

Abstract
Escherichia coli AraC is a well-described transcription activator of genes involved in arabinose metabolism. Using complementary genomic approaches, chromatin immunoprecipitation (ChIP)-chip, and transcription profiling, we identify direct regulatory targets of AraC, including five novel target genes: ytfQ, ydeN, ydeM, ygeA, and polB. Strikingly, only ytfQ has an established connection to arabinose metabolism, suggesting that AraC has a broader function than previously described. We demonstrate arabinose-dependent repression of ydeNM by AraC, in contrast to the well-described arabinose-dependent activation of other target genes. We also demonstrate unexpected read-through of transcription at the Rho-independent terminators downstream of araD and araE, leading to significant increases in the expression of polB and ygeA, respectively. AraC is highly conserved in the related species Salmonella enterica. We use ChIP sequencing (ChIP-seq) and RNA sequencing (RNA-seq) to map the AraC regulon in S. enterica. A comparison of the E. coli and S. enterica AraC regulons, coupled with a bioinformatic analysis of other related species, reveals a conserved regulatory network across the family Enterobacteriaceae comprised of 10 genes associated with arabinose transport and metabolism.
INTRODUCTION
Escherichia coli AraC is the founding member of a large family of transcription factors (TFs) found across a wide range of bacterial species (1). AraC was first identified in 1959 by virtue of the requirement of araC for the metabolism of l-arabinose (2) and is the first-described positive regulator of transcription (3, 4). E. coli AraC activates transcription of the araBAD, araFGH, araE, and araJ transcripts in the presence of its inducer, l-arabinose (5). AraC binds DNA as a dimer. Dimerization occurs between adjacent DNA sites when AraC binds arabinose. In the absence of arabinose, AraC represses transcription of araBAD and araC by forming a repression loop mediated by dimerization of distally bound AraC monomers (5, 6).
Chromatin immunoprecipitation (ChIP)-chip and ChIP sequencing (ChIP-seq) are widely used techniques for genome-wide mapping of protein-DNA interactions in vivo. Surprisingly, these methods have been used only sparingly to study bacterial systems (7). ChIP-chip and ChIP-seq studies of bacterial TFs have identified novel regulatory interactions, even for well-studied proteins (7, 8). Furthermore, TF binding sites have been identified in unexpected locations, such as inside genes (9), upstream of genes that are not detectably regulated by the TF, and in genomic regions that lack a canonical DNA sequence motif for the TF (7, 10).
Transcription profiling uses microarrays or RNA-seq to determine differences in genome-wide RNA levels between two growth conditions and/or strains (11). This approach is often used to identify regulatory targets of TFs by comparing RNA levels in wild-type cells and cells lacking the TF. In contrast to ChIP methods, transcription profiling identifies all genes regulated by a TF and the level and direction of regulation. However, transcription profiling identifies both direct and indirect regulatory targets. By combining ChIP methods and transcription profiling, it is possible to identify all direct regulatory targets of a TF for a given growth condition (11). We refer to the set of direct regulatory targets as a regulon.
Many TFs, including AraC, are highly conserved between E. coli and other species in the family Enterobacteriaceae. This suggests that DNA-binding specificity is the same for TF homologues across the family, and that TF regulon gene function is likely to be conserved. Most studies of regulon evolution have focused simply on whether regulon members (i.e., target genes) have homologues in related species. In contrast, very few studies have determined whether conserved genes are regulated by the TF (12). The best-studied TF in this regard is PhoP, a two-component regulator that is conserved across the family Enterobacteriaceae. Regulation of only three PhoP target genes is conserved across the family, although in any given species there are many more than three PhoP-regulated genes (13). Most PhoP target genes in any given species lack homologues in other species or the genes are conserved but are only regulated by PhoP in one or two species. The latter phenomenon is known as network rewiring (12). Most of the known AraC regulon members in E. coli are conserved across other Enterobacteriaceae members, but the extent of rewiring is unknown. Given that much of our understanding of regulon evolution is based on studies of a single TF, PhoP, it is important to experimentally compare regulons for additional TFs between related species (12).
Genome-scale approaches have not been previously used to identify AraC-regulated genes. We hypothesized that despite the extensive prior work on the AraC regulon, there are likely to be previously undescribed AraC-regulated genes and novel modes of regulation by AraC. In this work, we use a combination of ChIP-chip and transcription profiling with microarrays to identify all binding sites and all direct regulatory targets of E. coli AraC. In addition to identifying a novel mechanism of repression by AraC, our genomic approach reveals unexpected read-through of transcription terminators in AraC-activated transcripts and AraC-regulated genes with no connection to arabinose metabolism. We also identify all binding sites and all direct regulatory targets of AraC in the related species Salmonella enterica using a combination of ChIP-seq and RNA-seq. These targets include two novel, cotranscribed, AraC-activated genes (STM14_0178 and STM14_0177) that encode a putative arabinoside transporter and an α-l-arabinofuranosidase II precursor. We rename these genes araT and araU. Together with a bioinformatic analysis of other Enterobacteriaceae species, these data identify a conserved AraC regulon that includes 7 previously described AraC-regulated genes (araB, araA, araD, araE, araF, araG, and araH) as well as three novel targets identified in this work (ytfQ, araT, and araU). Moreover, our data indicate only limited rewiring of the AraC regulatory network in the Enterobacteriaceae.
MATERIALS AND METHODS
Strains and plasmids.
Bacterial strains and plasmids used in this work are listed in Table 1. Cells were grown in LB (1% NaCl, 1% tryptone, 0.5% yeast extract). All oligonucleotides used in this work are listed in Table S1 in the supplemental material. AMD054 was constructed using λ Red recombineering as described previously (14). The PCR product used for recombineering was generated with oligonucleotides JW464 and JW465, using pKD13 (14) as the template. SAC003 (MG1655 araC-TAP) was constructed by P1 transduction of the kanamycin resistance (Kanr) gene-linked araC-TAP from DY330 araC-TAP (15). The Kanr gene was removed using pCP20 as described previously (16). SAC001 (MG1655 ΔaraC) and AMD115 were constructed by P1 transduction of the Kanr-linked ΔaraC from BW25113 ΔaraC (17) into MG1655 and AMD054, respectively. The Kanr gene was removed using pCP20 as described previously (16). Note that SAC001 and AMD115 also contain the Δ(araD-araB)567 mutation that lacks the araBAD operon. AMD187 (E. coli MG1655 araC-3×FLAG), JTW010 (E. coli MG1655 with ytfQ AraC site mutation, araC-3×FLAG), and CB005 (S. enterica serovar Typhimurium 14028s araC-3×FLAG) were constructed using the FRUIT recombineering system (18). The PCR product used to generate the initial tagged strains was made using oligonucleotides JW1141 and JW1142 for E. coli and JW2895 and JW2901 for S. enterica, with pAMD135 as the template. For construction of JTW010, the thyA-containing PCR product for insertion upstream of ytfQ was amplified with oligonucleotides JW601 and JW602 using pAMD001 as the template. The PCR product for replacing thyA with mutated sequence was constructed using SOEing PCR (19) with oligonucleotides JW599, JW600, JW603, and JW604, using a colony of MG1655 as a template.
TABLE 1.
List of strains and plasmids
| Strain or plasmid | Genotype/description | Source |
|---|---|---|
| Escherichia coli | ||
| MG1655 | F− λ− ΔilvG rfb-50 rph-1 | 55 |
| SAC001 | MG1655 ΔaraC Δ(araD-araB)567 | This work |
| AMD054 | MG1655 ΔlacZ | This work |
| AMD115 | MG1655 ΔlacZ ΔaraC Δ(araD-araB)567 | This work |
| DY330 araC-TAP | W3110 ΔlacU169 gal490 λcI857 Δ(cro-bioA) araC-TAP::Kanr | 15 |
| SAC003 | MG1655 araC-TAP | This work |
| AMD187 | MG1655 araC-3× FLAG | This work |
| BW25113 | F− Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514 | 17 |
| BW25113 ΔydeN | F− Δ(araD-araB)567 lacZ4787(del)::rrnB-3 ΔLAM rph-1 Δ(rhaD-rhaB)568 hsdR514 ΔydeN::Kanr | 17 |
| BW25113 ΔydeM | F− Δ(araD-araB)567 lacZ4787(del)::rrnB-3 ΔLAM rph-1 Δ(rhaD-rhaB)568 hsdR514 ΔydeM::Kanr | 17 |
| BW25113 ΔaraC | F− Δ(araD-araB)567 lacZ4787(del)::rrnB-3 ΔLAM rph-1 Δ(rhaD-rhaB)568 hsdR514 ΔaraC::Kanr | 17 |
| JTW010 | MG1655 with ytfQ AraC binding site mutation, araC-3× FLAG | This work |
| S. enterica subsp. enterica serovar Typhimurium | ||
| 14028s | Wild type | 56 |
| CB005 | 14028s araC-3× FLAG | This work |
| AMD485 | 14028s ΔaraC::thyA | This work |
| Plasmids | ||
| pAMD-BA-lacZ | Single-copy lacZ expression vector, encodes chloramphenicol resistance | This work |
| pKD46 | Encodes λ recombinase system | 14 |
| pCP20 | Encodes Flp recombinase | 16 |
| pKD13 | Recombineering template vector | 14 |
| pAMD001 | FRUIT template vector | 18 |
| pAMD135 | FRUIT FLAG-tagging template vector | 18 |
| pAMD086 | pAMD-BA-lacZ with araE upstream sequence | This work |
| pAMD007 | pAMD-BA-lacZ with ytfQ upstream sequence | This work |
| pAMD124 | pAMD-BA-lacZ with ydeN upstream sequence from −371 to +1 (relative to transcription start site) | This work |
| pAMD132 | pAMD-BA-lacZ with ydeN upstream sequence from −371 to +14 (relative to transcription start site) | This work |
| pJTW064 | pAMD-BA-lacZ with constitutive promoter | This work |
| pJTW055 | pJTW064 with the araE terminator | This work |
| pJTW060 | pJTW064 with the ahpF terminator | This work |
| pJTW062 | pJTW064 with the tppB terminator | This work |
| pJTW061 | pJTW064 with the mutated tppB terminator | This work |
All lacZ reporter gene fusions were constructed in plasmid pAMD-BA-lacZ using the oligonucleotides listed in Table S1 in the supplemental material. PCR products were cloned as SphI-HindIII-digested fragments. pAMD-BA-lacZ has been described previously (20), but its construction has not been described in detail. pAMD-BA-lacZ is a derivative of pBAC-BA-lacZ (Addgene plasmid 13423) in which the NotI-HindIII fragment has been replaced with a PCR product (cut with NotI and HindIII) containing an intrinsic terminator from E. coli rrfB and additional restriction sites (BamHI, XhoI, and SphI). This PCR product was generated using oligonucleotides JW659 and JW660, with E. coli genomic DNA as the template (colony PCR). lacZ in this plasmid does not have a start codon or Shine-Dalgarno sequence, so fusions must be made translationally, as is the case for pAMD086 and pAMD007, or cloned fragments must include a Shine-Dalgarno sequence and start codon, as is the case (AGAAGGAGATATACATATG) for pAMD124 and pAMD132. Oligonucleotides used to generate PCR products for cloning of lacZ fusions for regions upstream of araE, ytfQ, and ydeN were JW679 and JW680 (araE), JW675 and JW678 (ytfQ), JW1438 and JW2391 (ydeN, −371 to +1), and JW1438 and JW1635 (ydeN, −371 to +14). The sequences of ytfQ and ydeN upstream sequences, indicating the pieces cloned into the lacZ fusion plasmid, are shown in Fig. S1 and S2, respectively, in the supplemental material. lacZ fusion plasmids to address transcription termination (pJTW064, pJTW055, pJTW060, pJTW062, and pJTW061) were cloned using SOEing PCR (19). A constitutive promoter was amplified from pAMD001 (18) using oligonucleotides JW3415 and JW3379. These were joined using SOEing PCR with PCR products amplified with oligonucleotides JW3381 and JW3416 (araE terminator), JW3476 and JW3478 (tppB terminator), or JW3424 and JW3425 (ahpF terminator). Final PCR products were cloned into pAMD-BA-lacZ using the In-Fusion method (Clontech). The mutant tppB terminator construct was isolated serendipitously as a result of a mutation introduced during the cloning of the wild-type construct.
Analysis of binding site conservation.
Sequences surrounding AraC binding sites upstream of E. coli araB, araF, araE, araJ, and ytfQ and within dcp (30 bp upstream sequence and 30 bp downstream sequence in addition to the 19-mer AraC site) were individually aligned with equivalent regions (i.e., the sequence 500 bp upstream of the homologous gene, or for the site within E. coli dcp, the entire homologous gene; for S. enterica araT, sequence was taken from −500 to +100 with respect to the gene start, since these genes may be misannotated) from S. enterica, Citrobacter rodentium ICC168, Enterobacter sp. strain 638, Klebsiella pneumoniae 342, and Cronobacter sakazakii ES15 using ClustalW (21). Similarly, the AraC site upstream of S. enterica araT was aligned with homologues from the same list of species. The number of matches to each position of each AraC site was determined, and the fraction of all species with a match to the reference sequence at each position was calculated. For each AraC binding site, the multispecies collection of aligned sites was used to compute the information content of each position (22) to generate conservation profiles.
β-Galactosidase assays.
Two to 3 ml cells was grown in LB or LB plus 0.2% arabinose at 37°C to an optical density at 600 nm (OD600) of 0.8 to 1.0, and the OD600 was recorded. Eight hundred μl cells was pelleted at full speed in a microcentrifuge for 1 min (80 μl was used for strongly active fusions, and this was corrected for at the final calculation step). Cell pellets were resuspended in 800 μl Z buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 0.001 M MgSO4) plus 50 mM β-mercaptoethanol (added fresh). Twenty μl chloroform and 10 μl 0.1% SDS was added to the cells, followed by vortexing for 5 s. Assays were started by addition of 160 μl o-nitrophenyl-β-d-galactopyranoside (ONPG; 4 mg/ml in distilled H2O) and stopped by addition of 400 μl 1 M Na2CO3 upon development of an appropriate yellow color. The reaction time was noted. Samples were centrifuged at full speed in a microcentrifuge to pellet the chloroform. The OD420 of the supernatant was recorded. Arbitrary assay units were calculated as 1,000 × [A420/(A600)(total time)].
RNA purification.
RNA was purified from cells using a modified version of the hot phenol method that has been described previously (11). Cells were grown in LB or LB plus 0.2% arabinose at 37°C to an OD600 of 0.6 to 0.8. One ml cells was mixed with 400 μl ice-cold 95% ethanol and 5% phenol-chloroform-isoamyl alcohol (25:24:21 mix). Cells were pelleted in a microcentrifuge for 1 min at full speed and washed once with Tris-buffered saline. Cell pellets were resuspended in 400 μl RNA lysis buffer (2% SDS, 4 mM EDTA) and boiled for 3 min. Four hundred μl acid phenol-chloroform-isoamyl alcohol mix (pH 4.3) was added and incubated at 65°C for 6 min and on ice for 5 min. Samples were centrifuged, and the aqueous layer was extracted once more with phenol-chloroform-isoamyl alcohol mix (pH 4.3). RNA was precipitated with 1 ml 100% ethanol and 40 μl 3 M sodium acetate. RNA was pelleted in a microcentrifuge for 10 min at full speed and washed once with room temperature 75% ethanol. RNA pellets were air dried and resuspended in water and treated with 10 U of DNase I (NEB) in 500 μl for 1 h at 37°C. RNA was then phenol extracted and ethanol precipitated as described above.
Transcription profiling using microarrays.
RNA was purified from MG1655 (wild-type) or SAC001 (ΔaraC) cells grown in LB with or without 0.2% arabinose at 37°C. cDNA synthesis, labeling, hybridization to Affymetrix GeneChip E. coli Genome 2.0 microarrays, washing, and scanning were performed according to the manufacturer's (Affymetrix) instructions. Triplicate data sets for each strain/condition pair were analyzed using GeneSpring software (Agilent) to calculate fold changes and P values. Only genes with >4-fold changes and P values of <0.1 are shown in Tables 1 and 2.
TABLE 2.
Arabinose-responsive genes in E. coli
| Genea | Fold change (log2) in mRNA level for: |
|
|---|---|---|
| Arabinosec | ΔaraCd | |
| araA | 9.6 | 7.6 |
| araB | 9.4 | 7.3 |
| araD | 7.8 | 7.0 |
| araE | 5.8 | 6.1 |
| araG | 4.8 | 4.9 |
| araJ | 4.1 | 4.7 |
| araH | 4.6 | 4.6 |
| araF | 4.8 | 4.3 |
| araHb | 4.8 | 4.0 |
| ygeA | 3.5 | 3.4 |
| isrB | 2.9 | 2.9 |
| cstA | −2.3 | −2.0 |
| melA | −2.2 | −2.0 |
| aldB | −2.5 | −2.1 |
| fucI | −2.0 | −2.2 |
| tdcF | −2.0 | −2.2 |
| tdcA | −2.1 | −2.2 |
| xylF | −2.6 | −2.5 |
| gudX | −2.6 | −2.5 |
| tdcE | −2.6 | −2.6 |
| garR | −2.7 | −2.8 |
| tdcC | −2.7 | −2.9 |
| tdcB | −3.3 | −3.1 |
| ydeN | −3.1 | −3.1 |
| tdcD | −2.4 | −3.1 |
| yjhA | −3.1 | −3.1 |
| tnaL | −3.5 | −3.2 |
| garD | −3.3 | −3.3 |
| garL | −3.9 | −3.4 |
| tnaA | −3.0 | −3.7 |
| garP | −3.2 | −3.8 |
| malG | −3.0 | −3.9 |
| malF | −3.8 | −4.1 |
| tnaB | −3.9 | −4.2 |
| gudP | −4.2 | −4.3 |
| malE | −3.6 | −4.5 |
| malM | −4.1 | −4.6 |
| malK | −4.5 | −5.1 |
| lamB | −4.6 | −5.2 |
Arabinose-responsive genes in E. coli were defined by a >4-fold change (significant difference) in growth with or without arabinose in wild-type (MG1655) cells and a >4-fold significant difference between wild-type (MG1655) and ΔaraC (SAC001) cells in the presence of arabinose. Direct regulatory targets of AraC are indicated by boldface. Previously described regulatory targets of AraC are shaded in gray.
araH is represented twice on the microarray.
Fold change in mRNA level for wild-type cells grown with or without arabinose.
Fold difference in mRNA level between wild-type and ΔaraC cells grown in the presence of arabinose.
5′ RACE.
RNA was purified from MG1655 cells grown in LB. 5′ Rapid amplification of cDNA ends (RACE) was performed using the FirstChoice RLM-RACE kit (Ambion) according to the manufacturer's instructions. Oligonucleotides JW1485 and JW1486, specific to ydeN, were used in conjunction with oligonucleotides provided by the manufacturer (GCTGATGGCGATGAATGAACACTG and CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG, respectively).
Northern blotting.
Ten μg RNA was run per lane on a 1% agarose, 1× 3-(N-morpholino)propanesulfonic acid (MOPS), 2% formaldehyde gel at 70 V for 4 h. RNA was blotted by capillary action onto Magna nylon transfer membrane (GE Water & Process Technologies) and fixed by UV irradiation. Membranes were incubated with ∼105 cpm PCR-generated double-stranded DNA (dsDNA) probe overnight in hybridization buffer (0.525 M Na2HPO4, 7% SDS, 1 mM EDTA, 10 mg/ml bovine serum albumin [BSA]) and washed twice with wash buffer 1 (40 mM Na2HPO4, 5% SDS, 1 mM EDTA), wash buffer 2 (40 mM Na2HPO4, 1% SDS, 1 mM EDTA), and wash buffer 3 (0.2% SDS, 0.2× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]) at 55°C (23). Blots were visualized by phosphorimaging. Oligonucleotides used to generate PCR products for probe labeling were JW243 and JW1399 for araE and JW2387 and JW2388 for ygeA.
RNA-seq.
RNA was purified from 1 ml cells grown in LB with or without 0.2% arabinose at 37°C to an OD600 of 0.6 to 0.8. Duplicate samples were prepared from independent biological replicates for each condition/strain. rRNA was removed using the RiboZero kit (Epicentre). Strand-specific DNA libraries for Illumina sequencing were prepared using the ScriptSeq 2.0 kit (Epicentre). Sequencing was performed using an Illumina HiSeq instrument (University at Buffalo Next Generation Sequencing Core Facility). Sequences were aligned to the 14028s genome using the CLC Genomics Workbench, and differences in expression between conditions/strains were determined using the Pathogen Portal RNA-seq Analysis Pipeline (24) that includes Bowtie (version 2.02; for aligning reads to reference genomes) (25), Cufflinks (version 2.02; for transcript mapping), and CuffDiff (for comparing expression of transcripts between samples) (26) with default settings.
Reverse transcription-PCR (RT-PCR).
To assess terminator read-through downstream of araE and araD, RNA was purified from MG1655 cells grown in LB plus 0.2% arabinose. RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen) with 100 ng random hexamer according to the manufacturer's instructions. A control reaction omitted the reverse transcriptase. One-twentieth of the cDNA (or negative control) was used as a template in a PCR with appropriate primers (see Table S1 in the supplemental material). Oligonucleotides used for PCR were JW435 and JW436 for araE-ygeA and JW1366 and JW1367 for araD-polB.
ChIP, ChIP-chip, and ChIP-seq.
ChIP methods are presented in the supplemental material.
Accession numbers.
Microarray and sequencing data sets are available in the supplemental material (E. coli ChIP-chip) or through the EBI/EMBL ArrayExpress repository under the following accession numbers: E. coli transcription profiling, E-MTAB-1916; S. Typhimurium ChIP-seq, E-MTAB-1915; S. Typhimurium RNA-seq, E-MTAB-1901. The Agilent microarray design used for E. coli ChIP-chip is available through ArrayExpress under accession number A-MEXP-2346.
RESULTS
Genome-wide mapping of AraC binding sites in E. coli.
E. coli AraC-regulated genes have been identified previously through a variety of genetic approaches (3, 27–29). Here, we used two complementary genomic approaches to comprehensively identify members of the AraC regulon. First, we mapped the genome-wide binding of TAP (tandem affinity purification)-tagged AraC (tagged at its native locus in an unmarked strain) using chromatin immunoprecipitation (ChIP) coupled with custom-designed oligonucleotide microarrays (ChIP-chip; see Table S2 in the supplemental material). We identified seven putative target loci for AraC: upstream of araB-araC, araE, araF, araJ, ytfQ, ydeN, and within dcp. These included all previously described AraC target loci, with the exception of xylA, which we believe is not a direct target of AraC under these growth conditions (see below). AraC association has not been previously described for ytfQ, ydeN, or dcp.
We validated the ChIP-chip data using ChIP coupled with quantitative real-time PCR (ChIP/qPCR). To demonstrate that ChIP signal was not an artifact of the TAP tag, we constructed an unmarked derivative of MG1655 that expresses a C-terminally 3× FLAG-tagged AraC from its native locus. ChIP/qPCR verified significant association of AraC with all regions tested in the presence of arabinose (Fig. 1A; araJ was not tested). Association of AraC with all regions was reduced in the absence of arabinose, with no association detected for ydeN (Fig. 1A). Thus, our data suggest that the overall affinity of AraC for its DNA sites is increased by association with arabinose. This is particularly important for AraC binding upstream of ydeN, since this interaction appears to be completely dependent upon arabinose.
FIG 1.

(A) Validation of putative E. coli AraC target regions identified by ChIP-chip. Data are from ChIP of FLAG-tagged AraC (from strain AMD187), followed by quantitative real-time PCR. Cells were grown in the absence (dark gray bars) or presence (light gray bars) of arabinose. Occupancy units represent background-subtracted fold enrichment relative to a control genomic region within the transcriptionally silent bglB. Error bars represent one standard deviation from the means based on three independent biological replicates. (B) Motif representing the AraC binding site, derived from the ChIP-chip data using MEME and displayed using WebLogo (54). The previously described motif (34) is also shown. (C) ChIP of FLAG-tagged AraC coupled with quantitative real-time PCR to measure binding upstream of ytfQ in a wild-type strain (AMD187) or a strain with a mutation in the putative AraC binding site (JTW010) for cells grown in the presence of arabinose. Data are normalized to binding upstream of araB. Error bars represent one standard deviation from the means based on three independent biological replicates.
The known consensus sequence for AraC (Fig. 1B) is based on extensive footprinting and mutagenesis studies of the araBAD, araC, araE, araFGH, and araJ promoters (30–34). From our validated AraC ChIP targets, we inferred a de novo position-specific weight matrix (PSWM) for AraC using MEME, a bioinformatic tool that identifies overrepresented motifs in multiple unaligned sequences (35). The top-scoring motif predicted by MEME is a good match to the known AraC motif (Fig. 1B). MEME identified many, but not all, of the known AraC binding sites. This is unsurprising, since cooperative interactions of AraC dimers stabilize binding to some nonconsensus DNA sites at previously described target loci (32).
Effects of AraC and arabinose on global gene expression in E. coli.
We used transcription profiling with Affymetrix high-density microarrays to determine the effects of AraC and arabinose on RNA levels genome wide. Wild-type or ΔaraC mutant cells were grown in the absence or presence of 0.2% l-arabinose. Table 2 lists the genes whose expression changed significantly by ≥4-fold in wild-type cells upon addition of arabinose and whose expression differed significantly by ≥4-fold between wild-type and ΔaraC cells in the presence of arabinose. As expected, expression of known AraC-regulated genes, i.e., araB, araA, araD, araE, araF, araG, araH, and araJ, increased substantially upon addition of arabinose in wild-type cells and was substantially higher in wild-type cells than ΔaraC cells in the presence of arabinose (Table 2). Novel AraC-regulated genes identified using this approach are discussed below. We did not detect significant AraC-dependent or arabinose-dependent regulation of xylA, a previously described AraC-regulated gene (36), nor did we detect binding of AraC upstream of xylA. Hence, we believe that xylA is not a direct regulatory target of AraC under the conditions tested here (cells were grown in tryptone broth in the other study).
Genes regulated indirectly by arabinose and AraC.
Many of the genes regulated by AraC/arabinose (Table 2) are not associated with binding of AraC, as determined by the ChIP-chip experiment. We conclude that these genes are indirectly regulated by arabinose and/or AraC. Almost all of these indirectly regulated genes are repressed by AraC/arabinose, and they include genes associated with maltose metabolism (malE, malF, malG, malK, malM, and lamB), threonine metabolism (tdcA, tdcB, tdcC, tdcD, and tdcE), d-glucarate/d-galactarate metabolism (garD, garL, garP, and garR), and tryptophan metabolism (tnaA, tnaB, and tnaL). Only one indirect target gene, isrB, is upregulated ≥4-fold by both AraC and arabinose. isrB was originally annotated as a small RNA but has more recently been shown to encode a small membrane protein (37). The mechanisms by which these genes are indirectly regulated by AraC and/or arabinose are unclear.
Arabinose-independent repression of ytfQ transcription by AraC.
The ChIP-chip analysis identified binding of AraC upstream of ytfQ and ppa (divergently transcribed genes). The MEME analysis identified a putative AraC binding site centered at positions −133.5 and −94.5 relative to the previously mapped transcription start sites of ytfQ and ppa, respectively (Fig. S1 in the supplemental material) (38). To determine experimentally whether this is the true AraC site upstream of ytfQ, we performed a ChIP experiment in a wild-type strain and in a strain in which the putative AraC binding site was mutated. Association of AraC, as determined by ChIP/qPCR, was significantly reduced by mutation of the putative DNA site (Fig. 1C). We conclude that this is a genuine DNA site for AraC. We did not detect significant regulation of ytfQ or ppa by AraC or arabinose in the transcription profiling experiment; however, ytfQ encodes a transporter that binds arabinose and galactose (39), consistent with ytfQ being a regulatory target of AraC. We constructed a translational fusion of ytfQ to a lacZ reporter gene and performed β-galactosidase assays with or without arabinose in a wild-type and a ΔaraC strain. We detected a small (∼1.5-fold) but significant increase in expression in the ΔaraC strain (see Fig. S3 in the supplemental material), suggesting that AraC directly represses transcription of ytfQ, albeit weakly. This apparent repression did not depend upon the addition of arabinose (see Fig. S3).
Arabinose-dependent repression of ydeNM transcription by AraC.
The ChIP-chip analysis identified binding of AraC upstream of ydeN (Fig. 1A). The relatively low resolution of ChIP-chip precluded precise identification of the binding site(s). We also showed in the transcription profiling experiment that expression of ydeN is reduced in the presence of arabinose and reduced in the presence of araC (Table 2). Similarly, expression of ydeM, the downstream gene, decreased 3.2-fold in the presence of arabinose and was reduced 7.3-fold by the presence of araC. This suggests that ydeN and ydeM are transcribed as a two-gene operon that is repressed by AraC. In the absence of arabinose, we did not detect AraC association upstream of ydeN (Fig. 1A), nor did we detect any significant difference in expression of ydeN or ydeM between wild-type and ΔaraC mutant cells in the absence of arabinose. ChIP/qPCR analysis of RNA polymerase (RNAP) at ydeN confirmed that transcription decreases in the presence of arabinose and that this decrease is dependent upon araC (Fig. 2). Thus, ydeNM is a novel AraC-regulated operon that is directly repressed by AraC in an arabinose-dependent manner.
FIG 2.
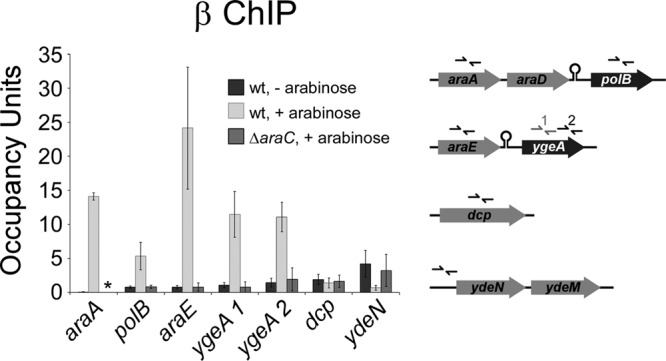
Association of RNA polymerase with members of the AraC regulon. ChIP of RNA polymerase (β subunit) coupled with quantitative real-time PCR to measure binding at various locations in a wild-type strain (MG1655) in the absence (dark gray) or presence (light gray) of arabinose or in a ΔaraC strain (SAC001) in the presence of arabinose (medium gray). The schematic indicates the positions of the primers used for the real-time PCR. The asterisk indicates that ChIP/qPCR could not be performed for araA in the ΔaraC strain (SAC001) due to the presence of the Δ(araD-araB)567 mutation. Error bars represent one standard deviation from the means based on three independent biological replicates.
We mapped the 5′ end of the ydeNM transcript using 5′ RACE and constructed transcriptional fusions to a lacZ reporter gene with fragments starting at position −371 and ending at position +1 or +14 with respect to the transcription start site. The longer fragment, from −371 to +14, showed ∼3-fold arabinose-dependent repression by AraC (Fig. 3). In contrast, the shorter fragment, from −371 to +1, showed no repression by AraC, suggesting association of AraC with the sequence around the transcription start site (Fig. S2 in the supplemental material), although no site matching the AraC motif could be identified in this region.
FIG 3.

Repression of ydeN by AraC. β-Galactosidase assay of fusions of the upstream region of ydeN fused to a lacZ reporter gene on a single-copy plasmid (pAMD124 and pAMD132). Fusions start at −371 and end at either +1 or +14 relative to the transcription start site. Assays were performed in AMD054 (ΔlacZ) and AMD115 (ΔaraC ΔlacZ) strains in the absence (dark gray bars) or presence (light/medium gray bars) of arabinose. Data are shown normalized to the wild-type values in the absence of arabinose. Error bars represent one standard deviation from the means based on at least three independent biological replicates.
ydeN and ydeM encode a predicted sulfatase and a predicted sulfatase maturase, respectively; thus, they have no apparent connection to arabinose metabolism. To determine whether either ydeN or ydeM is required for normal regulation of AraC-activated genes, we constructed a translational fusion of the araE upstream region to lacZ and measured β-galactosidase activity in a wild-type strain and in isogenic strains containing deletions of either ydeN or ydeM. We did not detect any substantial difference in β-galactosidase activity relative to the wild-type strain in either mutant (see Fig. S4 in the supplemental material).
AraC binding within dcp is not associated with detectable regulation of transcription.
We detected binding of AraC within dcp (Fig. 1A). The predicted binding site is located far from the 5′ end of any gene, including dcp itself (see Fig. S5 in the supplemental material), suggesting that it is not associated with regulation of an annotated gene. Intriguingly, association of AraC with the site in dcp, as measured by ChIP/qPCR, is the highest of all AraC-bound regions in the E. coli genome (Fig. 1A). To determine whether the AraC site within E. coli dcp is associated with transcription regulation, we used ChIP/qPCR to measure association of RNAP in the presence and absence of arabinose in a wild-type and a ΔaraC strain (Fig. 2). We did not detect any significant differences in RNAP association, suggesting that under these growth conditions, AraC does not regulate expression of a transcript that initiates within dcp.
RNAP reads through transcription terminators of AraC-activated transcripts.
In the transcription profiling experiment, we found that expression of ygeA is significantly induced by arabinose and is dependent on araC (Table 2). ygeA is located immediately downstream of araE, in the same orientation, suggesting that some RNAP reads through the terminator downstream of araE and transcribes ygeA. We tested this hypothesis using RT-PCR to detect RNA that spans the araE and ygeA genes. Despite the presence of a strong predicted terminator, we were able to detect RNA species that included both araE and ygeA, consistent with terminator read-through (Fig. 4A). ChIP/qPCR analysis of RNAP demonstrated high levels of RNAP association within ygeA at both the 5′ and 3′ ends, in the presence but not the absence of arabinose and dependent upon araC (Fig. 2). Northern blotting using probes specific to araE and ygeA also demonstrated read-through of the terminator downstream of araE (Fig. 4B), although the level of read-through transcript was lower than that of araE transcript. We also detected an araC-independent transcript by Northern blotting that is likely due to initiation of transcription immediately upstream of ygeA (Fig. 4B). Using densitometry analysis, we determined that the araE-ygeA read-through product is 11% as abundant as the araE transcript. In contrast, the ChIP/qPCR data (Fig. 2) indicate that ∼50% of RNAP complexes read through the terminator downstream of araE. Together, these data suggest that the read-through transcript is less stable than that for araE alone.
FIG 4.
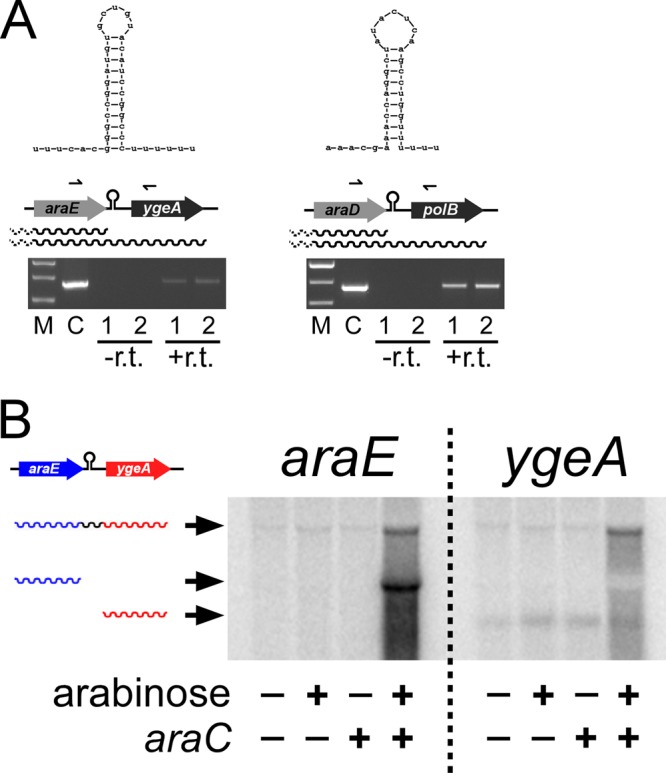
Read-through of the Rho-independent transcription terminators following araBAD and araE. (A) Ethidium bromide-stained agarose gel showing RT-PCR products generated from RNA (from MG1655) treated with reverse transcriptase (+r.t.), a control reaction without reverse transcriptase (−r.t.), or a colony (C). Lanes 1 and 2 show data from independent biological replicates. M, molecular size ladder. The predicted terminator structures are indicated above a schematic of the genes. Arrows represent the position of primers used in the PCR. (B) Northern blot hybridized with double-stranded DNA probes corresponding to araE or ygeA. RNA was purified from MG1655 or SAC001 (ΔaraC) grown in the absence or presence of arabinose. Arrowheads indicate the three relevant bands that correspond to araE-ygeA read-through RNA, araE RNA, and ygeA RNA. A cross-reacting band, probably due to rRNA, is seen in all lanes close to the band for araE-ygeA read-through RNA.
Using the transcription profiling data, we analyzed the differences in expression with or without arabinose and in the presence or absence of araC for the genes immediately downstream of araD, araH, and araJ. Only polB, the gene immediately downstream of araD, showed a >2-fold change in expression. Specifically, expression of polB increased 2.6-fold in the presence of arabinose and was 2.5-fold higher in wild-type cells than in ΔaraC cells. This suggests that RNAP also reads through the terminator downstream of araD. We confirmed this using RT-PCR (Fig. 4A) and ChIP/qPCR of RNAP (Fig. 2), as described above for araE-ygeA. From the ChIP/qPCR data, we estimate that ∼30% of RNAP complexes read through the terminator downstream of araD.
A recent study predicted sites of Rho-independent termination based on RNA sequence and structure (40). The sequence between araE and ygeA ranked 286th on the list of 1,058 predicted terminators, suggesting that it should function effectively to terminate transcription. To experimentally test the ability of this sequence to terminate transcription, we constructed a lacZ reporter fusion that includes the predicted terminator with limited flanking sequence downstream of a strong, constitutive promoter (Fig. 5A) (41). As controls, we constructed fusions with either no terminator sequence or predicted terminators and limited flanking sequence for the ahpF and tppB genes, ranked 293rd and 638th on the list of 1,058 predicted terminators, respectively (Fig. 5A) (40). While the ahpF and tppB terminators reduced expression by 98% and 99%, respectively, the araE terminator reduced β-galactosidase activity by only 56% (Fig. 5B). We also tested a mutant version of the tppB terminator that contains a point mutation in the upstream stem of the terminator stem-loop. This mutant terminator reduced β-galactosidase activity by 89% (Fig. 5B). Thus, the araE terminator is only weakly effective and does not even function as well as a mutant version of a terminator that has lower predicted strength.
FIG 5.
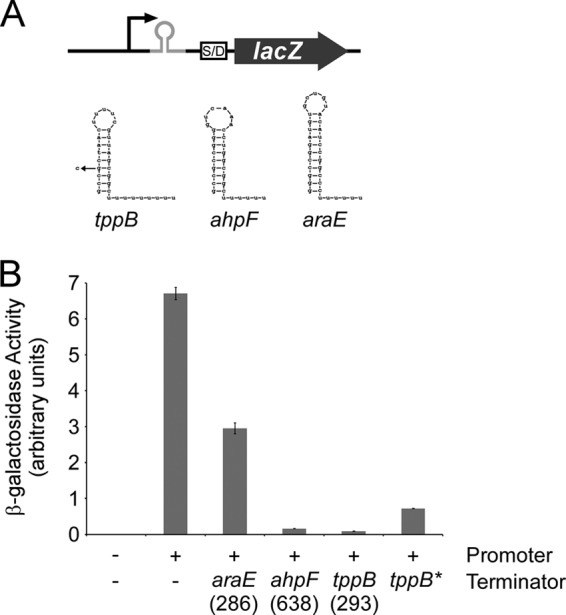
(A) Schematic of the lacZ reporter fusion used to assay terminator efficiency, including the sequences and predicted structures of the three terminators tested. The mutant tppB terminator is also indicated. (B) β-Galactosidase activity of an empty lacZ fusion plasmid (pAMD-BA-lacZ) and derivatives with a strong, constitutive promoter and either no terminator (pJTW064) or terminators from araE (pJTW055), ahpF (pJTW060), tppB (pJTW062), or a mutated tppB terminator (pJTW061). Numbers in parentheses indicate the rank of the terminator in a recent study (40). Assays were performed in AMD054 (ΔlacZ). Error bars represent one standard deviation from the means based on at least three independent biological replicates.
Genome-wide mapping of AraC binding sites in S. enterica.
We mapped the genome-wide binding of C-terminally FLAG-tagged AraC in S. enterica subsp. enterica serovar Typhimurium strain 14028s using ChIP coupled with deep sequencing (ChIP-seq). We identified five putative target loci for AraC: upstream of araB-araC, araE, araJ, STM14_0178 (araT), and within sseD. We validated the ChIP-seq data using ChIP/qPCR. Thus, we confirmed significant association of AraC with all regions identified by ChIP-seq (Fig. 6).
FIG 6.
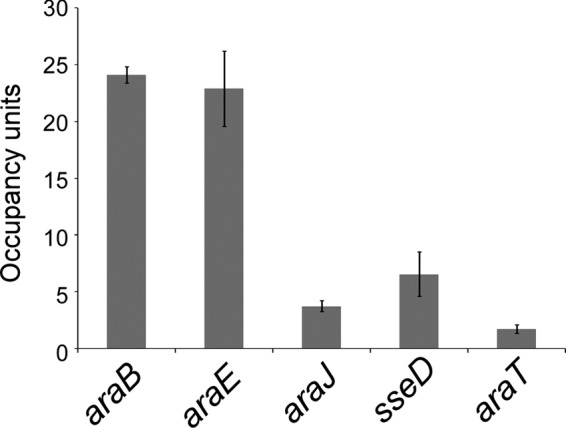
Validation of putative S. enterica AraC target regions identified by ChIP-seq. Data are from ChIP of FLAG-tagged AraC (from strain CB005) followed by quantitative real-time PCR. Cells were grown in the presence of arabinose. Occupancy units represent background-subtracted fold enrichment relative to a control genomic region upstream of sinR. Error bars represent one standard deviation from the means based on two or three independent biological replicates.
Effects of AraC and arabinose on global gene expression in S. enterica.
We used RNA-seq to determine the effects of AraC and arabinose on genome-wide RNA levels in S. enterica. Wild-type or ΔaraC mutant cells were grown in the presence or absence of 0.2% l-arabinose. Table 3 lists the 16 genes whose expression changed significantly (false discovery rate [FDR], <0.05) by ≥4-fold in wild-type cells upon addition of arabinose and whose expression differed significantly (FDR, <0.05) by ≥4-fold between wild-type and ΔaraC cells in the presence of arabinose. Of the 16 regulated genes, 9 are direct regulatory targets based on the association of AraC with regions upstream of these genes, as determined by ChIP-seq. All of the direct regulatory targets are positively regulated by AraC and arabinose. No direct targets were identified that are regulated by AraC in the absence of arabinose. We did not detect any significant change in expression of sseD or the surrounding genes, suggesting that, like E. coli dcp, this gene contains an AraC binding site that is not associated with regulation of transcription under the conditions tested. It is important to note, however, that sseD falls within Salmonella pathogenicity island 2 (SPI2), a region that is transcriptionally silenced by H-NS under the conditions used in our work (42). Thus, it is possible that AraC regulates transcription from the site within sseD under conditions that derepress SPI2.
TABLE 3.
Arabinose-responsive genes in S. enterica
| Genea | Fold change (log2) in mRNA level for: |
|
|---|---|---|
| Arabinoseb | araCc | |
| araC | 2.1 | 8.5 |
| araD | 9.4 | 8.5 |
| araA | 9.0 | 8.1 |
| araE | 8.4 | 8.0 |
| araB | 9.2 | 8.0 |
| araU | 5.6 | 5.7 |
| araT | 6.0 | 5.6 |
| STM14_0119 | 4.5 | 5.1 |
| ygeA | 4.1 | 4.3 |
| araJ | 3.8 | 4.1 |
| yjcB | 3.8 | 3.0 |
| ycfR | 2.7 | 2.9 |
| dctA | −2.4 | −3.7 |
| mglC | −2.4 | −3.9 |
| mglA | −3.2 | −4.7 |
| ygbM | −2.5 | −5.3 |
Arabinose-responsive genes in S. enterica were defined by >4-fold change in expression (significant difference) under growth with or without arabinose in wild-type (14028s) cells and >4-fold change (significant difference) between wild-type (14028s) and ΔaraC (AMD485) cells in the presence of arabinose. Direct regulatory targets of AraC are indicated by boldfaced text.
Fold change in mRNA level for wild-type cells grown with or without arabinose.
Fold difference in mRNA level between wild-type and ΔaraC cells grown in the presence of arabinose.
The direct regulatory targets of AraC include all classical ara genes that are conserved in S. enterica, with the exception of araH. Note that araH is part of the araFGH operon in E. coli but araF and araG are not conserved in S. enterica. As we have shown for E. coli, ygeA is a direct regulatory target of AraC in S. enterica (cotranscribed with araE). Lastly, STM14_0178 and STM14_0177 are direct regulatory targets of AraC. STM14_0178 and STM14_0177 do not have close homologues in E. coli and are predicted to encode an arabinoside transporter and an α-l-arabinofuranosidase II precursor, respectively. Thus, it is likely that S. enterica metabolizes arabinosides as a source of arabinose. Based on their predicted functions, we rename STM14_0178 and STM14_0177 araT (arabinoside transporter) and araU (arabinofuranosidase II precursor), respectively. The AraC site location upstream of araT can be estimated with <20-bp accuracy from the ChIP-seq data (predicted AraC sites upstream of araE and araJ are within 20 bp of the corresponding ChIP-seq peaks) (see Fig. S6 in the supplemental material). Two regions upstream of araT have sequences similar to the AraC consensus motif. The location of one of these regions is precisely aligned with the ChIP-seq peak, suggesting that this sequence is bound by AraC under the conditions tested. The more upstream conserved sequence that resembles an AraC binding site falls outside the region predicted by the ChIP-seq data; hence, it may bind AraC under other growth conditions, e.g., in the absence of arabinose. The end of the downstream putative AraC site is only 21 bp from the annotated gene start for araT, a distance inconsistent with activation of araTU transcription by AraC. However, the RNA-seq data strongly suggest that the transcription start site is downstream of the annotated gene start for araT. Hence, the translation start site of araT is likely to be incorrectly annotated, and the downstream putative AraC site is likely to be located upstream of position −40 with respect to the araTU transcription start site. This site position is consistent with transcription activation by AraC using a mechanism similar to that described for E. coli AraC-activated genes.
Conservation of the AraC regulon across the family Enterobacteriaceae.
AraC is highly conserved across the family Enterobacteriaceae, which includes E. coli and S. enterica. The two helix-turn-helix DNA-binding domains are particularly well conserved, e.g., they are 100% identical between E. coli and S. enterica. Hence, AraC likely binds with similar DNA sequence specificity across all Enterobacteriaceae species. To determine whether regulation of AraC target genes is conserved across the family Enterobacteriaceae, we aligned sequence surrounding E. coli and/or S. enterica AraC sites identified in this work with equivalent regions from four other Enterobacteriaceae species (Citrobacter rodentium, Enterobacter sp. strain 638, Klebsiella pneumoniae, and Cronobacter sakazakii; all alignments are shown in Fig. S7 in the supplemental material). S. enterica sseD is not conserved in any of the other species, and E. coli ydeN is only conserved in S. enterica; hence, these regions were not analyzed. Conservation of AraC sites was observed for araBAD, araFGH, araE, ytfQ, and araTU (Fig. 7; also see Fig. S6). No conservation of AraC sites was observed for araJ or dcp. Conservation was highest for two regions of the AraC binding site: positions 4 to 7 and 13 to 19. This is consistent with the information content of the motif derived from the E. coli AraC ChIP-chip data and with the known consensus sequence (Fig. 1B).
FIG 7.

Conservation of AraC binding sites in Enterobacteriaceae species. The position within the AraC motif is indicated on the x axis, and the different AraC targets are indicated on the y axis. The intensity of shading indicates the relative conservation of each position for each AraC target, measured as the information content of each subset of aligned sites in E. coli, S. enterica, C. rodentium, Enterobacter sp. strain 638, K. pneumoniae, and C. sakazakii. Darker shading indicates higher conservation, and the positional information content is indicated for each cell.
DISCUSSION
E. coli AraC is one of the best-studied TFs in any bacterial species and was the first described transcriptional activator (3, 4). With the exception of xylA, the last AraC-regulated gene to be identified was araJ, more than 30 years ago (27). We combined two complementary genomic approaches to expand the known E. coli AraC regulon. Specifically, we identified three novel binding targets of AraC (upstream of ytfQ and ydeN and within dcp) and five novel AraC-regulated genes (ytfQ, ydeN, ydeM, ygeA, and polB). Strikingly, regulation of four of the five novel target genes is mechanistically distinct from that observed previously for other AraC-regulated genes. Thus, our data demonstrate the power of integrating ChIP-chip/ChIP-seq and transcription profiling as an unbiased and comprehensive approach to identify regulatory networks.
ChIP-chip identifies noncanonical AraC binding sites.
Despite the extensive history of research on E. coli AraC, we identified several novel AraC-bound regions and several novel AraC-regulated genes. It is perhaps unsurprising that our unbiased, genomic approach identified AraC sites and AraC-regulated genes that differ functionally from those identified previously, as this would explain why they were missed in previous studies. Specifically, we identified AraC binding sites that (i) repress rather than activate transcription in an arabinose-dependent manner (ydeN), (ii) result in little or no observed regulation under standard laboratory growth conditions (ytfQ and dcp), and (iii) are located within a gene (dcp). We also identified AraC-regulated genes that are transcribed due to read-through of inefficient Rho-independent terminators (ygeA and polB).
Previous ChIP-chip studies in bacteria have identified many TF binding sites within genes (7). The most striking example in E. coli is RutR, for which 80% of binding sites are intragenic (9). With the exception of binding sites close to the 5′ end of genes (43), very few intragenic TF binding sites have a described function. We identified a binding site for AraC inside dcp, a gene that encodes dipeptidyl carboxypeptidase. Given the lack of conservation of this putative AraC binding site in other Enterobacteriaceae species and the lack of detectable regulation by AraC at this site, we conclude that the site is unlikely to have regulatory function under the tested growth conditions. We identified an analogous AraC site in S. enterica inside sseD. We propose that these binding sites have (i) regulatory function under a different growth condition, (ii) a function unrelated to transcription, or (iii) no function.
Novel E. coli AraC binding sites that repress transcription.
We identified two E. coli transcripts that are directly repressed by AraC: ytfQ and ydeNM. ytfQ encodes a galactose/arabinose transporter; thus, it has a clear connection to the established function of AraC in regulating arabinose metabolism. Repression of ytfQ by AraC is weak (∼1.5-fold; see Fig. S3 in the supplemental material), indicating that either AraC has only a minor effect on ytfQ expression or that more substantial regulation by AraC is associated with other growth conditions. AraC has previously been shown to repress its own transcription by binding to a region overlapping the araC promoter elements (32). This repression occurs independently of the addition of arabinose. The location of the AraC binding site upstream of ytfQ is too far upstream of the transcription start site to repress transcription by directly occluding RNAP. We propose that AraC bound at this site interacts with additional regulatory proteins, perhaps another monomer of AraC, bound closer to the transcription start site. GalR has been shown to regulate ytfQ (44) (Fig. S1 in the supplemental material). However, we detected no effect of GalR on regulation of ytfQ by AraC (data not shown).
Unlike AraC-dependent repression of araC and ytfQ, repression of ydeN occurs only in the presence of arabinose (Table 2 and Fig. 3). This is consistent with our ChIP data showing binding of AraC upstream of ydeN only in the presence of arabinose (Fig. 1A). Although arabinose-dependent repression by AraC has not been observed before, there are clear parallels with arabinose-dependent activation of araBAD transcription. Arabinose binding to AraC alters its DNA binding properties (5). At the araC-araBAD intergenic region, AraC forms a repression loop in the absence of arabinose due to the dimerization of distally bound AraC monomers. In the presence of arabinose, dimerization occurs at adjacent sites, breaking the repression loop and activating transcription of araBAD (6). This change in DNA binding is due to a rearrangement of the N-terminal arabinose-binding/dimerization domain and the C-terminal DNA-binding domain relative to one another (5). We propose that the DNA binding properties of AraC allow it to bind at ydeN only in the presence of arabinose. Our reporter fusions indicate that maximal repression by AraC requires sequence between +1 and +15 relative to the transcription start site (Fig. 3). This strongly suggests the presence of an AraC binding site overlapping the transcription start site, consistent with a role in transcriptional repression. We propose that AraC binds as a dimer to adjacent sites overlapping the transcription start site. Thus, arabinose-dependent repression of ydeNM by AraC would use the same mechanism as arabinose-dependent activation of araBAD.
Read-through of inefficient transcription terminators contributes to the E. coli AraC regulon.
ygeA and polB are positively regulated by AraC and arabinose due to partial read-through of Rho-independent terminators (Fig. 2, 4, and 5). We analyzed published microarray data from another group that used arabinose to induce overexpression of various proteins unrelated to AraC. Consistent with our own work, both ygeA and polB were in the top 5% of all genes when ranked by the level of arabinose induction (45). An equivalent analysis for ydeN showed that it is in the bottom 0.5% of all genes (45). From the Northern blot (Fig. 4B) it is clear that, in the presence of arabinose, the majority of ygeA mRNA is in the form of the read-through transcript, suggesting that read-through is physiologically relevant.
Many predictions have been made for intrinsic terminators in E. coli and other species (40, 46–50). Sequences downstream of araE and araD have been predicted to form terminators. This is especially true for the terminator downstream of araE, which has a long, G/C-rich stem-loop followed by a 10-mer sequence with 8 U's (Fig. 4). However, both the araE and araD terminators are only weakly effective. For the araE terminator this is unlikely to be due to alternative structures influenced by upstream sequence, since a minimal region is insufficient to terminate in the reporter assay we used (Fig. 5). Thus, our data suggest that terminator predictions are often inaccurate.
Regulatory functions for AraC beyond arabinose metabolism.
We have identified 7 novel AraC-regulated genes in E. coli and S. enterica. S. enterica araT and araU encode a likely transport/metabolism system for arabinosides. This suggests that S. enterica can use arabinosides as a carbon source by metabolizing them to arabinose. Only one other novel AraC-regulated gene identified in this work, E. coli ytfQ, has a known connection to arabinose metabolism (39). Furthermore, araJ is a long-established member of the AraC regulon but has no known connection to arabinose metabolism (51). It is possible that some or all of the novel AraC-regulated genes have as-yet-unidentified connections to arabinose metabolism, although this seems especially unlikely for polB, which encodes a well-characterized DNA polymerase. In addition, deletion of ydeN or ydeM did not substantially affect araE expression (see Fig. S4 in the supplemental material), suggesting that AraC and intracellular arabinose levels are unaffected by the absence of these genes.
Regulation of polB by AraC is particularly intriguing given the well-established function of polB in DNA replication and repair (52). A 6-fold increase in polB expression is sufficient to give a detectable increase in the spontaneous mutation rate independent of the SOS response (53). We were not able to detect a significant increase in the spontaneous mutation rate by growth in the presence of arabinose (data not shown), but polB expression increases only 2.6-fold. While it is likely that an increase in the spontaneous mutation rate would be below our detection threshold, the effect of arabinose on polB expression could contribute to genome variability during long-term growth.
Conservation of the AraC regulon.
The PhoP regulon is by far the best studied with respect to conservation. Only three genes are consistently regulated by PhoP across the family Enterobacteriaceae (13). In contrast, our data indicate that most members of the AraC regulon are conserved in this family. This “core” regulon is comprised of araBAD, araFGH, araE, ytfQ, and araTU. Three of these genes, ytfQ, araT, and araU, have not previously been described as AraC targets. The conservation of regulation of ygeA and polB by transcriptional read-through is more difficult to assess. araE-ygeA synteny is not well conserved, suggesting that ygeA is not a conserved AraC regulon member. We did not detect regulation of polB by AraC in S. enterica. However, there is a two-gene insertion between araD and polB in S. enterica. In contrast, most other Enterobacteriaceae species maintain the araD-polB synteny. Hence, polB regulation by AraC may be widely conserved.
Strikingly, one of the conserved regulatory targets of AraC, araTU, is absent from E. coli. This highlights the risk associated with making inferences on TF regulons if experimental data are only available for one species. An analysis of AraC regulon conservation based only on E. coli target genes would have missed araTU. Similarly, an analysis of AraC regulon conservation based only on S. enterica target genes would have missed araFGH. The importance of using experimental data from multiple species is especially high for TFs that have degenerate binding motifs, such as AraC, since binding sites cannot easily be predicted from DNA sequence alone.
Conclusions.
Our unbiased mapping of the AraC regulons of E. coli and S. enterica has revealed new functions and new mechanisms of action for this storied regulator. Our data suggest that AraC regulates functions beyond arabinose metabolism. Furthermore, unlike the PhoP regulon, most AraC regulatory targets are conserved across related species, although conservation is limited to genes required for the transport and metabolism of arabinose. Our work highlights the importance of genome-scale approaches in the study of bacterial gene expression.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Grainger, members of the Wade laboratory, Robert Schleif, and members of Keith Derbyshire and Todd Gray's group for helpful discussions. We thank David Grainger, Todd Gray, Keith Derbyshire, and Rick Wolf for comments on the manuscript. We thank Chunhong Mao for assistance with RNA-seq analysis. We thank the Wadsworth Center Bioinformatics Core, the Wadsworth Center Applied Genomic Technologies Core, and the University at Buffalo Next Generation Sequencing Core Facility for technical assistance.
This work was supported by National Institutes of Health (NIH) grant 1DP2OD007188 and Wadsworth Center start-up funds (J.W.), U.S. National Science Foundation grant MCB-1158056 (I.E.), and appointments (C.B. and B.P.) to the Emerging Infectious Diseases (EID) Fellowship Program administered by the Association of Public Health Laboratories (APHL) and funded by the Centers for Disease Control and Prevention (CDC).
Footnotes
Published ahead of print 22 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01007-13.
REFERENCES
- 1.Egan SM. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 184:5529–5532. 10.1128/JB.184.20.5529-5532.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gross J, Englesberg E. 1959. Determination of the order of mutational sites governing L-arabinose utilization in Escherichia coli B/r bv transduction with phage Plbt. Virology 9:314–331. 10.1016/0042-6822(59)90125-4 [DOI] [PubMed] [Google Scholar]
- 3.Englesberg E, Irr J, Power J, Lee N. 1965. Positive control of enzyme synthesis by gene C in the L-arabinose system. J. Bacteriol. 90:946–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenblatt J, Schleif R. 1971. Arabinose C protein: regulation of the arabinose operon in vitro. Nat. New Biol. 233:166–170 [DOI] [PubMed] [Google Scholar]
- 5.Schleif R. 2010. AraC protein, regulation of the L-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 34:779–796. 10.1111/j.1574-6976.2010.00226.x [DOI] [PubMed] [Google Scholar]
- 6.Dunn TM, Hahn S, Ogden S, Schleif RF. 1984. An operator at −280 base pairs that is required for repression of araBAD operon promoter: addition of DNA helical turns between the operator and promoter cyclically hinders repression. Proc. Natl. Acad. Sci. U. S. A. 81:5017–5020. 10.1073/pnas.81.16.5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wade JT, Struhl K, Busby SJ, Grainger DC. 2007. Genomic analysis of protein-DNA interactions in bacteria: insights into transcription and chromosome organization. Mol. Microbiol. 65:21–26. 10.1111/j.1365-2958.2007.05781.x [DOI] [PubMed] [Google Scholar]
- 8.Grainger DC, Aiba H, Hurd D, Browning DF, Busby SJ. 2007. Transcription factor distribution in Escherichia coli: studies with FNR protein. Nucleic Acids Res. 35:269–278. 10.1093/nar/gkl1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimada T, Ishihama A, Busby SJ, Grainger DC. 2008. The Escherichia coli RutR transcription factor binds at targets within genes as well as intergenic regions. Nucleic Acids Res. 36:3950–3955. 10.1093/nar/gkn339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonocora RP, Fitzgerald DM, Stringer AM, Wade JT. 2013. Non-canonical protein-DNA interactions identified by ChIP are not artifacts. BMC Genomics 14:254. 10.1186/1471-2164-14-254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhodius VA, Wade JT. 2009. Technical considerations in using DNA microarrays to define regulons. Methods 47:63–72. 10.1016/j.ymeth.2008.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez JC, Groisman EA. 2009. Evolution of transcriptional regulatory circuits in bacteria. Cell 138:233–244. 10.1016/j.cell.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez JC, Shin D, Zwir I, Latifi T, Hadley TJ, Groisman EA. 2009. Evolution of a bacterial regulon controlling virulence and Mg(2+) homeostasis. PLoS Genet. 5:e1000428. 10.1371/journal.pgen.1000428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A. 2005. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433:531–537. 10.1038/nature03239 [DOI] [PubMed] [Google Scholar]
- 16.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 17.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stringer AM, Singh N, Yermakova A, Petrone BL, Amarasinghe JJ, Reyes-Diaz L, Mantis NJ, Wade JT. 2012. FRUIT, a scar-free system for targeted chromosomal mutagenesis, epitope tagging, and promoter replacement in Escherichia coli and Salmonella enterica. PLoS One 7:e44841. 10.1371/journal.pone.0044841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horton RM, Cai Z, Ho SN, Pease L. 1990. Gene splicing by overlap extension: tailor made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 20.Dornenburg JE, DeVita AM, Palumbo MJ, Wade JT. 2010. Widespread antisense transcription in Escherichia coli. mBio 1:e00024–10. 10.1128/mBio.00024-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. 1986. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 188:415–431. 10.1016/0022-2836(86)90165-8 [DOI] [PubMed] [Google Scholar]
- 23.Church G, Gilbert W. 1984. Genomic sequencing. Proc. Natl. Acad. Sci. U. S. A. 81:1991–1995. 10.1073/pnas.81.7.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lew JM, Mao C, Shukla M, Warren A, Will R, Kuznetsov D, Xenarios I, Robertson BD, Gordon SV, Schnappinger D, Cole ST, Sobral B. 2013. Database resources for the tuberculosis community. Tuberculosis 93:12–17. 10.1016/j.tube.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28:511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosiba BE, Schleif R. 1982. Arabinose-inducible promoter from Escherichia coli. Its cloning from chromosomal DNA, identification as the araFG promoter and sequence. J. Mol. Biol. 156:53–66 [DOI] [PubMed] [Google Scholar]
- 28.Novotny CP, Englesberg E. 1966. The L-arabinose permease system in Escherichia coli B/r. Biochim. Biophys. Acta 117:217–230. 10.1016/0304-4165(66)90169-3 [DOI] [PubMed] [Google Scholar]
- 29.Brown CE, Hogg RW. 1972. A second transport system for L-arabinose in Escherichia coli B-r controlled by the araC gene. J. Bacteriol. 111:606–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoner C, Schleif R. 1983. The araE low affinity L-arabinose transport promoter. Cloning, sequence, transcription start site and DNA binding sites of regulatory proteins. J. Mol. Biol. 171:369–381 [DOI] [PubMed] [Google Scholar]
- 31.Hendrickson W, Stoner C, Schleif R. 1990. Characterization of the Escherichia coli araFGH and araJ promoters. J. Mol. Biol. 215:497–510. 10.1016/S0022-2836(05)80163-9 [DOI] [PubMed] [Google Scholar]
- 32.Hamilton EP, Lee N. 1988. Three binding sites for AraC protein are required for autoregulation of araC in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 85:1749–1753. 10.1073/pnas.85.6.1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee N, Francklyn C, Hamilton EP. 1987. Arabinose-induced binding of AraC protein to araI2 activates the araBAD operon promoter. Proc. Natl. Acad. Sci. U. S. A. 84:8814–8818. 10.1073/pnas.84.24.8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Flaherty C, Hendrickson W. 1992. AraC protein contacts asymmetric sites in the Escherichia coli araFGH promoter. J. Biol. Chem. 267:24848–24857 [PubMed] [Google Scholar]
- 35.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- 36.Desai TA, Rao CV. 2010. Regulation of arabinose and xylose metabolism in Escherichia coli. Appl. Environ. Microbiol. 76:1524–1532. 10.1128/AEM.01970-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. 2008. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 70:1487–1501. 10.1111/j.1365-2958.2008.06495.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho BK, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, Palsson BØ. 2009. The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol. 27:1043–1049. 10.1038/nbt.1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horler RS, Müller A, Williamson DC, Potts JR, Wilson KS, Thomas GH. 2009. Furanose-specific sugar transport: characterization of a bacterial galactofuranose-binding protein. J. Biol. Chem. 284:31156–31163. 10.1074/jbc.M109.054296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner PP, Barquist L, Bateman A, Nawrocki EP, Weinberg Z. 2011. RNIE: genome-wide prediction of bacterial intrinsic terminators. Nucleic Acids Res. 39:5845–5852. 10.1093/nar/gkr168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burr T, Mitchell J, Kolb A, Minchin S, Busby S. 2000. DNA sequence elements located immediately upstream of the −10 hexamer in Escherichia coli promoters: a systematic study. Nucleic Acids Res. 28:1864–1870. 10.1093/nar/28.9.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. 2006. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2:e81. 10.1371/journal.ppat.0020081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Irani MH, Orosz L, Adhya S. 1983. A control element within a structural gene: the gal operon of Escherichia coli. Cell 32:783–788. 10.1016/0092-8674(83)90064-8 [DOI] [PubMed] [Google Scholar]
- 44.Bulyk ML, McGuire AM, Masuda N, Church GM. 2004. A motif co-occurrence approach for genome-wide prediction of transcription-factor-binding sites in Escherichia coli. Genome Res. 14:201–208. 10.1101/gr.1448004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee K, Zhan X, Gao J, Qiu J, Feng Y, Meganathan R, Cohen SN, Georgiou G. 2003. RraA, a protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 114:623–634. 10.1016/j.cell.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 46.Lesnik EA, Sampath R, Levene HB, Henderson TJ, McNeil JA, Ecker DJ. 2001. Prediction of Rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 29:3583–3894. 10.1093/nar/29.17.3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Hoon MJ, Makita Y, Nakai K, Miyano S. 2005. Prediction of transcriptional terminators in Bacillus subtilis and related species. PLoS Comput. Biol. 1:e25. 10.1371/journal.pcbi.0010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.d'Aubenton-Carafa Y, Brody E, Thermes C. 1990. Prediction of Rho-independent Escherichia coli transcription terminators. A statistical analysis of their RNA stem-loop structures. J. Mol. Biol. 216:835–858 [DOI] [PubMed] [Google Scholar]
- 49.Kingsford CL, Ayanbule K, Salzberg SL. 2007. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 8:R22. 10.1186/gb-2007-8-2-r22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitra A, Kesarwani AK, Pal D, Nagaraja V. 2011. WebGeSTer DB–a transcription terminator database. Nucleic Acids Res. 39:D129–D135. 10.1093/nar/gkq971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reeder T, Schleif R. 1991. Mapping, sequence, and apparent lack of function of araJ, a gene of the Escherichia coli arabinose regulon. J. Bacteriol. 173:7765–7771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pham P, Rangarajan S, Woodgate R, Goodman MF. 2001. Roles of DNA polymerases V and II in SOS-induced error-prone and error-free repair in Escherichia coli. Proc. Nat. Acad. Sci. U. S. A. 98:8350–8354. 10.1073/pnas.111007198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al Mamun AA. 2007. Elevated expression of DNA polymerase II increases spontaneous mutagenesis in Escherichia coli. Mutat. Res. 625:29–39. 10.1016/j.mrfmmm.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 54.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome of Escherichia coli K-12. Science 277:1453–1474. 10.1126/science.277.5331.1453 [DOI] [PubMed] [Google Scholar]
- 56.Jarvik T, Smillie C, Groisman EA, Ochman H. 2010. Short-term signatures of evolutionary change in the Salmonella enterica serovar Typhimurium 14028 genome. J. Bacteriol. 192:560–567. 10.1128/JB.01233-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.