2013. Holding a grudge: persisting anti-phage CRISPR immunity in multiple human gut microbiomes. RNA Biol. 10:900–906. 10.4161/rna.23929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stern A, Mick E, Tirosh I, Sagy O, Sorek R. 2012. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 22:1985–1994. 10.1101/gr.138297.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190:1390–1400. 10.1128/JB.01412-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mills S, Griffin C, Coffey A, Meijer WC, Hafkamp B, Ross RP. 2010. CRISPR analysis of bacteriophage-insensitive mutants (BIMs) of industrial Streptococcus thermophilus—implications for starter design. J. Appl. Microbiol. 108:945–955. 10.1111/j.1365-2672.2009.04486.x [DOI] [PubMed] [Google Scholar]
- 44.Cong L, Ran F, Cox D, Lin S, Barretto R, Habib N, Hsu P, Wu X, Jiang W, Marraffini L, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang W, Fu Y, Reyon D, Maeder M, Tsai S, Sander J, Peterson R, Yeh J, Joung J. 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31:227–229. 10.1038/nbt.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang W, Bikard D, Cox D, Zhang F, Marraffini L. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31:233–239. 10.1038/nbt.2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mali P, Yang L, Esvelt K, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi L, Larson M, Gilbert L, Doudna J, Weissman J, Arkin A, Lim W. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, Accou-Demartin M, De Romans S, Lim C, Roux C, Passet V, Diancourt L, Guibourdenche M, Issenhuth-Jeanjean S, Achtman M, Brisse S, Sola C, Weill F-X. 2012. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One 7:e36995. 10.1371/journal.pone.0036995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG. 2011. Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp. enterica. Appl. Environ. Microbiol. 77:1946–1956. 10.1128/AEM.02625-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Embden J, van Gorkom T, Kremer K, Jansen R, van Der Zeijst B, Schouls L. 2000. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J. Bacteriol. 182:2393–2401. 10.1128/JB.182.9.2393-2401.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shariat N, DiMarzio MJ, Yin S, Dettinger L, Sandt CH, Lute JR, Barrangou R, Dudley EG. 2013. The combination of CRISPR-MVLST and PFGE provides increased discriminatory power for differentiating human clinical isolates of Salmonella enterica subsp. enterica serovar Enteritidis. Food Microbiol. 34:164–173. 10.1016/j.fm.2012.11.012 [DOI] [PubMed] [Google Scholar]
- 53.Groenen PM, Bunschoten AE, van Soolingen D, van Embden JDA. 1993. Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Mol. Microbiol. 10:1057–1065. 10.1111/j.1365-2958.1993.tb00976.x [DOI] [PubMed] [Google Scholar]
- 54.Kamerbeek J, Schouls LM, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten AE, Molhuizen H, Shaw R, Goyal M, Van Embden JDA. 1997. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35:907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Streicher EM, Victor TC, van der Spuy G, Sola C, Rastogi N, van Helden PD, Warren RM. 2007. Spoligotype signatures in the Mycobacterium tuberculosis complex. J. Clin. Microbiol. 45:237–240. 10.1128/JCM.01429-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cowan LS, Diem L, Brake MC, Crawford JT. 2004. Transfer of a Mycobacterium tuberculosis genotyping method, spoligotyping, from a reverse line-blot hybridization, membrane-based assay to the Luminex multianalyte profiling system. J. Clin. Microbiol. 42:474–477. 10.1128/JCM.42.1.474-477.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Zanden AGM, Kremer K, Schouls LM, Caimi K, Cataldi A, Hulleman A, Nagelkerke NJD, van Soolingen D. 2002. Improvement of differentiation and interpretability of spoligotyping for Mycobacterium tuberculosis complex isolates by introduction of new spacer oligonucleotides. J. Clin. Microbiol. 40:4628–4639. 10.1128/JCM.40.12.4628-4639.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Abadia E, Refregier G, Tafaj S, Boschiroli ML, Guillard B, Andremont A, Ruimy R, Sola C. 2010. Mycobacterium tuberculosis complex CRISPR genotyping: improving efficiency, throughput and discriminative power of “spoligotyping” with new spacers and a microbead-based hybridization assay. J. Med. Microbiol. 59(Pt 3):285–294. 10.1099/jmm.0.016949-0 [DOI] [PubMed] [Google Scholar]
- 59.Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuño L, Arora J, Baumanis V, Binder L, Cafrune P, Cataldi A, Cheong S, Diel R, Ellermeier C, Evans JT, Fauville-Dufaux M, Ferdinand S, Garcia de Viedma D, Garzelli C, Gazzola L, Gomes HM, Guttierez MC, Hawkey PM, van Helden PD, Kadival GV, Kreiswirth BN, Kremer K, Kubin M, Kulkarni SP, Liens B, Lillebaek T, Ho ML, Martin C, Martin C, Mokrousov I, Narvskaïa O, Ngeow YF, Naumann L, Niemann S, Parwati I, Rahim Z, Rasolofo-Razanamparany V, Rasolonavalona T, Rossetti ML, Rüsch-Gerdes S, Sajduda A, Samper S, Shemyakin IG, Singh UB, Somoskovi A, Skuce RA, van Soolingen D, Streicher EM, Suffys PN, Tortoli E, Tracevska T, Vincent V, Victor TC, Warren RM, Yap SF, Zaman K, Portaels F, Rastogi N, Sola C. 2006. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 6:23. 10.1186/1471-2180-6-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grimont P, Weill F. 2007. Antigenic formulae of the Salmonella serovars, 9th ed. WHO Collaborating Centre for Reference and Research; on Salmonella, Paris, France [Google Scholar]
- 61.Mokrousov I, Narvskaya O, Limeschenko E, Vyazovaya A. 2005. Efficient discrimination within a Corynebacterium diphtheriae epidemic clonal group by a novel macroarray-based method. J. Clin. Microbiol. 43:1662–1668. 10.1128/JCM.43.4.1662-1668.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mokrousov I, Limeschenko E, Vyazovaya A, Narvskaya O. 2007. Corynebacterium diphtheriae spoligotyping based on combined use of two CRISPR loci. Biotechnol. J. 2:901–906. 10.1002/biot.200700035 [DOI] [PubMed] [Google Scholar]
- 63.Mokrousov I, Vyazovaya A, Kolodkina V, Limeschenko E, Titov L, Narvskaya O. 2009. Novel macroarray-based method of Corynebacterium diphtheriae genotyping: evaluation in a field study in Belarus. Eur. J. Clin. Microbiol. Infect. Dis. 28:701–703. 10.1007/s10096-008-0674-4 [DOI] [PubMed] [Google Scholar]
- 64.Ginevra C, Jacotin N, Diancourt L, Guigon G, Arquilliere R, Meugnier H, Descours G, Vandenesch F, Etienne J, Lina G, Caro V, Jarraud S. 2012. Legionella pneumophila sequence type 1/Paris pulsotype subtyping by spoligotyping. J. Clin. Microbiol. 50:696–701. 10.1128/JCM.06180-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Honisch C, Mosko M, Arnold C, Gharbia SE, Diel R, Niemann S. 2010. Replacing reverse line blot hybridization spoligotyping of the Mycobacterium tuberculosis complex. J. Clin. Microbiol. 48:1520–1526. 10.1128/JCM.02299-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoe N, Nakashima K, Grigsby D, Pan X, Dou SJ, Naidich S, Garcia M, Kahn E, Bergmire-Sweat D, Musser JM. 1999. Rapid molecular genetic subtyping of serotype M1 group A Streptococcus strains. Emerg. Infect. Dis. 5:254–263. 10.3201/eid0502.990210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiMarzio MJ, Shariat N, Kariyawasam S, Barrangou R, Dudley EG. 2013. Antibiotic resistance in Salmonella enterica serovar Typhimurium associates with CRISPR sequence type. Antimicrob. Agents Chemother. 57:4282–4289. 10.1128/AAC.00913-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu F, Kariyawasam S, Jayarao BM, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG. 2011. Subtyping Salmonella enterica serovar enteritidis isolates from different sources by using sequence typing based on virulence genes and clustered regularly interspaced short palindromic repeats (CRISPRs). Appl. Environ. Microbiol. 77:4520–4526. 10.1128/AEM.00468-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shariat N, Kirchner MK, Sandt CH, Trees E, Barrangou R, Dudley EG. 2013. CRISPR-MVLST subtyping of Salmonella enterica serovar Newport outbreak isolates and determination of the relationship between CRISPR-MVLST and PFGE. J. Clin. Microbiol. 51:2328–2336. 10.1128/JCM.00608-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schouls LM, Reulen S, Duim B, Wagenaar JA, Willems RJL, Dingle KE, Colles FM, Van Embden JDA. 2003. Comparative genotyping of Campylobacter jejuni by amplified fragment length polymorphism, multilocus sequence typing, and short repeat sequencing: strain diversity, host range, and recombination. J. Clin. Microbiol. 41:15–26. 10.1128/JCM.41.1.15-26.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sebaihia M, Bocsanczy AM, Biehl BS, Quail MA, Perna NT, Glasner JD, DeClerck GA, Cartinhour S, Schneider DJ, Bentley SD, Parkhill J, Beer SV. 2010. Complete genome sequence of the plant pathogen Erwinia amylovora strain ATCC 49946. J. Bacteriol. 192:2020–2021. 10.1128/JB.00022-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smits TH, Rezzonico F, Kamber T, Blom J, Goesmann A, Frey JE, Duffy B. 2010. Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol. Plant Microbe Interact. 23:384–393. 10.1094/MPMI-23-4-0384 [DOI] [PubMed] [Google Scholar]
- 73.Jock S, Donat V, López MM, Bazzi C, Geider K. 2002. Following spread of fire blight in Western, Central and Southern Europe by molecular differentiation of Erwinia amylovora strains with PFGE analysis. Environ. Microbiol. 4:106–114. 10.1046/j.1462-2920.2002.00277.x [DOI] [PubMed] [Google Scholar]
- 74.McManus P, Jones A. 1995. Genetic fingerprinting of Erwinia amylovora strains isolated from tree-fruit crops and Rubus spp. Phytopathology 85:1547–1553. 10.1094/Phyto-85-1547 [DOI] [Google Scholar]
- 75.Kim W, Geider K. 1999. Analysis of variable short-sequence DNA repeats on the 29 kb plasmid of Erwinia amylovora strains. Eur. J. Plant Pathol. 105:703–713. 10.1023/A:1008723717211 [DOI] [Google Scholar]
- 76.Chen Y, Zhang W, Knabel SJ. 2007. Multi-virulence-locus sequence typing identifies single nucleotide polymorphisms which differentiate epidemic clones and outbreak strains of Listeria monocytogenes. J. Clin. Microbiol. 45:835–846. 10.1128/JCM.01575-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lomonaco S, Chen Y, Knabel SJ. 2008. Analysis of additional virulence genes and virulence gene regions in Listeria monocytogenes confirms the epidemiologic relevance of multi-virulence-locus sequence typing. J. Food Prot. 71:2559–2566 [DOI] [PubMed] [Google Scholar]
- 78.Verghese B, Schwalm ND, III, Dudley EG, Knabel SJ. 2012. A combined multi-virulence-locus sequence typing and staphylococcal cassette chromosome mec typing scheme possesses enhanced discriminatory power. Infect. Genet. Evol. 12:1816–1821. 10.1016/j.meegid.2012.07.026 [DOI] [PubMed] [Google Scholar]
- 79.Zhang W, Jayarao BM, Knabel SJ. 2004. Multi-virulence-locus sequence typing of Listeria monocytogenes. Appl. Environ. Microbiol. 70:913–920. 10.1128/AEM.70.2.913-920.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shariat N, Sandt CH, DiMarzio MJ, Barrangou R, Dudley EG. 2013. CRISPR-MVLST subtyping of Salmonella enterica subsp. enterica serovars Typhimurium and Heidelberg and application in identifying outbreak isolates. BMC Microbiol. 13:254. 10.1186/1471-2180-13-254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 26:2465–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Centers for Disease Control and Prevention 2009. National Salmonella surveillance annual summary 2009. Centers for Disease Control and Prevention, Washington, DC [Google Scholar]
- 83.Lior H. 1996. Classification of Escherichia coli, p 31–72 In Gyles CL. (ed), Escherichia coli in domestic animals and humans. CAB International, Wallingford, United Kingdom [Google Scholar]
- 84.Gyles C. 2007. Shiga toxin-producing Escherichia coli: an overview. J. Anim. Sci. 85(Suppl):E45–E62. 10.2527/jas.2006-508 [DOI] [PubMed] [Google Scholar]
- 85.Delannoy S, Beutin L, Fach P. 2012. Use of clustered regularly interspaced short palindromic repeat sequence polymorphisms for specific detection of enterohemorrhagic Escherichia coli strains of serotypes O26:H11, O45:H2, O103:H2, O111:H8, O121:H19, O145:H28, and O157:H7 by real-time PCR. J. Clin. Microbiol. 50:4035–4040. 10.1128/JCM.02097-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brooks J, Sowers E, Wells J, Greene K, Griffin P, Hoekstra R, Strockbine N. 2005. Non-O157 Shiga toxin-producing Escherichia coli infections in the United States, 1983–2002. J. Infect. Dis. 192:1422–1429. 10.1086/466536 [DOI] [PubMed] [Google Scholar]
- 87.Delannoy S, Beutin L, Burgos Y, Fach P. 2012. Specific detection of enteroaggregative hemorrhagic Escherichia coli O104:H4 strains by use of the CRISPR locus as a target for a diagnostic real-time PCR. J. Clin. Microbiol. 50:3485–3492. 10.1128/JCM.01656-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.European Food Safety Authority 11 July 2012. E. coli: rapid response in a crisis. http://www.efsa.europa.eu/en/press/news/120711.htm.
- 89.Price EP, Smith H, Huygens F, Giffard PM. 2007. High-resolution DNA melt curve analysis of the clustered, regularly interspaced short-palindromic-repeat locus of Campylobacter jejuni. Appl. Environ. Microbiol. 73:3431–3436. 10.1128/AEM.02702-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Le Flèche P, Hauck Y, Onteniente L, Prieur A, Denoeud F, Ramisse V, Sylvestre P, Benson G, Ramisse F, Vergnaud G. 2001. A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestis and Bacillus anthracis. BMC Microbiol. 1:2. 10.1186/1471-2180-1-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Driscoll JR. 2009. Spoligotyping for molecular epidemiology of the Mycobacterium tuberculosis complex. Methods Mol. Biol. 551:117–128. 10.1007/978-1-60327-999-4_10 [DOI] [PubMed] [Google Scholar]
- 92.Institut Pasteur Accessed 10 May 2013 Institut Pasteur CRISPR query page. http://www.pasteur.fr/recherche/genopole/PF8/crispr/CRISPRDB.html
- 93.Rousseau C, Gonnet M, Le Romancer M, Nicolas J. 2009. CRISPI: a CRISPR interactive database. Bioinformatics 25:3317–3318. 10.1093/bioinformatics/btp586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weinberger AD, Sun CL, Pluciński MM, Denef VJ, Thomas BC, Horvath P, Barrangou R, Gilmore MS, Getz WM, Banfield JF. 2012. Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput. Biol. 8:e1002475. 10.1371/journal.pcbi.1002475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Babu M, Beloglazova N, Flick R, Graham C, Skarina T, Nocek B, Gagarinova A, Pogoutse O, Brown G, Binkowski A, Phanse S, Joachimiak A, Koonin EV, Savchenko A, Emili A, Greenblatt J, Edwards AM, Yakunin AF. 2011. A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 79:484–502. 10.1111/j.1365-2958.2010.07465.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zegans ME, Wagner JC, Cady KC, Murphy DM, Hammond JH, O'Toole GA. 2009. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol. 191:210–219. 10.1128/JB.00797-08 [DOI] [PMC free article] [PubMed] [Google Scholar]