

Abstract
Chromosomal toxin-antitoxin (TA) systems are widespread among free-living bacteria and are supposedly involved in stress tolerance. Here, we report the first TA system identified in the soil bacterium Pseudomonas putida. The system, encoded by the loci PP1586-PP1585, is conserved in pseudomonads and belongs to the HigBA family. The new TA pair was named GraTA for the growth rate-affecting ability of GraT and the antidote activity of GraA. The GraTA system shares many features common to previously described type II TA systems. The overexpression of GraT is toxic to the antitoxin deletion mutants, since the toxin's neutralization is achieved by binding of the antitoxin. Also, the graTA operon structure and autoregulation by antitoxin resemble those of other TA loci. However, we were able to delete the antitoxin gene from the chromosome, which shows the unusually mild toxicity of innate GraT compared to previously described toxins. Furthermore, GraT is a temperature-dependent toxin, as its growth-regulating effect becomes more evident at lower temperatures. Besides affecting the growth rate, GraT also increases membrane permeability, resulting in higher sensitivity to some chemicals, e.g., NaCl and paraquat. Nevertheless, the active toxin helps the bacteria survive under different stressful conditions and increases their tolerance to several antibiotics, including streptomycin, kanamycin, and ciprofloxacin. Therefore, our data suggest that GraT may represent a new class of mild chromosomal regulatory toxins that have evolved to be less harmful to their host bacterium. Their moderate toxicity might allow finer growth and metabolism regulation than is possible with strong growth-arresting or bactericidal toxins.
INTRODUCTION
Toxin-antitoxin (TA) systems are widespread among bacteria. These gene cassettes consist of two small genes which code for a toxin that can influence bacterial life and an antitoxin that inhibits toxin action. Typically they have been divided into three types depending mainly on the nature and mode of action of the antitoxin. Type I and III antitoxins are RNAs that prevent toxin translation and inhibit the toxin protein by binding it, respectively (1, 2). In type II systems, which are the most abundant in prokaryotes (3), both the antitoxin and the toxin are small proteins, and toxin inactivation is achieved by complex formation between the two proteins (4). Recently, TA systems of new types have been reported, where the toxin neutralization mechanism involves stabilization of the toxin target (type IV) (5) or direct cleavage of the toxin mRNA by the antitoxin protein (type V) (6).
Toxins target various cellular processes, but the overwhelming majority of type II toxins impede translation (7). Inhibition of protein synthesis is achieved by different mechanisms, which include binding and inhibiting ribosomal subunits (8, 9) or elongation factor EF-Tu (10) and cleavage of rRNA, tRNA, and, most commonly, mRNA (11–13). Some toxins, such as RelE and HigB, associate with ribosomes and degrade only translated mRNAs (13, 14), while others, like MazF, cleave mRNA independently of ribosomes (12).
TA systems are coded on either plasmids or chromosomes. They were first identified on plasmids, where they contribute to plasmid maintenance in the bacterial population (15, 16). The functions of the chromosomal systems remain controversial—some seem to be just remnants of mobile elements (17), whereas others are involved in the regulation of crucial cellular functions (18). Still, accumulating evidence indicates that many TA systems are associated with a complex of functions that are important for survival under stressful conditions, including biofilm formation, bacterial persistence, growth regulation, protection against bacteriophages, and even programmed cell death (19–22). Activation of many TA systems in response to stresses, such as exposure to antibiotics or nutrient starvation (23–25), also indicates their role in stress survival. The hypothesis about TA systems as stress response elements is also supported by the finding that chromosomal TA systems are more abundant in free-living than in obligately intracellular bacteria (4, 26).
Members of the genus Pseudomonas are known for their versatility and adaptability, allowing their success in hostile and fluctuating habitats (27). Pseudomonas putida, for example, is common in polluted soil and aquatic environments and is thus extensively studied with respect to stress tolerance mechanisms (28). The high adaptability of P. putida relies on many features, including high genetic plasticity and broad metabolic, transport, signaling, and regulatory capabilities (29). Bioinformatic analyses suggest that the P. putida genome also codes for many TA systems. However, the number of predicted TA loci in the P. putida KT2440 chromosome differs between studies, varying from eight (26) to 12 (30), and only six of the TA systems are common to two predictions. To our knowledge, no information is available about the functionality of these putative TA systems. Still, we recently identified a transposon insertion in a putative antitoxin gene PP1585 of a predicted PP1586-PP1585 TA pair when we screened the suppressor mutants of colR-deficient P. putida (31).
ColR is a transcriptional regulator of the conserved ColRS two-component signal system, and it is conditionally essential for P. putida, as its deficiency results in a subpopulation lysis when bacteria grow on minimal glucose solid medium (32). Besides being important for cell survival on glucose medium, the ColRS system also contributes to the phenol and heavy metal tolerance of P. putida (33–35), the root colonization ability of Pseudomonas fluorescens (36, 37), and virulence of the human pathogen Pseudomonas aeruginosa and the plant pathogen Xanthomonas (38–40). Several lines of evidence suggest that all these phenotypes may be caused by impaired membrane homeostasis (31, 33, 37, 40) of colRS-deficient bacteria.
Recent selection of suppressors of lysis of the colR-deficient P. putida revealed among others an interruption of a predicted antitoxin gene PP1585 (31). Suppression of the lysis defect of a colR mutant by inactivation of an antitoxin gene seemed intriguing in several respects. The ability to disrupt the antitoxin gene is in itself uncommon among TA systems, as it results in the activity of the cognate toxin and normally leads to growth inability (25, 41). However, in the case of PP1586-PP1585, it was not only possible to disrupt the antitoxin gene of a putative TA system without significant growth impairment, but the antitoxin inactivation was even beneficial for the colR mutant, as it suppressed the lysis phenotype (31). Therefore, in this study, we addressed the question of whether the identified locus actually represents a bona fide TA system. We demonstrate that the PP1586-PP1585 locus of P. putida indeed codes for a functional TA system, which was named GraTA (growth rate-affecting TA system). GraT was shown to be an unusually mild toxin able to modulate the growth rate of bacteria. GraA is a very efficient antitoxin which neutralizes the innate GraT as well as overexpressed toxin. Interestingly, the growth-inhibitory effect of GraT increases significantly at lower temperatures, and its overexpression is toxic for graA deletion mutants. We present evidence that GraT can rescue the colR mutant from lysis and can also increase bacterial resistance to different antibiotics. This suggests that the GraTA system may be involved in stress adaptation of P. putida.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
The bacterial strains and plasmids used are listed in Table 1. All P. putida strains are derivatives of PaW85 (42), which is isogenic to the fully sequenced strain KT2440 (43). Bacteria were grown in lysogeny broth (LB) or in M9 minimal medium (44) containing either 0.2% glucose, 0.2% gluconate, 0.2% fructose, or 0.2% sodium benzoate. When selection was necessary, the growth medium was supplemented with ampicillin (100 μg ml−1) or kanamycin (50 μg ml−1) for E. coli and benzylpenicillin (1,500 μg ml−1) or kanamycin (50 μg ml−1) for P. putida. E. coli was incubated at 37°C and P. putida at 30°C if not specified otherwise. Bacteria were electrotransformed according to the protocol of Sharma and Schimke (45).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Genotype or characteristics | Source or reference |
|---|---|---|
| E. coli strains | ||
| DH5α | supE44 ΔlacU169 (ϕ80 lacZΔM15) recA1 endA1 hsdR17 thi-1 gyrA96 relA1 | 74 |
| DH5α λpir | λpir lysogen of DH5α | 46 |
| CC118 λpir | Δ(ara-leu) araD ΔlacX74 galE galK phoA20 thi-1 rpsE rpoB argE(Am) recA1 λpir lysogen | 75 |
| BL21(DE3) | hsdS gal (λcI ts857 ind-1 Sam7 nin-5 lacUV5-T7 gene 1) | 76 |
| P. putida strain or genotype | ||
| PaW85 | Wild type, isogenic to KT2440 | 42 |
| ΔgraA | PaW85 ΔgraA | This study |
| ΔgraAT(E80G) | PaW85 ΔgraA; graT contains an E80G substitution mutation | This study |
| ΔgraT | PaW85 ΔgraT | This study |
| ΔgraTA | PaW85 ΔgraTA | This study |
| colR | PaW85 colR::Kmr (Kmr) | 77 |
| colR ΔgraA | colR ΔgraA (Kmr) | This study |
| colR ΔgraAT(E80G) | colR ΔgraA; graT contains an E80G substitution mutation (Kmr) | This study |
| colR ΔgraT | colR ΔgraT (Kmr) | This study |
| colR ΔgraTA | colR ΔgraTA (Kmr) | This study |
| Plasmids | ||
| pEMG | Suicide plasmid containing lacZα with two flanking I-SceI sites (Kmr) | 46 |
| pSW(I-SceI) | Plasmid for I-SceI expression (Apr) | 78 |
| pEMG-ΔA | pEMG containing a PCR-designed 1.5-kb EcoRI-BamHI insert for deleting graA (Kmr) | This study |
| pEMG-ΔAT(E80G) | pEMG-ΔA; graT contains an E80G substitution mutation (Kmr) | This study |
| pEMG-ΔT | pEMG containing a PCR-designed 1.05-kb EcoRI-BamHI insert for deleting graT (Kmr) | This study |
| pEMG-ΔTA | pEMG containing a PCR-designed 1.2-kb EcoRI-BamHI insert for deleting graTA (Kmr) | This study |
| pGP704L/colR::Km | Suicide plasmid containing colR disrupted by Km (Apr Kmr) | 77 |
| pRK2013 | Helper plasmid for conjugal transfer of pGP704L derivatives (Kmr) | 79 |
| pKT240 | Cloning vector (Apr Kmr) | 80 |
| pBRlacItac | Expression vector containing Ptac promoter and lacIq repressor in pBR322 (Apr) | 81 |
| pSEVA-Km (RK2) | Cloning vector (Kmr) | 82 |
| pKTlacItac | Expression vector containing Ptac promoter and lacIq repressor from pBRlacItac and MCS from pSEVA-Km (RK2) (Apr) | This study |
| pKTlacItac-TA | pKTlacItac containing graTA operon under the control of lacI and Ptac promoter (Apr) | This study |
| pKTlacItac-T | pKTlacItac containing graT under the control of lacI and Ptac promoter (Apr) | This study |
| pKTlacItac-TE80G | pKTlacItac containing mutant graTE80G under the control of lacI and Ptac promoter (Apr) | This study |
| pKTlacItac-A | pKTlacItac containing graA under the control of lacI and Ptac promoter (Apr) | This study |
| p9TTBlacZ | Promoter probe plasmid (Cmr Apr) | 33 |
| p9TT1586 | p9TTBlacZ containing graTA promoter fused with lacZ (Cmr Apr) | This study |
| pET11c | Protein expression vector (Apr) | Stratagene |
| pET-Ahis | pET11c containing graA with C-terminal His6 tag (Apr) | This study |
| pET-hisTA | pET11c containing graT with N-terminal His6 tag and untagged graA (Apr) | This study |
| pET-A | pET11c containing graA (Apr) | This study |
Construction of plasmids and strains.
Oligonucleotides used in PCR amplifications are listed in Table S1 in the supplemental material. For generation of graTA deletion strains, the pEMG-based plasmids were constructed according to the protocol described elsewhere (46). The upstream and downstream regions (450 to 1,500 bp) of the gene(s) to be deleted were amplified separately and then joined into one fragment by overlap extension PCR. The obtained 1.05- to 1.5-kb DNA fragments were cut with EcoRI and BamHI and ligated into the corresponding sites of the plasmid pEMG, resulting in plasmids pEMG-ΔA, pEMG-ΔAT(E80G), pEMG-ΔT, and pEMG-ΔTA. Plasmid pEMG-ΔAT(E80G) with a mutant graT gene was obtained due to a PCR error. The pEMG plasmids were delivered to P. putida PaW85 by electroporation, and after 3 h of growth in LB medium, the bacteria were plated onto LB agar supplemented with kanamycin. Kanamycin-resistant cointegrates were selected and electrotransformed with the I-Sce-I expression plasmid pSW(I-SceI). In order to resolve the cointegrate, the plasmid-encoded I-SceI was induced with 1.5 mM 3-methylbenzoate overnight. Kanamycin-sensitive colonies were selected, and the deletion of graA, graT, or graTA was verified by PCR. Plasmid pSW(I-SceI) was eliminated from the deletion strains by growing them overnight in LB medium without antibiotics. Intactness of graT in the ΔgraA strain was verified by sequencing. For the construction of double mutant strains, the disrupted colR gene was inserted into the chromosome of TA system deletion strains by homologous recombination. The suicide plasmid pGP704L/colR::Km was conjugatively transferred from E. coli CC118 λpir into the ΔgraA, ΔgraAT(E80G), ΔgraT, and ΔgraTA strains using helper plasmid pRK2013. Kanamycin-resistant transconjugants were selected, and colR knockouts were verified by PCR analysis.
In order to construct the broad-host-range expression vector pKTlacItac, the 1.9-kb BamHI-HindIII fragment containing the lacI tac cassette from pBRlacItac was inserted into the corresponding sites of pKT240. The resulting plasmid was modified by replacing its HindIII-SacI region with a HindIII-SacI fragment containing a multicloning site of pSEVA-Km(RK2). The plasmid obtained, pKTlacItac, was used to construct plasmids pKTlacItac-TA, pKTlacItac-T, pKTlacItac-TE80G, and pKTlacItac-A. First, the whole graTA operon was amplified from the P. putida PaW85 chromosome with the primers 1585Acc and 1586Sal. The obtained DNA fragment was cut with Acc65I and SalI and cloned into Acc65I-SalI-opened pKTlacItac. The resulting plasmid, pKTlacItac-TA, was treated with SalI and XhoI and religated to obtain pKTlacItac-A. pKTlacItac-T was constructed by deleting graA from pKTlacItac-TA by the use of SacI and SacII. To construct pKTlacItac-TE80G, the mutant graT gene was amplified from the chromosome of the ΔgraAT(E80G) strain using primers 1586Sal and 1586Bam. The resulting PCR fragment was treated with BamHI, Klenow fragment, and XhoI and inserted into Ecl136II-XhoI-opened pKTlacItac-TA.
To construct the transcriptional fusion of the graTA promoter with lacZ, the graT gene and its upstream region was amplified from the P. putida PaW85 chromosome with primers 1585ATG and 1585Sac. The resulting PCR fragment was treated with Eco47III and Ecl136II and inserted into SmaI-opened p9TTBlacZ.
For purification of the antitoxin or the toxin-antitoxin complex, a hexahistidine tag was fused to the C terminus of GraA or to the N terminus of GraT, respectively. The graA- and graTA-containing fragments were amplified by using the oligonucleotide pair A-XhoNde and A-his and the pair T-Nhis and 1585Bam (see Table S1 in the supplemental material), respectively. For expression of untagged GraA, the graA gene was amplified by using oligonucleotides A-XhoNde and 1585Bam. All PCR fragments were treated with NdeI and BamHI and ligated into the corresponding sites of the plasmid pET11c, resulting in pET-Ahis, pET-hisTA and pET-A. All plasmids designed were sequenced in order to exclude PCR-generated errors in the cloned DNA fragments.
Determination of the generation time of bacteria.
Generation time was calculated for bacteria growing on microtiter plates. LB-grown overnight cultures were diluted 100-fold into 100 μl LB medium in microtiter plate wells. Microtiter plates were incubated with shaking at 37°C, 30°C, 25°C, or 20°C, and the optical density at 580 nm (OD580) was measured every 30 min to obtain the growth curve of bacteria. Generation time was calculated from the slope of the exponential growth curve according to the equation G = [t/3.3log(b/B)], where G is the generation time, t is the time interval (in minutes), and B and b are the numbers of bacteria (OD580) at the beginning and the end of the time interval, respectively.
Ectopic overexpression of GraTA proteins in P. putida.
In order to evaluate the effect of the overexpression of GraT, GraT(E80G), and GraA, the P. putida PaW85 and its graTA deletion derivatives were electrotransformed with plasmids pKTlacItac-T, pKTlacItac-TE80G, and pKTlacItac-A. After 1 h of growth in 1 ml LB medium, the bacterial cultures were serially diluted into LB, and 5 μl of diluted cultures was spotted onto LB agar plates supplemented with benzylpenicillin for plasmid selection. For the overexpression of TA proteins, the bacteria were also spotted onto LB medium containing 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Plates were incubated up to 48 h at 30°C or 25°C.
Purification of proteins.
Nickel affinity chromatography was used to purify the C-terminally histidine-tagged GraA and the GraT-GraA complex consisting of the N-terminally tagged GraT and the untagged GraA. The untagged GraA was also overexpressed and used as a negative control in protein purification. For the overexpression of proteins, the E. coli strain BL21(DE3) containing different expression plasmids was grown in LB medium at 30°C up to an OD580 of ∼0.5. Protein expression was induced with 0.5 mM IPTG. After 4 h of induction, cells were pelleted and sonicated in buffer A (50 mM phosphate buffer [pH 7.6], 1 M NaCl). Cellular debris was removed by centrifugation at 16,000 × g for 20 min at 4°C. The supernatant was filtered through a 0.22-μm filter before loading it on a 1-ml HisTrap HP (GE Healthcare Life Sciences) column equilibrated with buffer A. Protein purification was performed by fast protein liquid chromatography (FPLC) using an Äkta Prime chromatography system (GE Healthcare Life Sciences). The column was washed with buffer B (50 mM phosphate buffer [pH 7.6], 0.5 M NaCl, 50 mM imidazole, 10% glycerol) until the absorbance of the flowthrough at 280 nm approached baseline. Proteins were eluted from the column with linear imidazole gradient by using buffer C (50 mM phosphate buffer [pH 7.6], 0.5 M NaCl, 600 mM imidazole, 10% glycerol). Elution fractions with peak absorbance at 280 nm were collected and the purified proteins were dialyzed stepwise against the following buffers: (i) 10 mM Tris-HCl (pH 7.5), 300 mM KCl, 20% glycerol; (ii) 10 mM Tris-HCl (pH 7.5), 250 mM KCl, 35% glycerol; (iii) 10 mM Tris-HCl (pH 7.5), 200 mM KCl, 50% glycerol. Proteins were stored at −20°C.
Enzyme assay.
β-Galactosidase activities were measured from overnight cultures of LB-grown bacteria carrying plasmid p9TT1586 according to a previously described protocol (47).
5′ RACE analysis.
To determine the transcriptional start site of the graTA operon by 5′ RACE (random amplification of cDNA ends), the total RNA was isolated from exponential-phase cells of P. putida PaW85 using the NucleoSpin RNA II kit (Macherey-Nagel). RNA samples were additionally treated with DNase I (Thermo Scientific) to remove contaminating DNA. The first-strand cDNA was synthesized from total RNA using the primer graAstop and Maxima reverse transcriptase (Thermo Scientific). cDNA was purified with DNA Clean & Concentrator-5 kit (Zymo Research) and tailed with poly(dG) sequence by terminal deoxynucleotidyl transferase (Thermo Scientific). The second strand of the poly(dG)-tailed single-stranded cDNA was made using the primer C-ankur and Taq polymerase. Purified double-stranded cDNA was amplified by PCR using the primers graAsees and ARB2. The obtained PCR product was sequenced.
qRT-PCR.
Total RNA for graT mRNA quantification was isolated as described above for 5′ RACE. The quantitative reverse transcription-PCR (qRT-PCR) assay was performed on the Rotor-Gene Q system (Qiagen) using the SuperScript III Platinum SYBR green one-step qRT-PCR kit (Invitrogen) according to the manufacturer's protocol, except with double primer concentrations and a total reaction volume of 10 μl. A 10-ng portion of total RNA was used for each reaction. The graT gene was amplified using the primers graTqFw and graTqRev, and the rpoD gene was amplified using the primers rpoDqFw and rpoDqRev. Raw data were analyzed with the Rotor-Gene Q software version 2.02 (Qiagen), and mRNA amounts were calculated using the LinRegPCR software version 2013.0 (48). Data from three separate qRT-PCR experiments performed on two independently extracted RNAs were averaged and normalized against rpoD levels.
DNase I footprinting.
DNA fragments for DNase I footprinting assay were amplified from the plasmid p9TT1586 by PCR. Oligonucleotides used in PCRs are listed in Table S1 in the supplemental material. Prior to the PCR, one oligonucleotide was end labeled by phosphorylation with [γ-32P]ATP. The labeled DNA fragments were purified by native 5% polyacrylamide gel electrophoresis, eluted with buffer (0.5 M ammonium acetate [NH4Ac], 10 mM MgAc, 1 mM EDTA, 0.1% SDS) and resuspended in water. For the binding reaction, the purified His-tagged GraA or GraT-GraA complex (0.45, 0.9, 4.5, 9, and 45 pmol) was combined with 30,000 cpm of labeled DNA fragment, 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM CaCl2, 0.1 mM EDTA, 10 mM KCl, 5 μg of bovine serum albumin (BSA), 1 μg of salmon sperm DNA, and 5% glycerol in a final volume of 50 μl. Reactions with different volume of proteins were equalized with the addition of appropriate amount of GraA storage buffer. Proteins were allowed to bind to DNA during 30 min at room temperature before the start of digestion by DNase I (0.06 U; Thermo Scientific) for 3 min. Reactions were stopped by the addition of 50 μl of stop solution (20 mM Tris-HCl [pH 8.0], 20 mM EDTA, 0.8% sodium dodecyl sulfate, 100 μg of salmon sperm DNA per ml). The footprinting reaction mixtures were subsequently extracted once with phenol and chloroform (1:1, vol/vol) and once with chloroform, and finally, the DNA was precipitated with ethanol and NaCl. The DNA fragments were resuspended in 7 μl of sequence loading buffer (50% formamide, 10 mM EDTA, 0.3% bromophenol blue, and 0.3% xylene cyanol) and loaded onto a 6.5% polyacrylamide gel that contained 8 M urea. DNA sequencing reactions were performed with a Sequenase kit, version 2.0 (USB Corporation), and the products were loaded on a sequencing gel as size markers. After the run, the gels were dried, exposed to a PhosphorImager screen, scanned using a Typhoon Trio imager, and analyzed by ImageQuant TL v2005 software (GE Healthcare).
Flow cytometry analysis.
Different bacterial strains were grown on LB agar or on glucose or gluconate minimal plates as thin radial stripes. In order to enhance the glucose-dependent cell lysis of the colR mutant, the glucose plates were supplemented with 1 mM phenol. After 48 h of growth, the cells were scraped off the plates and suspended in M9 buffer. The cell suspension was diluted to an OD580 of 0.015. The two components of the LIVE/DEAD BacLight kit (Invitrogen), the red fluorescent dye propidium iodide (PI) and the green fluorescent dye SYTO9, were mixed in a 1:1 ratio and then diluted 17.6-fold into filter-sterilized M9 buffer. For staining of bacteria, the diluted cell suspension was mixed with the freshly prepared reagent mixture in a 20:1 ratio. Samples were incubated at 30°C in the dark for 30 min, and approximately 10,000 events from every sample were analyzed with the FACSAria flow cytometer (BD Biosciences). Fluorescent dyes were excited at 488 nm. Forward and side scatter (FSC and SSC) of the light and fluorescence emission at 530 and 616 nm were acquired for every event. Populations of intact, PI-permeable, and dead cells were defined as previously described (34). While PI-permeable cells differ from the intact subpopulation only by their PI-permeable membranes, the dead cells are more compromised, containing less DNA than other cells (34).
Stress tolerance assays.
Phenol sensitivity was evaluated on agar plates containing 0.2% glucose as a carbon source and 7 mM phenol. The cultures that had been grown overnight in LB were 10-fold serially diluted, spotted onto plates as 5-μl drops, and incubated at 30°C for up to 7 days.
In order to evaluate the stress tolerance of bacteria to different compounds, the serially diluted bacteria were spotted onto LB agar plates supplemented with different antibiotics and chemicals (specified in Results). Plates were incubated at 30°C for 24 or 48 h.
Streptomycin-mediated killing assay.
Bacterial cultures pregrown overnight in liquid LB medium at 37°C were diluted about 55-fold into fresh LB and grown at 25°C or 37°C until the optical density (580 nm) reached about 0.5. A 1.4-ml portion of each culture was transferred to an Eppendorf tube and treated with 300 μg/ml streptomycin with shaking at 25°C or 37°C. The number of CFU was determined before streptomycin addition and at various time points during treatment. For that, 200 μl of bacterial culture was centrifuged and resuspended in M9 buffer, and 10-fold serial dilutions were spotted onto solid LB medium as 5- to 10-μl spots. Plates were incubated at 30°C overnight.
RESULTS
Orthologs of PP1585 and PP1586 are present in the genomes of most pseudomonads.
It has been predicted that the Pseudomonas putida genes PP1586 and PP1585 code for a toxin and an antitoxin, respectively (26, 30). To determine whether this putative TA pair is conserved in 48 other completely sequenced pseudomonads, we performed a search in the Pseudomonas Genome Database (49). According to this analysis, the antitoxin protein has 30 and the toxin 17 annotated orthologs in other Pseudomonas species (see Table S2 in the supplemental material). Importantly, all 17 PP1586 orthologs constitute a pair with a putative antitoxin orthologous to PP1585. Moreover, the uncommon gene order, the toxin gene preceding the antitoxin, is conserved in these putative TA loci. The remaining 13 PP1585 orthologs were annotated as single gene loci, but careful sequence analysis revealed that nine of them possess a putative PP1586 toxin ortholog upstream of the antitoxin gene (see Table S2). Only four PP1585 orthologs in other pseudomonads did not have an obvious toxin partner. We also searched the genomes of the Pseudomonas species that at first glance did not contain any PP1585 and PP1586 orthologs and found three more TA loci orthologous to PP1586-PP1585. The loci most similar to PP1586-PP1585 were in the genomes of P. putida F1 and P. putida NBRC 14164 (97% and 92% identity of the toxin gene and 99% and 97% identity of the antitoxin gene, respectively), but well conserved TA loci were also present in all 13 P. aeruginosa strains, in two P. fluorescens strains, in one Pseudomonas stutzeri strain, in one Pseudomonas syringae strain, and in Pseudomonas denitrificans strains (64 to 72% identity of the toxin gene and 48 to 62% identity of the antitoxin gene) (see Table S2). Conclusively, of 48 completely sequenced pseudomonads, 30 contain a putative TA pair orthologous to the PP1586-PP1585 operon.
Using the NCBI BLAST tool, we searched for putative PP1586-PP1585 homologs in other bacteria and found that the protein most similar to PP1585 was a HigA family antitoxin protein from Nitrosococcus halophilus Nc4 (57% identity), and the one most similar to PP1586 was a putative killer protein of the plasmid maintenance system from Delftia sp. Cs1-4 (71% identity). The proteins most similar to PP1585 and PP1586 that have already been experimentally described are HigBA system proteins from Vibrio cholerae (41), with sequence identities of 38% for the antitoxin and 37% for the toxin (see Fig. S1 in the supplemental material). Given that the structure of the PP1586-PP1585 operon also resembles that of HigBA systems, where the toxin precedes the antitoxin gene and the antitoxin is larger than the toxin, we deduce that PP1585 and PP1586 belong to the HigBA family of TA systems.
PP1586 and PP1585 genes code for the growth rate-modulating TA system GraTA.
In order to examine whether PP1585 and PP1586 code for a functional TA system, we constructed three derivatives of P. putida PaW85 which had deletions of either PP1585 (ΔgraA), PP1586 (ΔgraT), or both genes (ΔgraTA) (Fig. 1A). Accidentally, we also obtained an antitoxin (PP1585) deletion strain [ΔgraAT(E80G)] which had a PCR-generated point mutation in the toxin (PP1586) coding region, resulting in the replacement of glutamic acid with glycine in the 80th position of the toxin (E80G). To investigate the effect of this substitution on the toxin activity, we included the antitoxin deletion strain with the mutant toxin [ΔgraAT(E80G)] in the experiments presented below.
FIG 1.
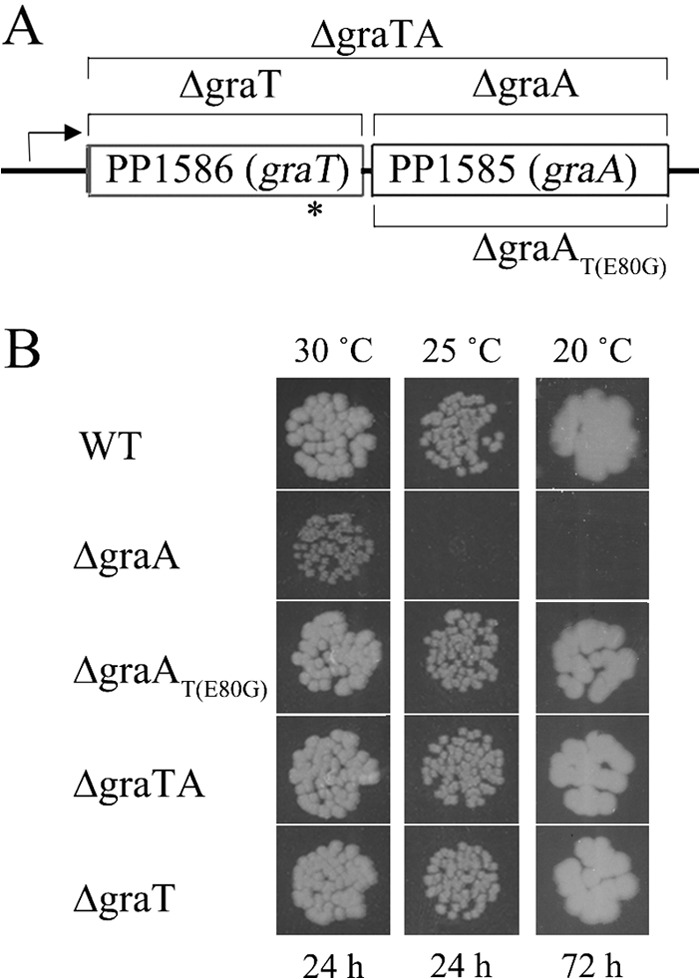
GraT-mediated growth inhibition of the ΔgraA strain. (A) Organization of genes in the P. putida graTA locus. DNA regions which are absent from the chromosomes of the deletion strains are indicated by brackets. The approximate location of the E80G substitution mutation in graT of the ΔgraAT(E80G) strain is indicated by an asterisk. (B) Growth of P. putida wild-type strain PaW85 (WT) and its graTA deletion derivatives on solid LB medium. Approximately 50 cells of each strain were inoculated onto LB agar plates. Incubation temperatures and growth times are indicated.
It is quite unusual that we could easily construct a stable antitoxin deletion strain. As sequencing evidenced that toxin gene was intact in the ΔgraA strain, we considered that the toxin is either very weak or entirely inactive. However, while constructing the deletion strains, we noticed that the PP1585 (antitoxin) deletion mutant had a somewhat lower growth rate than the parent strain and other deletion strains. This was the first hint of the functionality of the toxin in the absence of the antitoxin, and therefore we named the system GraTA, for growth rate-affecting toxin-antitoxin system.
We first noticed the GraT-mediated inhibition of growth on agar plates, where the colony formation of the ΔgraA strain was significantly delayed compared to that of the parent strain (Fig. 1B). This growth defect was obviously caused by GraT, because the colony formation rates of the ΔgraTA and ΔgraT mutants were equal to that of the wild-type strain. Moreover, the fact that the ΔgraAT(E80G) strain grows similarly to the parent strain clearly showed that GraT is responsible for the growth inhibition of the ΔgraA strain and that the mutation E80G in GraT reduces its activity. The slower growth of the GraA deletion strain was observed on different carbon sources, including glucose, gluconate, fructose, and sodium benzoate (data not shown). Interestingly, the GraT-mediated growth inhibition was stronger when the bacteria were grown at lower temperatures (Fig. 1B). While the outgrowth of the ΔgraA strain on LB solid medium was delayed for only a few hours at 30°C, this strain could not form colonies on LB plates at 20°C within 3 days. The temperature-sensitive growth defect of the ΔgraA strain is clearly caused by the activity of GraT, because the ΔgraAT(E80G) strain behaved similarly to the wild type (Fig. 1B), with only a slight hint of slower growth at lower temperatures (not observable in Fig. 1B). To investigate the effect of the toxin on growth rate more specifically, we determined the generation time of P. putida PaW85 and its graTA deletion derivatives grown in liquid LB medium. The generation times of wild-type and ΔgraA bacteria differed markedly at 30°C (64 min for the wild type versus 82 min for the mutant) but even more at 20°C (151 min for the wild type versus 804 min for the mutant) (Table 2). Surprisingly, the growth rate suppression effect of GraT was not observable at 37°C. Other deletion strains, including the ΔgraAT(E80G) mutant, revealed growth rates comparable to that of the wild-type strain at all temperatures. These results show that GraTA forms a functional TA system consisting of the temperature-dependent toxin GraT, which is able to inhibit the growth rate of bacteria, and the antitoxin GraA, capable of neutralizing the toxin's effect. Moreover, we show that the single E80G amino acid substitution suppresses the activity of GraT toxin.
TABLE 2.
Generation times of P. putida strains grown in LB medium
| Strain genotype | Generation time (P) ata: |
|||
|---|---|---|---|---|
| 20°C | 25°C | 30°C | 37°C | |
| WT | 151.0 ± 9.3 | 120.2 ± 2.8 | 64.7 ± 3.4 | 44.7 ± 1.6 |
| ΔgraA | 804.2 ± 77.7 (1.9 × 10−11) | 247.7 ± 9.6 (5.2 × 10−14) | 81.9 ± 3.1 (2.4 × 10−7) | 45.5 ± 2.7 (0.34) |
| ΔgraAT(E80G) | 153.4 ± 11.55 (0.52) | 122.8 ± 2.9 (0.02) | 61.8 ± 5.8 (0.53) | 43.8 ± 1.2 (0.31) |
| ΔgraTA | 149.7 ± 4.8 (0.63) | 117.6 ± 2.7 (0.03) | 62.7 ± 8.5 (0.81) | 45.3 ± 2.5 (0.43) |
| ΔgraT | 149.2 ± 9.3 (0.53) | 120.6 ± 4.1 (0.76) | 64.8 ± 1.4 (0.42) | 45.7 ± 1.6 (0.12) |
Values are means ± standard deviations. P values (in parentheses) are for comparison between each strain and the wild type (WT) at a given temperature.
Overexpression of GraT is toxic to graA deletion mutants.
The deletion of an antitoxin gene has been impossible for several other TA systems (41, 50), but we gained the graA deletion strain quite easily. This indicated that either the expression of graT is weak, the stability of the graT mRNA or of the GraT protein is low, or the activity of GraT is somehow hindered. To find out if the overexpression of GraT can be highly toxic to bacteria, plasmids pKTlacItac-T and pKTlacItac-TE80G, enabling overproduction of GraT and GraT(E80G), respectively, were constructed. The empty vector pKTlacItac was used as control plasmid (Fig. 2A). Data in Fig. 2B show that at 30°C, the ectopic expression of GraT was most toxic for the ΔgraA strain, which lacked the GraA antitoxin but retained the native graT gene in the genome. Notably, comparison of pKTlacItac-T-carrying bacteria with an empty vector control shows that the tac promoter used to overexpress the GraT is leaky, because the growth of the ΔgraA, ΔgraAT(E80G), and ΔgraTA strains was already retarded without induction with IPTG (Fig. 2A and B). Although the ΔgraTA and ΔgraAT(E80G) strains are devoid of graA like the ΔgraA strain, they were less affected by ectopic GraT, and the total growth arrest was observed only under IPTG-induced conditions. Interestingly, neither the wild-type nor the ΔgraT strain showed significant sensitivity to GraT overexpression at 30°C (Fig. 2A and B). This shows that overexpression of GraT can be toxic to bacteria but the native antitoxin can effectively inactivate the artificially overproduced toxin. As was expected, the overexpression of GraT(E80G) at 30°C was not toxic to bacteria (Fig. 2C), confirming the ability of the E80G substitution to reduce the GraT activity.
FIG 2.

Ectopic overexpression of GraT but not GraT(E80G) is toxic to strains devoid of graA. P. putida wild-type strain PaW85 (WT) and its graTA deletion derivatives were transformed with plasmids pKTlacItac (A and D), pKTlacItac-T (B), pKTlacItac-TE80G (C), and pKTlacItac-A (D), and series of 10-fold dilutions were spotted onto LB agar or onto LB with 0.5 mM IPTG (+IPTG). Plates were incubated either at 30°C for 24 h (A, B, and C) or at 25°C for 48 h (D).
Complementation of the ΔgraA strain with the GraA overexpression plasmid pKTlacItac-A eliminated the growth defect of the strain even at lower temperatures (Fig. 2D), indicating that ectopic expression of GraA can neutralize the effect of GraT toxin. Thus, these overexpression experiments confirmed that GraT is a toxin which is inactivated by GraA antitoxin, whether artificial or innate. Unfortunately, all plasmids containing the lacI tac cassette are unstable in P. putida. Therefore, the effect of the ectopic expression of GraT and GraA can be tested only immediately after transformation, and no long-term experiments are possible.
GraA and GraT form a complex.
In type II TA systems, the toxin is normally inactivated by a protein complex formation between the toxin and the antitoxin (51). To clarify if GraA and GraT form a complex, a pulldown assay was used. GraT with an N-terminal hexahistidine tag was overexpressed together with untagged GraA. As a negative control, the untagged GraA alone was also overexpressed. Affinity purification using Ni2+-coated agarose beads and subsequent SDS-PAGE revealed that while His6-GraT and GraA copurify in a 1:1 ratio, the untagged GraA alone cannot bind to the Ni2+ column (Fig. 3). This suggests specific binding between GraA and GraT, which can be considered the mechanism of toxin inactivation. Binding of GraT and GraA, despite the use of N-terminally His-tagged GraT, suggests that the N terminus of the toxin is not specifically important for antitoxin binding.
FIG 3.

GraT and GraA form a complex. His-tagged GraT and untagged GraA were coexpressed (lane 1) and copurified by nickel affinity chromatography (lane 2). The untagged GraA alone was also overexpressed (lane 3) and passed through the nickel affinity column (lane 4) as a negative control.
GraA represses the graTA promoter and binds just upstream of the graT start codon.
Commonly, the transcription of a type II TA operon is autorepressed by the antitoxin or toxin-antitoxin complex (52). Computational analysis of GraTA proteins predicted that GraA can putatively bind DNA, as it possesses a helix-turn-helix motif of XRE-family proteins. In order to test whether the expression of the graTA operon depends on the presence of the antitoxin, the graTA transcriptional fusion with the β-galactosidase gene was analyzed in wild-type and TA deletion mutants. The transcription from the graTA promoter was 5-fold upregulated in the ΔgraA and ΔgraTA strains compared to the parent strain, which indicated antitoxin-mediated autorepression (Fig. 4A). Promoter activity in the ΔgraT strain was even lower than in the wild-type strain. We also quantified the graT-specific mRNA in the wild type and the ΔgraA strain by qRT-PCR. As the ΔgraA strain displayed an approximately 85-fold-higher level of toxin mRNA than the wild type, GraA indeed acts as an autorepressor of graTA expression. Notably, this also indicates that the mild effect of the toxin cannot be attributed to the low expression level of the graT gene in the ΔgraA strain.
FIG 4.

Autorepression of graTA promoter by GraA. (A) β-Galactosidase activities measured in P. putida wild-type (WT) and graTA deletion strains carrying the transcriptional fusion of the graTA promoter with lacZ in plasmid p9TT1586. Bacteria were grown overnight in LB medium at 30°C. Data (means with 95% confidence intervals) of at least three independent experiments are presented. (B) Promoter region of graTA. The −10 and −35 elements of the promoter are boxed, the transcriptional start site is indicated by a black arrow, and the ATG of graT is underlined. Brackets indicate the regions protected by DNase I cleavage by binding of GraA, and the asterisk marks a hypersensitive cleavage site. The palindromic sequences of GraA binding site are designated by gray arrows. (C) DNase I footprint analysis for determining GraA and GraT-GraA binding sites in the graTA promoter region. Footprints on the upper strand of the promoter region are presented. Lane 0 represents the DNase I reaction carried out in the absence of TA proteins. Other lanes represent reactions which contain increasing amounts (in pmol) of either the GraA-His or the His-GraT+GraA complex. The line on the right designates the region of protection against DNase I cleavage, and the asterisk signifies a base which becomes hypersensitive to DNase I cleavage upon the formation of the DNA complex with TA protein.
To determine the transcription start site of graTA genes, we carried out 5′ RACE analysis with a primer complementary to the end of the operon. We detected a single transcription start site located 26 nt upstream of graT ATG codon (Fig. 4B), indicating that graTA genes are transcribed polycistronically. Consensus −10 and −35 hexamers of the sigma 70-dependent promoter were identified upstream of the transcriptional start site (Fig. 4B).
To confirm that the autorepression is caused by direct binding of GraA to the promoter region, a DNase I footprint analysis was performed with both the antitoxin and the toxin-antitoxin complex (Fig. 4C). The results show that both the GraA antitoxin alone and the GraTA complex bind a 35-nucleotide-long sequence directly in front of the toxin gene (Fig. 4B and C). The area protected from DNase I digestion is exactly the same for both GraA and the GraTA complex. The recognition site contains a perfect palindromic sequence (TTAACGAATAACGTTAA; underlining indicates the palindrome) and partially overlaps the −10 region of the graTA promoter. Such a location of the GraA binding site indicates that antitoxin binding may directly hinder the binding of RNA polymerase to the promoter.
GraT increases membrane permeability but nevertheless prevents the lysis of the colR mutant.
The data presented above show that GraTA is a functional TA system and that the toxin GraT is able to reduce bacterial growth rate. Slower growth has been shown to be beneficial for bacteria under stressful conditions (53). Considering that the disruption of graA can eliminate the glucose-specific subpopulation lysis phenotype of colR-deficient strain (31), we hypothesized that this may occur due to GraT-caused growth inhibition, which results in lower membrane stress of the colR mutant. To test this possibility, we decided to analyze the effects of GraT and GraT(E80G) on phenotypes of the colR-deficient strain. We presumed that only GraT could suppress the lysis defect of colR mutant, as only GraT and not GraT(E80G) can significantly affect the growth rate of bacteria.
The lysis-susceptible population of colR-deficient P. putida grown on glucose solid medium is highly heterogeneous, as judged by SYTO9 and propidium iodide (PI) staining and single-cell analysis by flow cytometry (32, 34). Besides a subpopulation of PI-impermeable cells similar to the wild type, the colR mutant culture also contains a large amount of PI-permeable cells and a characteristic subpopulation of dead cells (see Fig. S2 in the supplemental material) (34). In order to examine how GraT and GraT(E80G) can influence the population structure as well as membrane permeability of glucose-grown bacteria, we stained the wild type, colR-deficient strain, and TA deletion mutants with the LIVE/DEAD kit and assessed the populations at the single-cell level. Flow cytometry analysis revealed that the characteristic population structure with a dead subpopulation was present in colR-deficient bacteria and also in colR derivatives lacking either graT or the whole graTA operon (Fig. 5A). However, the subpopulation of dead cells had almost disappeared in the colR ΔgraA double mutant (P = 4.1e-04, Student's t test) and, contrary to our expectations, had also significantly decreased in the colR ΔgraAT(E80G) mutant (Fig. 5A) (P = 6.4e-04). The latter finding indicates that mutant GraT(E80G) retained part of its activity but also suggests that the growth rate reduction by GraT is not the only reason why the lysis of the colR mutant is suppressed.
FIG 5.
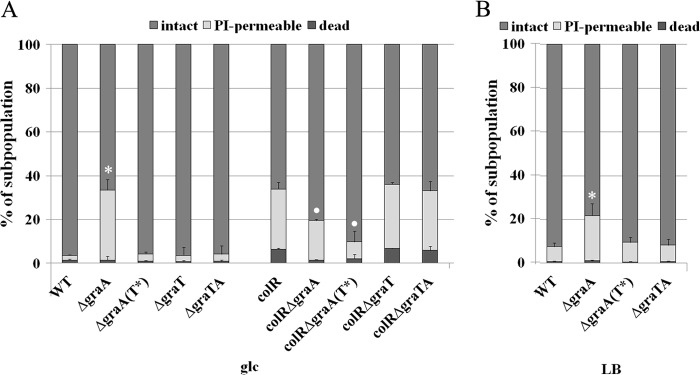
Cell population structure by flow cytometry analysis. (A) The P. putida wild type (WT), the colR mutant (colR) and their graTA deletion derivatives were grown for 48 h on solid glucose minimal medium supplemented with 1 mM phenol at 30°C. Inactive GraT(E80G) is designated T*. (B) The P. putida wild type and its graTA deletion derivatives were grown on LB agar at 30°C for 48 h. Cells were stained with PI and SYTO9 and analyzed by flow cytometry. Relative proportions of intact, PI-permeable, and dead subpopulations (specified in Fig. S1 in the supplemental material) are shown. Data (mean with 95% confidence intervals) of at least four independent determinations are presented. Asterisks indicate statistically significant differences (P < 0.001, Student's t test) between the PI-permeable populations of a particular strain and the wild type. Dots indicate that both the PI-permeable and the dead subpopulations of a particular strain differ significantly (P < 0.01) from that of the colR mutant.
Population analysis also revealed that deletion of graA essentially influenced the membrane permeability, because a large PI-permeable subpopulation was observed in ΔgraA bacteria (Fig. 5A; also, see Fig. S2 in the supplemental material). As this PI-permeable population was absent in the ΔgraAT(E80G) strain, we conclude that the toxin GraT is responsible for the membrane permeabilization. To test whether the higher membrane permeability of ΔgraA cells is specific to bacteria grown on glucose medium or whether this effect is also detectable on other carbon sources, we performed flow cytometry analyses with LB- and gluconate-grown bacteria. The membrane-permeabilizing effect of GraT also appeared on solid gluconate (data not shown) and LB media (Fig. 5B) (P = 1.6e-04). As bioinformatic analysis does not give any clue that GraT could be a membrane protein (data not shown), we conclude that the increased membrane permeability of the ΔgraA strain should be an indirect effect of the GraT toxin. Taken together, these data show that the toxin GraT increases membrane permeability to PI but can nevertheless eliminate the membrane stress and cell death of the colR mutant.
Opposite effects of GraT on survival under different stress conditions.
In addition to glucose-specific lysis phenotype, the colR-deficient P. putida also displays decreased phenol tolerance (33, 34). As phenol mostly affects the cell membrane and GraT increases membrane permeability (Fig. 5), it was interesting to find out how GraT affects the phenol tolerance of both wild-type and colR-deficient bacteria. Thus, we compared the growth of different strains on glucose solid medium supplemented with phenol. Bacteria with a disrupted colR gene tolerated 7.5 mM phenol only when graA was absent (Fig. 6), indicating that GraT can alleviate the phenol sensitivity of the colR-deficient strain. Also, GraT(E80G) can increase the phenol tolerance of the colR mutant, although to a lesser extent than wild-type GraT (Fig. 6). The tolerance had not changed for any of the deletion mutants in the wild-type background. Thus, despite the fact that GraT increases membrane permeability, it restores the phenol tolerance of the colR-deficient strain without affecting the tolerance of wild-type bacteria.
FIG 6.

Plate assay of phenol tolerance. The P. putida wild-type strain PaW85 (WT), the colR-deficient strain (colR), and their graTA deletion derivatives were grown on glucose solid medium supplemented with 5.5, 6.5, or 7.5 mM phenol for 7 days at 30°C. In each pair, the panels show serial dilutions of approximately 50 and 5 cells per spot.
To analyze whether GraT can increase the overall stress tolerance of P. putida, we tested the growth of the wild type and the graTA deletion strains on media with different compounds (Fig. 7) which inhibit transcription (rifampin), translation (streptomycin, kanamycin, and tetracycline), replication (mitomycin C and ciprofloxacin), or cell wall synthesis (ceftazidime and benzylpenicillin) or generate oxidative (paraquat, nitroquinoline, and H2O2) or osmotic stress (NaCl, LiCl, and urea). We observed that the ΔgraA strain had remarkably increased tolerance for the translation inhibitors streptomycin and kanamycin, the replication inhibitors mitomycin C and ciprofloxacin, and the cell wall inhibitor ceftazidime, whereas the ΔgraAT(E80G), ΔgraTA, and ΔgraT strains were as sensitive to these chemicals as the wild type (Fig. 7). However, the ΔgraA mutant showed an increased sensitivity to H2O2 (data not shown), paraquat, and tetracycline and particularly to NaCl (Fig. 7). The tolerance of the ΔgraA strain to other tested chemicals was not significantly changed. We also tested the effect of these chemicals on the graTA promoter, but no remarkable response of the graTA promoter was observed (data not shown). Therefore, GraT helps bacteria survive some stresses and increases the sensitivity to others, but the expression of the graTA operon is not responsive to these stress situations.
FIG 7.

Plate assays of stress tolerance. The P. putida wild-type strain PaW85 (WT) and its graTA deletion derivatives were grown on LB solid medium containing different chemicals at 30°C for 24 h (Sm and Km, 48 h). The final concentrations of tetracycline (Tet), paraquat, NaCl, streptomycin (Sm), kanamycin (Km), ciprofloxacin (Cipro), and mitomycin C (MMC) are indicated. In each pair, the panels show serial dilutions of approximately 5,000 and 500 cells per spot.
GraT increases the tolerance of bacteria to the killing activity of streptomycin.
TA loci are known to increase the tolerance of bacteria to the killing activity of antibiotics (54, 55). To test whether GraT can increase the survival of bacteria after antibiotic treatment, we exposed the exponentially growing cells of P. putida wild-type, ΔgraA, and ΔgraAT(E80G) strains to a lethal concentration of streptomycin. If the strains were pregrown at 25°C, i.e., under conditions in which the growth rate of the ΔgraA strain is decreased, clearly higher persistence of the ΔgraA strain was observed compared to the wild-type and ΔgraAT(E80G) strains (Fig. 8A). Bacteria growing at 37°C were killed much more rapidly, and no significant differences between killing curves of analyzed strains were recorded (Fig. 8B). These data suggest that a GraT-caused growth rate reduction is important for the increased streptomycin tolerance of the ΔgraA strain.
FIG 8.
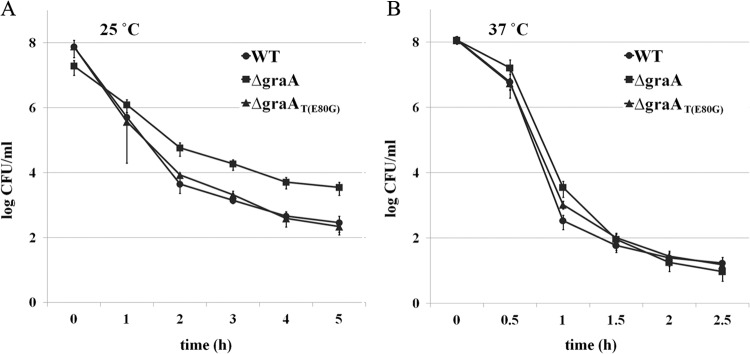
GraT increases bacterial tolerance to streptomycin-mediated killing. Exponentially growing cultures of P. putida PaW85 (WT) and its ΔgraA and ΔgraAT(E80G) derivatives were exposed to 300 μg/ml streptomycin either at 25°C for 5 h (A) or at 37°C for 2.5 h (B). The surviving cells were determined by plating and counting the CFU. Data (means with 95% confidence intervals) of at least four independent experiments are presented.
DISCUSSION
In this work, we identified the first TA system in P. putida, which was named GraTA, for the growth rate-affecting effect of the toxin GraT. The graTA system is homologous to the higBA system of Vibrio cholerae (41), and we show that it possesses several characteristic features of type II TA loci. First, GraT overexpression was toxic to cells in the absence of antitoxin GraA and resulted in severe growth inhibition (Fig. 2B). As the toxicity of GraT was inhibited by the substitution mutation E80G (Fig. 2C), the growth inhibition is clearly caused by the GraT protein. Second, chromosomally encoded GraA is an efficient antitoxin which can counteract the GraT-mediated growth inhibition even if GraT is ectopically overexpressed (Fig. 2B). Third, GraA directly associates with GraT (Fig. 3), suggesting that protein binding is the mechanism for toxin neutralization. Fourth, graT and graA form an operon and are cotranscribed in a single bicistronic mRNA. Fifth, graTA operon transcription is autorepressed by the antitoxin GraA (Fig. 4A).
Although the GraTA system possesses many features common to other TA loci, it is still unusual among TA pairs. While the antitoxin deletion is generally lethal for bacteria (41, 50), the deletion of the antitoxin gene graA not only was possible but was only modestly influential on the growth rate of P. putida. This shows that GraT is an unusually moderate growth inhibitor and even raises the question of whether it can actually be classified as a toxin. However, one should consider that, as with other TA toxins, the overexpression of GraT had a strong toxic effect on P. putida with graA deleted. At the same time, GraT seems not to be toxic to E. coli, as we never observed growth inhibition by different graT-containing plasmids in this bacterium (data not shown). This indicates that the activity of GraT might be host specific. Another interesting feature of GraT is that its effect depends on the growth conditions, as we observed significantly greater GraT-mediated growth rate suppression at 25°C and 20°C than at 30°C. Moreover, at 37°C, we did not detect a decrease in growth rate for P. putida with graA deleted. The temperature sensitivity of GraT raises the intriguing possibility that bacteria may benefit from GraT-mediated growth regulation during adaptation to lower temperatures. We tested this hypothesis in cocultivation experiments over 2 weeks, but as wild-type P. putida could not outcompete the ΔgraTA double mutant at either high or low temperatures (data not shown), the results did not support the possibility of the GraTA system influencing the fitness of bacteria at lower temperatures. Nevertheless, the temperature sensitivity of GraT deserves further study, especially considering that the temperature dependence of TA systems is not well documented. To our knowledge, there is only one recent study which reports a temperature-sensitive toxin of a type I system (56). The authors showed that the toxin's mRNA is less stable at 48°C, but the biological significance of the phenotype remained unclear.
Our data show that while ectopic overexpression of GraT is toxic to the ΔgraA and ΔgraTA strains, it is tolerated by wild-type P. putida (Fig. 2A), indicating that chromosomally encoded GraA can efficiently neutralize both the innate and the additionally expressed GraT. Such a high efficacy of the antitoxin would be difficult to achieve in the case of an unstable antitoxin. And indeed, we have seen indications of GraA's unusual stability, as His-tagged GraA was stable during the overexpression, lysate preparation, and protein purification processes, even though we did not use any protease inhibitors. Hence, the efficacy of GraA in counteracting artificially overproduced GraT possibly relies on its high stability.
It is common in type II TA systems that their operons are autorepressed by either the antitoxin or the toxin-antitoxin complex (52, 57, 58). In the latter case, the antitoxin is the DNA-binding moiety and the toxin acts as a corepressor if the ratio of toxin to antitoxin is low or as a derepressor if toxin is in excess (59, 60). Transcriptional coregulation of TA operons by toxin abundance is common among TA systems and is known as conditional cooperativity (61–63). However, TA operons can also be regulated by other mechanisms involving promoter repression by antitoxin alone or even activation by antitoxin (18, 57). Similarly to most TA systems, the graTA operon is regulated by autorepression (Fig. 4). We detected significantly higher repression of the graTA promoter in ΔgraT bacteria than in the wild type, which indicates that GraA antitoxin alone is an efficient repressor and the presence of the GraT causes partial derepression of the promoter activity. However, in vitro data show that both GraA alone and the GraA-GraT complex can bind to the promoter DNA, suggesting that the toxin GraT may also affect the promoter regulation. Still, we never observed that the protein complex could bind DNA more avidly than GraA alone, which shows that GraT cannot essentially influence the DNA-binding properties of GraA. Thus, our data do not support GraT-mediated corepression, and we conclude that the graTA operon is autorepressed by GraA only. Therefore, graTA regulation seems to resemble that of the mqsRA operon, as recent data demonstrate that the MqsR toxin is not a corepressor but rather alleviates MqsA-mediated repression (57).
The importance of chromosomal TA operons in bacterial physiology is highly debated, and different functions have been proposed for genomic TA systems, ranging from their participation in programmed cell death, persister formation, and stress tolerance to even considering them junk DNA and selfish modules (20, 22, 64). Still, accumulating evidence supports the idea that chromosomal TA systems are stress response loci which contribute to bacterial maintenance in unfavorable situations (4, 54, 65). Activation of the toxin leads to growth arrest, which facilitates bacterial survival under different stress conditions (66, 67). Our data also show that the GraT toxin can act as a stress-relieving factor, as it increased bacterial tolerance to several antibiotics. Given that antibiotics are more efficient against rapidly growing bacteria (53), the increased resistance of the ΔgraA strain is most obviously caused by its slower growth. This is supported by the finding that GraT(E80G), which is not able to decrease the growth rate of bacteria, is not able to increase antibiotic tolerance (Fig. 7 and 8). However, the observation that both GraT and GraT(E80G) can rescue the colR mutant from lysis indicates that the stress-relieving activity of GraT is more complicated and does not rely only on its ability to reduce growth rate. Furthermore, we also showed that GraT is not a universal stress-relieving factor, and its activity results in trade-offs between different types of resistance. While it increased resistance to streptomycin, kanamycin, ciprofloxacin, and mitomycin C, GraT decreased resistance to compounds such as tetracycline, paraquat, and NaCl (Fig. 7). Here, it is important to note that besides inhibiting the growth rate, GraT also compromised the barrier functions of membrane, as judged by the increased permeability of ΔgraA cells to propidium iodide (Fig. 5). This membrane defect is possibly responsible for the increased sensitivity of ΔgraA bacteria to tetracycline, paraquat, and NaCl. Thus, these data demonstrate that although GraT can in principle increase the tolerance of P. putida to particular stress factors, such stress protection is highly costly and results in harmful side effects. However, it should be emphasized that these data were obtained with the ΔgraA strain, and GraT most likely cannot exert such a severe effect in wild-type bacteria, as it is normally neutralized by GraA. Nevertheless, it would be worthwhile to find out if some stress conditions can lead to the destabilization of GraA and if the released GraT can have an impact on the survival of P. putida.
Bioinformatic analysis of the genomes in the Pseudomonas Genome Database (49) revealed that 30 out of 48 different Pseudomonas strains possess orthologs of graTA genes. Such a high occurrence of the GraTA system in the highly heterogeneous Pseudomonas genus indicates that this TA system should be beneficial for both human and plant pathogens like P. aeruginosa and P. syringae as well as plant-beneficial soil bacteria like P. fluorescens and P. putida. Interestingly, all P. aeruginosa genomes annotated in the Pseudomonas Genome Database possessed highly conserved graTA orthologs. In accordance with these findings, recent analysis of 46 clinical isolates of P. aeruginosa revealed that all of them contain graTA homologs and, furthermore, that all these operons are actively transcribed (68). Considering the rapid evolution of microbial pathogens (69), the maintenance of active GraTA modules indicates that they might contribute to the virulence of P. aeruginosa.
The activation of a TA system may be bactericidal (70, 71), but more commonly, it results in rapid growth arrest until favorable conditions return (41, 72, 73). Hence, in spite of the fact that TA systems were discovered as killing modules on plasmids, most chromosomal systems actually operate as components of a cellular regulatory network dedicated to optimizing the bacterial growth rate. Still, compared to other growth modulators, the participation of toxins in growth regulation seems to be rather strange, especially considering that most of the well-studied TA systems code for quite harmful proteins. Contrary to most toxins, the effect of GraT on bacterial growth is moderate, resembling that of the general cellular regulators. We suggest that GraT represents a new class of chromosomal toxins which have evolved to less toxic variants either to better suit the needs of bacterial regulatory network or just to lessen the harm they can cause to the host bacterium. We believe that further research on TA systems will discover more GraTA-like TA pairs in different bacteria.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Maia Kivisaar and Hanna Hõrak for critically reading the manuscript.
This work was supported by the grant 7829 from the Estonian Science Foundation and by Targeted Financing Project TLOMR0031 from the Estonian Ministry of Research and Education.
Footnotes
Published ahead of print 25 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00851-13.
REFERENCES
- 1.Fozo EM, Hemm MR, Storz G. 2008. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 72:579–589. 10.1128/MMBR.00025-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, Fineran PC, Luisi BF, Salmond GP. 2012. Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 40:6158–6173. 10.1093/nar/gks231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. 2011. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 39:5513–5525. 10.1093/nar/gkr131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerdes K, Christensen SK, Lobner-Olesen A. 2005. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3:371–382. 10.1038/nrmicro1147 [DOI] [PubMed] [Google Scholar]
- 5.Masuda H, Tan Q, Awano N, Wu KP, Inouye M. 2012. YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol. Microbiol. 84:979–989. 10.1111/j.1365-2958.2012.08068.x [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, Benedik MJ, Page R, Wood TK. 2012. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 8:855–861. 10.1038/nchembio.1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster CF, Bertram R. 2013. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340:73–85. 10.1111/1574-6968.12074 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Inouye M. 2011. RatA (YfjG), an Escherichia coli toxin, inhibits 70S ribosome association to block translation initiation. Mol. Microbiol. 79:1418–1429. 10.1111/j.1365-2958.2010.07506.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M, Zhang Y, Inouye M, Woychik NA. 2008. Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proc. Natl. Acad. Sci. U. S. A. 105:5885–5890. 10.1073/pnas.0711949105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, Brennan RG. 2009. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323:396–401. 10.1126/science.1163806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winther KS, Gerdes K. 2011. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U. S. A. 108:7403–7407. 10.1073/pnas.1019587108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M. 2003. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell 12:913–923. 10.1016/S1097-2765(03)00402-7 [DOI] [PubMed] [Google Scholar]
- 13.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. 2003. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112:131–140. 10.1016/S0092-8674(02)01248-5 [DOI] [PubMed] [Google Scholar]
- 14.Hurley JM, Woychik NA. 2009. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 284:18605–18613. 10.1074/jbc.M109.008763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen RB, Gerdes K. 1995. Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol. Microbiol. 17:205–210. 10.1111/j.1365-2958.1995.mmi_17020205.x [DOI] [PubMed] [Google Scholar]
- 16.Ogura T, Hiraga S. 1983. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. U. S. A. 80:4784–4788. 10.1073/pnas.80.15.4784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saavedra De Bast M, Mine N, Van Melderen L. 2008. Chromosomal toxin-antitoxin systems may act as antiaddiction modules. J. Bacteriol. 190:4603–4609. 10.1128/JB.00357-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nariya H, Inouye M. 2008. MazF, an mRNA interferase, mediates programmed cell death during multicellular Myxococcus development. Cell 132:55–66. 10.1016/j.cell.2007.11.044 [DOI] [PubMed] [Google Scholar]
- 19.Amitai S, Kolodkin-Gal I, Hananya-Meltabashi M, Sacher A, Engelberg-Kulka H. 2009. Escherichia coli MazF leads to the simultaneous selective synthesis of both “death proteins” and “survival proteins.” PLoS Genet. 5:e1000390. 10.1371/journal.pgen.1000390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerdes K, Maisonneuve E. 2012. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 66:103–123. 10.1146/annurev-micro-092611-150159 [DOI] [PubMed] [Google Scholar]
- 21.Hayes CS, Low DA. 2009. Signals of growth regulation in bacteria. Curr. Opin. Microbiol. 12:667–673. 10.1016/j.mib.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaguchi Y, Inouye M. 2011. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 9:779–790. 10.1038/nrmicro2651 [DOI] [PubMed] [Google Scholar]
- 23.Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. 2001. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. U. S. A. 98:14328–14333. 10.1073/pnas.251327898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen-Dalsgaard M, Jorgensen MG, Gerdes K. 2010. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 75:333–348. 10.1111/j.1365-2958.2009.06969.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6:53. 10.1186/1471-2180-6-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandey DP, Gerdes K. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33:966–976. 10.1093/nar/gki201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silby MW, Winstanley C, Godfrey SA, Levy SB, Jackson RW. 2011. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35:652–680. 10.1111/j.1574-6976.2011.00269.x [DOI] [PubMed] [Google Scholar]
- 28.Ramos JL, Krell T, Daniels C, Segura A, Duque E. 2009. Responses of Pseudomonas to small toxic molecules by a mosaic of domains. Curr. Opin. Microbiol. 12:215–220. 10.1016/j.mib.2009.02.001 [DOI] [PubMed] [Google Scholar]
- 29.Dos Santos VA, Heim S, Moore ER, Stratz M, Timmis KN. 2004. Insights into the genomic basis of niche specificity of Pseudomonas putida KT2440. Environ. Microbiol. 6:1264–1286. 10.1111/j.1462-2920.2004.00734.x [DOI] [PubMed] [Google Scholar]
- 30.Makarova KS, Wolf YI, Koonin EV. 2009. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 4:19. 10.1186/1745-6150-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Putrinš M, Ainelo A, Ilves H, Hõrak R. 2011. The ColRS system is essential for the hunger response of glucose-growing Pseudomonas putida. BMC Microbiol. 11:170. 10.1186/1471-2180-11-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Putrinš M, Ilves H, Kivisaar M, Hõrak R. 2008. ColRS two-component system prevents lysis of subpopulation of glucose-grown Pseudomonas putida. Environ. Microbiol. 10:2886–2893. 10.1111/j.1462-2920.2008.01705.x [DOI] [PubMed] [Google Scholar]
- 33.Kivistik PA, Putrinš M, Püvi K, Ilves H, Kivisaar M, Hõrak R. 2006. The ColRS two-component system regulates membrane functions and protects Pseudomonas putida against phenol. J. Bacteriol. 188:8109–8117. 10.1128/JB.01262-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Putrinš M, Ilves H, Lilje L, Kivisaar M, Hõrak R. 2010. The impact of ColRS two-component system and TtgABC efflux pump on phenol tolerance of Pseudomonas putida becomes evident only in growing bacteria. BMC Microbiol. 10:110. 10.1186/1471-2180-10-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu N, Zhao B. 2007. Key genes involved in heavy-metal resistance in Pseudomonas putida CD2. FEMS Microbiol. Lett. 267:17–22. 10.1111/j.1574-6968.2006.00505.x [DOI] [PubMed] [Google Scholar]
- 36.Dekkers LC, Bloemendaal CJ, de Weger LA, Wijffelman CA, Spaink HP, Lugtenberg BJ. 1998. A two-component system plays an important role in the root-colonizing ability of Pseudomonas fluorescens strain WCS365. Mol. Plant Microbe Interact. 11:45–56. 10.1094/MPMI.1998.11.1.45 [DOI] [PubMed] [Google Scholar]
- 37.de Weert S, Dekkers LC, Bitter W, Tuinman S, Wijfjes AH, van Boxtel R, Lugtenberg BJ. 2006. The two-component colR/S system of Pseudomonas fluorescens WCS365 plays a role in rhizosphere competence through maintaining the structure and function of the outer membrane. FEMS Microbiol. Ecol. 58:205–213. 10.1111/j.1574-6941.2006.00158.x [DOI] [PubMed] [Google Scholar]
- 38.Garvis S, Munder A, Ball G, de Bentzmann S, Wiehlmann L, Ewbank JJ, Tümmler B, Filloux A. 2009. Caenorhabditis elegans semi-automated liquid screen reveals a specialized role for the chemotaxis gene cheB2 in Pseudomonas aeruginosa virulence. PLoS Pathog. 5:e1000540. 10.1371/journal.ppat.1000540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramoni S, Pandey A, Vishnupriya MR, Patel HK, Sonti RV. 2012. The ColRS system of Xanthomonas oryzae pv. oryzae is required for virulence and growth in iron-limiting conditions. Mol. Plant Pathol. 13:690–703. 10.1111/j.1364-3703.2011.00777.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan Q, Wang N. 2011. The ColR/ColS two-component system plays multiple roles in the pathogenicity of the citrus canker pathogen Xanthomonas citri subsp. citri. J. Bacteriol. 193:1590–1599. 10.1128/JB.01415-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budde PP, Davis BM, Yuan J, Waldor MK. 2007. Characterization of a higBA toxin-antitoxin locus in Vibrio cholerae. J. Bacteriol. 189:491–500. 10.1128/JB.00909-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bayley SA, Duggleby CJ, Worsey MJ, Williams PA, Hardy KG, Broda P. 1977. Two modes of loss of the Tol function from Pseudomonas putida mt-2. Mol. Gen. Genet. 154:203–204. 10.1007/BF00330838 [DOI] [PubMed] [Google Scholar]
- 43.Regenhardt D, Heuer H, Heim S, Fernandez DU, Strömpl C, Moore ER, Timmis KN. 2002. Pedigree and taxonomic credentials of Pseudomonas putida strain KT2440. Environ. Microbiol. 4:912–915. 10.1046/j.1462-2920.2002.00368.x [DOI] [PubMed] [Google Scholar]
- 44.Adams MH. 1959. Bacteriophages, p 445–447, Interscience Publishers Inc., New York, NY [Google Scholar]
- 45.Sharma RC, Schimke RT. 1996. Preparation of electrocompetent E. coli using salt-free growth medium. Biotechniques 20:42–44 [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Garcia E, de Lorenzo V. 2011. Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ. Microbiol. 13:2702–2716. 10.1111/j.1462-2920.2011.02538.x [DOI] [PubMed] [Google Scholar]
- 47.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 48.Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37:e45. 10.1093/nar/gkp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winsor GL, Van Rossum T, Lo R, Khaira B, Whiteside MD, Hancock RE, Brinkman FS. 2009. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res. 37:D483–D488. 10.1093/nar/gkn861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fivian-Hughes AS, Davis EO. 2010. Analyzing the regulatory role of the HigA antitoxin within Mycobacterium tuberculosis. J. Bacteriol. 192:4348–4356. 10.1128/JB.00454-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamaguchi Y, Park JH, Inouye M. 2011. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45:61–79. 10.1146/annurev-genet-110410-132412 [DOI] [PubMed] [Google Scholar]
- 52.Kedzierska B, Lian LY, Hayes F. 2007. Toxin-antitoxin regulation: bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res. 35:325–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tuomanen E, Cozens R, Tosch W, Zak O, Tomasz A. 1986. The rate of killing of Escherichia coli by beta-lactam antibiotics is strictly proportional to the rate of bacterial growth. J. Gen. Microbiol. 132:1297–1304 [DOI] [PubMed] [Google Scholar]
- 54.Dörr T, Vulic M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8:e1000317. 10.1371/journal.pbio.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U. S. A. 108:13206–13211. 10.1073/pnas.1100186108 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Jahn N, Preis H, Wiedemann C, Brantl S. 2012. BsrG/SR4 from Bacillus subtilis—the first temperature-dependent type I toxin-antitoxin system. Mol. Microbiol. 83:579–598. 10.1111/j.1365-2958.2011.07952.x [DOI] [PubMed] [Google Scholar]
- 57.Brown BL, Lord DM, Grigoriu S, Peti W, Page R. 2013. The E. coli toxin MqsR destabilizes the transcriptional repression complex formed between the antitoxin MqsA and the mqsRA operon promoter. J. Biol. Chem. 288:1286–1294. 10.1074/jbc.M112.421008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li GY, Zhang Y, Inouye M, Ikura M. 2008. Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. J. Mol. Biol. 380:107–119. 10.1016/j.jmb.2008.04.039 [DOI] [PubMed] [Google Scholar]
- 59.Winther KS, Gerdes K. 2012. Regulation of enteric vapBC transcription: induction by VapC toxin dimer-breaking. Nucleic Acids Res. 40:4347–4357. 10.1093/nar/gks029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Afif H, Allali N, Couturier M, Van Melderen L. 2001. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 41:73–82. 10.1046/j.1365-2958.2001.02492.x [DOI] [PubMed] [Google Scholar]
- 61.Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, Magnuson RD, Charlier D, van Nuland NA, Loris R. 2010. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142:101–111. 10.1016/j.cell.2010.05.039 [DOI] [PubMed] [Google Scholar]
- 62.Overgaard M, Borch J, Jorgensen MG, Gerdes K. 2008. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 69:841–857. 10.1111/j.1365-2958.2008.06313.x [DOI] [PubMed] [Google Scholar]
- 63.Boggild A, Sofos N, Andersen KR, Feddersen A, Easter AD, Passmore LA, Brodersen DE. 2012. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 20:1641–1648. 10.1016/j.str.2012.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Melderen L, Saavedra De Bast M. 2009. Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet. 5:e1000437. 10.1371/journal.pgen.1000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang X, Kim Y, Hong SH, Ma Q, Brown BL, Pu M, Tarone AM, Benedik MJ, Peti W, Page R, Wood TK. 2011. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 7:359–366. 10.1038/nchembio.560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Wood TK. 2011. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77:5577–5583. 10.1128/AEM.05068-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Norton JP, Mulvey MA. 2012. Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLoS Pathog. 8:e1002954. 10.1371/journal.ppat.1002954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams JJ, Halvorsen EM, Dwyer EM, DiFazio RM, Hergenrother PJ. 2011. Toxin-antitoxin (TA) systems are prevalent and transcribed in clinical isolates of Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus. FEMS Microbiol. Lett. 322:41–50. 10.1111/j.1574-6968.2011.02330.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pallen MJ, Wren BW. 2007. Bacterial pathogenomics. Nature 449:835–842. 10.1038/nature06248 [DOI] [PubMed] [Google Scholar]
- 70.Hazan R, Sat B, Engelberg-Kulka H. 2004. Escherichia coli mazEF-mediated cell death is triggered by various stressful conditions. J. Bacteriol. 186:3663–3669. 10.1128/JB.186.11.3663-3669.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kolodkin-Gal I, Hazan R, Gaathon A, Carmeli S, Engelberg-Kulka H. 2007. A linear pentapeptide is a quorum-sensing factor required for mazEF-mediated cell death in Escherichia coli. Science 318:652–655. 10.1126/science.1147248 [DOI] [PubMed] [Google Scholar]
- 72.Pedersen K, Christensen SK, Gerdes K. 2002. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 45:501–510. 10.1046/j.1365-2958.2002.03027.x [DOI] [PubMed] [Google Scholar]
- 73.Fu Z, Tamber S, Memmi G, Donegan NP, Cheung AL. 2009. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J. Bacteriol. 191:2051–2059. 10.1128/JB.00907-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanahan D, Meselson M. 1983. Plasmid screening at high colony density. Methods Enzymol. 100:333–342. 10.1016/0076-6879(83)00066-X [DOI] [PubMed] [Google Scholar]
- 75.Herrero M, de Lorenzo V, Timmis KN. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 172:6557–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130. 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- 77.Hõrak R, Ilves H, Pruunsild P, Kuljus M, Kivisaar M. 2004. The ColR-ColS two-component signal transduction system is involved in regulation of Tn4652 transposition in Pseudomonas putida under starvation conditions. Mol. Microbiol. 54:795–807. 10.1111/j.1365-2958.2004.04311.x [DOI] [PubMed] [Google Scholar]
- 78.Wong SM, Mekalanos JJ. 2000. Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 97:10191–10196. 10.1073/pnas.97.18.10191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U. S. A. 76:1648–1652. 10.1073/pnas.76.4.1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bagdasarian MM, Amann E, Lurz R, Ruckert B, Bagdasarian M. 1983. Activity of the hybrid trp-lac (tac) promoter of Escherichia coli in Pseudomonas putida. Construction of broad-host-range, controlled-expression vectors. Gene 26:273–282 [DOI] [PubMed] [Google Scholar]
- 81.Ojangu EL, Tover A, Teras R, Kivisaar M. 2000. Effects of combination of different −10 hexamers and downstream sequences on stationary-phase-specific sigma factor sigma(S)-dependent transcription in Pseudomonas putida. J. Bacteriol. 182:6707–6713. 10.1128/JB.182.23.6707-6713.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silva-Rocha R, Martinez-Garcia E, Calles B, Chavarria M, Arce-Rodriguez A, de Las Heras A, Paez-Espino AD, Durante-Rodriguez G, Kim J, Nikel PI, Platero R, de Lorenzo V. 2013. The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 41:D666–D675. 10.1093/nar/gks1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.