

Abstract
Nitrogen fixation is a tightly regulated trait. Switching from N2 fixation-repressing conditions to the N2-fixing state is carefully controlled in diazotrophic bacteria mainly because of the high energy demand that it imposes. By using quantitative real-time PCR and quantitative immunoblotting, we show here how nitrogen fixation (nif) gene expression develops in Azotobacter vinelandii upon derepression. Transient expression of the transcriptional activator-encoding gene, nifA, was followed by subsequent, longer-duration waves of expression of the nitrogenase biosynthetic and structural genes. Importantly, expression timing, expression levels, and NifA dependence varied greatly among the nif operons. Moreover, the exact concentrations of Nif proteins and their changes over time were determined for the first time. Nif protein concentrations were exquisitely balanced, with FeMo cofactor biosynthetic proteins accumulating at levels 50- to 100-fold lower than those of the structural proteins. Mutants lacking nitrogenase structural genes or impaired in FeMo cofactor biosynthesis showed overenhanced responses to derepression that were proportional to the degree of nitrogenase activity impairment, consistent with the existence of at least two negative-feedback regulatory mechanisms. The first such mechanism responded to the levels of fixed nitrogen, whereas the second mechanism appeared to respond to the levels of the mature NifDK component. Altogether, these findings provide a framework to engineer N2 fixation in nondiazotrophs.
INTRODUCTION
The Mo nitrogenase is responsible for most biological nitrogen fixation (BNF) activity in the biosphere, and due to its great agronomic importance, it has been the subject of thorough genetic and biochemical studies. Although BNF is carried out by only a few species of bacteria and archaea denominated as diazotrophs, there is much interest in the possibility of engineering N2 fixation activity into nondiazotrophic organisms, including crop plants (1).
The Mo nitrogenase is composed of two oxygen-sensitive metalloproteins designated NifDK and NifH. The NifDK component is a tetramer of the nifD and nifK gene products that carries one iron-molybdenum cofactor (FeMo-co [7Fe-9S-Mo-C-homocitrate]) buried in the active site of each NifD subunit and one P cluster ([8Fe-7S] cluster) at each NifD-NifK dimer interface (2). NifH is a dimer of the nifH gene product that carries one [4Fe-4S] cluster at the subunit interface and serves as an obligate electron donor to the NifDK component (3).
A number of nitrogen fixation (nif) gene products work coordinately in the assembly of nitrogenase components in Azotobacter vinelandii and other diazotrophs. NifH biogenesis solely requires the activities of the NifU/NifS machinery to provide its [4Fe-4S] cluster (4) and NifM (5). In contrast, many Nif proteins, including NifH, are involved in NifDK biogenesis, mainly due to the structural complexity of FeMo-co (Fig. 1). NifU and NifS provide NifB with [Fe-S] cluster substrates to generate the NifB cofactor (NifB-co), a biosynthetic intermediate, not only for FeMo-co but also for the analogous FeV cofactor (FeV-co) and FeFe cofactor (FeFe-co) of the V and Fe nitrogenases, respectively (6–8). NifB-co is then transferred to NifEN, where additional Fe, Mo, and homocitrate are subsequently incorporated into the cofactor with the aid of NifQ, NifH, and NifV activities (9, 10). Completed FeMo-co is finally incorporated into a FeMo-co-deficient apo-NifDK via NafY to yield the holo-NifDK protein (11). Importantly, NafY has a second role as a chaperone that stabilizes FeMo-co-deficient apo-NifDK in preparation for FeMo-co insertion (12).
FIG 1.
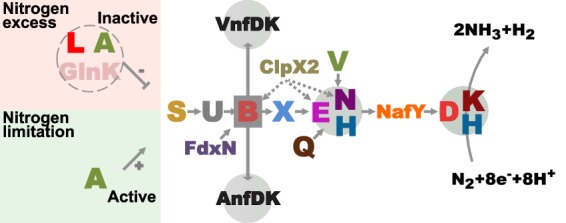
Schematic model of proteins involved in the formation of active nitrogenases in A. vinelandii. Nif proteins are indicated by using the last letter of their names. Gray circles, protein complexes containing NifB-co-derived cofactors (FeMo-co, FeV-co, and FeFe-co); dotted lines, regulation by proteolytic degradation. The function of Nif proteins has been reviewed elsewhere (9, 10).
Nitrogenase biosynthesis and N2 fixation activity are energy-demanding processes (13). Thus, nif gene expression is tightly controlled by the bacterium. The NifA activator is an enhancer-binding protein that binds to specific DNA sequences upstream of nif genes (upstream activator sequences [UASs]) and interacts with the σ54 subunit of the RNA polymerase to initiate transcription (14, 15). NifA is active under nitrogen-limiting conditions. The NifL antiactivator is a flavoprotein that senses cellular redox status and binds NifA to block its activity when conditions are not optimal for N2 fixation. Conditions of nitrogen excess also lead to inactive NifA protein through the formation of a GlnK/NifL/NifA complex. This regulatory mechanism also applies to changes in nitrogen status derived from endogenously fixed nitrogen. Thus, expression of the NifA regulon should respond to the level of nitrogenase activity, and Nif-minus mutants are likely to lack appropriate feedback regulatory mechanisms.
The nif regulon is defined as nifA and those genes under the control of NifA that are responsible for the production of a functional Mo nitrogenase. In A. vinelandii, nif genes are clustered in two chromosomal regions comprising at least eight operons (7, 16–18). The major nif cluster contains the nifHDKTYENX, iscAnif, nifUSV, cysE1nif, nifWZM, clpX2, and nifF genes and other interspersed open reading frames. The minor nif cluster contains the nifLAB, fdxN, nifOQ, rhdN, and grx5nif genes in one transcriptional direction and a divergent rnfABCDGEH and nafY gene cluster.
A genome-wide transcription profile analysis comparing steady-state gene expression levels in N2-fixing A. vinelandii cells versus NH4+-assimilating cells has recently been carried out and revealed that 400 genes are differentially expressed under Mo-dependent diazotrophic growth conditions (19). A synthetic biology approach to reorganize and simplify the regulation of nif genes in Klebsiella pneumoniae has recently been reported (20). With the goal of providing a framework to engineer N2 fixation in nondiazotrophs, we have investigated the time course of expression of nitrogenase components in A. vinelandii cells initiating diazotrophic growth, including the exact concentrations of Nif proteins and their variations over time.
MATERIALS AND METHODS
A. vinelandii strains and growth conditions.
Strains UW140 (ΔnifB) (21), DJ33 (ΔnifDK) (22), and UW356 (ΔnifA::spc) (23) have been previously described. Strain UW344, bearing an in-frame deletion of fdxN (ΔfdxN), was generated as described in the supplemental material.
A. vinelandii strains were cultivated in Burk's modified medium (24) at 30°C with or without nitrogen sources, as indicated. Antibiotics were added at standard concentrations (23).
For nitrogenase derepression experiments (diazotrophic growth conditions), NH4+-grown cells were diluted in fresh NH4+-containing medium at an optical density at 600 nm (OD600) of 0.005 and further cultivated for 17 h. Cells were then collected by centrifugation, washed with NH4+-free or NH4+-containing medium (control cultures), and resuspended in the same medium at a final OD600 of 0.5. Cells were then incubated at 30°C with shaking (200 rpm) in independent Erlenmeyer flasks. At different times, samples were collected and subjected to the following analyses: determination of in vivo acetylene reduction activity, cell volume determination, OD600 determination, quantitative real-time PCR (qRT-PCR), and quantitative immunoblotting.
Plasmid constructions and DNA manipulations.
Plasmid constructions, PCR, and transformation of Escherichia coli were carried out by standard methods (25). E. coli strains were cultivated in Luria-Bertani medium at 37°C with shaking (200 rpm). Antibiotics were added at standard concentrations (25).
Plasmid pRHB470 carries a synthetic DNA fragment containing the amplicons from the nifH, nifD, nifK, nifY, nifE, nifN, nifX, nifU, nifS, nifV, nifA, nifB, fdxN, nifQ, and nafY genes, tested in qRT-PCRs, cloned into the NotI site of pUC57. Plasmid pRHB506 carries a synthetic DNA fragment containing the amplicons from 16S rRNA and nifL genes cloned into the NotI site of pUC57. Plasmids pRHB470 and pRHB506, used as standards in qRT-PCRs, were generated by Proteogenix and GenScript, respectively.
To generate the ΔfdxN mutant, an in-frame fdxN deletion was introduced into the A. vinelandii chromosome. Flanking regions of the fdxN gene were generated by PCR using the oligonucleotides 5′-GCGGAATTCGCCTGATCAACCAGCACTTC-3′ and 5′-CCAGGATCCACCTCCACGCAATCCTCGAAT-3′ for the nifB region upstream of fdxN and 5′-CCCGGATCCGAGAGTCGCCATGACCAGCA-3′ and 5′-CTCGAATTCGAGGTGGTTGGCTCTGTCAT-3′ for the nifO region downstream of fdxN. The EcoRI (GAATTC) and BamHI (GGATCC) restriction sites incorporated into the oligonucleotides to facilitate vector construction are shown underlined in the oligonucleotide sequences. The resulting products were digested with EcoRI-BamHI and cloned into pGEM11Zf(+) (Promega) either by triple ligation reactions to generate plasmid pRHB312 or by quadruple ligations together with a BamHI-digested kanamycin resistance cassette to generate pRHB308.
The procedures used for A. vinelandii transformation and gene replacement have been described previously (26). Plasmid pRHB308 was introduced into the chromosome of the A. vinelandii DJ strain by homologous recombination to generate strain UW353. Strain UW344, bearing an in-frame ΔfdxN mutation, was isolated by cotransforming (congression) strain UW353 with plasmids pRHB312 and pDB303, which contains a rifampin resistance marker (kindly provided by D. Dean). Preliminary identification of ΔfdxN mutants was verified on rifampin-containing solid medium by the phenotypic loss of kanamycin resistance, which was further confirmed by PCR analysis and sequencing of the locus using genomic DNA of the mutant strains. Isolation of genomic DNA from A. vinelandii strains was performed with a DNeasy tissue kit (Qiagen).
All DNA constructions were confirmed by restriction analysis and by DNA sequencing. The A. vinelandii strains generated were confirmed by PCR analysis and, when possible, by immunoblot analysis with appropriate antibodies.
RNA isolation and absolute qRT-PCR analyses.
The A. vinelandii DJ (wild type), ΔfdxN, ΔnifA, ΔnifB, and ΔnifDK strains were grown as described above. Each biological replicate for qRT-PCR experiments consisted of a 20-ml cell culture harvested by centrifugation at 5,000 × g for 5 min at 4°C, quickly frozen in liquid N2, and stored at −80°C. Total RNA was isolated using a RiboPure-Bacteria kit (Ambion, Life Technologies) according to the manufacturer's instructions. Genomic DNA traces were digested using the Turbo DNA-free reagent (Ambion, Life Technologies). cDNA synthesis was carried out using a High Capacity cDNA reverse transcription kit (Ambion, Life Technologies) following the manufacturer's instructions. Each reaction mixture used 2 μg of total RNA. An aliquot of 1 μl of a 1:10 dilution of the cDNA (1:10,000 for the reference 16S rRNA gene) was used as the template for qRT-PCR amplification.
qRT-PCR experiments were performed using Power SYBR green PCR master mix (Ambion, Life Technologies). The final volume of the reaction mixture was 15 μl per well. A standard Applied Biosystems cycle protocol was used. All samples were run in an Eco real-time PCR system (Illumina). Gene-specific primers were designed with the Primer Express (v2.0) program (Ambion, Life Technologies). Primers with annealing temperatures of between 58°C and 62°C, which amplified products with lengths of ca. 100 bp, were selected and tested for specificity using the BLAST program. The gene-specific primers used for qRT-PCR are listed in Table S1 in the supplemental material. Data acquisition was performed using Eco real-time PCR system software (v4.1; Illumina). At least three independent biological replicates were used for each experiment.
In order to design DNA standards for absolute quantification calculations (27), the efficiency of amplification for each gene was determined using as the template cDNA from A. vinelandii DJ grown for 1 h under diazotrophic conditions. Only primers with optimal efficiency were used to design synthetic DNA standards. Plasmids pRHB470 and pRHB506, carrying DNA standards, were linearized using the HindIII restriction enzyme, purified, aliquoted, and stored at 10 pg/μl in diethyl pyrocarbonate-treated H2O (Ambion, Life Technologies). The dynamic range of the calibration curves was 1,000-fold (10 pg, 5 pg, 1 pg, 0.1 pg, and 0.01 pg).
Sample concentrations were determined by interpolating their quantification cycle (Cq) values into the corresponding standard curve. To compensate for intra- and interkinetic qRT-PCR variations (sample-to-sample and run-to-run variations), data normalization was performed using the endogenous unregulated gene transcript of 16S rRNA as a reference. The absolute abundance of each gene transcript was divided by the absolute value for 16S rRNA to obtain normalized absolute signal (NAS) values to allow direct comparison of different samples.
DNA motif analysis.
Five-hundred-base-pair sequences upstream of the 5′ ATG codon of A. vinelandii nif operons were retrieved with the Genome and Gene Information regulatory sequence analysis (RSA) tools to identify specific promoter consensus sequences using pattern-matching tools (one substitution allowed) (http://rsat.ulb.ac.be). Consensus sequences 5′-YTGGCACGR-(N)3-TTGCW for RpoN (28), 5′-WATCAANNNNTTR for the integration host factor (IHF) (29), and 5′-TGT-(N)10-ACA for the NifA-dependent UAS (14) were used to obtain frequency matrices of NifA binding sites upstream of A. vinelandii nif promoters. These motifs were assembled into frequency matrices using the pattern assembly tool with default parameters. Matrices were converted into consensus motifs using the convert matrix tool and were displayed using the WebLogo program (30). Positional weight matrices were used to determine the positions and numbers of NifA DNA binding sites in A. vinelandii by using matrix-scan RSA tools.
Controls were set with 10 in silico experiments in which 100 random sequences of 500 bp in length from the A. vinelandii genome were retrieved with build control RSA tools (http://rsat.ulb.ac.be) and analyzed using matrix-scan RSA tools.
Protein methods.
The protein concentration was determined by the bicinchoninic acid method with bovine serum albumin as the standard (31). Procedures for SDS-PAGE (32) and immunoblot analysis (33) have been described previously. Protein samples for immunoblot analyses were prepared by mixing pelleted cells with Tris-HCl (100 mM) at a final OD600 of 2 and then diluting the samples with 2× Laemmli sample buffer supplemented with 0.1 M dithiothreitol. Fifteen microliters of this mix was loaded per line. Absolute quantification of each protein was performed using a calibration curve obtained with known amounts of the same protein in a purified state. ImageJ software was used to quantify the protein levels in immunoblot membranes.
Purification of the NifB, NifEN, NifH, NifDK, and NifQ proteins from nif-derepressed A. vinelandii cells was performed under anaerobic conditions. NifB was purified from UW232 cells (21), NifEN was purified from UW243 cells (34), NifH and NifDK were purified from DJ cells (35), and NifQ was purified from UW300 cells (36). NifU was purified under anaerobic conditions from BL21 Rosetta(pRHB252) cells. NifX and NafY and were purified under aerobic conditions from BL21(pREP-4)(pRHB155) cells (34) and from BL21(pREP-4)(pRHB62) cells (11), respectively.
Nitrogenase activity.
In vivo nitrogenase activities were determined by the acetylene reduction assay at 30°C for 15 min using 1 ml of culture samples, as described previously (37).
Cell volume measurements.
A. vinelandii cell number and volume were measured with a ZM Coulter Counter with a 30-μm-diameter orifice connected to a C1000 Channelyzer apparatus (Coulter Electronics) after fixing the cells in 0.75% formaldehyde. For each sample, the median of the cell volume and cell number distribution was recorded. One hundred cells were analyzed for each time point.
RESULTS
Time course of nif gene expression upon nitrogen step-down.
The absolute levels of accumulation of nif-specific mRNAs were estimated by quantitative real-time PCR (qRT-PCR) by comparison with the results obtained with known amounts of synthetic DNA amplicons. mRNA levels were then compared to the 16S rRNA level present in each sample to yield normalized absolute signals (NASs). Figure 2 and Fig. S1 in the supplemental material show that A. vinelandii responded rapidly to nitrogen step-down by expressing nif genes and developing nitrogenase activity. The nifLA operon responded fast to nitrogen step-down, with nifA exhibiting a narrow wave of expression that peaked 30 min after NH4+ removal from the medium (Fig. 2D). Expression of genes within the iscAnifnifUSV-cysE1nif-nifWZM-clpX2 cluster also peaked after 30 min, exhibiting NAS values for nifU, nifS, nifV, and clpX2 of 10, 7.3, 3.6, and 1.8, respectively (Fig. 2C). Most genes involved in FeMo-co biosynthesis (the nifENX and nifB-fdxN-nifOQ operons and the nafY gene) showed maximum mRNA levels 1 h after derepression with NAS values ranging from 2 to 7 (Fig. 2B, E, and F). Similarly, nitrogenase structural genes showed maximum mRNA levels 1 h after derepression, but NAS values were much higher, ranging from 30 to 50 (Fig. 2A). In contrast, nifY showed the slowest response, with an accumulation peak at 2 h and a NAS value of 4.3. Two main patterns of mRNA accumulation occurred when nitrogenase activity was at steady state (4 to 7 h after derepression; see Fig. S1 in the supplemental material): nifH, nifD, nifK, nifE, nifN, nifX, and nafY levels were at least 25% of their maximum NAS levels 7 h after derepression, whereas the levels for all other genes returned to the basal NAS levels.
FIG 2.
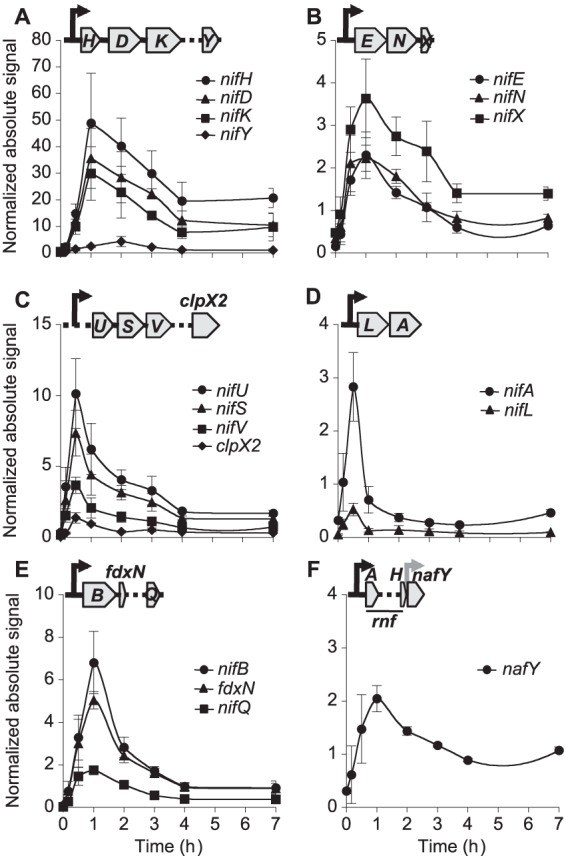
Time-dependent profile of induction of nif genes necessary for Mo nitrogenase in A. vinelandii. (A to C) Expression levels of genes within the major nif cluster, which contains nifHDKTYENX-iscAnif-nifUSV-cysE1nif-nifWZM-clpX2-nifF; (D to F) expression levels of genes within the minor nif cluster, which contains rnfABCDGEH-nafY in one DNA orientation and nifLAB-fdxN-nifOQ-rhdN-grx5nif in the opposite DNA orientation. In all cases, absolute expression levels were estimated by comparison to the levels on standard curves obtained with known amounts of synthetic DNA amplicons and were then normalized to 16S rRNA levels. Black arrows, predicted σ54-dependent promoter regions; gray arrow, a hypothetical constitutive promoter region; dotted lines, unstudied interspersed genes within each operon that have been omitted for clarity. Data are the averages of at least three biological replicates ± SEs.
No changes in nif mRNA levels were observed in control cultures of A. vinelandii growing in the presence of NH4+ 4 h after inoculation into fresh medium (see Table S2 in the supplemental material).
Operons belonging to the nif regulon can be identified by the presence of NifA-binding motifs or UASs (see Fig. S2 in the supplemental material). A correlation between the number of UASs present in nif promoter regions and accumulated mRNA levels was found. For example, the 500-bp upstream region controlling expression of nifHDKTY contains 3 UASs, whereas in silico control experiments determined a random frequency of 1.26 UASs for a 500-bp DNA region across the A. vinelandii genome. This correlation suggests positive cooperativity in NifA binding to UASs or in NifA activation of polymerase.
Time course of Nif protein accumulation upon nitrogen step-down.
The intracellular accumulation of a number of Nif proteins was estimated by quantifying the A. vinelandii whole-cell immunoblot signals against a standard curve generated by immunoblotting known amounts of the corresponding purified Nif protein (see Fig. S3 in the supplemental material) and dividing by the total cell volume in the sample. Consistent with its role in the general synthesis of [Fe-S] clusters for both nitrogenase structural and biosynthetic proteins, NifU accumulated rapidly, showing 90% of its maximum value of 23.7 μM 1 h after derepression (Fig. 3A). FeMo-co biosynthetic proteins showed relatively low accumulation levels, ranging from 0.4 μM for NifEN to 3.2 μM for NifB, 5.7 μM for NifQ, and 5.8 μM for NifX (Fig. 3B). On the other hand, nitrogenase structural proteins accumulated to very high levels 4 h after derepression: 108.5 μM for NifH, 54.8 μM for NifD, and 48.1 μM for NifK (Fig. 3C). The NafY chaperone that is involved in stabilizing FeMo-co-deficient apo-NifDK showed significant basal levels (8 μM) at the start of derepression, with levels increasing moderately over time (up to 19.9 μM).
FIG 3.
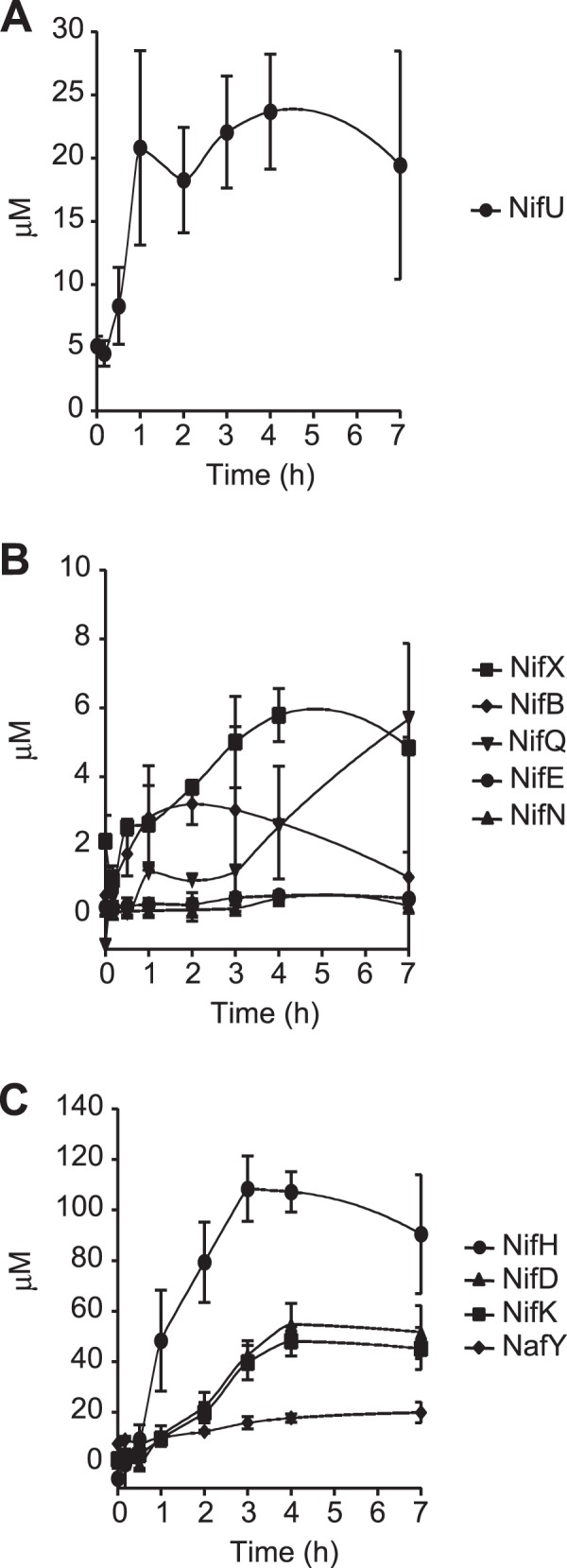
Time course of Nif protein accumulation in A. vinelandii. (A) NifU protein, required for the biosynthesis of [Fe-S] clusters for the nitrogenase components; (B) FeMo-co biosynthetic proteins; (C) nitrogenase structural proteins, including the NafY chaperone.
At the time of maximum in vivo nitrogenase activity (4 h after derepression), the nitrogenase proteins could be grouped into three main categories according to their levels: (i) low-concentration proteins involved in FeMo-co biosynthesis, NifE (0.4 μM), NifN (0.3 μM), NifB (2.7 μM), and NifQ (2.5 μM); (ii) midconcentration proteins dedicated to the transfer or storage of FeMo-co and its precursors, NifX (5.8 μM) and NafY (17.7 μM); and (iii) high-concentration proteins, NifD (54.8 μM), NifK (48.1 μM), and NifH (107.2 μM). Finally, NifU (23.7 μM) did not fit easily into the group of FeMo-co biosynthesis proteins, which is consistent with its role in providing [Fe-S] clusters to many different proteins.
The maximum mRNA concentration/maximum protein concentration ([mRNA]max/[protein]max) ratio was used as an index to understand the relationship between mRNA stability and protein accumulation (Table 1). Although this index varied between 0.3 and 0.6 in most cases, a few significant exceptions were found. The nafY mRNA/NafY index was 0.10, which suggested either a high degree of stability of the NafY chaperone or highly translated nafY mRNA. On the other hand, the nifN mRNA/NifN, nifE mRNA/NifE, and nifB mRNA/NifB indexes were 4.70, 4.16, and 2.12, respectively, consistent with targeted degradation of NifB and NifEN during diazotrophic growth by ClpX2 (38).
TABLE 1.
Comparison of [mRNA]max/[protein]max ratios between the wild-type and ΔfdxN A. vinelandii strains
| Protein | [mRNA]max/[protein]maxa |
|
|---|---|---|
| Wild type | ΔfdxN mutant | |
| NifU | 0.43 | 0.46 |
| NifB | 2.12 | 1.03 |
| NifX | 0.63 | 1.44 |
| NifE | 4.16 | 2.20 |
| NifN | 4.70 | 3.44 |
| NifQ | 0.31 | 0.11 |
| NafY | 0.10 | 0.08 |
| NifD | 0.65 | 1.58 |
| NifK | 0.62 | 1.36 |
| NifH | 0.45 | 0.62 |
Maximum levels of mRNA (NASs) and protein (μM) were used to calculate these ratios.
Gene expression in the ΔnifA mutant.
As expected, the ΔnifA mutant exhibited a Nif-minus phenotype and lacked in vivo nitrogenase activity. nifH, nifD, nifK, and nifY mRNA levels in the ΔnifA strain were negligible: 390-, 170-, 120-, and 130-fold lower than the level in the wild type, respectively (Fig. 4A). Similarly, the nifENX and nifB-fdxN-nifOQ operons showed large drops in expression compared to the levels in the wild type: 43-fold lower levels of mRNA for nifE, 75-fold lower levels for nifN, 20-fold lower levels for nifX, 270-fold lower levels for nifB, 44-fold lower levels for fdxN, and 17-fold lower levels for nifQ. Genes within the iscAnif-nifUSV-cysE1nif-nifWZM-clpX2 cluster were not as drastically dependent on NifA, since nifU, nifS, nifV, and clpX2 mRNA levels were 3, 4, 9, and 4 times lower than those in the in wild type, respectively. However, their time course expression profiles showed some response to nitrogen step-down, exhibiting mRNA induction ratios of 152, 66, 29, and 19 for nifU, nifS, nifV, and clpX2, respectively (see Fig. S4 in the supplemental material). Finally, nafY mRNA levels were reduced 10-fold with respect to the level for the wild type, a value lower than that previously reported (12).
FIG 4.

Different responses to the change toward diazotrophic growing conditions in A. vinelandii. (A) Wild type versus the ΔnifA mutant, which lacks the transcriptional activator NifA; (B) wild type versus the ΔfdxN mutant, which is partially impaired in FeMo-co biosynthesis; (C) wild type versus the ΔnifB mutant, which is completely impaired in FeMo-co biosynthesis; (D) wild type versus the ΔnifDK mutant, which lacks the nitrogenase structural genes NifDK. mRNA expression data were visualized with the MultiExperiment Viewer program (http:://www.tm4.org/). The scale color bars show mRNA-normalized absolute signals: black, a lack of mRNA; bright green, the maximum mRNA level in each experiment. Each data point represents the mean of at least three biological replicates.
Deletion of the fdxN gene alters nif gene expression and Nif protein accumulation.
The fdxN gene, located between nifB and nifQ in the minor nif cluster, encodes a ferredoxin with similarity to 2×[4Fe-4S]. Deletion of fdxN resulted in a 4-fold decrease in NifDK activity. The ΔfdxN strain consistently accumulated higher levels of nif-specific transcripts than the wild type, except for lower nifY and nifQ mRNA levels and the absence of fdxN expression (Fig. 4B). Seven hours after derepression, all genes tested presented higher mRNA levels in the ΔfdxN mutant than in the wild type (see Fig. S5 in the supplemental material).
Other fundamental differences in nif expression profiles were observed between the ΔfdxN and the wild-type strains. First, nifA and nifL mRNA levels did not decrease rapidly, as in the wild type, but, rather, stayed at maximum levels for 2 to 3 h after derepression (see Fig. S5 in the supplemental material). Second, the response to nitrogen step-down was generally delayed, with most mRNAs showing maximum accumulation levels 2 to 3 h after derepression and with the levels descending slowly over the 7-h period studied here (Fig. 4B).
The time course of Nif protein accumulation in the ΔfdxN mutant was different from that in the wild type (Fig. 5). The maximum concentration of NifH was 1.4-fold higher in the ΔfdxN mutant (144.5 μM) than in the wild type (107.2 μM). In contrast, NifD and NifK followed the opposite trend, with maximum concentrations being almost 2-fold lower in the mutant than in the wild type (33.2 μM and 29.4 μM, respectively). Importantly, the ΔfdxN strain accumulated as much NafY chaperone as NifD and NifK, suggesting the need to stabilize apo-NifDK, probably because of a defect in FeMo-co biosynthesis. ΔfdxN also accumulated higher concentrations of NifU (2.6-fold) and the FeMo-co biosynthetic proteins NifB (3.1-fold), NifEN (5.3-fold), and NifQ (2.3-fold) than the wild type. The levels of NifX, which has been proposed to function as a reservoir for FeMo-co precursors (34), were similar in the wild-type and ΔfdxN strains.
FIG 5.

Time-dependent profile of Nif protein accumulation upon nitrogen step-down in wild-type A. vinelandii (closed symbols) and the ΔfdxN mutant (open symbols). Proteins are organized by their role in nitrogenase biogenesis: proteins involved in early steps of FeMo-co synthesis (A), proteins involved in late steps of FeMo-co synthesis (B), proteins involved in FeMo-co insertion into apo-NifDK (C), and nitrogenase structural proteins (D). The total cell volume was used to calculate protein concentrations (μM). Data are the averages of three biological replicates ± SEs.
nif gene expression in ΔnifB and ΔnifDK mutants.
The marked effect of the ΔfdxN mutation on nif gene expression prompted us to investigate the response of A. vinelandii to nitrogen step-down in mutants unable to fix N2. Two different Nif-minus strains were selected: a ΔnifB mutant that was impaired at the early steps of FeMo-co biosynthesis and thus unable to accumulate cofactor biosynthetic intermediates and a ΔnifDK mutant that had no defect on FeMo-co biosynthesis but was unable to fix N2.
The ΔnifB and ΔnifDK mutants exhibited nifA and nifL mRNA levels (see Fig. S6 and S7 in the supplemental material) and expression profiles (Fig. 4C and D) similar to those in the wild type. However, this was not the case for the other nif genes, where mRNAs were observed to accumulate at very high levels in the mutants (Fig. 4C and D). Seven hours after switching to diazotrophic growth conditions, the ΔnifDK mutant accumulated 9-fold more nifH mRNA than the wild type (see Fig. S6 in the supplemental material), whereas the ΔnifB strain accumulated 10-fold more nifH mRNA and 4-fold more nifD mRNA than the wild type (see Fig. S7 in the supplemental material). The nifB, nifE, nifU, and nafY genes, whose products are involved in biosynthesis and insertion of FeMo-co, were also more highly expressed in both mutants (except for the lack of nifB mRNA in the ΔnifB mutant). Interestingly, clpX2 showed unusual peaks of expression over time, with a maximum 18-fold higher level of expression in the ΔnifB mutant and an 8-fold higher level of expression in the ΔnifDK mutant than in the wild type.
DISCUSSION
A. vinelandii responds in a highly coordinated manner to nitrogen step-down in preparation for diazotrophic growth. Expression from the nifLA operon, encoding the regulatory proteins NifL and NifA, was observed early after nitrogen step-down (14, 15). Maximum nifA mRNA accumulation occurred 10 to 30 min after NH4+ removal from the medium. However, at 1 h after derepression, nifA mRNA levels were barely detectable (Fig. 2). Expression of nifL followed an identical pattern, but nifL mRNA levels were considerably lower, even though nifL is located closer to the promoter of the nifLA operon. Since there are no apparent additional promoters upstream of nifA, a plausible scenario could be that nifL has a higher rate of mRNA degradation than nifA. A possible mechanism for this could be through specific noncoding RNAs that are capable of targeting nifL mRNA for degradation, such as the one described previously (39). The DNA region upstream of nifLA contains one RpoN (σ54)-binding motif and two NifA-binding UASs, suggesting that NifA autoregulates expression of the nifLA operon. Consistent with this, very low nifL mRNA levels were observed in the ΔnifA mutant.
Rapid accumulation of the nifU, nifS, nifV, and clpX2 mRNAs was also observed, but their pattern of disappearance was not as drastic as that for nifL and nifA (Fig. 2). The products of nifU and nifS are involved in [Fe-S] cluster biosynthesis for both nitrogenase components (18), including providing precursors for the early stages of FeMo-co biosynthesis (6). The product of clpX2 is part of a protease system that specifically targets the NifB and NifEN proteins for degradation (38). Expression of the nifU, nifS, and nifV genes, whose products are required for all three nitrogenases in A. vinelandii (40), was not completely eliminated in the ΔnifA mutant. One RpoN-binding motif and two putative AnfA-binding sites are located upstream of iscAnif and also between iscAnif and nifU, suggesting that expression of the iscAnif-nifUSV-cysE1nif-nifWZM-clpX2 operon is also under the control of AnfA. It has previously been shown that an A. vinelandii nifA mutant was able to express V or Fe nitrogenases when molybdate was not present in the growth medium (40) and that the VnfA and AnfA transcriptional activators (which regulate the expression of the vnf and anf genes of the V- and Fe-only nitrogenases, respectively) were able to modulate the expression of nifB (41) and nifM (42). However, it should be noted that both VnfA and AnfA are poorly expressed when molybdate is present in the growth medium (43).
This study provides accurate quantification of expression strength in terms of mRNA and Nif protein accumulation as well as the timing of expression required for N2 fixation. The expression of most of the genes studied here peaked 1 to 2 h after derepression. Importantly, expression of the nifB-fdxN-nifOQ-rhdN-grx5nif operon appears to end earlier than that of the nifHDKTY operon (Fig. 2), suggesting a correlation between expression timing and protein function. We found that, in general, genes required for the early stages of nitrogenase biosynthesis, i.e., nifU, nifS, fdxN, and nifB, were expressed and turned off earlier than genes required in later stages of the pathway. Our results suggest that proper expression dynamics could be important in the context of engineering nitrogen fixation in nondiazotrophs. The incorporation of genetic circuits adding temporal control to nif gene expression must be considered by the synthetic biologist. Recently, the K. pneumoniae nif gene cluster has been refactored by using synthetic biology tools that removed all native regulation (20). The resulting refactored strain showed 7% of wild-type nitrogenase activity and 3.5-fold lower diazotrophic growth rate. We argue that controlling the timing of metalloprotein maturation in this biosynthetic pathway could result in increased activities. For instance, initial synthesis of [Fe-S] clusters by NifS and NifU might be important to properly fold metalloproteins, such as NifEN or NifDK.
Although each nif operon yielded similar mRNA levels for the genes that it contains, clear exceptions were found in the cases of nifL (described above), nifY, and nifQ. The nifY mRNA levels were 10-fold lower than those of nifH, nifD, and nifK, while nifQ had 4-fold lower mRNA levels than nifB and fdxN. The presence of intergenic secondary structures within the nifHDKTY operon that affect mRNA levels has been described (19). Likewise, the lower level of expression of nifQ with respect to that of nifB and fdxN has been reported previously (44). We hypothesize that mRNA stability within nif operons may be modulated by the presence of uncharacterized noncoding RNAs.
The accumulation pattern of Nif proteins indicates that the diazotrophic cell is capable of maximizing its resources by logically arranging protein synthesis on the basis of protein functions. NifU was the first protein to reach maximum accumulation (Fig. 3), in accordance with its foundational role in [Fe-S] cluster biosynthesis for nitrogenase components, including the early steps in FeMo-co biosynthesis. However, NifH was the most prevalent protein 1 h after derepression and remained so during the next 6 h. It is possible that early NifH abundance is required to support P-cluster (45) and FeMo-co (46) syntheses. NifH achieved its maximum concentration 4 h after derepression, coinciding with the maximum NifDK concentration and the maximum in vivo nitrogenase activity. This was in line with the structural role of NifH in the nitrogenase complex. The observed in vivo NifH/NifDK molar ratio was 2 at the time when maximum nitrogenase activity occurred. This is in contrast to the optimum ratio in the in vitro assays, which was reported to be 40 NifH to 1 NifDK (47).
Once nitrogenase was fully operational, the demand for biosynthetic components, such as NifB and NifEN, decreased. NifB catalyzes the biosynthesis of NifB-co, an early intermediate in the FeMo-co biosynthetic pathway (8). Similar to NifU, the NifB concentration was the highest 1 h after derepression (3 μM), but it was observed to slowly decrease to 1 μM after 7 h of derepression. NifE and NifN polypeptide concentrations were similar throughout the experiment, consistent with the stoichiometric requirement to form the NifE2-NifN2 heterotetramer in which FeMo-co is assembled (9). The NifEN/NifDK molar ratio also decreased over time from 0.02 at 1 h after derepression to 0.007 after 7 h. The fact that NifB and NifEN are actively degraded during diazotrophic growth (38) may explain their very low concentrations. However, it is likely that NifB and NifEN are able to perform multiple enzymatic turnovers in vivo in order to provide sufficient FeMo-co for an excess of NifDK molecules.
In contrast to other biosynthetic proteins, the concentrations of NifQ and NafY steadily increased, suggesting that they are needed during and after the initial diazotrophic growth stages (Fig. 3). The roles of NafY in apo-NifDK stabilization and in FeMo-co insertion to form holo-NifDK (48) might be still required at later stages. The NifQ accumulation pattern is difficult to interpret, as its role is to donate the Mo atom to the NifEN/NifH complex for its incorporation into FeMo-co (36), and therefore, a pattern of accumulation similar to that of NifB or NifEN was expected.
New insights into the timing and regulation of nif expression were obtained from the study of the ΔfdxN mutant. The ΔfdxN strain has been suggested to be partially impaired in FeMo-co biosynthesis (44). The pattern of nif gene expression in this mutant was characterized by a lag in the expression of nif genes and increased nif mRNA accumulation (see Fig. S5 in the supplemental material). This delay may reflect the need to recycle proteins due to a low nitrogen-fixing activity. Importantly, nifL and nifA mRNA levels were increased, which might result in increased levels of NifA activity and, in turn, elevated levels of nif transcripts and of accumulated Nif proteins (with the exception of the NifDK protein, as discussed below). It is important to note that the increase of nifLA mRNA levels is not a general effect of mutants impaired in nitrogenase activity because the ΔnifB and ΔnifDK mutants showed wild-type profiles of nifLA expression. Thus, it is possible that FdxN has an additional regulatory role besides its role in FeMo-co biosynthesis.
Although nifD and nifK mRNA levels remained high during derepression in the ΔfdxN strain, the NifDK protein was much less abundant, and therefore, the corresponding [mRNA]max/[protein]max index was significantly higher than that in the wild type. In addition, NafY was more abundant in the ΔfdxN mutant, reaching an equimolecular concentration with NifDK. It is known that apo-NifDK is less stable than holo-NifDK and that NafY associates with and stabilizes the FeMo-co-deficient apo-NifDK (11, 48). Altogether, these observations suggest that the ΔfdxN mutant accumulates significant amounts of FeMo-co-deficient apo-NifDK. It also accumulated 3-fold the amount of FeMo-co biosynthetic proteins, including NifU, NifS, NifB, NifE, NifN, and NifQ, compared to wild type (the NifH concentration increased 1.4-fold), probably to compensate for its impairment in FeMo-co biosynthesis. The stoichiometries among the biosynthetic proteins remained almost identical, as if the fine-tuned regulation coordinating FeMo-co biosynthesis was not affected.
Finally, the ΔnifB and ΔnifDK mutants, which are unable to fix N2 and therefore remain under nitrogen starvation, responded to derepression by accumulating very high levels of nif mRNAs. It has previously been observed that cultures derepressed under argon showed exacerbated nitrogenase activity levels in a phenomenon termed hyperderepression (49). The mechanism by which A. vinelandii detects a defect in nitrogenase biosynthesis or activity and then overcompensates for this defect is not well understood, but it is possible that NifL could be less inhibitory in these situations, leading to increased NifA activity. This effect is consistent with the nitrogen feedback regulatory response mediated by NifL. Indeed, fixed nitrogen exerts negative feedback regulation over nif gene expression. In the wild-type strain, the initial burst of nif gene expression followed by a reduction toward steady-state levels is reminiscent of the behavior of other negative-feedback-regulated systems, such as the ornithine transcarbamylase activity in the arginine biosynthetic pathway of E. coli (50). Interestingly, nifA and nifL mRNA accumulation was unaltered in the ΔnifB and ΔnifDK mutants, suggesting that the inability to synthesize mature NifDK itself affects NifA activity by a mechanism different from (or in addition to) that involving the level of fixed nitrogen.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by ERC starting grant 205442 (to L.M.R.) and MICINN grant BIO2009-12661 (to L.M.R.)
We thank Ryan Kniewel for critically reading the manuscript.
Footnotes
Published ahead of print 15 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00942-13.
REFERENCES
- 1.Beatty PH, Good AG. 2011. Future prospects for cereals that fix nitrogen. Science 333:416–417. 10.1126/science.1209467 [DOI] [PubMed] [Google Scholar]
- 2.Spatzal T, Aksoyoglu M, Zhang L, Andrade SL, Schleicher E, Weber S, Rees DC, Einsle O. 2011. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 334:940. 10.1126/science.1214025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgiadis MM, Komiya H, Chakrabarti P, Woo D, Kornuc JJ, Rees DC. 1992. Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science 257:1653–1659. 10.1126/science.1529353 [DOI] [PubMed] [Google Scholar]
- 4.Dos Santos PC, Smith AD, Frazzon J, Cash VL, Johnson MK, Dean DR. 2004. Iron-sulfur cluster assembly: NifU-directed activation of the nitrogenase Fe protein. J. Biol. Chem. 279:19705–19711. 10.1074/jbc.M400278200 [DOI] [PubMed] [Google Scholar]
- 5.Howard KS, McLean PA, Hansen FB, Lemley PV, Koblan KS, Orme-Johnson WH. 1986. The Klebsiella pneumoniae nifM gene product is required for stabilization and activation of nitrogenase iron protein in Escherichia coli. J. Biol. Chem. 261:772–778 [PubMed] [Google Scholar]
- 6.Zhao D, Curatti L, Rubio LM. 2007. Evidence for nifU and nifS participation in the biosynthesis of the iron-molybdenum cofactor of nitrogenase. J. Biol. Chem. 282:37016–37025. 10.1074/jbc.M708097200 [DOI] [PubMed] [Google Scholar]
- 7.Joerger RD, Bishop PE. 1988. Nucleotide sequence and genetic analysis of the nifB-nifQ region from Azotobacter vinelandii. J. Bacteriol. 170:1475–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah VK, Allen JR, Spangler NJ, Ludden PW. 1994. In vitro synthesis of the iron-molybdenum cofactor of nitrogenase. Purification and characterization of NifB cofactor, the product of NifB protein. J. Biol. Chem. 269:1154–1158 [PubMed] [Google Scholar]
- 9.Rubio LM, Ludden PW. 2008. Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 62:93–111. 10.1146/annurev.micro.62.081307.162737 [DOI] [PubMed] [Google Scholar]
- 10.Schwarz G, Mendel RR, Ribbe MW. 2009. Molybdenum cofactors, enzymes and pathways. Nature 460:839–847. 10.1038/nature08302 [DOI] [PubMed] [Google Scholar]
- 11.Rubio LM, Singer SW, Ludden PW. 2004. Purification and characterization of NafY (apodinitrogenase γ subunit) from Azotobacter vinelandii. J. Biol. Chem. 279:19739–19746. 10.1074/jbc.M400965200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubio LM, Rangaraj P, Homer MJ, Roberts GP, Ludden PW. 2002. Cloning and mutational analysis of the gamma gene from Azotobacter vinelandii defines a new family of proteins capable of metallocluster-binding and protein stabilization. J. Biol. Chem. 277:14299–14305. 10.1074/jbc.M107289200 [DOI] [PubMed] [Google Scholar]
- 13.Seefeldt LC, Hoffman BM, Dean DR. 2009. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 78:701–722. 10.1146/annurev.biochem.78.070907.103812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon R, Kahn D. 2004. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2:621–631. 10.1038/nrmicro954 [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Argudo I, Little R, Shearer N, Johnson P, Dixon R. 2004. The NifL-NifA system: a multidomain transcriptional regulatory complex that integrates environmental signals. J. Bacteriol. 186:601–610. 10.1128/JB.186.3.601-610.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dos Santos PC, Dean DR. 2011. Co-ordination and fine-tuning of nitrogen fixation in Azotobacter vinelandii. Mol. Microbiol. 79:1132–1135. 10.1111/j.1365-2958.2011.07541.x [DOI] [PubMed] [Google Scholar]
- 17.Jacobson MR, Brigle KE, Bennett LT, Setterquist RA, Wilson MS, Cash VL, Beynon J, Newton WE, Dean DR. 1989. Physical and genetic map of the major nif gene cluster from Azotobacter vinelandii. J. Bacteriol. 171:1017–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson MR, Cash VL, Weiss MC, Laird NF, Newton WE, Dean DR. 1989. Biochemical and genetic analysis of the nifUSVWZM cluster from Azotobacter vinelandii. Mol. Gen. Genet. 219:49–57 [DOI] [PubMed] [Google Scholar]
- 19.Hamilton TL, Jacobson M, Ludwig M, Boyd ES, Bryant DA, Dean DR, Peters JW. 2011. Differential accumulation of nif structural gene mRNA in Azotobacter vinelandii. J. Bacteriol. 193:4534–4536. 10.1128/JB.05100-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Temme K, Zhao D, Voigt CA. 2012. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl. Acad. Sci. U. S. A. 109:7085–7090. 10.1073/pnas.1120788109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curatti L, Ludden PW, Rubio LM. 2006. NifB-dependent in vitro synthesis of the iron-molybdenum cofactor of nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 103:5297–5301. 10.1073/pnas.0601115103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson AC, Burgess BK, Dean DR. 1986. Activity, reconstitution, and accumulation of nitrogenase components in Azotobacter vinelandii mutant strains containing defined deletions within the nitrogenase structural gene cluster. J. Bacteriol. 166:180–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curatti L, Brown CS, Ludden PW, Rubio LM. 2005. Genes required for rapid expression of nitrogenase activity in Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A. 102:6291–6296. 10.1073/pnas.0501216102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah VK, Davis LC, Brill WJ. 1972. Nitrogenase. I. Repression and derepression of the iron-molybdenum and iron proteins of nitrogenase in Azotobacter vinelandii. Biochim. Biophys. Acta 256:498–511 [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26.Page WJ, von Tigerstrom M. 1979. Optimal conditions for transformation of A. vinelandii. J. Bacteriol. 139:1058–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfaffl MW. 2004. Quantification strategies in real-time PCR, p 87–112 In Bustin SA. (ed), A-Z of quantitative PCR. International University Line, La Jolla, CA [Google Scholar]
- 28.Barrios H, Valderrama B, Morett E. 1999. Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res. 27:4305–4313. 10.1093/nar/27.22.4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goosen N, van de Putte P. 1995. The regulation of transcription initiation by integration host factor. Mol. Microbiol. 16:1–7. 10.1111/j.1365-2958.1995.tb02386.x [DOI] [PubMed] [Google Scholar]
- 30.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76–85. 10.1016/0003-2697(85)90442-7 [DOI] [PubMed] [Google Scholar]
- 32.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 33.Brandner JP, McEwan AG, Kaplan S, Donohue TJ. 1989. Expression of the Rhodobacter sphaeroides cytochrome c2 structural gene. J. Bacteriol. 171:360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez JA, Igarashi RY, Soboh B, Curatti L, Dean DR, Ludden PW, Rubio LM. 2007. NifX and NifEN exchange NifB cofactor and the VK-cluster, a newly isolated intermediate of the iron-molybdenum cofactor biosynthetic pathway. Mol. Microbiol. 63:177–192. 10.1111/j.1365-2958.2006.05514.x [DOI] [PubMed] [Google Scholar]
- 35.Shah VK, Brill WJ. 1973. Nitrogenase. IV. Simple method of purification to homogeneity of nitrogenase components from Azotobacter vinelandii. Biochim. Biophys. Acta 305:445–454 [DOI] [PubMed] [Google Scholar]
- 36.Hernandez JA, Curatti L, Aznar CP, Perova Z, Britt RD, Rubio LM. 2008. Metal trafficking for nitrogen fixation: NifQ donates molybdenum to NifEN/NifH for the biosynthesis of the nitrogenase FeMo-cofactor. Proc. Natl. Acad. Sci. U. S. A. 105:11679–11684. 10.1073/pnas.0803576105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart WPD, Fitzgerald GP, Burris RH. 1967. In situ studies on N2 fixation using the acetylene reduction technique. Proc. Natl. Acad. Sci. U. S. A. 58:2071–2078. 10.1073/pnas.58.5.2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez-Noel G, Curatti L, Hernandez JA, Rubio LM. 2011. NifB and NifEN protein levels are regulated by ClpX2 under nitrogen fixation conditions in Azotobacter vinelandii. Mol. Microbiol. 79:1182–1193. 10.1111/j.1365-2958.2011.07540.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee E-J, Groisman A. 2010. An antisense RNA that governs the expression kinetics of a multifunctional virulence gene. Mol. Microbiol. 76:1020–1033. 10.1111/j.1365-2958.2010.07161.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy C, Dean D. 1992. The nifU, nifS and nifV gene products are required for activity of all three nitrogenases of Azotobacter vinelandii. Mol. Gen. Genet. 231:494–498 [DOI] [PubMed] [Google Scholar]
- 41.Drummond M, Walmsley J, Kennedy C. 1996. Expression from the nifB promoter of Azotobacter vinelandii can be activated by NifA, VnfA, or AnfA transcriptional activators. J. Bacteriol. 178:788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lei S, Pulakat L, Gavini N. 1999. Regulated expression of the nifM of Azotobacter vinelandii in response to molybdenum and vanadium supplements in Burk's nitrogen-free growth medium. Biochem. Biophys. Res. Commun. 264:186–190 [DOI] [PubMed] [Google Scholar]
- 43.Premakumar R, Pau RN, Mitchenall LA, Easo M, Bishop PE. 1998. Regulation of the transcriptional activators AnfA and VnfA by metals and ammonium in Azotobacter vinelandii. FEMS Microbiol. Lett. 164:63–68 [DOI] [PubMed] [Google Scholar]
- 44.Rodríguez-Quiñones F, Bosch R, Imperial J. 1993. Expression of the nifBfdxNnifOQ region of Azotobacter vinelandii and its role in nitrogenase activity. J. Bacteriol. 175:2926–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu Y, Fay AW, Lee CC, Ribbe MW. 2007. P-cluster maturation on nitrogenase MoFe protein. Proc. Natl. Acad. Sci. U. S. A. 104:10424–10429. 10.1073/pnas.0704297104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson AC, Dean DR, Burgess BK. 1987. Iron-molybdenum cofactor biosynthesis in Azotobacter vinelandii requires the iron protein of nitrogenase. J. Biol. Chem. 262:14327–14332 [PubMed] [Google Scholar]
- 47.Kim CH, Newton WE, Dean DR. 1995. Role of the MoFe protein alpha-subunit histidine-195 residue in FeMo-cofactor binding and nitrogenase catalysis. Biochemistry 34:2798–2808 [DOI] [PubMed] [Google Scholar]
- 48.Hernandez JA, Phillips AH, Erbil WK, Zhao D, Demuez M, Zeymer C, Pelton JG, Wemmer DE, Rubio LM. 2011. A sterile α-motif domain in NafY targets apo-NifDK for iron-molybdenum cofactor delivery via a tethered domain. J. Biol. Chem. 286:6321–6328. 10.1074/jbc.M110.168732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill S. 1992. Physiology of nitrogen fixation in free-living heterotrophs, p 87–134 In Stacey G, Burris RH, Evans HJ. (ed), Biological nitrogen fixation. Chapman-Hall, New York, NY [Google Scholar]
- 50.Gorini L, Maas WK. 1957. The potential for the formation of a biosynthetic enzyme in Escherichia coli. Biochim. Biophys. Acta 25:208–209 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.