

Abstract
Metronidazole (MTZ) is often used in combination therapies to treat infections caused by the gastric pathogen Helicobacter pylori. Resistance to MTZ results from loss-of-function mutations in genes encoding RdxA and FrxA nitroreductases. MTZ-resistant strains, when cultured at sub-MICs of MTZ (5 to 20 μg/ml), show dose-dependent defects in bacterial growth; depressed activities of many Krebs cycle enzymes, including pyruvate:ferredoxin oxidoreductase (PFOR); and low transcript levels of porGDAB (primer extension), phenotypes consistent with an involvement of a transcriptional regulator. Using a combination of protein purification steps, electrophoretic mobility shift assays (EMSAs), and mass spectrometry analyses of proteins bound to porG promoter sequences, we identified HP1043, an essential homeostatic global regulator (HsrA [for homeostatic stress regulator]). Competition EMSAs and supershift analyses with HsrA-enriched protein fractions confirmed specific binding to porGDAB and hsrA promoter sequences. Exposure to MTZ resulted in >10-fold decreases in levels of HsrA and in levels of the HsrA-regulated gene products PFOR and TlpB. Exposure to paraquat (PQ), hydrogen peroxide, or organic peroxides showed near equivalence with MTZ, revealing a common oxidative stress response pathway. Finally, direct superoxide dismutase (SOD) assays showed an inverse relationship between HsrA levels and SOD activity, suggesting that HsrA may serve as a repressor of sodB. As a homeostatic sentinel, HsrA appears to be ideally positioned to enable rapid shutdown of genes associated with metabolism and growth while activating (directly or indirectly) oxidative defense genes in response to low levels of toxic metabolites (MTZ or oxygen) before they reach DNA-damaging levels.
INTRODUCTION
Helicobacter pylori is a microaerobic bacterium that causes lifelong infections of the gastric mucosa of over 3 billion persons worldwide, and while most of these infections result in mild superficial gastritis, some infections progress to more serious diseases such as peptic and duodenal ulcers, mucosa-associated lymphoid tissue lymphoma, and gastric cancer (1–3). The remarkable ability of H. pylori to both colonize and persist in the gastric milieu is attributed to several systems that monitor local pH, including a novel pH-gated urea transporter (UreI) that modulates a powerful urease system (4, 5), a pH-sensing chemoreceptor (TlpB) that directs both colonization and avoidance of washout with mucus turnover (6), and a pH-sensing two-component regulatory system (ArsRS) that modulates expression of acid survival genes in sync with the daily cycling of stomach acid (7–10). In contrast to acid stress, little is known regarding the response to oxidative stress, yet H. pylori promotes gastric inflammation and the recruitment of neutrophils and macrophages, all of which produce reactive oxygen and nitrogen species (11, 12). While H. pylori responds to oxidative stress insults by increasing the expression levels of many genes, including catalase, alkyl hydroperoxide reductase, superoxide dismutase (SOD), and the quinone reductase MdaB (12–14), the underlying regulatory mechanisms orchestrating this response have not been elucidated.
Metronidazole (MTZ), a synthetic redox-active prodrug, is commonly included in triple and quadruple therapies (standard treatments) used to treat primary infections caused by H. pylori (15, 16). Interestingly, MTZ is often included at higher dosages in salvage therapies (17–19). The apparent therapeutic efficacy of MTZ in salvage therapies might indicate that higher drug doses might overcome drug resistance or perhaps that MTZ acts synergistically to enhance the efficiency of the other antimicrobials. Conceptually, where resistance to antimicrobials requires shifts in metabolic activities or in regulatory networks, these compensatory adaptations might themselves become attractive drug targets.
The basis for MTZ resistance in H. pylori remains somewhat controversial despite overwhelming evidence suggesting that loss-of-function mutations in rdxA, encoding a MTZ-reducing oxygen-insensitive nitroreductase, is both necessary and sufficient to confer clinically significant resistance, as defined by resistance greater than the breakpoint for the therapeutic (>8 μg/ml) (20–22). Additional mutations in a second nitroreductase (frxA) increase resistance in some strains (22–27). These enzymes, particularly RdxA, catalyze two 2-electron reductions of MTZ to DNA-damaging hydroxylamine adducts (20, 22). MTZ can also be reduced by single-electron reductions that are oxygen sensitive and generate superoxide anions (22). In this regard, sequential mutations in additional genes (mdaB, ribF, and fur) that are likely associated with one-electron reductions of MTZ, lead to even higher resistance levels (250 μg/ml), typically observed for aerobic bacteria and Escherichia coli (28). While expression of rdxA in E. coli produces a MTZ-sensitive phenotype (20, 22, 26), and complementation of an rdxA mutant of H. pylori with the wild-type (WT) allele restores full susceptibility (20), there appear to be additional chemistries associated with MTZ activation in H. pylori that are observed in E. coli only at very high drug concentrations (26, 28). The variable responses of MTZ-resistant (MTZr) strains of H. pylori to a range of MTZ concentrations suggest that the drug might be affecting metabolic activities. In this regard, MTZr H. pylori strains, when grown in the presence of sub-MICs of MTZ, showed dose-dependent defects in growth, manifested as extended lag times, lower growth rates, and diminished growth yields (29). When these strains were assayed for Krebs cycle enzymes, most were depressed, but activities for pyruvate:ferredoxin oxidoreductase (PFOR) and 2-oxoglutarate oxidoreductase (OOR) were below detectable levels (29). As little as 3.5 μg/ml of MTZ was sufficient to alter enzyme levels and bacterial growth rates, yet analysis of DNA damage by alkaline gel electrophoresis showed minimal DNA fragmentation at concentrations below 25 μg/ml (26, 29). Comparative proteomic studies also revealed gene expression changes resulting from MTZ exposure, including diminished levels of PFOR and apparent increases in levels of enzymes associated with oxidative stress, such as alkyl hydroperoxide reductase (AhpC) (13, 30, 31). Since PFOR in strictly anaerobic bacteria is considered the major enzyme associated with MTZ reduction, it seemed reasonable that H. pylori might downregulate enzymes, like PFOR, whose activities might contribute to cellular toxicity. However, direct enzyme assays with recombinant PFOR (purified from E. coli) under strictly anaerobic conditions, used to measure MTZ reductase activity of RdxA (22), revealed no pyruvate-dependent MTZ reduction under these assay conditions. The PFOR enzymes of both H. pylori and Campylobacter jejuni utilize flavodoxin in place of ferredoxin, and apparently, the redox potential is not sufficiently low to chemically reduce MTZ (32–35).
The apparent repression of genes encoding Krebs cycle enzymes coordinate with growth arrest suggests that responses to a synthetic drug like MTZ must be routed through an existing global regulatory system. However, H. pylori produces a paucity of transcriptional regulators compared with E. coli and lacks orthologues of the oxidative stress response regulatory genes rpoH, rpoS, soxRS, and oxyR (36, 37). Since a transcriptional regulator was not obvious, we used a protein purification-DNA electrophoretic mobility shift assay (EMSA) screening strategy to enrich for proteins that bind to the promoter region of porG of the PFOR operon as a first step in elucidating underlying regulatory mechanisms. Here we report that an essential atypical (orphan) response regulator, HP1043, renamed HsrA (homeostatic stress regulator), is a positive regulator of the porGDAB operon. Our studies show that MTZ exposure results in a dramatic decrease in HsrA protein levels coordinate with decreases in levels of PFOR as well as TlpB, which is regulated by HsrA (38). Exposure to paraquat (PQ) (a superoxide generator) or peroxides (hydrogen or organic) also resulted in depression of HsrA, suggesting that HsrA is also responsive to oxidative stress. In this regard, HsrA shares some similarities with CosR, an orthologue in C. jejuni that is also essential and was shown to regulate responses to oxidative stress (39, 40). While HsrA appears to be a negative regulator of sodB (superoxide dismutase activities increase in response to MTZ and PQ), regulation of katA and ahpC (catalase and alkyl hydroperoxide reductase, respectively) must be indirect, as these genes lack HsrA consensus DNA binding sequences (41). We previously proposed that HsrA functions as a global homeostatic regulator by syncing metabolic functions and virulence with availability of nutrients and cell division (41). Our studies reveal that the sub-MIC effects of MTZ on the growth rate of MTZr mutants of H. pylori are not due to extensive DNA damage but rather to activation of an existing oxidative defense pathway. Knowledge of these MTZ-induced shifts in metabolic pathways and other compensatory adaptations might be exploited in development of novel therapeutics.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in these studies are listed in Table 1. E. coli strains were routinely grown in LB medium (42) at 37°C, and H. pylori strains were grown under humid microaerobic conditions at 37°C on brucella-based medium (BA) supplemented with 7.5% newborn calf serum (Gibco Laboratories), 10 μg/ml vancomycin, 5 μg/ml trimethoprim, and 4 μg/ml amphotericin B (24). H. pylori HP1061 and HP1134R contain loss-of-function mutations in rdxA and frxA (20, 29). The HP1061fur::cat5 mutant was constructed as previously described (43).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristic(s) | Source or reference(s) |
|---|---|---|
| Bacterial strains | ||
| H. pylori | ||
| HP1061 | rdxA frxA | Laboratory collection |
| HP1061fur | rdxA frxA fur::Cm5 Camr | This study |
| HP1061CC11 | hsrA CC11 from G27CC11 | 41; this study |
| SS1 | Sydney 1 strain | Laboratory collection |
| HP1134R | MTZr clinical isolate | Laboratory collection |
| E. coli | ||
| DH5α | endA1 hsdR17 glnV44 thi-1 recA1 gyrA relA1 Δ(lac-argF)169 deoR [ϕ80dlacΔ(lacZ)M15] | Invitrogen |
| BL21-CodonPlus(DE3)-RIPL | Stratagene | |
| TOP10 | Invitrogen | |
| Plasmids | ||
| pCR2.1-TOPO | Vector for cloning of PCR fragments; Apr Kmr | Invitrogen |
| pTOPO-porGp | Derivative of pCR2.1-TOPO with porG promoter; Apr Kmr | This study |
| pET16b | Ap; T7 promoter | Novagen |
| pET16b-HP1043 | Ap; His10-HP1043-expressing plasmid | This study |
Growth studies, preparation of bacterial extracts for immunoblots, and enzyme assays.
MTZr strains and a fur mutant of H. pylori were grown in BA supplemented with various concentrations of MTZ (10 or 18 μg/ml), and the optical density at 660 nm (OD660) was measured spectrophotometrically as previously described (41). Bacteria were harvested at an OD600 of 0.9 (late log phase) by centrifugation (6,000 × g) at 4°C, and following suspension in phosphate-buffered saline (PBS), they were subjected to sonic disruption (29). Supernatants obtained from centrifugation at 10,000 × g to remove unbroken cells and debris were used for immunoblot or enzyme assays. The enzymatic activity of PFOR was determined by tracking reduction of benzyl viologen under anaerobic conditions (22, 29). Superoxide dismutase activity was determined by the xanthine oxidase, horse heart cytochrome c reduction method (44). One unit of SOD activity was determined as the protein concentration that inhibited the reduction of cytochrome c by 50% at 550 nm. Protein concentrations were estimated by the Bradford dye binding assay (Bio-Rad) with bovine serum albumin (BSA) as the standard. All antibodies used in these studies (HsrA, Hsp60, Hsp70, and TlpB) were described previously (6, 29, 41, 45, 46). SDS-PAGE and immunoblotting were performed according to general methods reported previously (47, 48).
Cloning of porG.
Cloning and expression of the porGDAB operon were described previously (48). The region of the porG promoter from positions −89 to +25 (Fig. 1) was amplified from H. pylori SS1 chromosomal DNA by PCR using Hot Star Taq DNA polymerase (Qiagen) and cloned into the pCR2.1-TOPO vector (Invitrogen) as specified by the manufacturers. The primers used to amplify the porG promoter were as follows: 5Por+50Sac2 (5′-TCCCCGCGGTCCTTGTGGGTATGCCCCCATTT) and +164PorR (5′-TCGTATAAAATAAATTGAATTTAGCCT). The pTOPO-porGp construct was verified by sequencing at GeneWIZ.
FIG 1.

Schematic version of the horH-porGDAB intergenic region. The alignment of the porGDAB promoters from different Helicobacter strains is shown below. The porGDAB −10 promoter hexamer and the HsrA DNA binding motif are boxed. A Fur box was identified upstream of porG but outside the depicted region and near the promoter region of horH.
Primer extension.
H. pylori strain HP1134R was grown in BA medium in the presence or absence of 18 μg/ml MTZ to an OD600 of 0.6 and harvested by centrifugation. Total RNA was extracted by the hot phenol method (49). RNA was suspended in 200 μl of diethyl pyrocarbonate-treated water, and the concentration was determined by using a Nanodrop instrument (Thermo Scientific). Oligonucleotide reverse primer 5′-ATAGGAAGCGAACGCTTGCA was end labeled with T4 polynucleotide kinase with [γ32P]ATP (3,000 Ci/mmol; ICN) and purified as previously described (49). Primer extension was performed on 50 μg of RNA samples mixed with purified labeled oligonucleotide (∼1.2 × 107 cpm) (50). Extensions were performed with reverse transcription (RT) buffer (1 M Tris-HCl [pH 8.3]; 1 M MgCl2; 1 M dithiothreitol [DTT]; 50 mM [each] dATP, dCTP, TTP, and dGTP; and 2 mg/ml actinomycin) and 200 U of Moloney murine leukemia virus (M-MLV) reverse transcriptase (Gibco/BRL, MD). Following incubation at 41°C for 1 h, extension was stopped by addition of 1 ml 0.5 M EDTA, and cDNA was prepared (49, 50). DNA sequencing was performed with the same primer. Samples were subjected to electrophoresis on a denaturing polyacrylamide sequencing gel and developed by autoradiography (50).
Electrophoretic mobility shift assay.
DNA fragments containing the region of the porG promoter from positions −89 to +25 were generated by PCR using plasmid pTOPO-porGp as the template and primers 5Por+50Sac2 and +164PorR. A 114-bp DNA fragment containing the hsrA promoter was generated by PCR using chromosomal DNA from H. pylori strain SS1 as the template and primers hp1043-p1R (5′-TCAGTAGAACGCGCATGGT) and hp1043-p1F (5′-AGGGCTTGATTTTAACCAAGC). The amplified DNA fragments were purified from a 1% agarose gel by using a QIAquick gel extraction kit (Qiagen) and end labeled by incubation with T4 polynucleotide kinase (New England BioLabs) and [γ-32P]ATP (3,000 Ci/mmol; ICN). The labeled DNA fragments were separated from unincorporated [γ-32P]ATP by gel filtration through Sephadex G-50 Quick Spin columns (Boehringer Mannheim). Radiolabeled DNA fragments (ca. 10,000 to 20,000 cpm per reaction) were incubated with cell extracts and fractions containing partially purified proteins and/or recombinant HsrA (rHsrA) protein at 25°C for 10 min in 0.5× TBE buffer (45 mM Tris, 45 mM boric acid, and 1 mM EDTA [pH 8.3]) with 2 ng of poly(dI-dC)/μl. For supershift experiments, polyclonal anti-HsrA serum was added to bind protein-DNA complexes. Samples were resolved by electrophoresis in 1.5-mm-thick, 6% nondenaturing polyacrylamide–0.5× TBE gels at 20 mA for 45 min at room temperature. The gels were dried, and the positions of radioactive DNA fragments were detected with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Identification of proteins bound to the porG promoter.
Two grams of H. pylori HP1061fur::Cm5 wet cells was resuspended to a total volume of 10 ml in 100 mM potassium phosphate buffer (pH 7.5) containing 14 mM β-mercaptoethanol (βME) and disrupted by sonication. Unbroken cells and particulate material were removed by centrifugation at 20,000 × g for 30 min at 4°C. Nucleic acids and some bound proteins were precipitated by addition of polyethyleneimine (final concentration, 0.5%) for 10 min on ice, followed by centrifugation at 10,000 × g for 5 min. After centrifugation, proteins of interest were precipitated at between 0 and 50% saturation of ammonium sulfate. The precipitate was dissolved in 4 ml of buffer B (30 mM Tris-HCl [pH 6.9], 0.5 mM EDTA, 14 mM βME), followed by dialysis against buffer B. The dialyzed sample was applied onto a UNO Q1 column (Bio-Rad Laboratories, Inc.). Proteins of interest were eluted with a 24-ml linear gradient of 0 to 500 mM NaCl in buffer B at a flow rate of 1 ml/min. Fractions were subjected to EMSA and analysis by SDS-polyacrylamide gel electrophoresis. The proteins from the EMSA-active fractions that bound porG sequences were identified at the W. M. Keck Biomedical Mass Spectrometry Laboratory at the University of Virginia.
Effects of MTZ, paraquat, and peroxides on the HsrA regulon.
Bacterial cultures (HP1061) were allowed to grow without additions to early logarithmic phase (OD600 of ∼0.3), at which time MTZ (10 μg/ml), PQ (10.5 μM), hydrogen peroxide (10 mM), cumene hydroperoxide (5 μM), or t-butyl hydroperoxide (5 μM) was added. As a control, one culture was untreated. Growth rates were measured optically, and aliquots were obtained at 12 and 24 h for MTZ-treated cultures and at hourly intervals (1 to 5 h) for cultures subjected to oxidative stress reagents. For aliquots obtained from MTZ treatment, bacteria were sonicated and subjected to SDS-PAGE. For these studies, lanes were loaded with 4, 8, 16, or 32 μg of protein. Following transfer of separated proteins onto nitrocellulose, blots were probed with antisera to HsrA, PorA, TlpB, Hsp60, and Hsp70. For oxidative stress-treated cultures, following determination of the OD, equal protein loadings were subjected to SDS-PAGE and immunoblotting with antisera to HsrA. Semiquantification of band intensities was determined by densitometry scanning and analysis with Image-Pro software (Media Cybernetics). All assays were performed at least twice, and representative results are depicted.
Direct effects of MTZ and PQ on HsrA.
Two terminal cysteine residues of HsrA were each replaced by serine (C215S and C221S) to produce single mutants and the double mutant (CC11), as previously described (41). PCR and DNA sequencing were used to confirm the mutant alleles in HP1061. The effects of MTZ and PQ were also assessed by disk diffusion. BA plates were uniformly streaked with 0.1 ml of H. pylori cell suspensions adjusted to an OD600 of 0.1. Sterile 7-mm filter paper disks saturated with 15 μl of MTZ (10 mg/ml) or PQ (3.3 mg/ml) were placed onto the plates in triplicate. The plates were incubated for 72 h before the zones of inhibition were measured (22).
RESULTS
The respiratory metabolism of H. pylori is highly dependent on the Krebs cycle for energy production; therefore, decreases in the specific activities of Krebs cycle enzymes would be expected to affect bacterial growth. We previously reported that sub-MICs of MTZ led to repression of PFOR, OOR, aconitase, isocitrate dehydrogenase, and succinate dehydrogenase (fumarate reductase) activities (29). While PFOR and OOR were similarly affected by MTZ exposure, we arbitrarily chose to focus our investigations on determining how PFOR was regulated in response to sub-MICs of MTZ. We first confirmed that growth of H. pylori strain HP1134R (MIC, 32 μg/ml) in medium supplemented with 18 μg/ml of MTZ depressed PFOR protein levels. As shown in Fig. 2, PorA protein levels (determined by immunoblotting with anti-PorA serum) were substantially decreased in bacteria grown in the presence of sub-MICs of MTZ. In contrast, Hsp70 levels were unaffected by MTZ, suggesting regulatory specificity. The decreased protein levels of PorA were consistent with previous work showing an absence of detectable enzymatic activity (29). Primer extension was then used to both identify the transcriptional start site of porGDAB and determine if mRNA levels were similarly depressed by MTZ. As shown in Fig. 3, a consensus −10 (TATAAT) hexamer sequence was located upstream of the transcription start site. The extension products generated from 50 μg of total RNA from HP1134R were so significantly depressed by MTZ to require overloading of the sequencing gel in order to detect transcript relative to the untreated control, thus confirming that the effect of MTZ on PFOR activity was due to strong repression of transcription. Since strong “on/off” repression of transcription is not typically observed in H. pylori (8, 9, 38, 41), the action of MTZ seemed to be associated with a novel regulatory mechanism. An examination of the porGDAB locus and the upstream region containing the transcription start site and promoter elements is shown in Fig. 1. A putative Fur DNA binding box is located outside the depicted region and in the promoter region of the upstream gene horH. The promoter region, like most in H. pylori, is highly AT rich. We previously identified a putative HrsA (HP1043) consensus DNA binding motif in sequences spanning the −35 region, as indicated in Fig. 1 (41).
FIG 2.
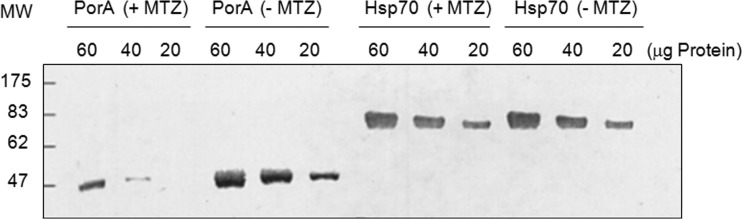
Effect of metronidazole on PFOR protein levels. Cell extracts from H. pylori strain Hp1134R grown in the presence (+ MTZ) or absence (− MTZ) of 18 μg/ml MTZ were subjected to SDS-PAGE and immunoblotting with antibodies prepared against the PorA subunit (A) and Hsp70 (B). MW, molecular weight (in thousands).
FIG 3.

Primer extension. Total RNA was obtained from MTZr strain HP1134R grown in the absence (−MTZ) or presence (+MTZ) of MTZ (18 μg/ml), and equal concentrations of RNA were extended. Extension products and DNA sequence were generated with the same primer. The sequencing gel was substantially overloaded in order to detect the extension product (porG arrow) for the +MTZ sample. In the depicted nucleotide sequence, the −10 element and the transcription start site are shown in boldface type.
Fur and MTZ-responsive repression of PFOR.
It was suggested by previous microarray studies that the ferric uptake regulator Fur regulated the expression of porGDAB under acidic conditions (51) and that Fur binds the promoter of oorDABC and contributes to its regulation (52). To test whether Fur is the MTZ-responsive regulator, we measured PFOR specific activities in cell extracts prepared from MTZr H. pylori strains HP1061 and HP1061Δfur, grown in the presence or absence of sub-MICs of MTZ. As shown in Fig. 4, in the absence of MTZ, PFOR specific activities were equivalent, at 150 to 160 nmol/min/mg protein. We then determined that 10 μg/ml of MTZ in the growth medium was sufficient to decrease the specific activity of PFOR in HP1061 by 50% compared to the controls (Fig. 4). Under similar growth conditions, the PFOR enzymatic activity in extracts prepared from the HP1061fur mutant strain was decreased by nearly 85% (Fig. 4). Since the putative fur box is located outside the porGDAB promoter region and near horH, we conclude that the regulatory effects attributed to Fur are most likely indirect. Since the MTZ-repressive effects on PFOR were not abolished by the fur mutation, we conclude that Fur is not the MTZ-activated repressor.
FIG 4.

Effects of MTZ and fur on PFOR activity. The specific activity of PFOR was determined for strains HP1061 (rdxA) and HP1061fur (rdxA fur) grown in the presence (+) or absence (−) of 10 μg/ml MTZ. The experiment was performed three times, and the means and standard deviations are depicted. The specific activities are reported in nmol of benzyl viologen reduced per minute per mg protein under anaerobic conditions.
Gel retardation identifies MTZ-responsive complexes.
We used EMSA to screen for proteins in cell extracts of HP1061 and HP1061fur that might bind to the porG promoter. The binding of proteins in extracts from bacteria grown in the presence or absence of MTZ to a 114-bp 32P-labeled porG promoter fragment (nucleotides −89 to +25) showed a common set of three retarded complexes, designated C-1, C-2, and C-3 (Fig. 5, lanes 2 and 3). The PCR-generated product used in EMSAs (see Materials and Methods for details) consisted of two DNA fragments. Cloning and sequencing of these fragments revealed that the upper band contained the porG promoter sequence (free DNA) (Fig. 5, lane 1). Thus, we decided to take advantage of the presence of the unspecific fragment in our EMSAs and use it as an additional internal control for nonspecific binding. As shown in Fig. 5, the porG promoter (free DNA band) was completely shifted in the presence of 5 μg of total protein from the HP1061 (lacking MTZ [MTZ−]) extract with formation of C-1, C-2, and C-3. A further 2-fold increase in the protein concentration (10 μg/ml) showed a dose response increase in C-3 but also shifted the control DNA. Importantly, band shifts with extracts from H. pylori HP1061 cells grown with MTZ showed reduced formation of all three complexes (Fig. 5, lanes 4 and 5), consistent with a MTZ-responsive transcriptional factor phenotype. Introduction of a fur deletion into strain HP1061 did not ablate any of the shifted complexes (Fig. 5, lanes 6 and 7), but most of the free DNA was unbound. Sub-MICs of MTZ further depressed the DNA binding pattern of HP1061fur (Fig. 5, lanes 8 and 9). The lack of definition of the shifted bands (lanes 8 and 9) might result from activation of DNases or more likely the action of proteases on protein-bound DNA complexes, since most of the free DNA had not shifted. To eliminate these competing effects as well as possible indirect effects of Fur, protein purification was conducted with the fur mutant in the absence of MTZ.
FIG 5.

Effect of MTZ on formation of gel-retarded complexes with the porG promoter. EMSA was performed by using the indicated amounts of cell extracts (μg/ml), which were prepared following growth of H. pylori HP1061 or HP1061fur in medium supplemented with 10 μg/ml MTZ (+ MTZ) or with no MTZ (− MTZ). The cell extracts were incubated with a 32P-labeled DNA amplicon representing a portion of the porGDAB promoter from positions −89 to +25 depicted in Fig. 1. Samples were resolved by nondenaturing polyacrylamide gel electrophoresis, and the positions of the radioactive bands (C-1, C-2, and C-3) were detected by use of a PhosphorImager. Note the near absence of shifted complexes, particularly band C-3 in extracts from MTZ-grown bacteria.
Identification of the transcriptional factor(s) mediating MTZ-responsive repression of PFOR.
Our purification strategy began with treatment of crude extracts with polyethyleneimine to precipitate out nucleic acids and some proteins. Second, proteins from the polyethyleneimine-treated extract were precipitated with ammonium sulfate and separated on a UNO Q1 column (see Materials and Methods). The collected fractions from UNO Q1 separation were subjected to EMSA and SDS-PAGE analysis. The set of three retarded complexes noted in Fig. 5 was detected in fractions 19 and 20 from HP1061fur (Fig. 6A). Analysis of these fractions by SDS-PAGE and silver staining revealed enrichment for proteins in the 18- and 60-kDa regions (data not shown). These regions were excised and subjected to mass spectrometry (MS) (liquid chromatography-tandem MS [LC-MS/MS]), which identified 69 proteins, 1 of which was the orphan response regulator HP1043. No other DNA binding proteins were present in this list, although it is possible that other DNA binding proteins exist in fractions 19 and 20 which were not detected by MS analysis. Ironically, we had generated a mutant variant of HP1043 (HP1043CC11) that enabled the identification of 70 new genes containing consensus DNA binding motifs in their promoter regions, including porGDAB, oorDABC, and sodB (41). Thus, the pleiotropic regulatory phenotypes reported for HP1043 seemed to overlap those generated by MTZ (13, 29, 38, 41). We named this gene hsrA (homeostatic stress regulator) to denote its function in regulating genes of central metabolism that would influence growth and growth cycle activities.
FIG 6.

Partial purification of proteins bound to the porG promoter. (A) Fractions eluted with an NaCl gradient from the UNO Q1 column (see Materials and Methods) were analyzed by EMSA. Note that in all EMSAs involving the porG amplicon, a control amplicon lacking HsrA binding sequences is included. (B) EMSA of porG and hsrA promoters in the presence of HsrA produced in E. coli (rHsrA-His10) or H. pylori (UNO Q1 column fraction 19). (C) EMSA of the porG promoter (114 bp) in the presence of competitor DNA (not-labeled hsrA promoter DNA; 114 bp; 70 ng). The amount of protein used for EMSA is depicted at the top. (D) Supershift with anti-HsrA serum and unbound DNA (lane 1). Protein concentrations (μg) and the presence or absence of anti-HsrA serum (lane 4) are indicated at the top of the gel. The supershift is indicated by an arrow. Note that the C-3 band in lane 4 was diminished compared to that in lane 3 and that the control amplicon had not shifted, a control for specificity.
DNA binding properties of native and recombinant HsrA proteins.
Since HsrA (HP1043) has been shown to bind its own promoter by DNase I footprinting (38, 41), we next determined whether the fraction containing the native HsrA protein (Q1 fraction 19) (Fig. 6A) also formed complexes with the hsrA promoter. As shown in Fig. 6B, the EMSA pattern for fraction 19 with the porG probe and a DNA probe containing the hsrA promoter region produced a similar band shift pattern (C-1, C-2, and C-3). As depicted in Fig. 6B, recombinant HsrA (rHsrA) bound poorly by EMSA (band denoted C-L), as previously reported (38, 41). The surprisingly weak DNA binding of rHsrA-His6 protein compared to fraction 19 might be due to the His tag. To test this possibility, we removed the His tag from rHsrA using thrombin protease. However, no changes in DNA binding properties of rHsrA versus rHsrA-His6 were detected (data not shown). The inability of rHsrA to strongly bind DNA might result from improper folding in E. coli or possibly to posttranslational modifications that are unique to H. pylori. The HsrA protein is denoted by an arrow in Fig. 6B, as this band showed a dose-dependent increase in intensity with both the porG and hsrA operator sequences.
To further investigate the specificity of HsrA in DNA binding, cold hsrA promoter competitor DNA was used to compete for binding with the radiolabeled porG promoter sequence. As shown in Fig. 6C, the gel shift pattern for binding of both fraction 19 and purified rHsrA-His protein to porG was abolished by competitor DNA, indicating that binding of HsrA to the porG promoter sequences was specific. It is also noteworthy that the addition of rHsrA protein to the fraction 19 extract led to an enhancement in DNA binding (Fig. 6C, lane 6) that was greater than the sum of fraction 19 and rHsrA combined (sum of lanes 2 and 4), suggesting the possibility that other components in fraction 19 may have improved the DNA binding efficiency of recombinant HsrA protein. To confirm that HsrA was in shifted complexes, HsrA antibody was added to the HsrA-DNA complexes, and as indicated in Fig. 6D, the supershifted labeled DNA complex (indicated by an arrow) confirms that HsrA is present in the complex. These studies confirm that HsrA binds the porGDAB promoter, as suggested previously (41).
HsrA and members of the HsrA regulon are repressed in the presence of MTZ.
We next tested the hypothesis that HsrA protein levels were responsive to MTZ exposure. HP1061 was grown in the presence or absence of 10 μg/ml of MTZ, cell extracts were diluted (range, 4 to 32 μg per well), and the extracts were probed with anti-HsrA serum following SDS-PAGE. As shown in Fig. 7, HsrA levels (compared with those of untreated controls) were decreased >10-fold by MTZ. Similar decreases were observed for PFOR (PorA) (4-fold) and TlpB (∼8-fold), which both contain consensus HsrA binding motifs (41). As a control, there was no change in Hsp60 protein levels. A bioinformatics search of the H. pylori genome with the HsrA consensus DNA binding sequence identified genes in which the consensus sequence was located between nucleotides 6 and 21 of the identifiable −10 hexamer, suggesting that HsrA is likely to be a positive regulator of hsrA, porGDAB, tlpB, oorD, fur, flhA, fliP, cag25, and cagA (41). Similarly, the presence of a consensus HsrA motif within the promoter of porG is confirmed in the present study. In the case of fur, the HsrA binding motif is within the high-affinity O-I Fur binding region of the gene (region from positions −35 to −60), which might affect transcription (53). Overall, our findings support the hypothesis that MTZ exposure leads to diminished levels of HsrA and account for the negative regulatory effects on porGDAB and other genes.
FIG 7.
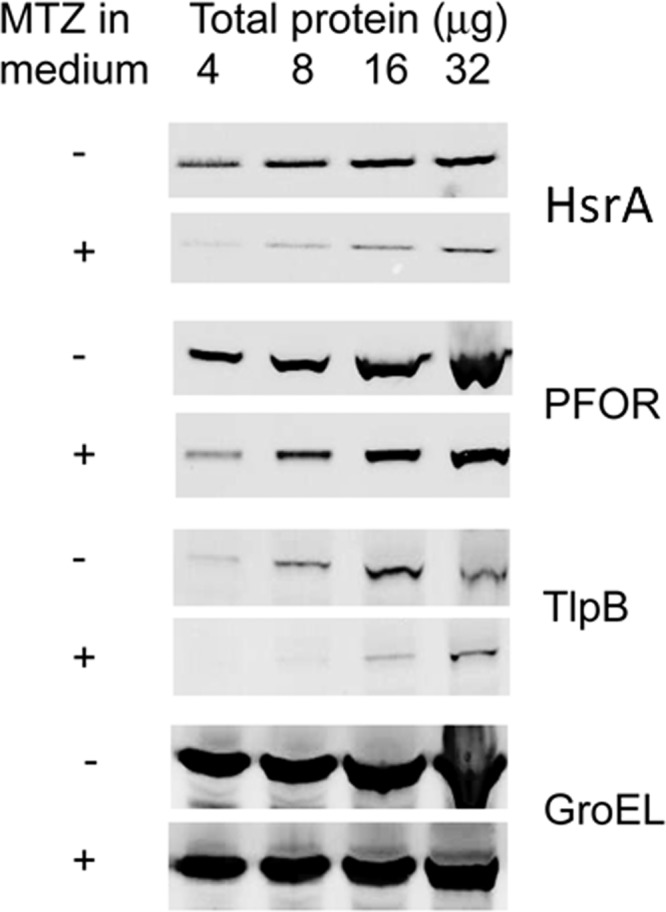
Effect of MTZ on the HsrA regulon. Shown is an immunoblot of MTZr strain HP1061 grown in the presence or absence of MTZ. Bacteria were grown to early logarithmic phase (OD600 of 0.3), at which time 10 μg/ml MTZ (+) was added to one flask, and the other served as a control (−). After 4 h, bacteria were collected, and cell extracts (4 to 32 μg/ml) were subjected to SDS-PAGE and probed with the indicated antibodies. GroEL (Hsp60) served as a control. The depicted immunoblot is representative of several replicates that also included different strains.
HsrA-repressed genes.
Since some studies have suggested that superoxide dismutase is activated in response to MTZ exposure (54), and a putative HsrA binding site was identified upstream of the promoter region (41), we examined SodB levels in extracts from MTZr bacteria grown in medium supplemented with MTZ. As shown in Fig. 8, SOD activity was nearly doubled in bacterial extracts from bacteria exposed to MTZ compared with controls. Since paraquat (PQ) (methyl viologen) has been reported to induce genes associated with oxidative stress, we determined whether SOD levels were increased in bacteria exposed to PQ. As shown in Fig. 8, SOD levels were similarly increased in extracts from bacteria grown in the presence of PQ. It is therefore conceivable that depression of HsrA levels in response to MTZ and PQ results in derepression of the sodB gene. These observations do not rule out indirect regulatory responses, such as those which might be mediated through Fur (11, 55).
FIG 8.

Superoxide dismutase activity of HP1061. Superoxide dismutase activity was measured in cell extracts of HP1061 grown in the absence or presence of MTZ (10 μg/ml) or paraquat (3.3 μg/ml). Units of activity per mg protein were determined based on 1 unit inhibiting the rate of cytochrome c reduction by 50%. The average data of two experiments are shown.
HsrA response to oxidative stress.
We next investigated the possibility that paraquat and hydrogen peroxide exposure might also activate the oxidative stress pathway by depressing levels of HsrA. Since peroxides and paraquat were toxic to H. pylori, we first optimized concentrations to enable bacterial growth, as depicted in Fig. 9A. Based on the accompanying immunoblot (Fig. 9B), HsrA levels were depressed at the 2- and 3-h time points and seemed to recover, as did microbial growth, by 5 h postchallenge. Cumene peroxide was toxic, and bacterial growth did not recover. The various peroxides were more toxic to H. pylori than was PQ, but in each case, levels of HsrA were depressed, as similarly observed with MTZ. The effect of oxidative stress on growth rates correlates with depression of HsrA levels, as was observed with MTZ.
FIG 9.

Effect of paraquat and peroxides on bacterial growth and HsrA protein levels. (A) Growth inhibition of HP1061 by paraquat (PQ) (3.3 μg/ml), hydrogen peroxide (Perox) (10 mM), cumene peroxide (CH) (5 μM), and t-butyl hydroperoxide (tBH) (5 μM). Bacterial growth was monitored spectrophotometrically at 600 nm. (B) Aliquots were taken at the indicated times and subjected to SDS-PAGE and immunoblotting with anti-HsrA serum. The results depicted are representative of two independent experiments.
HsrA is not directly affected by oxidative stress.
In examining the amino acid sequence of HsrA, we noticed two C-terminal cysteine residues that might be susceptible to oxidation by peroxides or superoxide anions generated by PQ. To evaluate a direct role for HsrA, we used hsrA mutants in which the two terminal cysteine residues were each changed to serine and a double mutant (HsrAC215S, HsrAC221S, and HsrACC11), as previously described (41). A disk diffusion assay was used to evaluate susceptibility of these mutants and wild-type HP1061 to MTZ and PQ. As shown in Fig. 10, there was essentially no difference in the diameters of zones of inhibition by MTZ or PQ between the WT and the mutants. These results suggest that the cysteine residues of HsrA are not directly involved in sensing oxidative stress and that additional levels of regulation modulate the cellular levels of HsrA in response to MTZ and to toxic oxygen species.
FIG 10.

Cysteine residues of HsrA are not inactivated by MTZ or PQ. A disk diffusion assay was used to evaluate susceptibility of HP1061 and the HP1061 HsrA C215S (Ser215), HP1061 HsrA C221S (Ser221), and HP1061 HsrA C215S,C221S (CC11) mutants to MTZ and PQ. BA plates were uniformly streaked with 0.1 ml of H. pylori cell suspensions adjusted to an OD600 of 0.1. Sterile 7-mm filter paper disks saturated with 15 μl of MTZ (10 mg/ml) or PQ (3.3 mg/ml) were placed onto the plates in triplicate. The plates were incubated for 72 h before the zones of inhibition were measured (22). The means and standard deviations of three experiments are depicted.
DISCUSSION
Atypical or orphan response regulators like HsrA of H. pylori are members of the OmpR/PhoB family of transcription factors, whose activation does not require phosphorylation and whose regulatory functions are poorly understood (41, 56–59). We recently proposed that HsrA (HP1043) serves as a homeostatic marker whose steady-state level is strictly maintained by unknown regulatory mechanisms operating on transcriptional, posttranscriptional, and posttranslational levels (31, 38, 41, 60). Here we show that exposure of H. pylori to sub-MICs of MTZ, a synthetic redox-active prodrug, perturbs this regulation, resulting in a ≥10-fold decrease in HsrA protein levels that correlated with similar decreases in expression levels of HsrA-regulated genes. We identified HsrA as the transcriptional regulator of porGDAB in a functional screen of H. pylori cell extracts for proteins that bind the porG promoter region. HsrA binding to porG and to hsrA promoter sequences was validated by EMSA, competition with hsrA and porG competitor DNA, and supershifting with antibody specific for HsrA. We mapped the transcriptional start site for porG and identified the HsrA binding motif in the −35 area of the promoter. While not depicted, the HsrA DNA binding motif in the oorDABC promoter region also spanned the −35 hexamer sequence (positions −52 to −21 relative to the transcription start site). By binding near or overlapping the −35 sequences of regulated genes, HsrA might function like a bacterial class II transcriptional activator by interacting with the housekeeping sigma factor RpoD or with the α-C-terminal domain (αCTD) of RNA polymerase to initiate transcription (61). While our studies do not rule out the possibility that other DNA binding proteins may participate in regulation of porGDAB or oorDABC, they do support a prominent role for the homeostatic regulator HsrA as a positive regulator of transcription and establish a strong correlation between HsrA levels and those of PFOR, TlpB (Fig. 7), and OOR (29).
Precisely how MTZ or its toxic metabolites mediate repression of HsrA levels is not known, but the demonstration that exposure to the superoxide anion-generating drug paraquat or to various peroxides similarly depressed HsrA protein levels suggests a common pathway associated with the response to oxidative stress. We previously noted that for some MTZr strains, as little as 3.5 μg/ml of MTZ was sufficient to depress PFOR and OOR activities and bacterial growth rates (29), while drug concentrations of >25 μg/ml were required to produce detectable DNA damage (26). This difference suggests that H. pylori may be wired to respond to reactive oxygen species before serious DNA and cellular damage occurs. The notion seems to fit with the general view that H. pylori plays a game of “cat and mouse” with the host gastric mucosa, both promoting nutrient-bearing inflammation and responding to the host oxidative inflammatory response (11, 62, 63). Our studies with a CC11 mutant tended to diminish the possibility that C-terminal cysteine residues of HsrA were chemically modified by reduced forms of MTZ or oxygen. We did not determine how reduced products of MTZ and oxygen were detected or what regulatory systems became activated to control HsrA levels.
Our previous studies also suggested that autoregulation may not play a strong role in regulating HsrA gene expression, since the CC11 mutant expressed half the HsrA protein level (also predicted to bind more tightly to DNA) of the wild-type strain and 40-times more transcript (41). Similarly, Muller et al. used a promoter swap strategy (Pcag, Ppfr, and PfecA for PhsrA) and hsrA gene duplication to alter HsrA protein levels, but while primer extension studies showed that large amounts of transcript were produced, the protein levels of HsrA were unaltered (60). These results seem to suggest that transcription control is less important than translation control in maintaining steady-state levels of HsrA. Posttranslational control systems are also largely unexplored, and we envision enzyme modifications to HsrA that improve DNA binding efficiency (suggested from gel shift experiments with native and recombinant HsrA) and perhaps proteases that become activated in response to MTZ and toxic oxygen species that might accelerate protein turnover. Given the tight on/off phenotype generated by MTZ and toxic oxygen species and in the absence of tight gene regulation reported for this species (8, 9, 38, 52, 53), these alternatives seem plausible. Studies are in progress to explore these alternative regulatory mechanisms and how they are activated.
Several studies have indicated that HsrA is highly conserved among members of the epsilonproteobacteria and that CosR and HsrA are functionally interchangeable (38–40, 60). In C. jejuni, CosR has been characterized as an oxidative stress regulator that directly controls the expression of catalase and SOD (39). Since we had identified consensus HsrA binding motifs in the divergent promoter regions of tagD (thiol peroxidase) and sodB (41) and determined experimentally that MTZ exposure led to a 2-fold increase in SOD activity over untreated controls, perhaps MTZ metabolism also contributes to oxygen toxicity. In this regard, single-electron reductions of MTZ produce the 5-nitro anion radical that in the presence of molecular oxygen is restored to the 5-nitro group with formation of superoxide anions (64). Our finding that exposure of H. pylori to sublethal concentrations of PQ or various organic and hydrogen peroxides similarly depressed levels of HsrA seems to suggest that MTZ toxicity is mediated through a preexisting oxidative stress response pathway. Here our findings differ from those reported previously for C. jejuni, where only PQ was found to affect CosR levels (39). Interpretation of PQ results for both species is complicated by PQ (methyl viologen) being a substrate of PFOR and an inhibitor of the PFOR-Fld-FqrB pathway that produces NADPH from the oxidation of pyruvate (33). Alternatively, changes in cellular energy charge could feed into the HsrA regulatory circuit. We offer two lines of evidence to support the oxidative stress regulatory route from MTZ. First, exposure to peroxides resulted in depressed levels of HsrA, and second, MTZ is not a substrate of PFOR (11, 29, 33, 48).
An HsrA orthologue was recently characterized in Helicobacter pullorum (HPMG439) and was found to be part of a two-component regulatory system and phosphorylated by a cognate histidine kinase (65). In this system, HPMG439 is associated with nitrogen regulation and not oxidative stress (65). As found with CosR and HsrA, HPMG439 could also replace hsrA in H. pylori (65). In reviewing the consensus DNA binding motifs reported for these species and inferences from superimpositions of the crystal structures of CosR and HsrA, it is reasonable to suggest that the DNA binding domains of HsrA and CosR are highly conserved, while the consensus DNA binding motifs are rather degenerate (56). Ordinarily, degenerate promoters are associated with weak binding by transcription factors, so the phenotypes of tight on/off regulation observed with PFOR and OOR seem contradictory. Since HsrA is a positive regulator of the porGDAB and oorDABC operons, perhaps posttranslational modifications to HsrA in response to MTZ or oxidative stress, such as activation of proteases, might lead to rapid depletion of HsrA and the appearance of a strong repression phenotype. We favor this possibility over transcriptional regulation of hsrA because previous efforts to manipulate gene expression through controllable promoters and gene duplication, while achieving increases in hsrA mRNA levels, did not result in changes in the protein level (38, 60, 65). Proteolytic degradation of transcription factors is considered a common mechanism involving proteases such as ClpX and Lon (66–68). In this regard, the expression level of the serine protease HtrA has been shown to increase upon exposure of H. pylori to hydrogen peroxide (69).
Overlapping regulatory networks and oxidative stress.
Gastric species of Helicobacter reside in an extreme environment with few microbial competitors and a rather restricted range of environmental stimuli to contend with. Moreover, the strongly acidic environment both influences growth and concentrates metals, which, together with reactive oxygen and nitrogen species produced in the chronic inflammatory response, contribute generally to oxidative stress (reviewed in reference 11). The importance of acid is underscored by the number of regulatory factors associated with the acid-protective urease system, including Fur, NikR, and the acid response two-component regulator ArsRS (7, 8, 10, 60, 70–72). The effect of MTZ on these regulators has not been extensively studied, although overexpression of sodB, a Fur-regulated gene, has been shown to contribute to MTZ resistance (54). In this regard, we showed that mutations of hsrA (CC11 mutant) lowered transcript levels of fur by 1.75-fold in exponential phase and had a similar effect on flhA (regulator of flagellum biogenesis) (41). In the present study, we found PFOR enzymatic activity to be repressed by 85% in fur mutants exposed to MTZ, compared to 50% for WT strains (Fig. 4). We identified an HsrA motif in the upstream region of fur from positions −35 to −60 in an area containing a Fur DNA binding motif (53). It is conceivable that HsrA and Fur compete for binding (autoregulation) and consequently affect transcription of this important global regulator. Further study will be required to explore the effects of MTZ on Fur and other downstream regulatory consequences associated with altered levels of HsrA.
In summary, our studies identified HsrA as a positive transcriptional regulator of the porGDAB operon in H. pylori. HsrA is a master regulator that coordinates the expression of genes associated with central metabolism and virulence with those of cell division and bacterial growth. Cellular levels of HsrA are tightly controlled throughout the growth cycle, and genetic efforts to manipulate HsrA levels have not been successful (31, 38, 41). Our studies show that the dose-dependent effects of MTZ on bacterial growth and the activities of core enzymes of central metabolism are mediated through HsrA. While it was unexpected to find MTZ toxicity routed through an existing oxidative defense pathway, we believe that this response pathway may be unique to those mucosal pathogens that promote inflammation to acquire nutrients. Our findings might partly explain perceived beneficial effects of inclusion of MTZ in salvage therapies to treat infections caused by drug-resistant strains. Perhaps, further elucidation of the metabolic and regulatory shifts associated with MTZ exposure and depressed levels of HsrA will lead to the identification of potentially new drug targets.
ACKNOWLEDGMENT
This work was supported by NIH grant R01-DK073823 to P.S.H.
Footnotes
Published ahead of print 2 December 2013
REFERENCES
- 1.Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315 [DOI] [PubMed] [Google Scholar]
- 2.Peek RM, Jr, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2:28–37. 10.1038/nrc703 [DOI] [PubMed] [Google Scholar]
- 3.Dunn BE, Cohen H, Blaser MJ. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bury-Moné S, Skouloubris S, Labigne A, De Reuse H. 2001. The Helicobacter pylori UreI protein: role in adaptation to acidity and identification of residues essential for its activity and for acid activation. Mol. Microbiol. 42:1021–1034. 10.1046/j.1365-2958.2001.02689.x [DOI] [PubMed] [Google Scholar]
- 5.Sachs G, Kraut JA, Wen Y, Feng J, Scott DR. 2006. Urea transport in bacteria: acid acclimation by gastric Helicobacter spp. J. Membr. Biol. 212:71–82. 10.1007/s00232-006-0867-7 [DOI] [PubMed] [Google Scholar]
- 6.Croxen MA, Sisson G, Melano R, Hoffman PS. 2006. The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J. Bacteriol. 188:2656–2665. 10.1128/JB.188.7.2656-2665.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Müller S, Götz M, Beier D. 2009. Histidine residue 94 is involved in pH sensing by histidine kinase ArsS of Helicobacter pylori. PLoS One 4:e6930. 10.1371/journal.pone.0006930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pflock M, Finsterer N, Joseph B, Mollenkopf H, Meyer TF, Beier D. 2006. Characterization of the ArsRS regulon of Helicobacter pylori involved in acid adaptation. J. Bacteriol. 188:3449–3462. 10.1128/JB.188.10.3449-3462.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pflock M, Kennard S, Delany I, Scarlato V, Beier D. 2005. Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73:6437–6445. 10.1128/IAI.73.10.6437-6445.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2006. Involvement of the HP0165-HP0166 two-component system in expression of some acidic-pH-upregulated genes of Helicobacter pylori. J. Bacteriol. 188:1750–1761. 10.1128/JB.188.5.1750-1761.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Alamuri P, Maier RJ. 2006. The diverse antioxidant systems of Helicobacter pylori. Mol. Microbiol. 61:847–860. 10.1111/j.1365-2958.2006.05302.x [DOI] [PubMed] [Google Scholar]
- 12.Ernst PB, Gold BD. 2000. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 54:615–640. 10.1146/annurev.micro.54.1.615 [DOI] [PubMed] [Google Scholar]
- 13.McAtee CP, Hoffman PS, Berg DE. 2001. Identification of differentially regulated proteins in metronidazole resistant Helicobacter pylori by proteome techniques. Proteomics 1:516–521. [DOI] [PubMed] [Google Scholar]
- 14.Morishita K, Takeuchi H, Morimoto N, Shimamura T, Kadota Y, Tsuda M, Taniguchi T, Ukeda H, Yamamoto T, Sugiura T. 2012. Superoxide dismutase activity of Helicobacter pylori per se from 158 clinical isolates and the characteristics. Microbiol. Immunol. 56:262–272. 10.1111/j.1348-0421.2012.00433.x [DOI] [PubMed] [Google Scholar]
- 15.Glupczynski Y, Burette A. 1990. Drug therapy for Helicobacter pylori infection: problems and pitfalls. Am. J. Gastroenterol. 85:1545–1551 [PubMed] [Google Scholar]
- 16.Graham DY, Opekun AR, Belson G, El-Zimaity HM, Carlson MR. 2005. Novel bismuth-metronidazole-tetracycline triple-layer tablet for treatment of Helicobacter pylori. Aliment. Pharmacol. Ther. 21:165–168. 10.1111/j.1365-2036.2005.02322.x [DOI] [PubMed] [Google Scholar]
- 17.Venerito M, Krieger T, Ecker T, Leandro G, Malfertheiner P. 2013. Meta-analysis of bismuth quadruple therapy versus clarithromycin triple therapy for empiric primary treatment of Helicobacter pylori infection. Digestion 88:33–45. 10.1159/000350719 [DOI] [PubMed] [Google Scholar]
- 18.Dore MP, Marras L, Maragkoudakis E, Nieddu S, Manca A, Graham DY, Realdi G. 2003. Salvage therapy after two or more prior Helicobacter pylori treatment failures: the super salvage regimen. Helicobacter 8:307–309. 10.1046/j.1523-5378.2003.00150.x [DOI] [PubMed] [Google Scholar]
- 19.Kuo CH, Hu HM, Kuo FC, Hsu PI, Chen A, Yu FJ, Tsai PY, Wu IC, Wang SW, Li CJ, Weng BC, Chang LL, Jan CM, Wang WM, Wu DC. 2009. Efficacy of levofloxacin-based rescue therapy for Helicobacter pylori infection after standard triple therapy: a randomized controlled trial. J. Antimicrob. Chemother. 63:1017–1024. 10.1093/jac/dkp034 [DOI] [PubMed] [Google Scholar]
- 20.Goodwin A, Kersulyte D, Sisson G, Veldhuyzen van Zanten SJO, Berg DE, Hoffman PS. 1998. Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol. Microbiol. 28:383–393. 10.1046/j.1365-2958.1998.00806.x [DOI] [PubMed] [Google Scholar]
- 21.Martínez-Júlvez M, Rojas AL, Olekhnovich I, Espinosa Angarica V, Hoffman PS, Sancho J. 2012. Structure of RdxA—an oxygen-insensitive nitroreductase essential for metronidazole activation in Helicobacter pylori. FEBS J. 279:4306–4317. 10.1111/febs.12020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olekhnovich IN, Goodwin A, Hoffman PS. 2009. Characterization of the NAD(P)H oxidase and metronidazole reductase activities of the RdxA nitroreductase of Helicobacter pylori. FEBS J. 276:3354–3364. 10.1111/j.1742-4658.2009.07060.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerrits MM, van der Wouden EJ, Bax DA, van Zwet AA, van Vliet AH, de Jong A, Kusters JG, Thijs JC, Kuipers EJ. 2004. Role of the rdxA and frxA genes in oxygen-dependent metronidazole resistance of Helicobacter pylori. J. Med. Microbiol. 53:1123–1128. 10.1099/jmm.0.45701-0 [DOI] [PubMed] [Google Scholar]
- 24.Jeong JY, Mukhopadhyay AK, Dailidiene D, Wang Y, Velapatiño B, Gilman RH, Parkinson AJ, Nair GB, Wong BCY, Lam SK, Mistry R, Segal RI, Yuan Y, Gao H, Alarcon HT, Brea ML, Ito Y, Kersulyte D, Lee HK, Gong Y, Goodwin A, Hoffman PS, Berg DE. 2000. Sequential inactivation of rdxA (HP0954) and frxA (HP0642) nitroreductase genes causes moderate and high-level metronidazole resistance in Helicobacter pylori. J. Bacteriol. 182:5082–5090. 10.1128/JB.182.18.5082-5090.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon DH, El-Zaatari FA, Kato M, Osato MS, Reddy R, Yamaoka Y, Graham DY. 2000. Analysis of rdxA and involvement of additional genes encoding NAD(P)H flavin oxidoreductase (FrxA) and ferredoxin-like protein (FdxB) in metronidazole resistance of Helicobacter pylori. Antimicrob. Agents Chemother. 44:2133–2142. 10.1128/AAC.44.8.2133-2142.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sisson G, Jeong JY, Goodwin A, Bryden L, Rossler N, Lim-Morrison S, Raudonikiene A, Berg DE, Hoffman PS. 2000. Metronidazole activation is mutagenic and causes DNA fragmentation in Helicobacter pylori and in Escherichia coli containing a cloned H. pylori rdxA+ (nitroreductase) gene. J. Bacteriol. 182:5091–5096. 10.1128/JB.182.18.5091-5096.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong JY, Mukhopadhyay AK, Akada JK, Dailidiene D, Hoffman PS, Berg DE. 2001. Roles of FrxA and RdxA nitroreductases of Helicobacter pylori in susceptibility and resistance to metronidazole. J. Bacteriol. 183:5155–5162. 10.1128/JB.183.17.5155-5162.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albert TJ, Dailidiene D, Dailide G, Norton JE, A. Kalia A, Richmond TA, Molla M, Singh J, Green RD, Berg DE. 2005. Mutation discovery in bacterial genomes: metronidazole resistance in Helicobacter pylori. Nat. Methods 2:951–953. 10.1038/nmeth805 [DOI] [PubMed] [Google Scholar]
- 29.Hoffman PS, Goodwin A, Johnsen J, Magee K, Veldhuyzen van Zanten SJO. 1996. Metabolic activities of metronidazole-sensitive and -resistant strains of Helicobacter pylori: repression of pyruvate oxidoreductase and expression of isocitrate lyase activity correlate with resistance. J. Bacteriol. 178:4822–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaakoush NO, Asencio C, Megraud F, Mendz GL. 2009. A redox basis for metronidazole resistance in Helicobacter pylori. Antimicrob. Agents Chemother. 53:1884–1891. 10.1128/AAC.01449-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croxen MA, Ernst PB, Hoffman PS. 2007. Antisense RNA modulation of alkyl hydroperoxide reductase (AhpC) levels in Helicobacter pylori correlates with organic peroxide toxicity but not infectivity. J. Bacteriol. 189:3359–3368. 10.1128/JB.00012-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes NJ, Clayton CL, Chalk PA, Kelly DJ. 1998. Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J. Bacteriol. 180:1119–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.St. Maurice M, Cremades N, Croxen MA, Sisson G, Sancho J, Hoffman PS. 2007. Flavodoxin:quinone reductase (FqrB): a redox partner of pyruvate:ferredoxin oxidoreductase that reversibly couples pyruvate oxidation to NADPH production in Helicobacter pylori and Campylobacter jejuni. J. Bacteriol. 189:4764–4773. 10.1128/JB.00287-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galano JJ, Alías M, Pérez R, Velázquez-Campoy A, Hoffman PS, Sancho J. 2013. Improved flavodoxin inhibitors with potential therapeutic effects against Helicobacter pylori infection. J. Med. Chem. 56:6248–6258. 10.1021/jm400786q [DOI] [PubMed] [Google Scholar]
- 35.Daucher JA, Krieg NR. 1995. Pyruvate:ferredoxin oxidoreductase in Campylobacter species. Can. J. Microbiol. 41:198–201. 10.1139/m95-027 [DOI] [Google Scholar]
- 36.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176-180 [DOI] [PubMed] [Google Scholar]
- 37.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. 10.1038/41483 [DOI] [PubMed] [Google Scholar]
- 38.Delany I, Spohn G, Rappuoli R, Scarlato V. 2002. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 184:4800–4810. 10.1128/JB.184.17.4800-4810.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang S, Kim M, Ryu S, Jeon B. 2011. Regulation of oxidative stress response by CosR, an essential response regulator in Campylobacter jejuni. PLoS One 6:e22300. 10.1371/journal.pone.0022300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang S, Shang Q, Ryu S, Jeon B. 2012. Transcriptional regulation of the CmeABC multidrug efflux pump and the KatA catalase by CosR in Campylobacter jejuni. J. Bacteriol. 194:6883–68891. 10.1128/JB.01636-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olekhnovich IN, Vitko S, Chertihin O, Hontecillas R, Viladomiu M, Bassaganya-Riera J, Hoffman PS. 2013. Mutations to essential orphan response regulator HP1043 of Helicobacter pylori result in growth-stage regulatory defects. Infect. Immun. 81:1439–1449. 10.1128/IAI.01193-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 43.Chalker AF, Minehart HW, Hughes NJ, Koretke KK, Lonetto MA, Brinkman KK, Warren PV, Lupas A, Stanhope MJ, Brown JR, Hoffman PS. 2001. Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183:1259–1268. 10.1128/JB.183.4.1259-1268.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCord JM, Fridovich I. 1969. Superoxide dismutase: an enzymatic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244:6049–6055 [PubMed] [Google Scholar]
- 45.Mamelak D, Mylvaganam M, Whetstone H, Hartmann E, Lennarz W, Wyrick PB, Raulston J, Han H, Hoffman PS, Lingwood CA. 2001. Hsp70s contain a specific sulfogalactolipid binding site. Differential aglycone influence on sulfogalactosyl ceramide binding by recombinant prokaryotic and eukaryotic hsp70 family members. Biochemistry 40:3572–3582. 10.1021/bi001643u [DOI] [PubMed] [Google Scholar]
- 46.Huesca M, Borgia S, Hoffman P, Lingwood CA. 1996. Acidic pH changes receptor binding specificity of Helicobacter pylori: a binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect. Immun. 64:2643–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helsel LO, Bibb WF, Butler CA, Hoffman PS, McKinney RM. 1988. Recognition of a genus-wide antigen of Legionella by a monoclonal antibody. Curr. Microbiol. 16:201–208. 10.1007/BF01568530 [DOI] [Google Scholar]
- 48.Sisson G, Goodwin A, Raudonikiene A, Birks NJ, Han H, Mukhopadhyay A, Berg DE, Hoffman PS. 2002. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob. Agents Chemother. 46:2116–2123. 10.1128/AAC.46.7.2116-2123.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toptchieva A, Sisson G, Bryden LJ, Taylor DE, Hoffman PS. 2003. An inducible tellurite-resistance operon in Proteus mirabilis. Microbiology 149:1285–1295. 10.1099/mic.0.25981-0 [DOI] [PubMed] [Google Scholar]
- 50.Hoffman PS, Ripley M, Weeratna R. 1992. Cloning and nucleotide sequence of a gene (ompS) encoding the major outer membrane protein of Legionella pneumophila. J. Bacteriol. 174:914–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gancz H, Censini S, Merrell DS. 2006. Iron and pH homeostasis intersect at the level of Fur regulation in the gastric pathogen Helicobacter pylori. Infect. Immun. 74:602–614. 10.1128/IAI.74.1.602-614.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilbreath JJ, West AL, Pich OQ, Carpenter BM, Michel S, Merrell DS. 2012. Fur activates expression of the 2-oxoglutarate oxidoreductase genes (oorDABC) in Helicobacter pylori. J. Bacteriol. 194:6490–6497. 10.1128/JB.01226-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delany I, Spohn G, Rappuoli R, Scarlato V. 2003. An anti-repression Fur operator upstream of the promoter is required for iron-mediated transcriptional autoregulation in Helicobacter pylori. Mol. Microbiol. 50:1329–1338. 10.1046/j.1365-2958.2003.03757.x [DOI] [PubMed] [Google Scholar]
- 54.Tsugawa H, Suzuki H, Satoh K, Hirata K, Matsuzaki J, Saito Y, Suematsu M, Hibi T. 2011. Two amino acids mutation of ferric uptake regulator determines Helicobacter pylori resistance to metronidazole. Antioxid. Redox Signal. 14:15–23. 10.1089/ars.2010.3146 [DOI] [PubMed] [Google Scholar]
- 55.Carpenter BM, Gancz H, Gonzalez-Nieves RP, West AL, Whitmire JM, Michel SL, Merrell DS. 2009. A single nucleotide change affects fur-dependent regulation of sodB in H. pylori. PLoS One 4:e5369. 10.1371/journal.pone.0005369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong E, Lee HM, Ko H, Kim DU, Jeon BY, Jung J, Shin J, Lee SA, Kim Y, Jeon YH, Cheong C, Cho HS, Lee W. 2007. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 282:20667–20675. 10.1074/jbc.M609104200 [DOI] [PubMed] [Google Scholar]
- 57.Rotter C, Muhlbacher S, Salamon D, Schmitt R, Scharf B. 2006. Rem, a new transcriptional activator of motility and chemotaxis in Sinorhizobium meliloti. J. Bacteriol. 188:6932–6942. 10.1128/JB.01902-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hickey JM, Weldon L, Hefty PS. 2011. The atypical OmpR/PhoB response regulator ChxR from Chlamydia trachomatis forms homodimers in vivo and binds a direct repeat of nucleotide sequences. J. Bacteriol. 193:389–398. 10.1128/JB.00833-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fraser JS, Merlie JP, Jr, Echols N, Weisfield SR, Mignot T, Wemmer DE, Zusman DR, Alber T. 2007. An atypical receiver domain controls the dynamic polar localization of the Myxococcus xanthus social motility protein FrzS. Mol. Microbiol. 65:319–332. 10.1111/j.1365-2958.2007.05785.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muller S, Pflock M, Schar J, Kennard S, Beier D. 2007. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol. Res. 162:1–14. 10.1016/j.micres.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 61.Decker KB, James TD, Stibitz S, Hinton DH. 2012. The Bordetella pertussis model of exquisite gene control by the global transcription factor BvgA. Microbiology 158:1665–1676. 10.1099/mic.0.058941-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berg DE, Hoffman PS, Appelmelk BJ, Kusters JG. 1997. The Helicobacter pylori genome sequence: genetic factors for long life in the gastric mucosa. Trends Microbiol. 5:468–474 [DOI] [PubMed] [Google Scholar]
- 63.Choi SS, Chivers PT, Berg DE. 2011. Point mutations in Helicobacter pylori's fur regulatory gene that alter resistance to metronidazole, a prodrug activated by chemical reduction. PLoS One 6:e18236. 10.1371/journal.pone.0018236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreno NJ, Docampo R. 1985. Mechanism of toxicity of nitro compounds used in the chemotherapy of trichomoniasis. Environ. Health Perspect. 64:199–208. 10.1289/ehp.8564199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bauer S, Endres M, Lange M, Schmidt T, Schumbrutzki C, Sickmann A, Beier D. 2013. Novel function assignment to a member of the essential HP1043 response regulator family of epsilon-proteobacteria. Microbiology 159:880–889. 10.1099/mic.0.066548-0 [DOI] [PubMed] [Google Scholar]
- 66.Gora KG, Cantin A, Wohlever M, Joshi KK, Perchuk BS, Chien P, Laub MT. 2013. Regulated proteolysis of a transcription factor complex is critical to cell cycle progression in Caulobacter crescentus. Mol. Microbiol. 87:1277–1289. 10.1111/mmi.12166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takaya A, Kubota Y, Isogai E, Yamamoto T. 2005. Degradation of the HilC and HilD regulator proteins by ATP-dependent Lon protease leads to downregulation of Salmonella pathogenicity island 1 gene expression. Mol. Microbiol. 55:839–852. 10.1111/j.1365-2958.2004.04425.x [DOI] [PubMed] [Google Scholar]
- 68.Heuveling J, Possling A, Hengge R. 2008. A role for Lon protease in the control of the acid resistance genes of Escherichia coli. Mol. Microbiol. 69:534–547. 10.1111/j.1365-2958.2008.06306.x [DOI] [PubMed] [Google Scholar]
- 69.Huang CH, Chiou SH. 2011. Proteomic analysis of upregulated proteins in Helicobacter pylori under oxidative stress induced by hydrogen peroxide. Kaohsiung J. Med. Sci. 27:544–553. 10.1016/j.kjms.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beier D, Frank R. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182:2068–2076. 10.1128/JB.182.8.2068-2076.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Contreras M, Thiberge JM, Mandrand-Berthelot MA, Labigne A. 2003. Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol. Microbiol. 49:947–963. 10.1046/j.1365-2958.2003.03621.x [DOI] [PubMed] [Google Scholar]
- 72.Danielli A, Roncarati D, Delany I, Chiarini V, Rappuoli R, Scarlato V. 2006. In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. J. Bacteriol. 188:4654–4662. 10.1128/JB.00120-06 [DOI] [PMC free article] [PubMed] [Google Scholar]