

Abstract
Major efforts in bioenergy research have focused on producing fuels that can directly replace petroleum-derived gasoline and diesel fuel through metabolic engineering of microbial fatty acid biosynthetic pathways. Typically, growth and pathway induction are conducted under aerobic conditions, but for operational efficiency in an industrial context, anaerobic culture conditions would be preferred to obviate the need to maintain specific dissolved oxygen concentrations and to maximize the proportion of reducing equivalents directed to biofuel biosynthesis rather than ATP production. A major concern with fermentative growth conditions is elevated NADH levels, which can adversely affect cell physiology. The purpose of this study was to identify homologs of Escherichia coli FabG, an essential reductase involved in fatty acid biosynthesis, that display a higher preference for NADH than for NADPH as a cofactor. Four potential NADH-dependent FabG variants were identified through bioinformatic analyses supported by crystallographic structure determination (1.3- to 2.0-Å resolution). In vitro assays of cofactor (NADH/NADPH) preference in the four variants showed up to ∼35-fold preference for NADH, which was observed with the Cupriavidus taiwanensis FabG variant. In addition, FabG homologs were overexpressed in fatty acid- and methyl ketone-overproducing E. coli host strains under anaerobic conditions, and the C. taiwanensis variant led to a 60% higher free fatty acid titer and 75% higher methyl ketone titer relative to the titers of the control strains. With further engineering, this work could serve as a starting point for establishing a microbial host strain for production of fatty acid-derived biofuels (e.g., methyl ketones) under anaerobic conditions.
INTRODUCTION
A recent trend in biofuel research is to produce fuels that can directly replace petroleum-derived products such as gasoline and diesel fuel. Many of these advanced biofuels can be produced by altering native fatty acid biosynthetic pathways in microorganisms such as Escherichia coli and Saccharomyces cerevisiae and take the form of fatty alcohols, esters, and alkanes (1). Studies of these biofuel pathways have typically been conducted under aerobic conditions. It is desirable, however, to establish a microbial host strain that can function under fully anaerobic conditions. A major concern with aerobic culture conditions is that molecular oxygen, being the preferred terminal electron acceptor, consumes reducing equivalents derived from catabolic pathways to support growth and ATP production (2). This diverts reducing power away from metabolic pathways that lead to reduced biofuels. Also, aeration and oxygen-level control on an industrial scale represent additional operating expenses that detract from the profitability of biofuel production and undermine cost competitiveness relative to petroleum-based fuels.
There have been studies of obligate anaerobic production of higher alcohols through fermentation (3). However, in the case of producing fatty acid-based biofuels, fermentative pathways may not be desired, since carbon and electrons are lost through mixed acid fermentation (4). Another issue that arises with anaerobic microbial growth is redox imbalance (e.g., a relatively high NADH/NAD+ ratio). This can have detrimental effects on cell physiology, both at a regulatory level and at the enzyme function level (5–9).
Several strategies to address redox imbalance focus on enzyme cofactor usage, including altering the global availability of cofactors through metabolic engineering of the pentose phosphate pathway or overexpression of NADH oxidases, kinases, or transhydrogenases (10). Another strategy involves engineering pathway-specific enzymes that utilize cofactors, either to relax selectivity or change cofactor specificity. These cofactor-based approaches can lead to increased titers of target compounds and productivity (11, 12).
The E. coli fatty acid synthetic pathway involves two reductases that utilize NAD cofactors, FabG and FabI. Whereas FabI displays activity with both NADH and NADPH (13), FabG, which belongs to the short-chain dehydrogenase/reductase (SDR) enzyme family, is active only with NADPH in characterized versions (14). In the production of one palmitic acid (C16) molecule, FabG oxidizes seven NADPH molecules. FabG is essential, as deletion of the corresponding gene is lethal (15). The vast majority of E. coli FabG homologs have been characterized as displaying either absolute specificity for NADPH or a much higher preference for NADPH than NADH. Our concept is that a FabG homolog displaying a higher preference for NADH as a cofactor could be heterologously expressed in E. coli to increase fatty acid production under anaerobic conditions. A precedent for the potential of a NADH-specific FabG homolog is the study of mevalonate production using variant 3-hydroxy-3-methylglutaryl (HMG)-coenzyme A (CoA) reductases, in which it was observed that NADH-utilizing reductases produced higher levels of mevalonate during anaerobic growth than variants that preferred NADPH (16).
The goal of this study was to identify FabG homologs with a high preference for NADH that could be expressed in E. coli to increase fatty acid production under anaerobic conditions. We report several FabG homologs that, when overexpressed, led to increased production of free fatty acids and methyl ketones (fatty acid-derived biofuels) in an E. coli host under anaerobic conditions. With further engineering, this work could serve as a starting point for establishing a microbial host strain for production of fatty acid-derived biofuels under anaerobic conditions.
MATERIALS AND METHODS
Reagents, synthesized genes, and oligonucleotides.
Acetoacetyl coenzyme A (acetoacetyl-CoA), NADH, NADPH, and fatty acid methyl and ethyl esters were purchased from Sigma-Aldrich (St. Louis, MO) and were of the highest purity available. Genes were synthesized by GenScript (Piscataway, NJ) and codon optimized for expression in E. coli with the proprietary GenScript OptimumGene algorithm. The synthesized gene sequences are listed in Table S1 in the supplemental material. Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). All organic solvents were purchased from Sigma-Aldrich and were pesticide residue analysis grade or better. Highly purified water (18-MΩ resistance) obtained from a Barnstead Nanopure system (Thermo Scientific, Waltham, MA) was used to prepare the growth medium and all other aqueous solutions described in this article.
Bioinformatics.
Structural alignments of E. coli FabG bound to NADP+ (17) and SDRs found in the Protein Data Bank were performed with 3D-Hit (18). The aligned structures were then separated into two groups, based on whether NAD(H) or NADP(H) was bound, and the sequences were analyzed for patterns at residues near the 2′ position of the cofactor adenine. Putative FabG homologs possessing residues consistent with patterns found in NAD(H)-binding SDRs were identified by BLAST searching (19), and the respective sequences were aligned to that of E. coli FabG using MUSCLE (20). Homologs from organisms that have been isolated from environments at near-neutral pH and annotated as mesophilic in the Integrated Microbial Genomes system (Joint Genome Institute) (Table 1) were selected for biochemical characterization and fatty acid production experiments to help ensure that the enzymes would function properly in E. coli.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Reference or source |
|---|---|---|
| E. coli strains | ||
| BL21(DE3) | F− ompT gal dcm lon hsdSB(rB− mB−) λ(DE3) | 40 |
| DH1 | endA1 recA1 gyrA96 thi-1 glnV44 relA1 hsdR17(rK− mK+) λ− | 41 |
| DH1ΔfadE | DH1 ΔfadE | 22 |
| DH10B | F− mcrA endA1 recA1 galE15 galK16 nupG rpsL ΔlacX74 ϕ80lacZΔM15 araD139 Δ(ara leu)7697 Δ(mrr-hsdRMS-mcrBC) λ− | 42 |
| EGS522 | DH1 ΔfadE ΔfadA | 25 |
| EGS895 | EGS522 with pEG530 and pEG855 | 25 |
| EGS1670 | EGS522 with pEG530 and pEG1635 | This study |
| PJ001 | BL21(DE3) with pPJ100 | This study |
| PJ002 | BL21(DE3) with pPJ101 | This study |
| PJ003 | BL21(DE3) with pPJ102 | This study |
| PJ004 | BL21(DE3) with pPJ104 | This study |
| PJ005 | BL21(DE3) with pPJ103 | This study |
| PJ010 | DH1ΔfadE with pPJ133 | This study |
| PJ011 | DH1ΔfadE with pPJ134 | This study |
| PJ012 | DH1ΔfadE with pPJ135 | This study |
| PJ013 | DH1ΔfadE with pPJ136 | This study |
| PJ014 | DH1ΔfadE with pPJ137 | This study |
| Plasmids | ||
| pBbE8k-RFP | Kmr; ColE1 plasmid; PBAD promoter with araC repressor | 24 |
| pEG530 | Cmr; ′tesA, acyl-CoA oxidase (Mlut_11700) and fadB under lacUV5 promoter | 25 |
| pEG855 | Kmr; fadM under Ptrc promoter | 25 |
| pEG1600 | Kmr; C. taiwanensis fabG cloned into pBbE8k-RFP at NdeI and BamHI sites | This study |
| pEG1635 | Kmr; araC-PBAD-fabG fragment from pEG1600 cloned into pEG855 at the NdeI site | This study |
| pET-24a(+) | Kmr | Novagen |
| pKS1 | Cmr; p15a vector containing ′tesA under lacUV5 promoter | 22 |
| pPJ100 | Kmr; E. coli fabG, with N-terminal 6× His tag sequence, cloned into pET-24a(+) at NdeI and BamHI sites | This study |
| pPJ101 | Kmr; Acholeplasma laidlawii PG-8A ACL-0504 fabG, with N-terminal 6× His tag sequence, in pET24a(+) | This study |
| pPJ102 | Kmr; Bacillus sp. SG-1 BSG1-13201 fabG, with N-terminal 6× His tag sequence, in pET24a(+) | This study |
| pPJ104 | Kmr; Cupriavidus taiwanensis LMG 19424 RALTA_A2639 fabG, with N-terminal 6× His tag sequence, in pET24a(+) | This study |
| pPJ103 | Kmr; Plesiocystis pacifica SIR-1 PPSIR1_02236 fabG, with N-terminal 6× His tag sequence, in pET24a(+) | This study |
| pPJ132 | Cmr; lacI repressor cloned into pKS1 | This study |
| pPJ133 | Cmr; codon-optimized E. coli fabG, with N-terminal 6× His tag sequence, in pPJ132 | This study |
| pPJ134 | Cmr; Acholeplasma laidlawii PG-8A ACL-0504 fabG, with N-terminal 6× His tag sequence, in pPJ132 | This study |
| pPJ135 | Cmr; Bacillus sp. SG-1 BSG1-13201 fabG, with N-terminal 6× His tag sequence, in pPJ132 | This study |
| pPJ136 | Cmr; Cupriavidus taiwanensis LMG 19424 RALTA_A2639 fabG, with N-terminal 6× His tag sequence, in pPJ132 | This study |
| pPJ137 | Cmr; Plesiocystis pacifica SIR-1 PPSIR1_02236 fabG, with N-terminal 6× His tag sequence, in pPJ132 | This study |
DNA assembly of fabG variants.
Plasmid pPJ100 (Table 1) consists of the wild-type E. coli fabG gene and sequence encoding an N-terminal 6×His tag cloned into the pET-24 expression vector (Novagen Biosciences, Madison, WI) for protein purification. Genes for the other fabG homologs were synthesized with sequences encoding N-terminal 6×His tags and cloned into pET-24. For fatty acid production, each fabG variant was PCR amplified from its respective pET-24 vector and then cloned, using the sequence- and ligation-independent cloning (SLIC) method (21), into pPJ132, which was adapted from plasmid pKS1 (22) to express the lacI repressor as well as the leaderless thioesterase ′tesA (22, 23). Plasmids were amplified in E. coli strain DH10B during cloning, and protein was expressed in strain BL21(DE3). The fatty acid production host strain was E. coli DH1ΔfadE, which was disabled in β-oxidation in order to accumulate fatty acids (22). For methyl ketone production, the C. taiwanensis fabG gene was PCR amplified and cloned, as described above, into pBbE8k-RFP (24), resulting in pEG1600 (Table 1). Subsequently, the araC-PBAD-fabG fragment from pEG1600 was cloned into pEG855 (25) using the SLIC method (21), resulting in pEG1635. pEG1635 was transformed into E. coli EGS522 (Table 1) with pEG530 (25) for analysis of anaerobic methyl ketone production in vivo.
fabG expression and protein purification.
An overnight culture of E. coli BL21(DE3) expressing a fabG variant in pET-24 was used to inoculate 1 liter of lysogeny broth (LB) supplemented with 50 μg/ml kanamycin, which was then grown at 37°C to an optical density at 600 nm (OD600) of 0.4 to 0.6. At that point, protein expression was induced by addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and cells were grown overnight at 18°C. The cells were harvested by centrifugation, washed with 50 ml of buffer F (50 mM Tris-Cl [pH 7.4], 250 mM NaCl, and 10% glycerol), and stored as pellets at −80°C. The cells were lysed with an EmulsiFlex-C3 high-pressure homogenizer (Avestin, Ottawa, ON, Canada) after resuspension in 100 ml of buffer F. The lysate was clarified by centrifugation (15,000 × g for 1 h at 4°C) followed by binding of lysate supernatant to 3 ml of Profinity immobilized metal affinity chromatography (IMAC) Ni resin (Bio-Rad, Hercules, CA) at 4°C for at least 30 min. Bound protein was sequentially washed with 20, 40, and 80 mM imidazole in buffer F. Pure FabG was eluted at 150, 250, and 400 mM imidazole in buffer F. Pure fractions were pooled together, concentrated with an Amicon Ultra-15 10,000-nominal-molecular-weight-limit (NMWL) concentrator (Millipore, Billerica, MA), and exchanged into buffer F using a PD-10 desalting column (GE Healthcare Life Sciences, Pittsburgh, PA).
In vitro assay for FabG specific activity.
Activity was determined by monitoring the reduction of acetoacetyl-CoA in the presence of either NADH or NADPH over 10 min using a 96-well plate on a SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA). Although the true FabG substrate is a β-ketoacyl–acyl carrier protein (ACP), acetoacetyl-CoA was chosen as an in vitro substrate due to its commercial availability and amenability for assay preparation. Although the absolute specific activity of FabG is considerably higher for acetoacetyl-ACP than for acetoacetyl-CoA (26), assays with acetoacetyl-CoA were sufficiently sensitive for the purposes of this study. The change in 340-nm absorbance from the conversion of NAD(P)H to NAD(P)+ was used to determine reaction rate, based on a standard curve of NADH concentration versus absorbance. Assays were performed in 100 mM NaPO4 buffer (pH 7.4) containing 0.5 to 8 μg FabG and 240 μM NADH or NADPH, and were initiated with the addition of 500 μM acetoacetyl-CoA in a total volume of 250 μl. For the control reactions, acetoacetyl-CoA was replaced with an equal volume of water.
Crystallization of fabG homologs in complex with NADH.
The Acholeplasma laidlawii FabG (Al_FabG), Bacillus sp. strain SG-1 FabG (Bs_FabG), Cupriavidus taiwanensis FabG (Ct_FabG), and Plesiocystis pacifica FabG (Pp_FabG) solutions were concentrated to 10 mg/ml and screened using the sparse matrix method (27) with a Phoenix Robot (Art Robbins Instruments, Sunnyvale, CA) using the following crystallization screens: Crystal Screen I and II, Index, PEG/Ion, SaltRx, and PEGRx (Hampton Research, Aliso Viejo, CA) and Berkeley Screen. The optimum conditions for crystallization of the different homologs were as follows: Al_FabG, 0.2 M ammonium fluoride and 20% polyethylene glycol 3350 (PEG 3350); Bs_FabG, 0.2 M sodium citrate and 20% PEG 3350; Ct_FabG, 0.1 M sodium malonate (pH 7.0), 0.03 M citric acid, 0.07 M Bis-Tris propane (pH 7.6), and 20% PEG 3350; and Pp_FabG, 0.2 M ammonium tartrate dibasic (pH 7.0) and PEG 3350. Crystals were obtained after 2 days by the sitting-drop vapor diffusion method with drops consisting of a mixture of 0.5 μl protein solution and 0.5 μl reservoir solution.
X-ray data collection and structure determination.
Crystals were placed in a reservoir solution containing 15 to 20% glycerol and then flash frozen in liquid nitrogen. Data sets for the Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG were collected at the Berkeley Center for Structural Biology beamlines 8.2.1 and 8.2.2 of the Advanced Light Source at Lawrence Berkeley National Laboratory (LBNL). The diffraction data were recorded using an ADSC-Q315r detector. The data sets were processed using the program HKL-2000 (28).
Crystal structures of Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG were determined by the molecular-replacement method with the program PHASER (29) within the Phenix suite (30), using the structure of E. coli FabG (PDB accession no. 1Q7B) as a search model. The atomic positions obtained from molecular replacement and the resulting electron density maps were used to build all the FabG homolog structures and initiate crystallographic refinement and model rebuilding. Structure refinement was performed using the phenix.refine program (31). Translation-liberation-screw (TLS) refinement with each protein chain as a single TLS group was used in the process. Manual rebuilding using COOT (32) and addition of water molecules allowed for construction of the final models. Five percent of the data were randomly selected for cross validation. Root mean square deviation differences from ideal geometries for bond lengths, angles, and dihedrals were calculated with PHENIX (30). The overall stereochemical quality of the final models was assessed using the program MOLPROBITY (33).
Anaerobic growth conditions for fatty acid production.
Sample handling was primarily conducted in an anaerobic glove box (Coy Laboratory Products, Grass Lake, MI) under an atmosphere of 85% N2, 10% CO2, and 5% H2. Anaerobic reagent water was prepared by autoclaving and then cooling under an atmosphere of ultrahigh-purity nitrogen gas for at least 20 min. EZ Rich medium components (Teknova, Hollister, CA) were mixed, filtered, and then stored in the anaerobic glove box for at least 16 h before being mixed with the appropriate volume of anaerobic water. Sterile 30-ml glass tubes and Mininert caps (VICI, Baton Rouge, LA) were also stored in the anaerobic glove box for at least 16 h before use. Anaerobic cultures, consisting of 20 ml of EZ Rich medium containing 0.2% glucose and supplemented with 30 μg/ml chloramphenicol, were inoculated inside the anaerobic glove box with aerobically grown 10-ml overnight cultures at a 1:100 dilution and sealed with Mininert caps. Sealed cultures were incubated outside the anaerobic glove box at 37°C without shaking, and samples for OD600 measurements were collected inside the glove box. Protein expression was induced in the glove box by addition of 0.2 mM IPTG in anaerobic water.
Extraction and analysis of fatty acids.
Fatty acids were extracted from anaerobically grown cells using high-purity methanol and hexane as described previously (34). Based on preliminary experiments, more than 80% of total (intracellular plus extracellular) fatty acids are recovered using this cell pellet extraction method. The hexane phase was concentrated to 100 μl under a stream of ultrahigh-purity nitrogen gas and derivatized with ethereal diazomethane prepared with a small-scale diazomethane generator (Sigma-Aldrich) (34). The derivatized extract was concentrated to 100 μl for gas chromatography analysis. Care was taken to ensure that organic solvents contacted only glass or polytetrafluoroethylene (PTFE) that were precleaned with high-purity acetone.
Fatty acid extract samples were applied to a Thermo Scientific Focus gas chromatograph (GC) equipped with a TriPlus autosampler, Trace TR-5 column (5-m length, 0.25-mm inner diameter, 0.25-μm film thickness), and a flame ionization detector (FID). The GC oven was programmed from 45°C (held for 2.25 min) to 300°C at 40°C/min; the FID temperature was 350°C. The injection port temperature was 250°C, and the carrier gas, ultrahigh-purity helium, flowed at a constant pressure of 65 kPa. Injections were splitless with the split turned on after 1 min. Fatty acid concentrations were determined by external standard quantification using C8 to C20 fatty acid methyl ester standards.
Anaerobic growth conditions for methyl ketone production.
Preparation of media and cultures for anaerobic growth conditions were performed as described above. Briefly, 120-ml serum bottles with 60 ml of EZ Rich medium containing 0.2% glucose supplemented with 25 μg/ml chloramphenicol and 50 μg/ml kanamycin were inoculated with overnight aerobic cultures at starting OD600 values of ∼0.02. Serum bottles were then sealed with 20-mm butyl rubber stoppers and crimp seals to maintain an anaerobic environment. Protein expression was induced in the anaerobic glove box by addition of 0.5 mM IPTG and 1 mM arabinose at 6 h. Two milliliters of decane amended with 40 ng/μl of perdeuterated tetracosane (C24D50) internal standard was also added at 6 h. Sealed cultures were incubated outside the anaerobic glove box at 37°C with mild shaking (120 rpm) to allow partitioning of methyl ketones into the decane overlay, and samples for OD600 measurements were collected inside the glove box via syringe.
Extraction and analysis of methyl ketones.
After 96 h of anaerobic growth, the decane overlay was removed for direct gas chromatography-mass spectrometry (GC-MS) analysis as previously described (25). Briefly, GC-MS analyses were performed with a model 7890A GC (Agilent, Santa Clara, CA) with a DB-5 fused silica capillary column (30-m length, 0.25-mm inner diameter, 0.25-μm film thickness; J & W Scientific) coupled to an HP 5975C series quadrupole mass spectrometer; 1-μl injections were performed by a model 7683B autosampler. The GC oven was programmed from 40°C (held for 3 min) to 295°C at 15°C/min; the injection port temperature was 250°C, and the transfer line temperature was 280°C. The carrier gas, ultrahigh-purity helium, flowed at a constant rate of 1 ml/min. Injections were splitless, with the split turned on after 0.5 min. Quantification of methyl ketones was carried out with external standards as previously described (25).
PDB accession numbers.
Coordinates for Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG have been deposited in the Protein Data Bank with accession numbers 4NBT, 4NBU, 4NBV, and 4NBW, respectively.
RESULTS
FabG homolog selection strategy.
In order to identify potential NADH-utilizing FabG homologs, structural and sequence alignments were made between E. coli FabG bound to NADP+ (17) and SDRs from the Protein Data Bank. The Asp36 residue (E. coli FabG numbering) is well conserved among NAD-binding SDRs, whereas in the NADP-binding enzymes, there appear to be only slight preferences for either a tyrosine or serine at that position (Fig. 1). Moreover, there is a preference for a basic amino acid (arginine or lysine) at residue 15. Using these residue motifs as a basis, FabG homologs from other microbial organisms with molecular motifs consistent with NAD-binding SDRs were identified. On the basis of annotations in the Integrated Microbial Genomes system (Joint Genome Institute), we focused on organisms that are mesophilic and found in environments at near-neutral pH to help ensure that the homologous enzymes would function properly in E. coli. These fabG homologs, encoded in plasmids pPJ134-137, are listed in Table 1.
FIG 1.

Alignments of short-chain dehydrogenase/reductase (SDR) enzymes with structures deposited in the PDB (identified by species name followed by the PDB accession number in parentheses) that bind either NADH or NADPH. Amino acid numbering refers to the native E. coli FabG sequence. Enzymes that prefer NADH possess a conserved aspartate residue (see the text and arrows in figure).
In vitro assay indicates FabG homologs that have preference for NADH over NADPH.
FabG homologs were purified with N-terminal 6×His tags and assayed in vitro with the substrate analog acetoacetyl-CoA in the presence of either NADH or NADPH. The specific activities of E. coli FabG and four homologs are shown in Fig. 2. As expected from previous reports (14), E. coli FabG displayed a specific activity of approximately 2 μmol/min/mg with NADPH as a cofactor and was essentially inactive in the presence of NADH. On the other hand, the selected FabG homologs displayed higher activity with NADH as a cofactor than with NADPH, ranging from a 2- to 36-fold-higher preference (Fig. 2). Also, with the exception of the P. pacifica variant, each homolog displayed higher absolute specific activity toward the substrate than E. coli FabG did. Interestingly, the C. taiwanensis variant had almost 7-fold-higher activity than E. coli FabG and displayed the highest preference for NADH of all the enzymes assayed.
FIG 2.
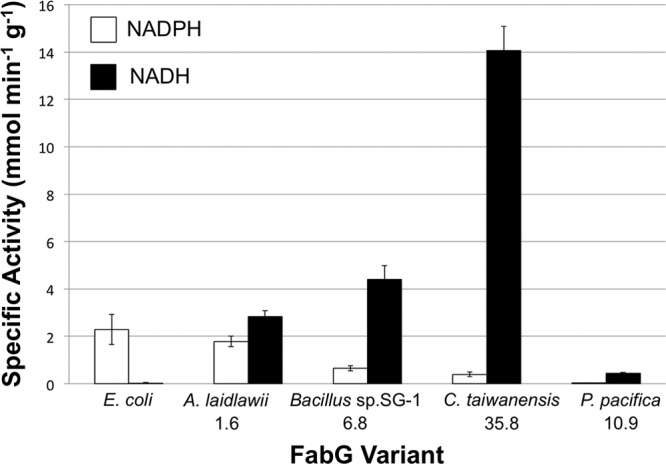
Specific activity of FabG homologs from E. coli and other species toward acetoacetyl-CoA. The number below each species name is the ratio of activity with NADH to activity with NADPH. E. coli FabG was essentially inactive with NADH. Bar heights represent average activities from 3 replicates, and error bars represent 1 standard deviation.
Structural analyses of FabG homolog cofactor-binding sites.
Crystals of the four FabG homologs diffracted between 1.3- and 2.0-Å resolution. The statistics for X-ray data collection and structure refinement are summarized in Table 2. The highly conserved tetrad of Asn110/Ser138/Tyr151/Lys155 residues (E. coli numbering) are essential for FabG catalytic activity (35). A structural comparison between NADPH-utilizing E. coli FabG and the NADH-utilizing homologs Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG showed that residues surrounding the adenosine ribose moiety of the cofactor differ significantly in these enzymes. The most obvious structural difference between E. coli FabG and the NADH-utilizing homologs is the presence of a negatively charged aspartate (corresponding to the Ala36 position in E. coli FabG) interacting with the 2′- and 3′-hydroxyl groups of the adenosine ribose (Fig. 3). NADPH binding is unfavorable as a result of electrostatic repulsion between this aspartate residue and the NADPH 2′-phosphate group. A similar conclusion was previously reported for other SDR family enzymes (35, 36).
TABLE 2.
Data collection and refinement statistics
| Parameter | Value(s) for FabG homologa: |
|||
|---|---|---|---|---|
| Al_FabG | Bs_FabG | Ct_FabG | Pp_FabG | |
| Resolution range (Å) | 39.49–1.48 (1.533–1.48) | 23.33–1.34 (1.387–1.34) | 37.33–1.645 (1.703–1.645) | 44.83–2.00 (2.072–2.00) |
| Space group | P 1 | P 1 | P 43 21 2 | P 21 |
| Unit cell dimensions (a, b, and c [Å]; α, β, and γ [°]) | 61.96, 62.36, 66.76; 66.03, 76.63, 79.93 | 63.41, 68.77, 70; 67.85, 88.65, 62.64 | 111.76, 111.76, 79.19; 90, 90, 90 | 77.73, 75.14, 97.72; 90, 113.43, 90 |
| No. of unique reflections | 142,273 (13,758) | 201,633 (17,136) | 60,813 (5,545) | 61,547 (6,103) |
| Completeness (%) | 96.44 (93.39) | 93.71 (79.62) | 99.19 (91.94) | 88.02 (87.64) |
| Mean I/sigma(I) | 14.21 (3.43) | 11.30 (2.65) | 12.64 (1.7) | 5.53 (2.30) |
| Wilson B-factor | 12.29 | 12.59 | 19.78 | 8.35 |
| R-merge | 0.039 | 0.044 | 0.102 | 0.127 |
| R-work | 0.1282 (0.1678) | 0.1606 (0.2304) | 0.1443 (0.2277) | 0.2047 (0.2394) |
| R-free | 0.1540 (0.1824) | 0.1735 (0.2604) | 0.1600 (0.2741) | 0.2541 (0.3018) |
| No. of: | ||||
| Atoms | 16,056 | 8,420 | 7,814 | 8,164 |
| Macromolecules | 4 | 4 | 2 | 4 |
| Water | 1,328 | 962 | 542 | 1,368 |
| NADH | 4 | 4 | 4 | |
| Protein residues | 958 | 962 | 492 | 1,008 |
| RMS(bonds [Å])b | 0.017 | 0.016 | 0.009 | 0.016 |
| RMS(angles [°]) | 1.59 | 1.54 | 1.15 | 1.19 |
| Ramachandran favored (%) | 97 | 98 | 98 | 98 |
| Ramachandran outliers (%) | 0 | 0.1 | 0 | 0.1 |
| Average B-factor | 15.90 | 20.40 | 24.80 | 11.10 |
Al_FabG, Acholeplasma laidlawii FabG; Bs_FabG, Bacillus sp. SG-1 FabG; Ct_FabG, Cupriavidus taiwanensis FabG; Pp_FabG, Plesiocystis pacifica FabG. Statistics for the highest-resolution shell are shown in parentheses.
RMS, root mean square.
FIG 3.
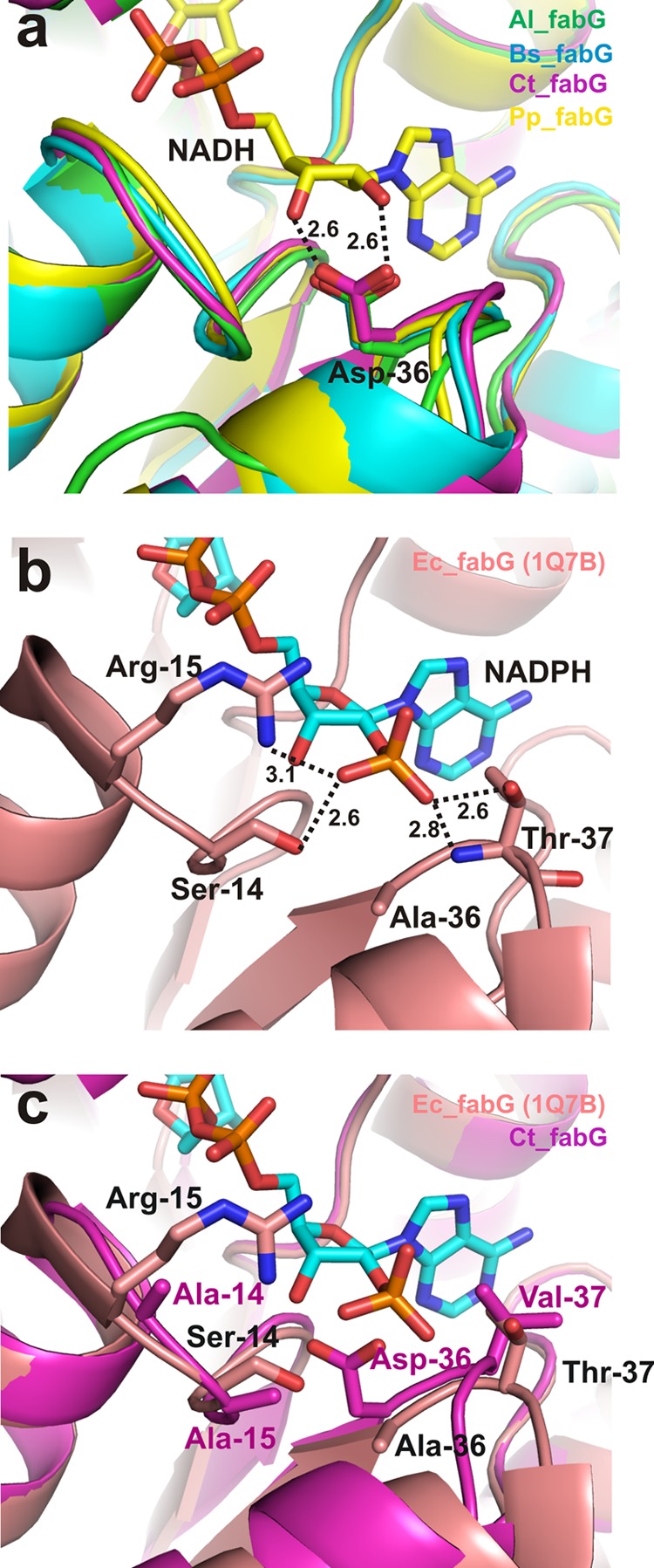
Comparison of cofactor-binding sites in FabG homologs. (a) Structural overlay of the Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG homologs with NADH. (b) Residues interacting with NADPH in the E. coli FabG (Ec_FabG) crystal structure. (c) Overlay of E. coli and Ct_FabG, depicting the clash that occurs between the NADPH 2′-phosphate and Asp36 (E. coli numbering) of Ct_FabG. Dotted lines represent ionic and polar interactions. Numbers represent distances between atoms in angstroms.
To understand the structural basis for the differences in FabG homolog activity with NADH versus NADPH (Fig. 2), residues making direct interactions with the NADPH 2′-phosphate from the E. coli FabG structure were compared against the respective residues in the NADH-utilizing homologs. The E. coli FabG structure in complex with NADPH shows residues Ser14, Arg15, and Thr37 interacting with the NADPH 2′-phosphate through polar and ionic interactions (17) (Fig. 3b). None of these residues is conserved in Al_FabG, Bs_FabG, Ct_FabG, or Pp_FabG (Table 3). Ser14 is replaced by Ala residues in all the NADH-utilizing FabGs. Arg15 is replaced by Lys, Asn, Ala, and Asn in Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG, respectively. The substitution of a positive-charged Lys residue in Al_FabG for Arg15 of E. coli FabG may be responsible for the higher activity observed with NADPH for the Al_FabG homolog (Fig. 2). Thr37 of E. coli FabG is replaced by the apolar residues Leu, Phe, Val, and Leu in Al_FabG, Bs_FabG, Ct_FabG, and Pp_FabG, respectively.
TABLE 3.
Sequence comparison of the E. coli FabG residues that interact with the NADPH 2′-phosphate with the respective residue positions in NADH-utilizing homologs
| FabG variant | Residue interacting with NADPH 2′-phosphatea |
||
|---|---|---|---|
| Ser14 | Arg15 | Thr37 | |
| Al_FabG | Ala15 | Lys16 | Leu38 |
| Bs_FabG | Ala22 | Asn23 | Phe45 |
| Ct_FabG | Ala14 | Ala15 | Val37 |
| Pp_FabG | Ala21 | Asn22 | Leu44 |
Ser14, Arg15, and Thr37 refer to E. coli FabG residues.
Anaerobic fatty acid production.
It is possible that because lipid production correlates with cell division (37), and E. coli grows at a relatively lower rate under anaerobic conditions, fatty acid biosynthesis as a whole would also proceed more slowly than during aerobic growth. If so, then the effect of overexpression of fabG may not be experimentally detectable. To address this concern, we chose E. coli DH1ΔfadE expressing the ′tesA thioesterase (22) as the host strain for fatty acid production in this study (Table 1). The thioesterase activity would enable overproduction of fatty acids via hydrolysis of acyl-ACP thioesters and consequent deregulation of fatty acid biosynthesis by precluding acyl-ACP-mediated inhibition. The DH1ΔfadE strain was also utilized for the overproduction of fatty acid-based fuels, including fatty acid ethyl esters (FAEEs), alcohols, and waxes, and thus serves as an established model system (22). In order to control gene expression, the lacI repressor was introduced into the pKS1 plasmid, which carries the ′tesA gene, and the resulting vector was used to express each respective fabG homolog under the control of a lacUV5 promoter (24) (Table 1). The control strain consisted of DH1ΔfadE expressing a codon-optimized version of the native E. coli fabG from the pKS1 vector, so that all tested strains would be under similar stress from gene overexpression. Codon-optimized fabG was chosen to create a sequence sufficiently distinct from the chromosomal fabG to help prevent possible integration of plasmid sequence into the chromosome via homologous recombination.
Each strain reached a final OD600 of approximately 0.45 after 24 h, and there were no significant differences in growth among the strains (see Fig. S1 in the supplemental material). Fatty acids were extracted from pelleted cultures at the end of the 24-h incubation, the extracts were derivatized with ethereal diazomethane to form fatty acid methyl esters (FAMEs) and then subjected to GC-FID analysis.
The absolute titers of free fatty acids produced by the strains were much lower than the ∼1 g/liter previously reported under aerobic conditions, although the latter cultures were grown to a higher final cell density (22) (Fig. 4). The distribution of fatty acids was as expected, based on the preference of the TesA thioesterase for C14 and C16 acyl groups (38). The major fatty acids produced anaerobically by the strains were saturated C14, C16, and C18, with C14 accounting for approximately 60% of the total free fatty acids. The Bacillus sp. SG-1 and C. taiwanensis strains also produced small amounts of monounsaturated C16 fatty acid (see Table S2 in the supplemental material). Interestingly, whereas all strains displayed similar growth characteristics, free fatty acid levels differed. Expression of fabG variants corresponded with increased fatty acid levels relative to the E. coli fabG control strain, except for the A. laidlawii variant, which produced a concentration similar to that of the control strain. The strains engineered with P. pacifica, Bacillus sp. SG-1, and C. taiwanensis FabGs produced, respectively, 27, 52, and 59% more free fatty acids than the control strain under anaerobic conditions.
FIG 4.
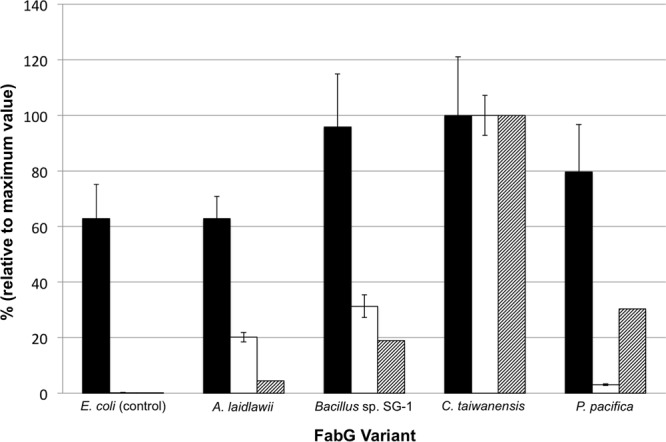
Summary of total free fatty acid titer and in vitro assay data, normalized to the respective values for C. taiwanensis FabG, which produced 11.1 mg/liter total free fatty acids. Titers are from separate E. coli DH1ΔfadE strains overexpressing ′tesA and a fabG variant under anaerobic conditions. The control strain overexpressed a codon-optimized sequence of the native E. coli fabG. Total free fatty acid titer (black bars), NADH specific activity (white bars), and ratio of NADH:NADPH specific activities (hatched bars) are shown. Bar heights represent average titers from 3 replicates, and error bars represent 1 standard deviation.
Anaerobic methyl ketone production.
After observing that expression of the various FabG variants resulted in an increase in free fatty acid titers under anaerobic conditions, we wanted to examine whether these improvements also translated to fatty acid-derived biofuels, such as methyl ketones. The top-performing FabG (C. taiwanensis) was cloned into an E. coli host expressing the methyl ketone pathway, resulting in strain EGS1670 (Table 1). Methyl ketone production in this strain was compared with that of the control strain, EGS895 (25). Production was measured for biological triplicates of each strain.
Methyl ketone production was measured after 96 h by removing the decane overlay for GC-MS analysis. OD600 values of each culture were approximately 0.2 after 96 h, and as observed for the free fatty acid strains, there were no significant differences in growth among the methyl ketone-producing strains.
The methyl ketone titer for strain EGS1670 was 75% higher than that of the control strain (EGS895) under anaerobic conditions (Fig. 5), but titers for both strains were several orders of magnitude lower than the 380 mg/liter reported for EGS895 under aerobic conditions (25). Note that O2-limited conditions will adversely affect the methyl ketone pathway as it is currently constituted, since the acyl-CoA oxidase used in this pathway requires O2 to function (25). Regardless, the relative improvements in methyl ketone titers were consistent with those observed for free fatty acids.
FIG 5.

Summary of total methyl ketone titer in strain EGS895 (without C. taiwanensis FabG) and strain EGS1670 (with C. taiwanensis FabG) (Table 1). Bar heights represent average titers from 3 biological replicates, and error bars represent 1 standard deviation.
DISCUSSION
The goal of this study was to identify a homolog of E. coli FabG with a much higher preference for NADH than for NADPH, which would presumably give rise to higher fatty acid levels during anaerobic growth. The in vitro assay identified several candidate variants that had higher activity with NADH than with NADPH. Of these, three gave rise to higher free fatty acid titers than the control strain under anaerobic culture conditions. It appears that a key molecular determinant of cofactor specificity in FabG is the presence of a negatively charged residue interacting with the 2′-hydroxyl of the NADH adenine ribose, instead of a basic residue that would participate in ionic interactions with the 2′-phosphate moiety of NADPH. A similar phenomenon was reported in investigations of changing the cofactor specificity of E. coli glutathione reductase (39). In that study, a mutant with increased NADH activity was engineered, in part, by substituting residues that interact with the NADPH 2′-phosphate with negatively charged residues (39).
The in vitro activity assay and fatty acid titer data are compared in Fig. 4. These data are normalized to values for the C. taiwanensis FabG, as this FabG homolog displayed the greatest in vitro specific activity, preference for NADH, and fatty acid titer. Although the data do not reflect a linear relationship between fatty acid titer and FabG in vitro specific activity or NADH preference, it appears that a FabG homolog having both higher absolute activity with NADH and preference for NADH facilitates increased fatty acid production under the tested conditions. The second highest fatty acid-producing strain, expressing the Bacillus sp. SG-1 FabG, also displayed high preference and activity with NADH. In contrast, the A. laidlawii FabG led to a fatty acid titer comparable to that of the control strain. Moreover, this variant displayed less than a 2-fold preference for NADH and an in vitro specific activity comparable to that of E. coli FabG (Fig. 2). The C. taiwanensis FabG, which led to a 60% higher fatty acid production, also led to a 75% higher production of fatty acid-derived methyl ketones. In a study of cofactor dependence in the mevalonate pathway, it was similarly observed that strains expressing the NADH-preferring HMG-CoA reductase with the highest forward reaction rate led to increased amorphadiene production (16).
Given the results of this study, a promising future step would be to substitute the chromosomal fabG in E. coli with a variant that led to improved free fatty acid titer under anaerobic conditions, namely, the C. taiwanensis homolog. This host strain could then be further engineered and evolved for a more robust growth phenotype under anaerobic conditions and serve as a base strain for biofuel pathways that are fatty acid based, such as for production of fatty acid ethyl esters, fatty alcohols, or methyl ketones. Thus, the identification of FabG homologs that contribute to improved fatty acid titers under anaerobic conditions serves as a preliminary step toward engineering a host strain for anaerobic production of advanced biofuels.
Supplementary Material
ACKNOWLEDGMENTS
This work conducted by the Joint BioEnergy Institute was supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under contract DE-AC02-05CH11231. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract DE-AC02-05CH11231.
We are grateful to the staff of the Berkeley Center for Structural Biology at the Advanced Light Source of Lawrence Berkeley National Laboratory.
J.D.K. has a financial interest in LS9, Amyris, and Lygos.
Footnotes
Published ahead of print 8 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03194-13.
REFERENCES
- 1.Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. 2012. Microbial engineering for the production of advanced biofuels. Nature 488:320–328. 10.1038/nature11478 [DOI] [PubMed] [Google Scholar]
- 2.Unden G, Bongaerts J. 1997. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320:217–234. 10.1016/S0005-2728(97)00034-0 [DOI] [PubMed] [Google Scholar]
- 3.Trinh CT, Li J, Blanch HW, Clark DS. 2011. Redesigning Escherichia coli metabolism for anaerobic production of isobutanol. Appl. Environ. Microbiol. 77:4894–4904. 10.1128/AEM.00382-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark DP. 1989. The fermentation pathways of Escherichia coli. FEMS Microbiol. Rev. 5:223–234 [DOI] [PubMed] [Google Scholar]
- 5.Singh A, Lynch MD, Gill RT. 2009. Genes restoring redox balance in fermentation-deficient E. coli NZN111. Metab. Eng. 11:347–354. 10.1016/j.ymben.2009.07.002 [DOI] [PubMed] [Google Scholar]
- 6.de Graef MR, Alexeeva S, Snoep JL, Teixeira de Mattos MJ. 1999. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 181:2351–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong SH, Lee SY. 2002. Importance of redox balance on the production of succinic acid by metabolically engineered Escherichia coli. Appl. Microbiol. Biotechnol. 58:286–290. 10.1007/s00253-001-0899-y [DOI] [PubMed] [Google Scholar]
- 8.Berrios-Rivera SJ, Sanchez AM, Bennett GN, San KY. 2004. Effect of different levels of NADH availability on metabolite distribution in Escherichia coli fermentation in minimal and complex media. Appl. Microbiol. Biotechnol. 65:426–432. 10.1007/s00253-004-1609-3 [DOI] [PubMed] [Google Scholar]
- 9.Futai M. 1974. Stimulation of transport into Escherichia coli membrane vesicles by internally generated reduced nictotinamide adenine dinucleotide. J. Bacteriol. 120:861–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, San KY, Bennett GN. 2013. Cofactor engineering for advancing chemical biotechnology. Curr. Opin. Biotechnol. 24:994–999. 10.1016/j.copbio.2013.03.022 [DOI] [PubMed] [Google Scholar]
- 11.Bastian S, Liu X, Meyerowitz JT, Snow CD, Chen MM, Arnold FH. 2011. Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab. Eng. 13:345–352. 10.1016/j.ymben.2011.02.004 [DOI] [PubMed] [Google Scholar]
- 12.Ma C, Zhang L, Dai J, Xiu Z. 2010. Relaxing the coenzyme specificity of 1,3-propanediol oxidoreductase from Klebsiella pneumoniae by rational design. J. Biotechnol. 146:173–178. 10.1016/j.jbiotec.2010.02.005 [DOI] [PubMed] [Google Scholar]
- 13.Bergler H, Fuchsbichler S, Hogenauer G, Turnowsky F. 1996. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur. J. Biochem. 242:689–694. 10.1111/j.1432-1033.1996.0689r.x [DOI] [PubMed] [Google Scholar]
- 14.Toomey RE, Wakil SJ. 1966. Studies on the mechanism of fatty acid synthesis. XV. Preparation and general properties of beta-ketoacyl acyl carrier protein reductase from Escherichia coli. Biochim. Biophys. Acta 116:189–197 [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Cronan JE., Jr 1998. Transcriptional analysis of essential genes of the Escherichia coli fatty acid biosynthesis gene cluster by functional replacement with the analogous Salmonella typhimurium gene cluster. J. Bacteriol. 180:3295–3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma SM, Garcia DE, Redding-Johanson AM, Friedland GD, Chan R, Batth TS, Haliburton JR, Chivian D, Keasling JD, Petzold CJ, Lee TS, Chhabra SR. 2011. Optimization of a heterologous mevalonate pathway through the use of variant HMG-CoA reductases. Metab. Eng. 13:588–597. 10.1016/j.ymben.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 17.Price AC, Zhang YM, Rock CO, White SW. 2004. Cofactor-induced conformational rearrangements establish a catalytically competent active site and a proton relay conduit in FabG. Structure 12:417–428. 10.1016/j.str.2004.02.008 [DOI] [PubMed] [Google Scholar]
- 18.Plewczynski D, Pas J, von Grotthuss M, Rychlewski L. 2002. 3D-Hit: fast structural comparison of proteins. Appl. Bioinformatics 1:223–225. 10.1007/s11590-013-0697-3 [DOI] [PubMed] [Google Scholar]
- 19.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li MZ, Elledge SJ. 2007. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4:251–256. 10.1038/nmeth1010 [DOI] [PubMed] [Google Scholar]
- 22.Steen EJ, Kang Y, Bokinsky G, Hu Z, Schirmer A, McClure A, Del Cardayre SB, Keasling JD. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559–562. 10.1038/nature08721 [DOI] [PubMed] [Google Scholar]
- 23.Cho H, Cronan JE., Jr 1995. Defective export of a periplasmic enzyme disrupts regulation of fatty acid synthesis. J. Biol. Chem. 270:4216–4219. 10.1074/jbc.270.9.4216 [DOI] [PubMed] [Google Scholar]
- 24.Lee TS, Krupa RA, Zhang F, Hajimorad M, Holtz WJ, Prasad N, Lee SK, Keasling JD. 2011. BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J. Biol. Eng. 5:12. 10.1186/1754-1611-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goh EB, Baidoo EE, Keasling JD, Beller HR. 2012. Engineering of bacterial methyl ketone synthesis for biofuels. Appl. Environ. Microbiol. 78:70–80. 10.1128/AEM.06785-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang YM, Wu B, Zheng J, Rock CO. 2003. Key residues responsible for acyl carrier protein and beta-ketoacyl-acyl carrier protein reductase (FabG) interaction. J. Biol. Chem. 278:52935–52943. 10.1074/jbc.M309874200 [DOI] [PubMed] [Google Scholar]
- 27.Jancarik J, Kim SH. 1991. Sparse-matrix sampling - a screening method for crystallization of proteins. J. Appl. Crystal. 24:409–411. 10.1107/S0021889891004430 [DOI] [Google Scholar]
- 28.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystal. 40:658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66(Part 2):213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. 2012. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68:352–367. 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 33.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375–W383. 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beller HR, Goh EB, Keasling JD. 2010. Genes involved in long-chain alkene biosynthesis in Micrococcus luteus. Appl. Environ. Microbiol. 76:1212–1223. 10.1128/AEM.02312-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlieben NH, Niefind K, Muller J, Riebel B, Hummel W, Schomburg D. 2005. Atomic resolution structures of R-specific alcohol dehydrogenase from Lactobacillus brevis provide the structural bases of its substrate and cosubstrate specificity. J. Mol. Biol. 349:801–813. 10.1016/j.jmb.2005.04.029 [DOI] [PubMed] [Google Scholar]
- 36.Fan F, Lorenzen JA, Plapp BV. 1991. An aspartate residue in yeast alcohol dehydrogenase-I determines the specificity for coenzyme. Biochemistry 30:6397–6401. 10.1021/bi00240a008 [DOI] [PubMed] [Google Scholar]
- 37.Carty CE, Ingram LO. 1981. Lipid synthesis during the Escherichia coli cell cycle. J. Bacteriol. 145:472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonner WM, Bloch K. 1972. Purification and properties of fatty acyl thioesterase I from Escherichia coli. J. Biol. Chem. 247:3123–3133 [PubMed] [Google Scholar]
- 39.Scrutton NS, Berry A, Perham RN. 1990. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343:38–43. 10.1038/343038a0 [DOI] [PubMed] [Google Scholar]
- 40.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130. 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- 41.Meselson M, Yuan R. 1968. DNA restriction enzyme from E. coli. Nature 217:1110–1114. 10.1038/2171110a0 [DOI] [PubMed] [Google Scholar]
- 42.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179–207. 10.1016/0022-2836(80)90283-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.