

Abstract
The sal lantibiotic locus plays an important role in the virulence of Streptococcus pyogenes. Our transcriptional analysis of the sal locus provides new information on the complex regulation of this operon. Transcription of the operon is regulated by a promoter upstream of the operon and by a second internal promoter upstream of the salKRZ genes. Here we identify the location of the internal promoter and provide information on how this promoter is autoregulated by proteins within the locus. We determined by primer extension that the salKR promoter is located within the salY gene and identified several regulatory regions important for expression. The higher activity of the promoter in a salKR deletion strain indicates a role in repression by the SalR response regulator. Further, this promoter had higher activity in a salA deletion strain, implicating corepression or a signaling role for the SalA peptide. Finally, we demonstrate that this promoter can be controlled by host factors. Analysis of transcriptional regulation of this locus provides a better understanding of the function of the sal locus in S. pyogenes pathogenesis.
INTRODUCTION
Many bacterial species can be found to be inhabitants of polymicrobial environments. Only those microbes that have evolved effective strategies to outcompete their coinhabitants survive in this setting. One such strategy is the production of a lantibiotic. Lantibiotics are bacteriocin-like inhibitory peptides that are ribosomally synthesized and are produced exclusively by Gram-positive bacteria. Their unique structure contains lanthionine or methyl-lanthionine residues formed by the cross-linking of cysteine to either dehydrolamine or dehydrobutyrine (1, 2). The lanthionine peptides act as bactericidal pore-forming toxins targeted against closely related Gram-positive bacteria, providing an ecological advantage to the producing bacterium, especially those inhabiting competitive environments, such as mucosal surfaces (1, 3).
There are common genetic components found in most lantibiotic loci, including a gene encoding the prelantibiotic peptide, a gene encoding the enzyme to modify the peptide into the lanthionine form, and a gene encoding a transport protein to transport the modified peptide to the extracellular environment. To prevent the bacterium from being subjected to the bactericidal effect of its own lantibiotic, these loci also commonly encode an immunity protein or a specialized ABC transporter providing an immunity function (2, 4–7). Regulation of lantibiotic production occurs at the transcriptional level through a regulatory protein that is activated by its cognate signaling kinase as part of a two-component regulatory system (TCS). The cognate lantibiotic also functions as a cell density-signaling molecule for the TCS, similar to a quorum-sensing system (6, 8). The promoters that regulate gene expression are commonly located at the beginning of the operon, upstream of the TCS, and, in some cases, upstream of the immunity proteins (2, 9). Transcription of these loci needs to be tightly regulated to ensure production of the peptides along with the self-immunity proteins at the appropriate times.
The salivaricin (sal) locus of Streptococcus salivarius encodes proteins that produce and regulate a type 1 lantibiotic, salivaricin A (10). The salivaricin A lantibiotic was first identified in this commensal bacterium, predominantly found in oral and pharyngeal sites. Salivaricin A has an inhibitory function against several streptococci, such as S. mutans and S. pyogenes, which can inhabit the same host sites (11, 12). Paradoxically, S. pyogenes also harbors the sal locus, which has ∼85% homology to the sal locus of S. salivarius. However, S. pyogenes, other than the M4 strain, does not produce an active salivaricin A and is not immune to the salivaricin A produced by S. salivarius (13, 14). Despite the lack of lantibiotic and immunity functions, the sal locus is highly conserved, with >92% homology in all S. pyogenes serotypes sequenced. The retention and conservation of this locus, even in the absence of lantibiotic production, suggest that the function of the S. pyogenes sal locus has evolved to serve another purpose, perhaps a function toward bacterial fitness in a different ecological niche.
Many of the Gram-positive bacteria that harbor lantibiotic loci are commensal organisms. However, some pathogenic bacteria encode lantibiotic loci that also function in virulence. The cytolysin locus in Enterococcus faecalis can also function as a lantibiotic locus (15). The activity of the cytolysin is targeted against both bacterial and eukaryotic cells, including phagocytic cells, such as neutrophils and macrophages (15). This scenario suggests that bacteria can evolve to use the product of a lantibiotic locus to achieve a competitive advantage from the toxic effects of the host's innate immunity.
A previous transposon mutagenesis screen identified a mutant with a transposon insertion in the salK gene of the S. pyogenes sal locus (16). The salK gene encodes the putative histidine kinase of the sal locus TCS. The salK insertion mutant, as well as a mutant with an in-frame deletion of salK, is highly attenuated in the zebrafish infection model (13; P. Namprachan-Frantz, unpublished data). Moreover, this strain is attenuated for survival in human whole blood and an in vitro neutrophil survival assay (Namprachan-Frantz, unpublished). These results suggest a role for the sal locus in the pathogenesis of S. pyogenes. Therefore, there is a need to determine the factors influencing regulation of this locus and the conditions under which these genes are expressed in order to broaden the understanding of S. pyogenes pathogenesis during host-pathogen interactions.
In the present study, we analyzed the transcriptional regulation of the sal locus in S. pyogenes and the function of proteins encoded by this locus that are involved in this regulation. This is the first study to describe the exact salKR promoter (salKR-pro) location of the sal locus in S. pyogenes. Furthermore, this is the first study to demonstrate that SalR is a transcriptional repressor toward the expression of the salKR promoter, as well as data suggesting that SalA may be acting as an intracellular signaling molecule.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
All plasmids were maintained in either Escherichia coli DH5α cells or Escherichia coli TOP10 cells (Invitrogen) and cultured aerobically in Luria-Bertani medium (BBL) supplemented with 25 μg/ml kanamycin (Kan) or with 20 μg/ml chloramphenicol (Cam) at 37°C. The Streptococcus pyogenes serotype M14 HSC5 strain (17) used in this study was cultured anaerobically in Todd-Hewitt medium (BBL) supplemented with 0.2% yeast extract (THY medium) or THY medium supplemented with 2% proteose peptone (TP medium) and incubated in 15-ml conical tubes at 37°C under static conditions. Plasmids in S. pyogenes were maintained with medium supplemented with 500 μg/ml kanamycin and 3 μg/ml chloramphenicol. S. pyogenes cultured on solid medium (THY medium supplemented with 1.4% agar) was incubated in an anaerobic gas chamber with GasPak (BBL) cartridges at 37°C. Mid-log-phase cultures were grown by diluting overnight cultures of S. pyogenes 1:50 into fresh THY medium. When necessary, S. pyogenes mutants were selected on THY medium containing Cam (3 μg/ml), Kan (500 μg/ml), or erythromycin (Erm; 1 μg/ml), as appropriate.
Manipulation of DNA.
Chromosomal DNA was purified from S. pyogenes as described previously (18). Plasmid DNA was isolated using a PureYield plasmid miniprep kit (Promega) and transformed into E. coli and S. pyogenes by electroporation, as described previously (17, 18). Restriction endonucleases, ligases, and polymerases were used according to the manufacturer's recommendations. When required, DNA fragments were purified using a gel extraction kit (Fermentas).
RNA isolation.
Ten milliliters of mid-log-phase bacterial cultures was centrifuged, washed twice with phosphate-buffered saline (PBS), and transferred into a 1.5-ml microcentrifuge tube. The pellet was resuspended in 100 μl TE (Tris-EDTA) buffer with 25 mg/ml lysozyme and 10 mg/ml mutanolysin and incubated at 37°C for 1 h. One milliliter of TRIzol reagent (Invitrogen) was added, followed by incubation at room temperature for 5 min. Two hundred microliters chloroform was added, and the mixture was vortexed and incubated for 3 min at room temperature, followed by centrifugation at 13,000 × g for 15 min at 4°C. The aqueous layer was then transferred into a new microcentrifuge tube containing 500 μl of isopropanol to precipitate the nucleic acid. The samples were then centrifuged at 13,000 × g for 15 min at 4°C. The pellet was washed with 70% ethanol and air dried to evaporate the residual alcohol. The nucleic acid pellets were resuspended in 100 μl RNase-free water and subsequently treated with DNase and purified with an RNeasy MinElute cleanup kit (Qiagen).
Reverse transcription-PCR (RT-PCR) analysis of expression of genes in the sal locus.
RNA was isolated from mid-log-phase S. pyogenes cultures grown in THY medium as described above. The concentration of RNA was determined using a NanoDrop spectrophotometer (Thermo Scientific). The RNA was first subjected to PCR amplification to check for possible DNA contamination. Initial cDNA synthesis was carried out using avian myeloblastosis virus (AMV) reverse transcriptase (Fermentas) with 1 μg of RNA sample and a single reverse primer complementary to the 3′ end of the sal locus in the salZ gene (primer SalR-hyp-rev, 5′-CGT TCT GGG AGT TGT GAA GC-3′), to produce a cDNA of any transcript that read through the end of the sal locus. Following incubation at 60°C for 1 h, the cDNA was purified using a chromatin immunoprecipitation (ChIP) DNA Clean & Concentrator kit (Zymo Research). The cDNA was then used as a template for PCR amplification to confirm the read-through transcripts between genes using the primers listed in Table 1. The PCR products were visualized on an electrophoresis gel stained with SYBR safe stain (Invitrogen).
TABLE 1.
Primers for RT-PCR
| PCR no. | Primer name | Sequence |
|---|---|---|
| 1 | 5′-SalA-N | GAC TAA TGC TAT CGA AGA AGT TTC TG |
| 3′-SalA-N2 | GTA TCT AAT ATG TCG TAA TC | |
| 2 | 5′-SalT-end | CTT TTG TAT CTC AAG ATT CTC C |
| 3′-SalX-mid-new | GAT TGC TCT GAT AAT ACT GAC C | |
| 3 | 5′-Kan-insert-check | GAT GGG AAA CTT CAC ATG GAG |
| 3′-SalY-N | GAT AAG TCC ATT TGT GAT GAC | |
| 4 | 5′-SalK-pro-del3 | CGC GGA TCC CAG TTT AGT ATG GTT CCT G |
| 3′-SalK-del-seg1-SalI | ACG CGT CGA CAG CTG CTG TAT CAA TAA GCG GTC | |
| 5 | 5′-SalR-del-seg1 | GAT ACT TAC CAC CCA ATA G |
| 3′-SalR-del-seg1 | ACG CGT CGA CTG CAA ATA GTC TGT GGT CAT C | |
| 6 | 5′-SalR-del-seg2 | ACG CGT CGA CGT TGT TAT GTT GAC AGG ATA TG |
| 5′-SalR-hyp-rev | CGT TCT GGG AGT TGT GAA GC |
Promoter reporter construct.
To measure the expression of the salKR promoter, a transcriptional fusion was constructed using an alkaline phosphatase (AP) gene (phoZ) placed downstream of each putative promoter. The promoter regions for salKR were amplified using the primers listed in Table 2. The PCR products were gel isolated and digested with BamHI and EcoRI (Invitrogen). The digested DNA fragments were then ligated using ligase (Invitrogen) with the pMNN1 plasmid (19), which was also digested with BamHI and EcoRI. The ligated products were subjected to butanol precipitation, resuspended in 10 μl of nuclease-free water, and transformed into E. coli DH5α cells. Transformants were selected on LB agar plates containing 25 μg/μl kanamycin and 20 μg/μl chloramphenicol. Each plasmid construct was isolated and transformed into S. pyogenes wild-type (WT) and mutant strains and maintained in THY medium containing 500 μg/μl kanamycin and 3 μg/μl chloramphenicol. Promoter sequences were confirmed by sequencing.
TABLE 2.
Primers for pMNN1-phoZ reporter constructs
| Plasmid construct | Primer name | Sequence |
|---|---|---|
| pMNN1-salA-pro | 5′-salA-pro-BamHI | CGC GGA TCC GAA TGA TTT TGA GAT ACT TC |
| 3′-salA-pro-EcoRI | CCG GAA TTC CTT CTT CGA TAG CAT TAG TC | |
| pMNN1-salK-pro | 5′-salK-pro-BamHI | CGC GGA TCC CAT TGC GGT CAG TGA CAT CC |
| 3′-salK-pro-EcoRI | CCG GAA TTC GCC AGC CAA ATA AAA GCT G | |
| pMNN1-salKR-pro | 5′-salY-BamHI | CGC GGA TCC GAT AGA GGA GAG TAA TAT G |
| 3′-salY-EcoRI | CCG GAA TTC CAT ATT ATT TAC TTA ATC G | |
| pMNN1-salKR-pro-del1 | 5′-salK-pro-del1 | CGC GGA TCC GTT ATC GCT AAT GGG TCA TTG |
| 3′-salY-EcoRI | CCG GAA TTC CAT ATT ATT TAC TTA ATC G | |
| pMNN1-salKR-pro-del2 | 5′-salK-pro-del2 | CGC GGA TCC GAC TCC ATC TGC CAT AG |
| 3′-salY-EcoRI | CCG GAA TTC CAT ATT ATT TAC TTA ATC G | |
| pMNN1-salKR-pro-del3 | 5′-salK-pro-del3 | CGC GGA TCC CAG TTT AGT ATG GTT CCT G |
| 3′-salY-EcoRI | CCG GAA TTC CAT ATT ATT TAC TTA ATC G | |
| pMNN1-salKR-pro-short | 5′-salK-pro-del2 | CGC GGA TCC GAC TCC ATC TGC CAT AG |
| 3′-salY-pro-short-EcoRI | CCG GAA TTC GTT TTT CCA AGG CTA ATT TCT G | |
| pMNN1-salKR-pro-short-flip | 5′-salKR-pro-del2-EcoRI | CCG GAA TTC GAC TCC ATC TGC CAT AG |
| 3′-salK-pro-short-BamHI | CGC GGA TCC GTT TTT CCA AGG CTA ATT TCT G | |
| pMNN1-salKR-pro-del2A | 5′-salYK-pro-del2A | CGC GGA TCC GAG TTA ATG AAA GCT CAA GG |
| 3′-salY-EcoRI | CCG GAA TTC CAT ATT ATT TAC TTA ATC G |
Alkaline phosphatase liquid assay.
S. pyogenes strains that contained the alkaline phosphatase reporter constructs were grown in THY medium with 500 μg/ml kanamycin and 3 μg/ml chloramphenicol overnight. Subcultures were made by transfer into fresh THY medium with the same antibiotics and grown to mid-log phase. One milliliter of each log-phase culture was grown with or without treatment for 3 h at 37°C. After treatment, all cultures were normalized to an optical density at 600 nm (OD600) of 0.75 in PBS, and 50 μl of culture was then added, in triplicate, into the wells of a 96-well plate containing 200 μl of 1-mg/ml p-nitrophenylphosphate (Sigma) suspended in 1 M Tris (pH 8). Following incubation in the dark for 1 h at room temperature, three optical densities (OD405, OD550, and OD600) were measured using a spectrophotometric plate reader (VersaMax). Alkaline phosphatase activity was determined using the following formula: {[OD405 − (1.75 × OD550)]/(volume × time × OD600)} × 1,000. Each assay was performed in triplicate and repeated a minimum of three times.
DIG-labeled sequencing reaction.
The fragment for the digoxigenin (DIG)-labeled sequencing template was PCR amplified using primers 5′-salK-pro-del2 (CGC GGA TCC GAC TCC ATC TGC CAT AG) and 5′-phoZ-rev (GTT GCC TTC GCT TCA GCA ACC TCT G) with the reporter construct pMNN1-salKR-pro-del2 as the template. The fragment was gel purified, and the concentration was determined using a NanoDrop spectrophotometer (Thermo Scientific). The sequencing reaction was carried out using VentR (exo-) DNA polymerase (NEB) with DIG-labeled primer 5′-phoZ-rev (DIG-5′-GTT GCC TTC GCT TCA GCA ACC TCT G-3′). The ratios of the deoxynucleoside triphosphates/dideoxynucleoside triphosphates used in the reaction mixtures were as follows: 0.5 mM dATP/100 mM ddATP, 0.5 mM dGTP/25 mM ddGTP, 0.5 mM dCTP/50 mM ddCTP, and 0.5 mM dTTP/150 mM ddTTP. Following the PCR, the sequencing products were loaded directly onto an 8% urea-acrylamide gel, and the reactions were run at 1,500 V and 45 mA for 3 h. The DIG-labeled DNA was transferred onto a nylon membrane using the capillary transfer method (20). The labeled products were detected using anti-DIG-AP antibody 32 (Roche) and CDP-Star (Roche) as the substrate following the manufacturer's instructions and subsequently exposed to autoradiograph film.
Primer extension.
An overnight culture of the S. pyogenes wild type expressing pMNN1-salKR-pro-del2 was transferred into fresh THY medium supplemented with 500 μg/μl kanamycin and 3 μg/μl chloramphenicol and grown to mid-log phase. The cell pellet was collected for RNA isolation (see above for the protocol). Total purified RNA (5 μg) was used for primer extension with DIG-labeled primer 5′-phoZ-rev (DIG-5′-GTT GCC TTC GCT TCA GCA ACC TCT G-3′). RNA was heated at 90°C for 5 min and immediately cooled on ice. Reverse transcription was carried out using AMV reverse transcriptase (Fermentas), and the reaction mixture was incubated at 60°C for 1 h. RNase A (Invitrogen) was added to the reaction mixture, and the mixture was incubated at 37°C for 1 h to remove the remaining RNA due to its ability to interfere with the electrophoresis mobility of labeled cDNA. The primer extension products were purified using a ChIP DNA Clean & Concentrator kit (Zymo Research) and eluted with 10 μl of nuclease-free water. The corresponding DNA sequencing reactions were performed using the same DIG-labeled primer and a PCR fragment containing the expected promoter region with sequencing-grade Taq DNA polymerase (Promega). The extension product and sequencing ladder were resolved on an 8 M urea–8% polyacrylamide gel. Electrophoresis was performed at 1,500 V and 45 mA for 3 h. Transfer of labeled DNA from the gel onto a nylon membrane (Magna Charge) was done using the capillary transfer method (20). The labeled products were detected using anti-DIG-AP antibody (Roche) and CDP-Star (Roche) as the substrate following the manufacturer's instructions and subsequently exposed to autoradiograph film.
Disruption of salKR.
An in-frame deletion of the salKR region was constructed as follows. A 660-bp fragment containing the 5′ end of the salK gene along with ∼500 bp of sequence upstream was amplified by PCR using the primers 5′-SalK-del-seg1 (CTT CGA TTA GGT CAA GTG AAC C) and 3′-SalK-del-seg1-SalI (ACG CGT CGA CAG CTG CTG TAT CAA TAA GCG GTC). A 583-bp fragment containing the 3′ end of the salR gene was amplified with primers 5′-SalKR-del-seg2-SalI (ACG CGT CGA CGT ATC CAA CTG TTA TTC CAA CAG) and 3′-SalKR-del-seg2 (GTC GTT TGA TTA TCT GCA ACT CAG). The PCR amplification products were digested with the SalI restriction enzyme and ligated together into the PCR 8/GW/TOPO vector (Invitrogen), and the vector was transformed into E. coli TOP10 cells (Invitrogen). The vector containing the sal sequences with the in-frame deletion of salKR was then digested with SalI (cut between the upstream and downstream fragments) and ligated with a fragment containing a kanamycin cassette containing SalI sites isolated from vector pABG-5 (21). The ligation product was transformed into E. coli cells to propagate a plasmid vector containing the kanamycin resistance cassette between the 5′ end of salK and the 3′ end of salR, resulting in psalKR-del. The plasmid was then used as a template in PCR amplification of the salK-kan-salR sequence using primers 5′-salK-BamHI (CGC GGA TCC GAG AGA ACC TGT CTC TTC) and 3′-SalKR-del-seg2-PstI (AAA ACT GCA GGT CGT TTG ATT ATC TGC AAC TCA G). The PCR fragment was digested with BamHI and PstI, ligated into the suicide streptococcal vector pJRS233 (22, 23), and transformed into E. coli cells for plasmid propagation, creating pΔsalKR-kan. The plasmid was then isolated and transformed into the WT strain S. pyogenes HSC5. The recombination of the temperature-sensitive vector pΔsalKR-kan resulted in replacement of the WT salKR alleles during cycling of the permissive to the nonpermissive temperature, as described previously (23). The salKR deletion was confirmed by PCR.
Construction of a salA in-frame deletion strain.
An in-frame deletion of the salA gene (salA-IFD) was constructed by amplifying a 1,270-bp fragment using S. pyogenes HSC5 chromosomal DNA as the template and the primers 5′-salA-IFD (CAC CAA GTG AAA AGG CAA CAT TGC) and 3′-salA-IFD (CCT AAG AAG AGT GCC TAC TGG). This fragment was ligated into the pENTR/SD/DTOPO vector (Invitrogen). Inverse PCR was used to amplify the entire vector minus the salA gene sequences using primers SalA-IFD-fwd (TCC CCC GGG TTT CAT AAA ACTY CAT TCT CCT TTC C) and SalA-IFD-rev (TCC CCC GGG TGT TGT TAA TTA AAA CAA TCT GGA C) with SmaI restriction sites (underlined) incorporated into the ends of the primers. This fragment was subsequently cut with SmaI, ligated to circularize the fragment, and transformed into E. coli TOP10 cells, effectively removing 130 bp inside the salA coding sequence. Using the original primers described above (5′-salA-IFD and 3′-salA-IFD), the now 1,140-bp fragment (containing sequences upstream and downstream of salA but with an in-frame deletion of the salA gene) was amplified from the new plasmid and cloned into pJRS233, and chromosomal replacement proceeded as described above for the ΔsalKR deletion.
RESULTS
Analysis of sal locus transcripts.
To determine whether genes in the sal operon of the S. pyogenes serotype M14 HSC5 strain are cotranscribed, we performed RT-PCR on mRNA isolated from the wild-type strain during mid-log-phase growth. A single primer that annealed to a region at the 3′ end of the sal locus in the reverse orientation was used to make cDNA of the sal operon transcript by RT-PCR. Using this cDNA as the template, fragments were then amplified by PCR using primer pairs (Table 1) that overlap the 3′ and 5′ regions of 5 adjacent sal locus genes to confirm the cotranscription of genes in the sal locus (Fig. 1A). Using this technique, PCR fragments were detected for the regions in the beginning, middle, and end of the sal locus, including the newly identified salZ gene, encoding a hypothetical protein, at the 3′ end of the sal operon (Fig. 1B). These results confirmed that the entire sal operon from the salA gene through the end of the operon encodes a single transcript, since the cDNA was made from a single primer at the 3′ end of the operon. However, they do not rule out the possibility that additional transcripts may also be made starting at internal promoters reading through to the 3′ end or that transcripts may terminate before the 3′ end of the operon.
FIG 1.

RT-PCR analysis of genes in the sal locus expressed in S. pyogenes. (A) Locations of overlapping primer pairs 1 to 6 on the sal locus. (B) RT-PCR products from primers 1 to 3 and 4 to 6 with a DNA ladder in the leftmost lane. Lanes a, negative controls consisting of the RNA sample without reverse transcriptase; lanes b, positive controls with genomic DNA; lanes c, cDNA from the RNA sample. (C) Transcripts identified in the salA operon. RT-PCR analysis of WT strain and salY deletion strain RNA with primers for the salK gene. Lane 1, DNA ladder; lanes 2 and 3, negative controls consisting of RNA samples without reverse transcriptase; lane 4, WT RNA; lane 5, salY deletion strain RNA.
Promoter identification.
In order to analyze the regulation of the sal locus, it was first necessary to identify the promoter region(s). Examination of the common organization of genes and promoters found in other lantibiotic loci, as well as our RT-PCR data presented above, suggested that a promoter existed upstream of the salA gene at the 5′ end of the operon (2). In addition, a promoter is usually found upstream of the genes encoding the two-component system, which in the sal operon would be the salKR genes (2). A region ∼500 bp upstream of the salA gene (the entire intergenic region between salA and the upstream lacG gene) and another region from the 3′ end of the salY gene and overlapping the start of the salK gene were amplified by PCR. These regions were cloned individually into a reporter vector upstream of the E. faecalis alkaline phosphatase gene, phoZ, creating pPN-salA-pro and pPN-salK-pro, and then transformed into the wild-type HSC5 S. pyogenes strain. Mid-log-phase cultures of the wild-type strain expressing the reporter constructs were tested for alkaline phosphatase activity. Neither of these constructs showed any activity above the background (data not shown). In an attempt to identify a region that may contain a promoter for regulation of salKR expression, a region ∼2,000 bp upstream of the salK gene encompassing the entire open reading frame of salY was cloned into the salKR-pro reporter plasmid (pPN-salKR-pro). Alkaline phosphatase activity of 720 ± 28 units was detected in the wild-type strain expressing pPN-salKR-pro, indicating that an additional promoter expressing the downstream genes of the sal locus may be located within the salY gene (Fig. 2). We also constructed several reporter vectors containing regions further upstream of the salA gene in an effort to determine the upstream operon promoter. The highest activity came from a region ∼1,500 bp upstream of salA within the lacG gene (300 ± 45 units). However, the focus of this report is on salKR promoter expression, and an analysis of the salA promoter will be reported elsewhere.
FIG 2.

Analysis of the salKR promoter. Alkaline phosphatase activities were produced by S. pyogenes WT strain HSC5 expressing various truncations of the salKR promoter reporter plasmid. Alkaline phosphatase activity was measured using a colorimetric assay. Boxes with dashed-line borders, DNA regions with possible repression sites; boxes with bold borders, possible promoter regions. Error bars represent SEMs. *, P < 0.001.
Analysis of the salKR promoter.
To further define the sequences required for expression of the salKR promoter, 5′ end DNA truncations of the salKR promoter region from plasmid pPN-salKR-pro were tested for promoter activity (Fig. 2). Deletion of 187 bp from the 5′ end (deletion 1) resulted in only a slight increase in salKR promoter activity that was not significant (803 ± 37 units). Further truncation of another 806 bp from the 5′ end (deletion 2) resulted in a significant increase in the alkaline phosphatase activity compared to that achieved with the full-length salKR promoter region (2,106 ± 194 units). The increased promoter activity after deletion of the 5′ sequences suggests possible regions of repression. However, an additional deletion of 393 bp (deletion 3) demonstrated a total loss of promoter activity, suggesting that promoter elements are contained within this region (Fig. 2).
To determine if there were additional regulatory sites downstream of the promoter, a deletion of the 3′ end of the deletion 2 construct was also made, leaving only the 393-bp region (deletion 2-short). This construct showed an ∼3-fold increase in expression compared to that achieved with the deletion 2 construct and a greater than 10-fold increase compared to that achieved with the full-length promoter construct (7,619 ± 234 units), suggesting that another possible repression site or, alternatively, a terminator site is located downstream of the promoter region. The presence of terminator elements in the region from bp 7282 to 7869 was ruled out by cloning this fragment between the strong hasA promoter from the capsule synthesis operon in S. pyogenes and the phoZ reporter gene. If this DNA segment contained a terminator, then alkaline phosphatase activity would be greatly reduced compared to the activity observed in the absence of this segment. No significant difference in activity was observed between constructs with and without the sal locus segment (data not shown). However, we cannot rule out the possibility that modifying the 3′ end of the construct may affect reporter activity by alternative RNA folding. To confirm that the 393-bp region between bp 6888 and 7181 contained the −10 and −35 promoter elements, this segment was cloned into the reporter plasmid in the reverse direction (3′ to 5′; deletion 2-short-reverse), which resulted in no activity. Therefore, the sequences required for the salKR promoter lie within the 393-bp region from bp 6888 to 7181 of the salY gene (Fig. 2).
The identification of the salKR promoter downstream of the salA gene suggested that salKRZ genes could be transcribed from both an upstream sal operon promoter and the downstream salKR promoter, leading to the production of two transcripts. To further confirm that the upstream operon promoter provides a read-through transcript, RT-PCR was also performed with an RNA sample isolated from a strain carrying an in-frame deletion of the entire salY gene, which would result in a deletion of the salKR promoter. The results revealed that the salK transcript could still be detected using primers for the salK gene in the absence of the salKR promoter, indicating a read-through transcript from the upstream operon promoter (Fig. 1C).
SalR regulates salKR promoter expression.
On the basis of homology to other two-component regulators, the SalK and SalR proteins comprise a putative two-component regulatory system. The putative regulator SalR has a conserved DNA binding domain that would allow it to bind directly to DNA, resulting in regulation of gene expression. Most lantibiotic regulators control expression of their own promoters (6, 8, 24). Therefore, to determine whether SalR has an effect on salKR promoter expression, the reporter plasmid containing the salKR promoter was transformed into a strain containing an in-frame deletion of the salKR genes. The activity of the promoters was compared between the wild type and the ΔsalKR deletion strain. The results showed that salKR promoter activity in the ΔsalKR strain was approximately two times higher than that in the wild-type strain that expressed SalKR (Fig. 3). These results suggest that the SalR protein can act as an autoregulator to repress the expression of its own promoter.
FIG 3.

Alkaline phosphatase activities measured in the WT strain and the salKR and the salA-IFD strains expressing various truncations of the salKR promoter reporter plasmid. The gray regions represent the salKR promoter location. The values in the table are in alkaline phosphatase activity units with standard errors. *, P < 0.05 compared to the WT full-length construct; **, P < 0.01 compared to the WT full-length construct.
SalA affects salKR promoter expression.
Most promoters from lantibiotic loci are autoregulated, and expression is induced when the sensor kinase senses the presence of the cognate lantibiotic peptide (2, 8). While the S. pyogenes serotype M14 HSC5 strain does not produce detectable SalA lantibiotic activity (25), we did observe salA transcripts by RT-PCR. To investigate if the SalA peptide plays a role in expression of the downstream salKR promoter, the salKR promoter reporter plasmid was transformed into the wild type and a salA in-frame deletion (IFD) strain. Expression analysis revealed that salKR promoter activity in the salA-IFD strain was significantly (P < 0.005) increased (Fig. 4), suggesting that SalA can also play a role in salKR promoter repression.
FIG 4.
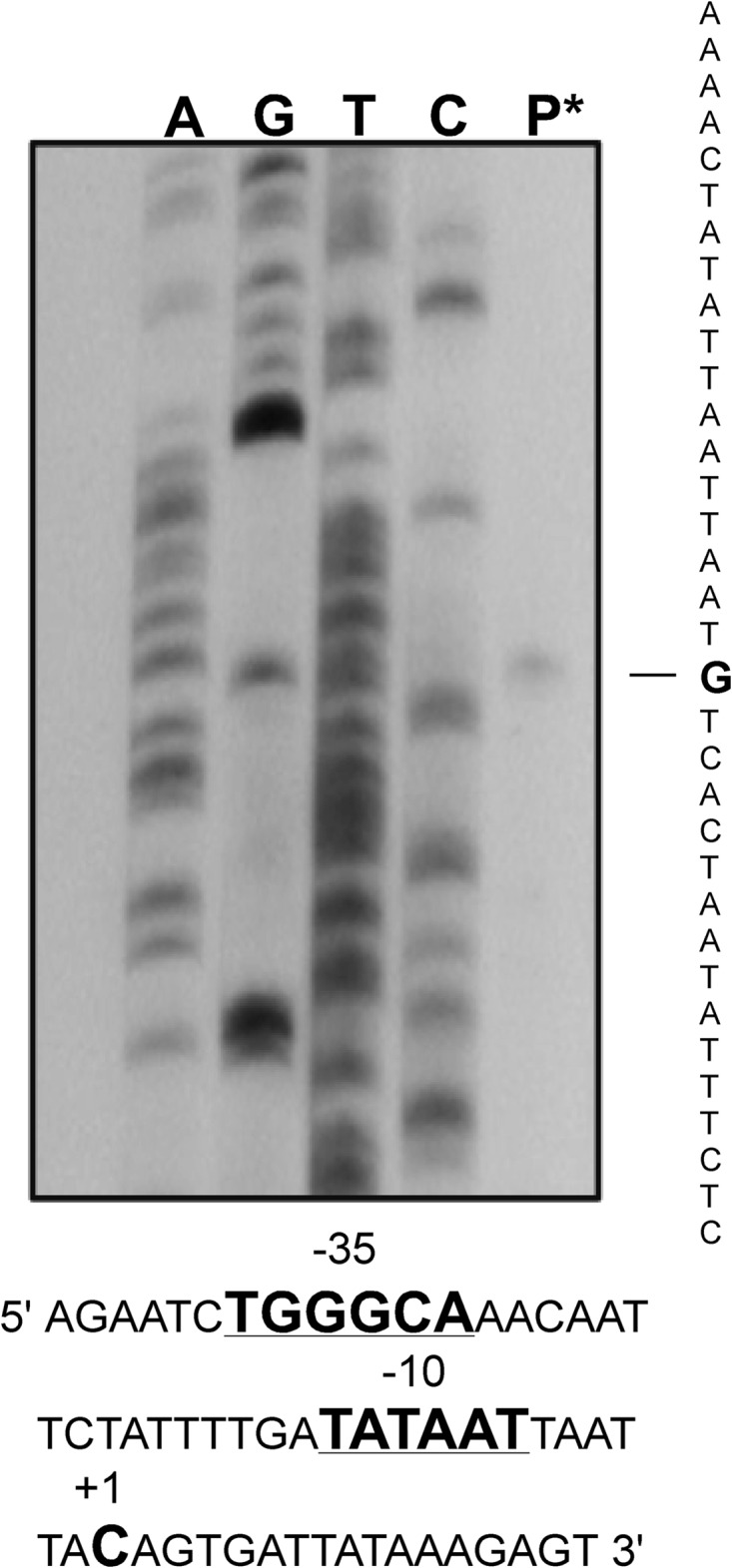
Identification of the 5′ end of the salKR transcript expressed from the salKR promoter. Primer extension analysis was used to identify a transcriptional start site of salKR transcripts. The primer extension product is shown in lane P*. The nucleotide start site (indicated by the boldface G) was compared to the sequencing ladder. The promoter regions identified from the transcription start site (position +1), the −10 consensus region, and a degenerate −35 region are shown in bold.
Analyses were then performed with the salKR promoter deletion constructs to determine promoter expression in the salA-IFD and ΔsalKR mutants compared to that in the wild-type strain. As shown in Fig. 4, the construct with the full-length promoter had higher alkaline phosphatase activity in both mutants. Furthermore, the deletion 2 construct also showed higher levels of expression in the salA-IFD mutant but not the ΔsalKR mutant than in the wild-type strain. However, expression from the construct with the shortest promoter, deletion 2-short, was the highest of that from all constructs and was not significantly different between any of the strains (Fig. 3). Therefore, these results strongly suggest that both SalKR and SalA play a role in the repression of the salKR transcript, as loss of either component results in higher expression, except when regulatory regions are removed, as in the deletion 2-short construct.
Mapping of the salKR transcription start sites.
The exact location of the salKR promoter elements was identified using a nonradioactive primer extension technique. The protocol utilized a DIG-labeled universal primer that annealed to the 5′ end of the phoZ gene in the reverse orientation using RNA isolated from the strain carrying the pPN-salKR-pro deletion 2 construct, as described in Materials and Methods. Use of the RNA transcribed off the plasmid reporter construct instead of the chromosome eliminated any interference from the long mRNA that was transcribed from the upstream operon promoter.
The −10 and −35 promoter region was determined from identification of the transcription start site (position +1) of the salKR transcript. A well-conserved consensus −10 region (TATAAT) was identified, as was a degenerate −35 site (TgGgCA, where the lowercase nucleotides indicate the nonconserved positions) (Fig. 4).
The salKR promoter responds to human serum.
Genes in the sal locus are important for the full virulence of S. pyogenes strain HSC5 (16). To determine if the identified salKR promoter was affected by host factors, we tested the expression of the salKR promoter (full length with all regulatory sequences) in the wild-type strain in the presence of human serum compared to that in the presence of fetal bovine serum. A significant (P < 0.05) increase in expression of the salKR promoter was detected when it was exposed to human serum (Fig. 5). In an effort to determine what factors in the human serum were responsible for the increase in promoter activity, the serum was inactivated by heat, which destroys the complement factors found in the serum, or charcoal stripped, which results in removal of hormones. Even under these conditions promoter activity was higher than that in THY medium alone or with fetal bovine serum, suggesting that factors in human serum other than complement or hormones are responsible for the increased promoter activity.
FIG 5.

salKR promoter expression is controlled by host factors. Alkaline phosphatase activities produced by WT S. pyogenes expressing full-length pMNN1-salKR-pro after 3 h of treatment under several conditions: with THY broth (ThyB), fetal bovine serum (FBS), human serum, heat-inactivated human serum (HiS), and charcoal-stripped human serum. Error bars indicate SEMs. *, P < 0.05.
DISCUSSION
This study demonstrated that there are two promoters controlling the expression of the sal locus of S. pyogenes: one located upstream of the salA gene controlling the expression of the entire locus and another promoter located within the salY gene controlling the expression of the downstream salKRZ genes. This organization is similar to that found in other lantibiotic loci (14, 25–28). As shown by RT-PCR, the upstream operon promoter provides a read-through transcript, demonstrating that the salAMTXYKRZ genes can be cotranscribed as a single transcript. The presence of the internal promoter suggests that the expression of salKRZ could be regulated independently from the upstream operon promoter under different physiological conditions.
Analysis of promoter activity in truncated versions of the salKR promoter suggested possible repression sites located both upstream and downstream of the promoter region. Deletion of both putative repression sites resulted in a 6-fold increase in salKR promoter expression. These results suggest that the salKR promoter requires tight regulation, indicating the importance of regulated expression of the downstream genes encoding the two-component signaling system salKR. Previous studies demonstrated that the SalKR two-component system is required for the full virulence of pathogenic bacteria like S. suis (29) and S. pyogenes (16).
As a transcriptional regulator, SalR represses its own salKR promoter, as demonstrated by the increased expression of the promoter in the absence of the SalR protein. This feedback-loop repression suggests that this promoter is expressed transiently only under certain conditions. Our finding correlates with the microarray data from S. pyogenes growth in human blood reporting that the salR transcript is increased 50-fold after only 30 min of exposure to blood but reduced to 1.28-fold after 60 min and then upregulated again to 30-fold after 90 min of exposure (30). The complex regulation of the salKR promoter may be to ensure that the downstream genes, salKRZ, are expressed only at the appropriate time. Further investigation is required to identify possible global regulatory functions of SalR. We propose that SalR may also regulate genes other than those in the sal operon. This hypothesis is supported by the microarray data from S. suis showing that several genes encoding transporters and recombination proteins are downregulated in the absence of salKR (29). In addition, a recent mariner-based transposon mutagenesis screen designed to identify S. pyogenes factors required for survival in human blood identified a SalK mutant that was deficient for hemolysin expression in an M1T1 strain (31). However, the HSC5 serotype M14 strain used in the current analysis did not show a requirement for SalK in hemolysin expression, suggesting diversity in virulence requirements between S. pyogenes M types.
While most lantibiotic regulatory proteins perform a transcriptional activation role during lantibiotic production, the Enterococcus faecalis cytolysin regulator (CylR2) acts in transcriptional repression on both the cylL and cylR promoters of the cytolysin operon (32). The helix-turn-helix (H-T-H) domain of CylR2 was shown to bind specifically to the two inverted repeats located in the intergenic region between the cylL and cylR promoters to repress transcription of the cytolysin genes (15, 33). The accumulation of an active cytolysin, CylLs, signals the derepression of CylR2, resulting in increased production of cytolysin. The CylLs cytolysin is a lantibiotic-family toxin that has activity against both prokaryotic and eukaryotic cells. Several Gram-positive bacteria are highly sensitive to the bactericidal activity of the cytolysin, giving E. faecalis a competitive advantage in its ecological niche (34). The cytolysin also contributes to the virulence of E. faecalis, as it is active against human erythrocytes and innate immune cells, such as mouse neutrophils and macrophages (35).
Previous studies demonstrated that none of the S. pyogenes serotypes, except M4, produce an active salivaricin A lantibiotic, even though a salA transcript was present (12, 14, 36), suggesting that the peptide may be produced but not modified to the active lantibiotic. This is most likely because of the mutations found in the salM gene, which encodes the lantibiotic-modifying enzyme in S. salivarius. However, our data suggest a role for the SalA peptide in S. pyogenes. As demonstrated above, SalR represses its own promoter, and generally, feedback-loop repression, such as in the Lac operon, involves a cofactor or a corepressor (37). Our study suggests that the SalA peptide may play a role in corepression at the salKR promoter, as determined by the promoter expression analysis, which showed increased salKR promoter expression in the absence of SalA. This effect appeared to be additive, as a strain with a double deletion of salA and salKR showed the highest promoter expression (data not shown). For this to occur, localization of SalA may be intracellular or acting extracellularly through a sensor outside the salivaricin locus. We have not yet been able to determine whether SalA is intracellular or extracellular using an antibody against a synthesized SalA peptide (data not shown), suggesting that SalA may be modified by S. pyogenes and therefore cannot be recognized by the antibody. Another possibility for the role of SalA would be as a quorum-sensing regulator, and therefore, promoter activities in late-log-phase and stationary-phase cultures of the wild-type strain were also tested (data not shown). However, there was no strong evidence suggesting that quorum sensing is playing a role at this locus.
Mapping of the transcription start site of the salKR promoter revealed a well-conserved −10 region (TATAAT) with a degenerate −35 region (TgGgCA, where the lowercase nucleotides indicate the mutated positions). The promoter also contained a sequence for an extended −10 promoter region (TNTGNTATAAT) with one nucleotide mismatch to the sequence found in the S. mutans mutacin II locus but an exact match to the sequence in the S. pneumoniae DpnII operon (38, 39). The extended −10 promoter sequence is also found in front of other lantibiotic promoters, including the mutR promoter of the S. mutans mutacin II operon and the nisR promoter from the nisin locus in Lactococcus lactis (25, 40). Interestingly, while the nisR promoter is constitutively expressed, the mutR promoter is controlled by an upstream repeat sequence that is a proposed consensus binding site for a LytR-family regulatory protein (39). The complex transcriptional regulation of lantibiotic two-component systems is also in evidence in the subtilin locus of Bacillus subtilis. In this locus, the spaKR promoter is regulated by an alternative sigma factor, sigma factor H, which is in turn inhibited by a transition state regulator, ArbB (6). Therefore, the regulation of lanKR promoter regions appears to be specific for each operon. Notably, the regulation of the salKR promoter is not affected by the S. pyogenes global regulator CovR (41), Mga (42), or Rgg (43), as salKR promoter expression did not change when analyzed in strains carrying mutations in these regulators under the conditions tested (data not shown). However, this does not rule out a role for these regulators in vivo.
The salKR promoter demonstrated increased promoter activity when grown in medium containing human serum. The signaling molecule(s) responsible for the increased expression is a heat-stable, nonhormonal molecule that is unique to human serum, as the same result was not observed with fetal bovine serum. This response suggests that the sal locus in S. pyogenes is involved in sensing and responding to host-pathogen interactions in vivo, which is supported by the increased expression of the sal locus genes observed during an acute and chronic infection in a macaque pharyngitis model (44) as well as during growth in human blood (30).
In conclusion, this study demonstrated that even in the absence of production of a functional lantibiotic, the sal locus genes of S. pyogenes are expressed and differentially regulated. The complex regulation observed for the salKR promoter suggests the importance of the appropriate expression of the downstream genes salKRZ. Several bacteriocin-like loci found in bacterial pathogens have been implicated in virulence, such as the sag operon in S. pyogenes (45), the salKR locus in S. suis (29), the enterococcal cytolysin in E. faecalis (15, 46), and the Bsa bacteriocin locus in Staphylococcus aureus (47). Therefore, further study of the S. pyogenes sal locus in vivo is necessary to fully understand its role in pathogenesis during host-pathogen interactions.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI078147 from the National Institute of Allergy and Infectious Diseases (NIAID).
Special thanks go to M.D. fellow Ramesh Bharadwaj and undergraduate researcher Mouhammadou Seydi for their valuable technical assistance.
Footnotes
Published ahead of print 15 November 2013
REFERENCES
- 1.Jack RW, Sahl HG. 1995. Unique peptide modifications involved in the biosynthesis of lantibiotics. Trends Biotechnol. 13:269–278. 10.1016/S0167-7799(00)88962-3 [DOI] [PubMed] [Google Scholar]
- 2.Willey JM, van der Donk WA. 2007. Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 61:477–501. 10.1146/annurev.micro.61.080706.093501 [DOI] [PubMed] [Google Scholar]
- 3.Sahl HG, Jack RW, Bierbaum G. 1995. Biosynthesis and biological activities of lantibiotics with unique post-translational modifications. Eur. J. Biochem. 230:827–853. 10.1111/j.1432-1033.1995.tb20627.x [DOI] [PubMed] [Google Scholar]
- 4.Draper LA, Ross RP, Hill C, Cotter PD. 2008. Lantibiotic immunity. Curr. Protein Pept. Sci. 9:39–49. 10.2174/138920308783565750 [DOI] [PubMed] [Google Scholar]
- 5.Stein T, Heinzmann S, Kiesau P, Himmel B, Entian KD. 2003. The spa-box for transcriptional activation of subtilin biosynthesis and immunity in Bacillus subtilis. Mol. Microbiol. 47:1627–1636. 10.1046/j.1365-2958.2003.03374.x [DOI] [PubMed] [Google Scholar]
- 6.Stein T, Borchert S, Kiesau P, Heinzmann S, Kloss S, Klein C, Helfrich M, Entian KD. 2002. Dual control of subtilin biosynthesis and immunity in Bacillus subtilis. Mol. Microbiol. 44:403–416. 10.1046/j.1365-2958.2002.02869.x [DOI] [PubMed] [Google Scholar]
- 7.Corvey C, Stein T, Dusterhus S, Karas M, Entian KD. 2003. Activation of subtilin precursors by Bacillus subtilis extracellular serine proteases subtilisin (AprE), WprA, and Vpr. Biochem. Biophys. Res. Commun. 304:48–54. 10.1016/S0006-291X(03)00529-1 [DOI] [PubMed] [Google Scholar]
- 8.Kleerebezem M. 2004. Quorum sensing control of lantibiotic production; nisin and subtilin autoregulate their own biosynthesis. Peptides 25:1405–1414. 10.1016/j.peptides.2003.10.021 [DOI] [PubMed] [Google Scholar]
- 9.Siezen RJ, Kuipers OP, de Vos WM. 1996. Comparison of lantibiotic gene clusters and encoded proteins. Antonie Van Leeuwenhoek 69:171–184. 10.1007/BF00399422 [DOI] [PubMed] [Google Scholar]
- 10.Ross KF, Ronson CW, Tagg JR. 1993. Isolation and characterization of the lantibiotic salivaricin A and its structural gene salA from Streptococcus salivarius 20P3. Appl. Environ. Microbiol. 59:2014–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wescombe PA, Heng NC, Burton JP, Chilcott CN, Tagg JR. 2009. Streptococcal bacteriocins and the case for Streptococcus salivarius as model oral probiotics. Future Microbiol. 4:819–835. 10.2217/fmb.09.61 [DOI] [PubMed] [Google Scholar]
- 12.Wescombe PA, Upton M, Dierksen KP, Ragland NL, Sivabalan S, Wirawan RE, Inglis MA, Moore CJ, Walker GV, Chilcott CN, Jenkinson HF, Tagg JR. 2006. Production of the lantibiotic salivaricin A and its variants by oral streptococci and use of a specific induction assay to detect their presence in human saliva. Appl. Environ. Microbiol. 72:1459–1466. 10.1128/AEM.72.2.1459-1466.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phelps HA, Neely MN. 2005. Evolution of the zebrafish model: from development to immunity and infectious disease. Zebrafish 2:87–103. 10.1089/zeb.2005.2.87 [DOI] [PubMed] [Google Scholar]
- 14.Upton M, Tagg JR, Wescombe P, Jenkinson HF. 2001. Intra- and interspecies signaling between Streptococcus salivarius and Streptococcus pyogenes mediated by SalA and SalA1 lantibiotic peptides. J. Bacteriol. 183:3931–3938. 10.1128/JB.183.13.3931-3938.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox CR, Coburn PS, Gilmore MS. 2005. Enterococcal cytolysin: a novel two component peptide system that serves as a bacterial defense against eukaryotic and prokaryotic cells. Curr. Protein Pept. Sci. 6:77–84. 10.2174/1389203053027557 [DOI] [PubMed] [Google Scholar]
- 16.Phelps HA, Neely MN. 2007. SalY of the Streptococcus pyogenes lantibiotic locus is required for full virulence and intracellular survival in macrophages. Infect. Immun. 75:4541–4551. 10.1128/IAI.00518-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caparon MG, Geist RT, Perez-Casal J, Scott JR. 1992. Environmental regulation of virulence in group A streptococci: transcription of the gene encoding M protein is stimulated by carbon dioxide. J. Bacteriol. 174:5693–5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caparon MG, Scott JR. 1991. Genetic manipulation of the pathogenic streptococci. Methods Enzymol. 204:556–586. 10.1016/0076-6879(91)04028-M [DOI] [PubMed] [Google Scholar]
- 19.Neely M, Pfeifer J, Caparon MG. 2002. Streptococcus-zebrafish model of bacterial pathogenesis. Infect. Immun. 70:3904–3914. 10.1128/IAI.70.7.3904-3914.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21.Granok AB, Parsonage D, Ross RP, Caparon MG. 2000. The RofA binding site in Streptococcus pyogenes is utilized in multiple transcriptional pathways. J. Bacteriol. 182:1529–1540. 10.1128/JB.182.6.1529-1540.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King KY, Horenstein JA, Caparon MG. 2000. Aerotolerance and peroxide resistance in peroxidase and PerR mutants of Streptococcus pyogenes. J. Bacteriol. 182:5290–5299. 10.1128/JB.182.19.5290-5299.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz N, Wang B, Pentland A, Caparon MG. 1998. Streptolysin O and adherence synergistically modulate proinflammatory responses of keratinocytes to group A streptococci. Mol. Microbiol. 27:337–346. 10.1046/j.1365-2958.1998.00681.x [DOI] [PubMed] [Google Scholar]
- 24.Kuipers OP, Beerthuyzen MM, de Ruyter PG, Luesink EJ, de Vos WM. 1995. Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J. Biol. Chem. 270:27299–27304. 10.1074/jbc.270.45.27299 [DOI] [PubMed] [Google Scholar]
- 25.de Ruyter PG, Kuipers OP, Beerthuyzen MM, van Alen-Boerrigter I, de Vos WM. 1996. Functional analysis of promoters in the nisin gene cluster of Lactococcus lactis. J. Bacteriol. 178:3434–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altena K, Guder A, Cramer C, Bierbaum G. 2000. Biosynthesis of the lantibiotic mersacidin: organization of a type B lantibiotic gene cluster. Appl. Environ. Microbiol. 66:2565–2571. 10.1128/AEM.66.6.2565-2571.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung YJ, Hansen JN. 1992. Determination of the sequence of spaE and identification of a promoter in the subtilin (spa) operon in Bacillus subtilis. J. Bacteriol. 174:6699–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staron A, Finkeisen DE, Mascher T. 2011. Peptide antibiotic sensing and detoxification modules of Bacillus subtilis. Antimicrob. Agents Chemother. 55:515–525. 10.1128/AAC.00352-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li M, Wang C, Feng Y, Pan X, Cheng G, Wang J, Ge J, Zheng F, Cao M, Dong Y, Liu D, Lin Y, Du H, Gao GF, Wang X, Hu F, Tang J. 2008. SalK/SalR, a two-component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS One 3:e2080. 10.1371/journal.pone.0002080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham MR, Virtaneva K, Porcella SF, Barry WT, Gowen BB, Johnson CR, Wright FA, Musser JM. 2005. Group A Streptococcus transcriptome dynamics during growth in human blood reveals bacterial adaptive and survival strategies. Am. J. Pathol. 166:455–465. 10.1016/S0002-9440(10)62268-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Breton Y, Mistry P, Valdes KM, Quigley J, Kumar N, Tettelin H, McIver KS. 2013. Genome-wide identification of genes required for fitness of group A Streptococcus in human blood. Infect. Immun. 81:862–875. 10.1128/IAI.00837-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haas W, Shepard BD, Gilmore MS. 2002. Two-component regulator of Enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature 415:84–87. 10.1038/415084a [DOI] [PubMed] [Google Scholar]
- 33.Rumpel S, Razeto A, Pillar CM, Vijayan V, Taylor A, Giller K, Gilmore MS, Becker S, Zweckstetter M. 2004. Structure and DNA-binding properties of the cytolysin regulator CylR2 from Enterococcus faecalis. EMBO J. 23:3632–3642. 10.1038/sj.emboj.7600367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jett BD, Gilmore MS. 1990. The growth-inhibitory effect of the Enterococcus faecalis bacteriocin encoded by pAD1 extends to the oral streptococci. J. Dent. Res. 69:1640–1645. 10.1177/00220345900690100301 [DOI] [PubMed] [Google Scholar]
- 35.Miyazaki S, Ohno A, Kobayashi I, Uji T, Yamaguchi K, Goto S. 1993. Cytotoxic effect of hemolytic culture supernatant from Enterococcus faecalis on mouse polymorphonuclear neutrophils and macrophages. Microbiol. Immunol. 37:265–270 [DOI] [PubMed] [Google Scholar]
- 36.Johnson DW, Tagg JR, Wannamaker LW. 1979. Production of a bacteriocine-like substance by group-A streptococci of M-type 4 and T-pattern 4. J. Med. Microbiol. 12:413–427. 10.1099/00222615-12-4-413 [DOI] [PubMed] [Google Scholar]
- 37.Wilson CJ, Zhan H, Swint-Kruse L, Matthews KS. 2007. The lactose repressor system: paradigms for regulation, allosteric behavior and protein folding. Cell. Mol. Life Sci. 64:3–16. 10.1007/s00018-006-6296-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabelnikov AG, Greenberg B, Lacks SA. 1995. An extended −10 promoter alone directs transcription of the DpnII operon of Streptococcus pneumoniae. J. Mol. Biol. 250:144–155. 10.1006/jmbi.1995.0366 [DOI] [PubMed] [Google Scholar]
- 39.van der Ploeg JR. 2005. Regulation of bacteriocin production in Streptococcus mutans by the quorum-sensing system required for development of genetic competence. J. Bacteriol. 187:3980–3989. 10.1128/JB.187.12.3980-3989.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi F, Chen P, Coufield PW. 1999. Functional analyses of the promoters in the lantibiotic mutacin II biosynthetic locus in Streptococcus mutans. Appl. Environ. Microbiol. 65:652–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. U. S. A. 99:13855–13860. 10.1073/pnas.202353699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McIver KS, Thurman AS, Scott JR. 1999. Regulation of mga transcription in the group A streptococcus: specific binding of Mga within its own promoter and evidence for a negative regulator. J. Bacteriol. 181:5373–5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smoot LM, Smoot JC, Graham MR, Somerville GA, Sturdevant DE, Migliaccio CA, Sylva GL, Musser JM. 2001. Global differential gene expression in response to growth temperature alteration in group A Streptococcus. Proc. Natl. Acad. Sci. U. S. A. 98:10416–10421. 10.1073/pnas.191267598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virtaneva K, Porcella SF, Graham MR, Ireland RM, Johnson CA, Ricklefs SM, Babar I, Parkins LD, Romero RA, Corn GJ, Gardner DJ, Bailey JR, Parnell MJ, Musser JM. 2005. Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc. Natl. Acad. Sci. U. S. A. 102:9014–9019. 10.1073/pnas.0503671102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, de Azavedo JCS. 2000. Genetic locus for streptolysin S production by group A streptococcus. Infect. Immun. 68:4245–4254. 10.1128/IAI.68.7.4245-4254.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilmore MS, Segarra RA, Booth MC, Bogie CP, Hall LR, Clewell DB. 1994. Genetic structure of the Enterococcus faecalis plasmid pAD1-encoded cytolytic toxin system and its relationship to lantibiotic determinants. J. Bacteriol. 176:7335–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daly KM, Upton M, Sandiford SK, Draper LA, Wescombe PA, Jack RW, O'Connor PM, Rossney A, Gotz F, Hill C, Cotter PD, Ross RP, Tagg JR. 2010. Production of the Bsa lantibiotic by community-acquired Staphylococcus aureus strains. J. Bacteriol. 192:1131–1142. 10.1128/JB.01375-09 [DOI] [PMC free article] [PubMed] [Google Scholar]