

Abstract
To understand the energy conversion activities of the anaerobic sulfate-reducing bacteria, it is necessary to identify the components involved in electron flow. The importance of the abundant type I tetraheme cytochrome c3 (TpIc3) as an electron carrier during sulfate respiration was questioned by the previous isolation of a null mutation in the gene encoding TpIc3, cycA, in Desulfovibrio alaskensis G20. Whereas respiratory growth of the CycA mutant with lactate and sulfate was little affected, growth with pyruvate and sulfate was significantly impaired. We have explored the phenotype of the CycA mutant through physiological tests and transcriptomic and proteomic analyses. Data reported here show that electrons from pyruvate oxidation do not reach adenylyl sulfate reductase, the enzyme catalyzing the first redox reaction during sulfate reduction, in the absence of either CycA or the type I cytochrome c3:menaquinone oxidoreductase transmembrane complex, QrcABCD. In contrast to the wild type, the CycA and QrcA mutants did not grow with H2 or formate and sulfate as the electron acceptor. Transcriptomic and proteomic analyses of the CycA mutant showed that transcripts and enzymes for the pathway from pyruvate to succinate were strongly decreased in the CycA mutant regardless of the growth mode. Neither the CycA nor the QrcA mutant grew on fumarate alone, consistent with the omics results and a redox regulation of gene expression. We conclude that TpIc3 and the Qrc complex are D. alaskensis components essential for the transfer of electrons released in the periplasm to reach the cytoplasmic adenylyl sulfate reductase and present a model that may explain the CycA phenotype through confurcation of electrons.
INTRODUCTION
Environmental impacts of the sulfate-reducing bacteria (SRB), such as the formation of toxic sulfide and corrosion of metals, stem from SRB sulfate respiration and from vigorous metal metabolism, including both reduction and oxidation. The involvement of these bacteria in microbially influenced metal corrosion (1, 2) and their possible application for the remediation of toxic metal-contaminated environments (3, 4) have driven a systems biology investigation of their metabolism. To predict or control metabolic capabilities for beneficial environmental purposes, the bioenergetic pathways of the SRB need to be elucidated in more detail.
Typically, SRB of the genus Desulfovibrio use H2, organic acid substrates, formate, or short-chain alcohols as electron donors for sulfate reduction, a process that occurs in the cytoplasm and is mediated by soluble enzymes. With hydrogen as the source of electrons, at an environmentally relevant partial pressure of 10 Pa (reduction potential [E′] = −300 mV) (5), the first two-electron reduction of sulfate to sulfite (E0′ = −516 mV) cannot be accomplished without an additional energy input. Sulfate is activated at the expense of two ATP equivalents through the activity of sulfate adenylyltransferase (equation 1), producing adenosine phosphosulfate (APS), which increases the redox potential of the APS/HSO3− couple to −60 mV (5, 6), allowing reduction by electrons from H2 or organic electron donors. The APS is reduced by APS reductase, generating sulfite and AMP (equation 2). Finally, the six-electron reduction of sulfite to sulfide (E0′ = −116 mV) is carried out by dissimilatory sulfite reductase (equation 3) and is apparently coupled to electron transport-linked phosphorylation. Although the immediate electron donor for this reaction is not proven, electrons from H2 can support this reduction.
| (1) |
| (2) |
| (3) |
Members of the genus Desulfovibrio have been widely used as experimental models for biochemical and molecular studies of sulfate reduction because these strains have been easy to cultivate and were the first to be genetically manipulated (7, 8). Desulfovibrio strains incompletely oxidize the preferred substrates lactate and pyruvate (9–11), as shown in equation 4 for lactate.
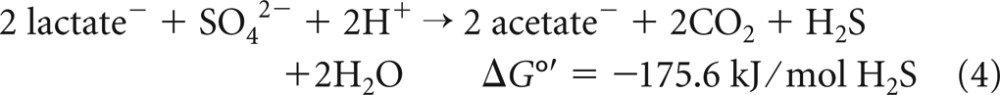 |
The enzyme for the oxidation of lactate is a cytoplasm-facing membrane-associated pyridine nucleotide-independent lactate dehydrogenase (11, 12), but the gene encoding the enzyme has yet to be unequivocally identified. Oxidation of pyruvate to acetyl coenzyme A (acetyl-CoA) and CO2 is carried out by the soluble pyruvate:ferredoxin oxidoreductase. In addition to growth by sulfate reduction, many of the Desulfovibrio species can ferment lactate (11) or pyruvate (10), with substrate-level phosphorylation conserving the energy of acetyl-CoA (equations 5 and 6).
| (5) |
| (6) |
For electrons from hydrogen to be used for sulfate reduction, it is presumed that oxidation of H2 takes place in the periplasm, where there are multiple isozymes of hydrogenase (13, 14). If true, pathways must exist for electron flow from the periplasm to the cytoplasm, where sulfate reduction occurs. In contrast, oxidation of organic acids proceeds in the cytoplasm. The energy obtained from oxidation of lactate and pyruvate (15) has been proposed to involve a hydrogen cycling mechanism that contributes to the metabolic energy available during sulfate reduction (16). In this model, electrons from the oxidized organic acids would be used for the production of hydrogen by the activity of cytoplasmically located hydrogenases. The hydrogen could diffuse to the periplasm, where oxidation by one or more of the various hydrogenase isozymes would recapture the electrons for sulfate reduction and leave the protons to contribute to the proton motive force. Even though evidence for the functioning of such a process was obtained with cells growing on pyruvate (16), sequencing of SRB genomes (14, 17, 18) has not revealed conserved genes for cytoplasmic hydrogenases that might be expected if this metabolism were essential to energy conversion during sulfate respiration. It is significant that among the sulfate-reducing bacteria there are two conserved transmembrane complexes (TMCs), QmoABC and DsrMKJOP, which have been predicted to provide membrane conduits for the electrons required for APS reductase and bisulfite reductase, respectively (5, 13, 14, 19). Although the QmoABC complex has been shown to be essential for sulfate reduction (20), electron delivery to and from this TMC has not been established.
Recently, a third TMC, QrcABCD, has been described in two Desulfovibrio spp. and has been shown to be specifically associated with sulfate respiration (21). It appears that the Qrc complex accepts electrons from periplasmic hydrogenases and formate dehydrogenases via type I tetraheme cytochrome c3 (TpIc3), acting as a TpIc3:menaquinone oxidoreductase (14, 21–23). All three TMCs, Qmo, Dsr, and Qrc, contain subunits binding b-type hemes, from which we infer their involvement in electron transfer to or from the menaquinone pool, thus potentially participating in the energy-conserving redox loop that may operate during sulfate reduction (24).
With the availability of complete genome sequences for a number of SRB, the use of a systems biology approach for exploring electron pathways is now possible. The work here focuses on Desulfovibrio alaskensis G20 (17) (formerly Desulfovibrio desulfuricans G20) for which mutants lacking the abundant TpIc3 (Dde_1382; Mr, 13,000), CycA mutants, are available (25, 26). This tetraheme cytochrome is the most abundant c-type cytochrome in the periplasm of Desulfovibrio spp. (27) and has been suggested to be an electron reservoir or function as a periplasmic capacitor (18) providing electrons to (or receiving electrons from) a number of transmembrane complexes. Here we explore the alterations in electron flow in G20 resulting from the elimination of TpIc3 or QrcA. We have found physiological evidence that both the cytochrome and Qrc complex are essential for growth by sulfate respiration with H2 or formate and play important roles in the flow of electrons from pyruvate to APS reductase for sulfate reduction. An absence of TpIc3 may also affect gene transcription by altering the redox status of the cell.
MATERIALS AND METHODS
Bacterial strains.
Table 1 lists the strains and plasmids used in this study. Strain G20 (originally considered D. desulfuricans but now renamed D. alaskensis G20 [17]) was derived from a Desulfovibrio desulfuricans G100A strain isolated from a petroleum well corrosion site (28). The G20 strain is a spontaneously nalidixic acid-resistant isolate that is also cured of the cryptic endogenous, 2.3-kb plasmid pBG1 (29, 30). This small plasmid has provided an SRB replicon for use in construction of shuttle vectors for SRB (29). I2 is a mutant derived from G20 through plasmid insertion and interruption of the gene encoding the type I tetraheme c-type cytochrome TpIc3 (cycA, Dde_3182) (26). Recombination events have been documented in I2 maintained under plasmid-encoded antibiotic selection that restored a complete cycA gene without removal of the plasmid insert from the genome (26). Throughout these studies, care was taken to monitor and minimize the suppressor population by maintaining antibiotic selection and avoiding extensive exposure to electron donor-limiting conditions.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristicsa | Source or reference(s) |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| One Shot TOP10 competent cells | F− ϕ80lacZΔM15 ΔlacX74 recA1 endA1 mcrA Δ(mrr-hsdRMS-mcrBC) araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) nupG | Invitrogen |
| Alpha-Select Bronze and Silver Efficiency | deoR endA1 recA1 relA1 gyrA96 hsdR17(rK− mK+) supE44 thi-1 Δ(lacZYA-argFV169) ϕ80lacZΔM15 F− | Bioline |
| Desulfovibrio alaskensis G20 | ||
| Wild type | Desulfovibrio alaskensis G20 (formerly Desulfovibrio desulfuricans G20); cured of pBG1; Nxr | 17, 28 |
| I2 | G20 with (Dde_3182) cycA encoding type I tetraheme cytochrome c3 interrupted by plasmid insertion; Kmr | 26 |
| CycA mutant | G20 cycA::mini-Tn10 with (Dde_3182) cycA encoding type I tetraheme cytochrome c3 interrupted by a mini-Tn10 transposon; Kmr | 39 |
| QrcA mutant | G20 qrcA::mini-Tn5 with (Dde_2932) qrcA encoding a subunit of type I cytochrome c3:menaquinone oxidoreductase interrupted by a mini-Tn5 transposon; Kmr | A. Deutschbauer |
| Plasmids | ||
| pCR4Blunt-TOPO | Cloning vector; Kmr Ampr | Invitrogen |
| pCR8/GW/TOPO | Cloning vector; Spr | Invitrogen |
| pMO719 | Desulfovibrio shuttle vector; SRB replicon pBG1; Spr | 52 |
| pMO736 | A 763-bp fragment containing the 393-bp type I tetraheme cytochrome c3 (Dde_3182) and 343 bp of upstream region cloned into pCR4Blunt-TOPO; Kmr | This study |
| pMO738 | Complementing plasmid for the type I tetraheme cytochrome c3 (Dde_3182) fragment removed from pMO736 via PmeI/SnaBI digestion and ligated into the EcoRV site of pMO719; Spr | This study |
| pMO756 | Complementing plasmid for QrcA derived from pMO9075b with Paph(3′)-II::RBS::qrcA (Dde_2932); Spr | This study |
| pMO9072 | Modified from pMO719 with Paph(3′)-II and MCS; no RBS; for complementation constructs; Sprb | 20 |
| pMO9075 | Modified from pMO9072 with three fragments removed via restriction digests; for complementation constructs; Sprb | This study |
| pSC27 | Desulfovibrio shuttle vector; source of SRB replicon pBG1, mob; Kmr | 29 |
Strr, streptomycin resistant; Nxr nalidixic acid resistant; Kmr, kanamycin resistant; Ampr, ampicillin resistant; Spr, spectinomycin resistant; aph(3′)-IIp, promoter from the kanamycin resistance gene aph(3′)-II; RBS, ribosomal binding site (TGCAGTCCCAGGAGGTACCAT); MCS, multicloning site.
See Fig. S1 in the supplemental material.
The G20 cycA::mini-Tn10 (Dde_3182) mutant was graciously provided by L. Krumholz (25), and the qrcA::mini-Tn5 mutant (Dde_2932) was generously made available to us by A. Deutschbauer from a G20 transposon library constructed as described by Oh et al. (31).
Media and culture conditions.
Cultures of G20 and the constructed mutants were routinely grown anaerobically in a defined medium designated MO basal salts (20). MO basal salts were amended to contain various electron donors and acceptors at the following concentrations: lactate-sulfate medium contained sodium lactate at 60 mM and Na2SO4 at 30 mM, and pyruvate-sulfate medium contained sodium pyruvate at 60 mM and Na2SO4 at 15 mM. In lieu of Na2SO4, 30 mM Na2SO3 was used in medium designated lactate-sulfite and 15 mM Na2SO3 replaced Na2SO4 to make pyruvate-sulfite medium. Culture medium used for growth not dependent on the reduction of sulfur oxides was MO basal salts with 60 mM sodium pyruvate (for fermentation) or 60 mM sodium fumarate (for disproportionation). The formate growth medium contained 20 mM acetate to supplement 50 mM formate and 30 mM Na2SO4. H2 growth medium contained 30 mM Na2SO4 and 20 mM acetate with 80% H2–20% CO2 in the headspace. The Na2SO3, sodium pyruvate, and sodium fumarate were prepared immediately before use, filter sterilized, and added to sterile medium. The pH values of all media were adjusted to 7.0 to 7.2 before use. Prior to inoculation, all liquid media were reduced with Na2S at a final concentration of 0.5 mM from an anoxically prepared and maintained 100 mM Na2S stock solution, unless stated otherwise. In media that lacked sulfur oxides as terminal electron acceptors, the added Na2S also provided a sulfur source for growth. When deviations from these medium compositions were used, they are indicated.
Rich medium was any medium modified by addition of yeast extract to a final concentration of 0.1% (wt/vol). For plating, agar was added to 1.5% (wt/vol) to solidify the medium that was reduced with 7.5 ml per liter of 2.5% (wt/vol) cysteine-HCl, pH 7.0. Culture manipulations were carried out in an anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI) that was kept at ∼34°C and that had an atmosphere of ca. 5% (vol/vol) hydrogen in nitrogen.
To determine growth rates and final culture densities, 5-ml cultures were grown at 34°C in 27-ml glass tubes (17 by 150 mm; Bellco Glass, Vineland, NJ), and growth was monitored as increases in culture turbidity (optical density at 600 nm [OD600]). These tubes had crimp-sealed stoppers and were flushed with N2 to establish a 100% N2 headspace. For fermentation of cultures, the headspace-to-culture volume was maintained at a ratio of 4:1. To test growth on H2, 10-ml cultures were incubated at 37°C in tubes placed horizontally to increase surface area gas exchange. The final extent of growth was established by Bradford protein determinations (32) with bovine serum albumin as the standard.
Escherichia coli strains were cultured aerobically in LC medium (10 g tryptone, 5 g yeast extract, and 5 g NaCl per liter) at 37°C. Following transformations, E. coli cultures were recovered in SOC medium (20) and plated on LC medium solidified with 1.5% (wt/vol) agar containing 50 μg kanamycin/ml or 100 μg spectinomycin/ml.
Complementation of I2, cycA::mini-Tn10, and qrcA::mini-Tn5 mutants.
Complementing plasmids for the cycA and qrcA mutants were constructed either by the use of restriction digests with ligation or by the sequence- and ligation-independent cloning method (33), as outlined in the methods described in the supplemental material and Fig. S1, and designated pMO738 and pMO756, respectively (Table 1).
HPLC methods.
Identification and quantification of metabolites produced by respiring or fermenting cultures were performed by high-pressure liquid chromatography (HPLC) (Table 2). Cell-free filtrate samples (10 μl) were injected onto an Aminex HPX-76H column (300 by 7.8 mm; Bio-Rad, Hercules, CA) maintained at 50°C. All HPLC components and data analysis software were from Shimadzu Scientific Instruments, Inc. (Columbia, MD). The mobile phase was 2.5 mM H2SO4 and was maintained at a constant flow rate of 0.6 ml/min and at approximately 2.2 MPa. Quantification of metabolites was calculated from standard curves of pure compounds established for both a UV detector (at 210 nm) and a refractive index monitor. The reproducibility of technical replicates was within ±5%.
TABLE 2.
Analysis of metabolites in spent culture medium of D. alaskensis G20 wild type and CycA mutant I2a
| Medium and strain | Concn of substrate or accumulated metabolite remainingb |
Protein yield (mg/liter) | Recovery (%) |
Initial electron donor/acceptor concn (mM)i | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pyruvate | Malate | Succinate | Lactate | Fumarate | Formate | Acetate | CO | H2e | H2Sf | Carbong | Reductanth | |||
| Lactate-sulfate | ||||||||||||||
| G20 | NDc | 0.2 | 0.7 | 27.4 | ND | ND | 24.1 | NQd | 0.6 | 13.7 | 102.1 | 102 | 112 | 51.9/30.0 |
| I2 | ND | ND | ND | 32.0 | ND | 1.6 | 20.3 | ND | 1.0 | 8.8 | 95.3 | 101 | 101 | 52.0/30.0 |
| Pyruvate-sulfate | ||||||||||||||
| G20 | 34.5 | 0.3 | 0.4 | ND | ND | 1.9 | 16.4 | NQ | 0.4 | 4 | 85.5 | 95 | 103 | 54.5/15.0 |
| I2 | 47.6 | ND | ND | ND | ND | 0.6 | 5.8 | ND | 0.9 | ND | 12.0 | 100 | 99 | 53.4/15.0 |
| Pyruvate | ||||||||||||||
| G20 | 41.0 | ND | 0.2 | ND | 0.1 | 3.2 | 7.3 | ND | 0.7 | ND | 31.3 | 102 | 102 | 48.7 |
| I2 | 40.4 | ND | ND | ND | ND | 0.7 | 7.6 | ND | 0.9 | ND | 32.4 | 99 | 95 | 48.0 |
Duplicate 200-ml lactate-sulfate and pyruvate-sulfate cultures were harvested when their growth reached an OD600 of ∼0.5, with the exception of I2 on pyruvate-sulfate, which was harvested at an OD600 of 0.17. Cells fermenting pyruvate were harvested at an OD600 of ∼0.2.
The soluble metabolite concentrations in the filtrates of harvested cultures. Typically, from the inoculum, there was measurable carryover of acetate to the experimental cultures. This carryover amount was subtracted from the concentration reported in the table. Data are in millimolar unless indicated otherwise and are averages of measurements from duplicate cultures.
ND, not detected.
NQ, detected but not quantified. H2 interfered with the precise measurement of CO with the gas chromatograph.
The H2 was collected, and the amounts (mmol) produced in a 200-ml culture were calculated by use of the ideal gas law.
No H2S was detected in the collected gaseous end products, and dissolved H2S was quantified (34). The 0.5 mM sulfide added at the time of inoculation, to reduce the medium and to provide a sulfur source for fermenting cultures, was subtracted from the sulfide concentration measured at harvest. The lower limit of detection in this experiment was 0.5 mM.
The carbon recovered in culture filtrates was the ratio of the concentration of carbon present in measured end products and the remaining unused substrate to the initial concentration of carbon provided as the electron donor. For the purpose of this calculation, it was assumed that as a primary end product of organic acid oxidation, the CO2 concentration was essentially equal to that of the acetate generated during growth minus the concentration of any other C1 or C4 end products, such as formate, fumarate, and succinate, which were identified and quantified by HPLC. The amount of carbon present in the biomass produced was not taken into account in these calculations.
Reductant recovery was the ratio of the number of mole equivalents of electrons in the measured end products as well as the electrons remaining in unused substrate to the total number of moles of electrons available for consumption in the substrate added.
Measured concentrations of electrons and the carbon donor determined by HPLC, corrected for inoculum carryover, and calculated concentrations of sulfate in the growth media for G20 and I2 at the initiation of growth.
Gas detection.
Gases from the headspace of sealed cultures were identified with an SRI Instruments (Torrance, CA) model 8610C gas chromatograph fitted with two detectors: a reduced gas detector (RGD) with an Alltek (Fisher Scientific) molecular sieve 5A 80/100 column and a flame photometric detector (FPD) on a Restek (Bellefote, PA) 60-m MXT-1 column. The injector-detector oven was kept at 70°C, and helium was the carrier gas for both detectors.
H2 and CO were measured on the RGD with 500-μl sample injections from the culture headspace. Generally, the H2 produced prevented accurate quantification of the CO being produced; therefore, a notation of not detected or detected but not quantified was made for CO (Table 2). The FPD was used to detect H2S. The chromatography acquisition and integration software used was PeakSimple (version 3.56) from SRI instruments.
Cultures for metabolite analysis.
For end product analysis of cultures at the same optical densities as those of the fermentor cultures used for omics studies, duplicate 200-ml cultures for each medium were grown in modified stoppered and crimp-sealed 225-ml glass bottles at 34°C. Stoppers contained a bleed line allowing the gases generated by the cultures to be collected by water displacement in graduated cylinders as they were produced. The gaseous end products were predicted to be a mix of H2, CO2, CO, and H2S. As cultures grow by sulfate respiration, they become basic (about pH 7.3 at an OD600 of 0.5). The pKa1 of H2S is 7.04, and that of H2CO3 is 6.37; therefore, the H2S and CO2 would be about 65 and 89% in solution, respectively, and the collected gas would be predominantly H2 and CO. CO2 and H2S were scrubbed from the evolved gas by bubbling through 5 N NaOH. Because preliminary experiments had shown that CO concentrations were low, the amount of H2 produced was calculated by use of the ideal gas law. Curiously, no H2S gas was present in the nonscrubbed culture headspace; therefore, only dissolved H2S was quantified according to the method of Cord-Ruwisch (34). The CO2 concentration was assumed to be that of the acetate produced by the oxidation of the lactate or pyruvate minus the concentration of any measured C4 products. The sulfate-grown cultures were harvested when their growth reached an OD600 of ∼0.5, with the exception of I2 on pyruvate-sulfate, which was harvested when its growth reached an OD600 of 0.17. Cells fermenting pyruvate were harvested at an OD600 of ∼0.2. Calculations for mass balance were made from the concentrations of all remaining substrates and products in the cultures.
Cultures for transcriptomic and proteomic analyses.
D. alaskensis G20 biomass for use in transcriptomic and proteomic analyses was generated at Oak Ridge National Laboratory in BioFlo110 7.5-liter fermentors (New Brunswick Scientific, Edison, NJ). The medium contained resazurin (0.00003%, wt/vol) and cysteine-HCl (0.35 mg/ml, added after sterilization). Prior to inoculation, the fermentors were sparged with filter-sterilized prepurified N2 gas for about 15 h. Gas flow was continued throughout cultivation to maintain a positive pressure in the fermentor and to prevent possible growth inhibition by gaseous end products. Gases were vented from the fermentors via chilled condensers. Fermentor pHs were maintained at 7.2 by automatic titration of 2 M NaOH or 2 M HCl, cultures were agitated at 200 rpm, and temperatures were held at 34°C with a Neslab Merlin M-150 refrigerated recirculator (Thermo Fisher Scientific, Newington, NH). Inocula were grown to mid-exponential growth phase in the medium of the experiment. Initial cell densities were an OD600 of approximately 0.03 to 0.05. Replicate cultures, four for G20 and three for the CycA mutant, were grown in lactate-sulfate medium, and duplicate cultures of both strains were grown in pyruvate medium. Lactate-sulfate cultures were harvested at an OD600 of 0.5, and pyruvate cultures were harvested at an OD600 of 0.2.
The microarray samples were 50-ml culture aliquots which were harvested by centrifugation at 14,500 × g for 2 min at 4°C. Cell pellets were subsequently rapidly frozen in liquid nitrogen and stored at −80°C until used for microarray analysis. Proteomic samples were 250-ml culture aliquots similarly harvested, except that centrifugation was at 10,400 × g for 4 min.
Microarray analysis.
Total cellular RNA isolation, genomic DNA extraction, and subsequent Cy5-dUTP or Cy3-dUTP labeling of cDNA or genomic DNA, respectively, were performed as described previously (35). Oligonucleotide probes (70-mers) had been designed for 3,532 of the predicted 3,775 protein-coding sequences in the first annotation of the G20 genome (NCBI accession number CP000112) with CommOligo software (36). Oligonucleotide probes were synthesized without modification (Invitrogen, Carlsbad, CA) and were spotted onto UltraGAPS glass slides (Corning Life Sciences, Corning, NY) in duplicate with a BioRobotics Microgrid II microarrayer (Genomic Solutions, Ann Arbor, MI). DNA microarray hybridizations were carried out as described previously (35), with the exceptions that the hybridization solutions contained 30% (vol/vol) formamide and were carried out at 45°C with active mixing by a Hybridization System 12 mixer (Roche NimbleGen, Inc., Madison, WI). The G20 DNA microarrays were washed according to the slide manufacturer's instructions (Corning Life Sciences) and dried with an automated wash system (BioMicro Systems, Inc., Salt Lake City, UT). DNA microarrays were scanned and quantified as reported previously (35). JMP Genomics software (version 4.0; SAS Institute, Cary, NC) was used for statistical analyses with the JMP Genomics standard normalization procedure. An analysis of variance (ANOVA) was performed to determine significant differential gene expression by the false discovery rate method. Genes displaying a statistically significant change (P < 0.01) in expression with a change in magnitude of ≥2-fold were considered significant in this study. The genes presented in Tables 3 and 4 included only the current 3,258 protein-coding sequences annotated in 2011 (17) with additional modifications by M. Price (personal communication; see Table S2 in the supplemental material).
TABLE 3.
Comparison of gene expression and relative protein abundance differences between D. alaskensis G20 and CycA mutant I2a
| Enzyme and locus | Protein | Annotationb | Lactate-sulfatec |
Pyruvated |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Microarray Log2 R | Proteomics |
Microarray log2 R | Proteomics |
|||||||
| I2 relative abundance | G20 relative abundance | Log2 R | I2 relative abundance | G20 relative abundance | Log2 R | |||||
| Cytoplasmic carbon metabolism enzymes | ||||||||||
| Dde_1250 | NfnA-2 | Electron-bifurcating transhydrogenase 2, subunit A | −1.61 | 4.61 | 6.67 | −0.53 | −3.16 | NRe | 3.77 | Gf |
| Dde_1251 | NfnB-2 | Electron-bifurcating transhydrogenase 2, subunit B | −1.79 | 5.13 | 13.07 | −1.35 | −3.12 | NDg | 3.07 | G |
| Dde_1252 | COG-CitT | Malate/fumarate uptake transporter or antiporter with succinate | −2.54 | 1.33 | 2.08 | −0.62 | −3.43 | ND | ND | ND |
| Dde_1253 | COG-SfcA | Malate:NADPH oxidoreductase, decarboxylating | −2.74 | 0.40 | 40.38 | −6.67 | −3.71 | 2.41 | 55.18 | −4.52 |
| Dde_1254 | COG-FumA | Fumarate hydratase, β subunit | −2.39 | ND | 30.30 | G | −3.30 | ND | 28.20 | G |
| Dde_1255 | COG-TtdA | Fumarate hydratase, α region | −2.41 | ND | 22.59 | G | −3.25 | NR | 30.78 | G |
| Dde_1256 | FrdB | Fumarate reductase, FeS protein | −0.90 | ND | 26.64 | G | −2.46 | NR | 22.28 | G |
| Dde_1257 | FrdA | Fumarate reductase, flavoprotein unit | −1.98 | 6.38 | 27.15 | −2.09 | −2.84 | NR | 27.28 | G |
| Dde_1258 | FrdC | Fumarate reductase, cytochrome b subunit | −2.47 | ND | 12.55 | G | −3.01 | ND | 34.69 | G |
| Dde_3028 | CooS | Carbon monoxide dehydrogenase | 0.07 | 4.88 | 1.21 | 2.02 | 2.24 | 97.48 | 5.38 | 4.18 |
| Dde_3029 | CooC | Carbon monoxide dehydrogenase | 0.11 | 4.66 | 0.94 | 2.32 | 2.03 | 26.27 | 2.12 | 3.63 |
| Periplasmic formate dehydrogenases | ||||||||||
| Dde_0717 | FdhA-1 | Formate dehydrogenase, α subunit | 0.72 | 46.97 | 2.04 | 4.53 | 3.76 | 119.82 | 0.33 | 8.57 |
| Dde_0718 | FdhB-1 | Formate dehydrogenase, β subunit | 1.03 | 41.10 | 1.80 | 4.51 | 3.33 | 143.51 | NR | Ih |
| Dde_0719 | COG-MobB | Molydopterin-guanine dinucleotide biosynthesis protein | 0.58 | 19.39 | 2.83 | 2.78 | 1.61 | 66.07 | 3.07 | 4.42 |
| Periplasmic hydrogenases | ||||||||||
| Dde_2134 | HysB | [NiFeSe] hydrogenase, small subunit | 0.13 | 17.86 | 18.67 | −0.06 | ND | 29.76 | 4.86 | 2.62 |
| Dde_2135 | HysA | [NiFeSe] hydrogenase, large subunit | 0.19 | 20.31 | 20.31 | 0.00 | 0.82 | 43.34 | 20.09 | 1.11 |
| Dde_2136 | HysBA | Maturation protease for hydrogenase | 0.05 | 2.64 | 1.94 | 0.45 | 0.22 | NR | NR | ND |
| Dde_2137 | HynB-1 | [NiFe] hydrogenase, small subunit | 0.18 | 43.00 | 33.83 | 0.35 | ND | NR | NR | ND |
| Dde_2138 | HynA-1 | [NiFe] hydrogenase, large subunit | 0.12 | 16.97 | 19.67 | −0.21 | 0.15 | 32.02 | 9.38 | 1.77 |
| Sulfate-reducing enzymes | ||||||||||
| Dde_2265 | Sat | Sulfate adenylyltransferase | 0.10i | 347.34 | 427.86 | −0.30 | NRj | 272.06 | 740.47 | −1.44 |
| Dde_1778 | PpaC | Pyrophosphatase/inorganic diphosphatase | −0.02 | 351.10 | 504.34 | −0.52 | −1.13 | 331.91 | 408.52 | −0.30 |
| Dde_1109 | ApsB | Adenylyl sulfate reductase, β subunit | −0.06 | 707.53 | 988.72 | −0.48 | NR | 226.75 | 333.56 | −0.56 |
| Dde_1110 | AspA | Adenylyl sulfate reductase, α subunit | 0.50i | 508.68 | 600.23 | −0.24 | NRj | 340.12 | 534.26 | −0.65 |
| Dde_1111 | QmoA | Quinone-interacting membrane-bound oxidoreductase | 0.23 | 72.80 | 78.51 | −0.11 | −0.35 | 30.16 | 37.62 | −0.32 |
| Dde_1112 | QmoB | Quinone-interacting membrane-bound oxidoreductase | 0.20 | 65.97 | 71.95 | −0.13 | −0.25 | 31.01 | 23.18 | 0.42 |
| Dde_1113 | QmoC | Quinone-interacting membrane-bound oxidoreductase | 0.05 | 54.56 | 58.67 | −0.10 | −0.28 | 16.57 | 21.76 | −0.39 |
| Dde_2932 | QrcA | Type I cytochrome c3:menaquinone oxidoreductase hexaheme cytochrome c | 0.38 | NR | NR | ND | 0.05 | NR | NR | ND |
| Dde_2933 | QrcB | Type I cytochrome c3:menaquinone oxidoreductase, molybdopterin-containing subunit | 0.29 | NR | 204.68 | G | 0.41 | 42.57 | 13.59 | 1.65 |
| Dde_2934 | QrcC | Type I cytochrome c3:menaquinone oxidoreductase, periplasmic molybdopterin-containing subunit | 0.25 | 45.16 | 44.34 | 0.03 | 0.21 | 52.63 | 21.10 | 1.32 |
| Dde_2935 | QrcD | Type I cytochrome c3:menaquinone oxidoreductase, integral membrane protein subunit | −0.02 | ND | ND | ND | 0.31 | NR | ND | ND |
Log2 R, where R is the transcript expression measured by microarray analysis for I2 protein-coding genes compared with that obtained for G20 or relative protein abundance values obtained from proteomic data. Log2 (I2/G20) values in bold are for those genes or proteins with at least a 2-fold difference in expression or abundance between I2 and G20. Relative abundance is derived from the number of spectra acquired for a protein.
Gene annotations were was obtained from http://www.microbesonline.org, http://genome.ornl.gov/microbial/ddes/, and M. Price (personal communication; see Table S2 in the supplemental material).
Lactate-sulfate was MO basal salts with 60 mM lactate and 30 mM sulfate providing medium for sulfate respiration.
Pyruvate medium was MO basal salts containing 60 mM pyruvate for growth by fermentation.
NR, data do not meet the criteria for reproducibility.
G, log2 R was not meaningful since protein was detected only in the G20 culture.
ND, no protein was detected in either G20 or I2.
I, log2 R was not meaningful since protein was detected only in the I2 culture.
Microarray data for Dde_2265 and Dde_1110 were reevaluated by qRT-PCR, and log2 ratios of I2/G20 transcripts from lactate-sulfate (60 mM/30 mM) cultures were found to be −0.004 and 0.017, respectively, indicating no change.
Microarray data for Dde_2265 and Dde_1110 were reevaluated by qRT-PCR, and log2 ratios of I2/G20 transcripts from pyruvate (60 mM) cultures were found to be −0.083 and −0.077, respectively, indicating no change.
TABLE 4.
Relative protein abundances of the enzymes metabolizing pyruvate during sulfate respiration and pyruvate fermentation by D. alaskensis G20 and CycA mutant I2a
| Reaction | Locus | Protein name | Annotationc | Lactate-sulfated |
Pyruvatee |
||||
|---|---|---|---|---|---|---|---|---|---|
| I2 abundance | G20 abundance | Log-2 Rf (I2/G20) | I2 abundance | G20 abundance | Log2 R (I2/G20) | ||||
| Ab | Dde_0182 | GlcD | (S)-2-Hydroxy acid oxidase | 7.30 | 6.52 | 0.16 | 0.42 | NRg | Ih |
| Dde_0312 | COG-GlcD | FAD/FMN-containing dehydrogenase | 11.13 | 9.23 | 0.27 | 2.46 | 6.74 | −1.45 | |
| Dde_0750 | LdlD | Lactate dehydrogenase | 14.58 | 11.82 | 0.30 | 20.09 | 11.74 | 0.77 | |
| Dde_1087 | COG-GlcD | FAD/FMN-containing dehydrogenase | NR | NR | NDi | NR | ND | ND | |
| Dde_3238 | LldP | l-Lactate permease | 4.23 | 4.12 | 0.04 | 1.04 | 1.32 | −0.32 | |
| Dde_3239 | GlcD | Glycolate oxidase | 445.77 | 441.43 | 0.01 | 407.13 | 508.48 | −0.32 | |
| Dde_3240 | LdhB | Lactate dehydrogenase | 34.22 | 31.51 | 0.12 | 19.34 | 11.13 | 0.79 | |
| B | Dde_2081 | Pyc | Pyruvate carboxylase | 21.66 | 21.05 | 0.04 | 6.70 | 6.32 | 0.08 |
| C | Dde_1253 | COG-SfcA | Malate:NADPH oxidoreductase, decarboxylating | 0.40 | 40.38 | −6.67 | 2.41 | 47.73 | −4.31 |
| Dde_3637 | Tme | Malic enzyme | 8.93 | 11.63 | −0.38 | ND | 53.20 | Gj | |
| D | Dde_0240 | FumC | Fumarate hydratase | ND | ND | ND | ND | ND | ND |
| Dde_1254 | COG-FumA | Fumarate hydratase, β subunit | ND | 30.30 | G | ND | 28.20 | G | |
| Dde_3638 | COG-FumA | Fumarate hydratase, β subunit | ND | NR | ND | NR | ND | ND | |
| Dde_3639 | COG-TtdA | Fumarate hydratase, α region | ND | 0.26 | G | NR | ND | ND | |
| E | Dde_1256 | FrdB | Fumarate reductase, FeS protein | ND | 26.64 | G | NR | 22.28 | G |
| Dde_1257 | FrdA | Fumarate reductase, flavoprotein subunit | 6.38 | 27.15 | −2.09 | NR | 27.28 | G | |
| Dde_1258 | FrdC | Fumarate reductase, cytochrome b subunit | ND | 12.55 | G | ND | 34.69 | G | |
| F | Dde_0473 | FhcA | Putative cytoplasmic formate:hydrogen lyase, formate dehydrogenase subunit | ND | ND | ND | ND | ND | ND |
| Dde_0474 | FhcB | Putative cytoplasmic formate:hydrogen lyase, FeS subunit 1 | ND | ND | ND | ND | ND | ND | |
| Dde_0475 | FhcC | Putative cytoplasmic formate:hydrogen lyase, hydrogenase, small and large subunits | ND | ND | ND | ND | ND | ND | |
| Dde_0476 | FhcD | Putative cytoplasmic formate:hydrogen lyase, FeS subunit 2 | ND | ND | ND | ND | ND | ND | |
| Dde_0679 | FdoG | Formate dehydrogenase, sulfur transferase | 0.68 | 0.74 | −0.11 | 11.37 | 4.34 | 1.39 | |
| Dde_0680 | FdoI | Formate dehydrogenase, cytochrome B556 | ND | ND | ND | ND | ND | ND | |
| Dde_0681 | FdoH | Formate dehydrogenase, FeS cluster | 2.85 | 2.57 | 0.15 | 3.07 | ND | I | |
| Dde_0716 | Formate dehydrogenase, TAT pathway signal | 35.29 | 0.48 | 6.21 | 114.55 | ND | I | ||
| Dde_0717 | FdhA-1 | Formate dehydrogenase, α subunit | 46.97 | 2.04 | 4.53 | 119.83 | 0.33 | 8.57 | |
| Dde_0718 | FdhB-1 | Formate dehydrogenase, β subunit | 41.10 | 1.80 | 4.51 | 143.51 | NR | I | |
| Dde_0812 | FdhB-2 | Formate dehydrogenase, β subunit | ND | ND | ND | ND | ND | ND | |
| Dde_0813 | FdhA-2 | Formate dehydrogenase, α subunit | 22.38 | NR | I | 128.51 | NR | I | |
| Dde_3513 | FdhA-3 | Formate dehydrogenase, α subunit | NR | 0.36 | G | 9.62 | ND | I | |
| Dde_3514 | FdhB-3 | Formate dehydrogenase, β subunit | ND | ND | ND | ND | ND | ND | |
| G | Dde_3028 | CooS | Carbon monoxide dehydrogenase | 4.88 | 1.21 | 2.02 | 97.48 | 5.38 | 4.18 |
| Dde_3029 | CooC | Carbon monoxide dehydrogenase | 4.66 | 0.94 | 2.32 | 26.27 | 2.12 | 3.63 | |
| H | Dde_1792 | PorA | Pyruvate-ferredoxin- oxidoreductase | NR | 1.44 | G | 11.71 | 6.47 | 0.86 |
| Dde_1793 | PorB | Pyruvate-ferredoxin- oxidoreductase | 3.08 | 3.04 | 0.02 | 8.17 | 7.74 | 0.08 | |
| Dde_3237 | Por | Pyruvate-ferredoxin- oxidoreductase | 232.69 | 224.03 | 0.05 | 118.14 | 116.82 | 0.02 | |
| I | Dde_3241 | Pta | Phosphotransacetylase | 69.06 | 67.97 | 0.02 | 21.88 | 26.32 | −0.34 |
| J | Dde_3242 | AckA | Acetate kinase | 70.66 | 88.75 | −0.33 | 27.02 | 27.07 | 0.00 |
The data presented here are supporting data for Fig. 3. The protein abundance values given in Fig. 3 were chosen for the most abundant isozyme for that reaction, and the locus, protein name, annotation, and data are highlighted in bold.
A through J designate the reactions illustrated in Fig. 3.
Protein annotation was obtained from http://www.microbesonline.org. FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; TAT pathway, twin-arginine translocation pathway.
Lactate-sulfate was MO basal salts with 60 mM sodium lactate and 30 mM sodium sulfate for sulfate respiration.
Pyruvate was MO basal salts with 60 mM sodium pyruvate for growth by fermentation.
R, I2/G20 relative protein abundance values.
NR, data did not meet the criteria for reproducibility.
I, log2 R was not meaningful since protein was detected only in the I2 culture.
ND, no protein was detected in either G20 or I2 or the data did not meet the confidence criteria.
G, log2 R was not meaningful since protein was detected only in the G20 culture.
Validation of the microarray data was carried out by comparison of the results to those obtained by quantitative reverse transcription-PCR (qRT-PCR) analysis of the transcripts of nine genes representing the range of expression levels in the microarray data (see Table S5 in the supplemental material) (37). Values of log2 R, where R is the ratio of CycA mutant transcripts/G20 wild-type transcripts of those genes from cells grown in lactate-sulfate or pyruvate medium, were generated. The microarray log2 R values were plotted against those from qRT-PCR. Comparison of the two methods indicated a correlation coefficient for transcripts from lactate-sulfate-grown cells of 0.91 and a correlation coefficient for transcripts from pyruvate-grown cells of 0.99 (see Fig. S2 in the supplemental material) (37).
Accurate mass tag and time proteome analysis.
Proteomic analyses of cell pellets were performed by the Environmental Molecular Science Laboratory of the Pacific Northwest National Laboratory (38) according to the following protocol. Whole-cell lysis of quick-frozen cell pellets from D. alaskensis G20 was achieved by bead beating as previously described (39). Tryptic digestion of the released proteins utilized sequencing-grade-modified trypsin (Promega, Madison, WI) in a 5-h digestion at 37°C with a 1:50 (wt/wt) trypsin-to-protein ratio (40). Samples were then quick-frozen in liquid N2 and stored at −80°C until analyzed. Protein concentrations were determined by use of a bicinchoninic acid assay kit (Pierce, Rockford, IL).
The capillary liquid chromatography system used for peptide analysis was described previously (40). Briefly, peptide samples were chromatographed on 5,000-psi reversed-phase packed capillaries at ∼1.8 μl/min (150 μm [inner diameter] by 360 μm [outer diameter]; Polymicro Technologies, Phoenix, AZ) (41) with two mobile-phase solvents, with the first consisting of 0.2% (vol/vol) acetic acid and 0.05% (vol/vol) trifluoroacetic acid (TFA) in water and the second consisting of 0.1% (vol/vol) TFA in 90% acetonitrile–10% water. The linear ion trap Fourier transform mass spectrometer (LTQ-FT) data were processed by the Prism data analysis system as described previously for the liquid chromatography (LC)-Fourier transform ion cyclotron resonance (FTICR) mass spectrometry (MS) data (40). Since the separation systems for both the LTQ-FT and the LC-FTICR-MS analyses were identical, peptide confirmation was based on both the calculated mass (from the mass tag database) and the measured mass (from the FTICR analysis) of the peptide matching to within 6 ppm and the elution times matching to within 5%. The mass spectrometer measurements were analyzed with the SEQUEST program (42) and the D. alaskensis G20 database (39), and all data tables used the 2011 reannotation (17) with additional reannotations by M. Price (personal communication; see Table S2 in the supplemental material). Mass spectra were acquired with a resolution of approximately 105 (the instrument detected signals with ±0.005 m/z precision).
The peptide identifications were made with the spatially localized confidence scoring (SLiC) algorithm (38, 40), which incorporates a number of constraints and estimates the confidence of each peptide identification by yielding a score of 0 to 1. In this work, a minimum SLiC score of 0.3 (default) or higher was required for confident identification (38, 40). Further, at least one high-confidence unique peptide (i.e., a peptide mapping to only one possible parent protein) and a total of two peptides were required for protein identification in each technical replicate.
Each peptide preparation was injected onto the mass spectrometer three times to yield triplicate MS data sets for each biological replicate. These data sets were then interrogated at the peptide level with the confidence measures described above applied and then rolled up to the protein level. To meet our standard for a high level of confidence, at least two of the biological replicates must have identified the protein, and the variation for the protein abundance in biological replicates had to be within 1 standard deviation of the average.
To compare protein abundance data from cultures grown with lactate-sulfate medium with those from pyruvate-fermenting cells, we made the assumption that the majority of protein concentrations would be similar under the two conditions. Therefore, we averaged the abundances of all proteins meeting our identification criteria under each growth condition and found that the protein abundances from cells grown on lactate-sulfate were an average of 4.716-fold higher than those from fermenting cells. We then normalized the data from pyruvate-grown cultures by multiplying all protein abundance numbers by 4.716. Normalized abundances are reported in all tables presented here, with the exception of the complete lists of original data (see Tables S6, S7, S8, and S9 in the supplemental material).
The normalized data were used to generate ratios of relative protein abundance between the G20 and CycA mutant (I2) strains and between different cultivation conditions with each strain. Log2 values of the ratio of I2/G20 abundance of >|1.00| are reported (Tables 3 and 4; see Table S4 in the supplemental material). It should be noted that four biological samples were analyzed for the lactate-sulfate cultures of G20, whereas three biological samples were analyzed for the lactate-sulfate cultures of I2. Only two biological samples were obtained for fermenting cultures of each strain; therefore, a protein had to be confidently determined in both biological replicates of the fermenting cultures to be included in the ratios for relative abundances.
For malic enzyme isozymes (Dde_1253 and Dde_3637) (Table 4), two identical peptides were found in both. One of the shared peptides was found in abundances that were higher by 3 to 14 times the standard deviation of the average abundances of other peptides identifying these proteins. This shared peptide was removed from the abundance data calculation for both isozymes, and new averages were determined. Corrected values were reported.
Microarray data accession number.
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus (GEO) database http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO series accession number GSE21287.
RESULTS
We previously reported the construction of a D. alaskensis G20 mutant, I2, that had a plasmid insertion in the cycA gene causing a loss of TpIc3 (26). To examine the consequences of the loss of TpIc3 on energy conversion, we explored the rates and extents of growth by respiration versus those by fermentation (Fig. 1A). When respiring sulfate with electrons from lactate, I2 had a generation time that was slower than that of G20 by about 60% (5.0 ± 0.4 h for I2 versus 3.1 ± 0.3 h for G20), but the final cell densities obtained were essentially the same. When fermentative capacity on pyruvate was tested, limited growth of both strains was observed, but growth was at similar rates for the two strains and the cell densities obtained were nearly the same (Fig. 1C; Table 2). Thus, TpIc3 is not required for sulfate respiration with lactate or for pyruvate fermentation by G20.
FIG 1.

Growth of D. alaskensis G20 (●) and I2 (○) on lactate-sulfate (60 mM/30 mM) (A), pyruvate-sulfate (60 mM/30 mM) (B), and pyruvate (60 mM) (C). Each point is the average of triplicate 5-ml cultures, and the error bars are within the data symbols in most cases.
The most dramatic growth effect caused by the elimination of TpIc3 was the limited growth on pyruvate with sulfate (Fig. 1B). On that medium, I2 grew much slower than G20 and yielded only 14% of the cell protein of G20 (Table 2). When a plasmid carrying the wild-type TpIc3 was introduced into I2, growth on pyruvate sulfate was restored to wild-type G20 levels (data not shown). The differences in sulfate respiration with lactate versus pyruvate were not anticipated since pyruvate is the oxidation product of lactate. In addition, I2 grew on lactate-sulfate medium, producing acetate and generating sulfide to a concentration that necessitated electrons from pyruvate. The growth of I2 that did occur in pyruvate-sulfate medium was not accompanied by the blackening of the cultures expected when sulfide was generated. Measurements of dissolved sulfide when I2 was grown on pyruvate in the presence of sulfate confirmed that I2 did not produce sulfide (Table 2). From these observations, we deduced that I2 might be involved in oxidizing the pyruvate but not respiring the sulfate.
Analysis of metabolic end products during growth.
If TpIc3 were a significant component of electron transfer pathways in G20, we predicted that its absence might alter the array of reduced end products made during either respiration or fermentation or perhaps during both. Therefore, we examined the consumption of substrates and metabolites released to identify any major changes in reduced products (Table 2). Recoveries of carbon varied from 95 to 102%, and reductant recoveries were 95 to 112%. As expected, some of the electrons released from organic acids accumulated as hydrogen or formate. During lactate-supported respiration of sulfate by either G20 or I2, only 5 to 12% of the electrons were released as hydrogen. In contrast, G20 fermentative cultures produced almost equal quantities of electrons in hydrogen and formate (47 and 43%, respectively). Cultures of I2 grown with pyruvate-sulfate medium produced about 88% of the electrons from oxidized pyruvate as hydrogen, with the remaining 12% of the electrons from oxidized pyruvate as formate, quite similar to the distribution of electrons found during fermentation of pyruvate by the mutant (87% hydrogen and 13% formate) (Table 2). On this medium, no sulfide was produced and no sulfite accumulated with an assay that was sensitive to 20 μM (43). I2 grown with lactate or pyruvate with sulfite produced sulfide (data not shown). These results were consistent with the lack of sulfate (or APS) reduction by the mutant when pyruvate was the electron donor.
One additional feature distinguished the lack of TpIc3. No C4 intermediates from the reductive branch of the tricarboxylic acid (TCA) pathway were observed in cultures of the mutant. Small amounts of malate, fumarate, or succinate were generally observed in culture filtrates of G20, regardless of the growth mode or substrate.
From the growth and end product comparison, we concluded that TpIc3 is not required for sulfate reduction when lactate is the electron donor but that the flow of electrons from pyruvate to sulfate is disrupted in D. alaskensis G20 in the absence of this cytochrome. The growth by pyruvate fermentation of the mutant lacking TpIc3 was equal to that documented for the wild type, but its absence may influence the reduced end products that accumulate.
Confirmation of the I2 phenotype with cycA::mini-Tn10.
Because the phenotype of I2 was rather surprising, we obtained a second cycA mutant induced in G20 by a mini-Tn10 transposon (25) to test. If the growth phenotypes differed, this result might indicate that I2 had secondary mutations contributing to the growth characteristics. We characterized the physiology of the transposon mutant like we did that of I2, and in all respects, the two CycA mutants were similar in phenotype (data not shown).
Growth on H2 and formate by G20 and CycA mutants.
Metabolite analysis indicated that G20 accumulated a smaller percentage of available reductant in H2 than I2 in all media tested (Table 2). In contrast, formate accumulation was decreased in the mutant under pyruvate-only conditions. Both H2 and formate are known to be oxidized by most Desulfovibrio species (13, 14), and therefore, the differential accumulation of these reduced products might indicate an alteration in their oxidation. Both G20 and I2 were tested for their growth rates and extents with H2 or formate as the electron donor. G20 grew well when supplied with either H2 or formate supplemented with acetate and Na2SO4 (Fig. 2A and B). To test the effect of the loss of TpIc3 on growth on H2 or formate, the more stable cycA::mini-Tn10 mutant was used. That mutant did not grow on formate or H2 (Fig. 2A and B), which confirmed that the electrons generated in the periplasm could not reenter the cytoplasm for sulfate reduction without TpIc3.
FIG 2.

Growth of D. alaskensis G20 and cycA::mini-Tn10 mutant on formate-acetate-sulfate (50 mM/20 mM/30 mM) (A) and H2-acetate-sulfate (80%/20 mM/30 mM) (B). The CycA transposon mutant (the cycA::mini-Tn10 mutant) was used instead of I2 in these growth experiments. Each point is the average of triplicate 5-ml cultures, and the error bars are included.
Growth on fumarate by G20 and the I2 mutant.
Since C4 compounds of the reductive TCA branch did not accumulate in I2 cultures, regardless of growth mode or electron donor (Table 2), we considered that I2 had altered fumarate metabolism. The growth of SRB by fumarate disproportionation has been described previously (53), where 3 fumarate → 1 acetate + 2 succinate + 2 CO2. When G20 grown in lactate-sulfate was inoculated into fumarate medium, robust growth occurred after a lag (data not shown) and the expected primary end products of succinate and acetate accumulated in the predicted 2:1 ratio as the cells entered stationary phase (data not shown). The CycA mutants, both I2 and the transposon mutant, were not able to adapt to growth by fumarate disproportionation. Omics analyses (Table 3, discussed later) indicated decreases in both transcription of the genes and translation of the mRNAs encoding the putative pathway for fumarate disproportionation in the TpIc3 mutant. We hypothesize that these decreases result from an alteration in redox balance across the membrane.
Phenotype of qrcA::mini-Tn5 mutant.
The transmembrane complex QrcABCD has been proposed to accept electrons from the periplasm via TpIc3 (21, 23). If this is true and if this complex forms a unique conduit of electrons to APS reductase, a mutant lacking a functional Qrc complex should have a phenotype like that of the null mutant of cycA. We tested a QrcA transposon mutant (A. Deutschbauer, unpublished data) generated by insertion of a modified mini-Tn5 (22, 44) and found that it exhibited growth inhibition on H2 and impaired growth on formate with sulfate as the electron acceptor (data not shown), confirming similar observations by Krumholz and coworkers (22). Our results are consistent with the suggested interaction of the periplasmic TpIc3 and the QrcABCD transmembrane complex providing electrons for sulfate reduction (21). An additional similarity with the phenotype of the CycA mutant was that the QrcA mutant was found to be unable to grow by disproportionation of fumarate.
Transcriptomic analysis of differentially expressed genes in I2.
To evaluate the causes of the altered growth properties of D. alaskensis G20 lacking TpIc3, changes in the gene expression and protein content of I2 respiring sulfate or fermenting pyruvate were identified. Microarrays were performed with transcripts from four biological replicates of G20 cells and three biological replicates of I2 cells grown in temperature- and pH-controlled fermentors on lactate-sulfate medium. Twenty-two genes meeting our criteria for significance were differentially transcribed by at least 2-fold (log2 R ≥ |1.0|) in I2 versus G20 (see Table S3 in the supplemental material). Eight of the 13 genes that were upregulated in I2 (in the region from Dde_0899 to Dde_0930) appeared to be in a region encoding a bacteriophage. The remaining five were hypothetical genes in regions lacking functionally annotated genes. Among the genes downregulated in I2 was a prominent group of six genes encoding enzymes involved in energy metabolism, the reductive branch of the TCA cycle, fumarate hydratase, fumarate reductase, and a malic enzyme (Dde_1258 to Dde_1253) (see Table S3 in the supplemental material). This decrease in transcription correlated well with the observation that I2 did not appear to produce these C4 organic acids while reducing sulfate with lactate as the electron donor or to grow by fumarate disproportionation. Also downregulated were two genes of a predicted three-gene operon (Dde_1252 to Dde_1250) encoding a malate-fumarate uptake transporter (or antiporter with succinate) and two subunits of an electron-bifurcating transhydrogenase 2, NfnA-2 and NfnB-2 (45) (reannotated by M. Price, personal communication; see Table S2 in the supplemental material).
Results of the examination of transcriptional changes showed that many more genes were transcribed differentially in I2 grown by pyruvate fermentation than in sulfate-respiring I2 cells compared with the results for G20. Forty-eight genes were upregulated in I2 at least 2-fold, whereas 57 were downregulated (see Table S3 in the supplemental material). The operon with the largest increased expression in I2 encodes a periplasmic formate dehydrogenase complex (Dde_0717 to Dde_0719) (see Table S3 in the supplemental material). Surprisingly, there was not an increased accumulation of formate in fermenting I2 cultures relative to fermenting G20 cultures (Table 2); however, product accumulation may not reflect changes in flux. Also during pyruvate fermentation, there was increased transcription in I2 of the two-gene operon (Dde_3028-Dde_3029) encoding CooS, the carbon monoxide dehydrogenase, and CooC, the nickel insertion accessory protein for CO dehydrogenase (see Table S3 in the supplemental material). However, no CO was detected in either G20 or I2 cultures that were growing on pyruvate (Table 2).
Showing the greatest decrease in transcription in fermenting I2 cells were the nine genes that apparently encode the functions necessary for fumarate disproportionation and succinate production from pyruvate, Dde_1250 to Dde_1258 (Table 3; see Table S3 in the supplemental material). This expression change is apparently not dependent on the growth mode of the cells but is a result of the mutation in cycA eliminating TpIc3.
Proteomic analyses of differentially accumulated proteins in I2.
Accurate mass tag and time proteomics analyses were performed on culture samples obtained at the same ODs as those used for the transcriptome determinations. The total numbers of proteins identified and those meeting our criteria for confident identification are given in Table S10 in the supplemental material. Similar numbers of proteins were identified in G20 and I2, whereas 20 to 25% more proteins were identified in cultures from the lactate-sulfate medium than pyruvate-grown cultures. In the sulfate-respiring cultures, nearly 85% of the proteins identified met our criteria for confident identification; only 53% did so in the samples of pyruvate-fermenting cells, where fewer biological replicates were processed.
Among proteins with 2-fold or greater putative abundance differences in sulfate-respiring I2 versus sulfate-respiring G20 (Table 3; see Table S4 in the supplemental material), prominent increases were observed for a formate dehydrogenase encoded in three genes (Dde_0717 to Dde_0719). An additional protein for formation of formate dehydrogenases (Dde_0706) was also increased. These enzymes could be involved in the production of the small amounts of formate that appeared to accumulate in cultures of I2 (Table 2) or be functioning to consume formate. Since apparent increases in the same proteins were found in I2 fermenting pyruvate, we again suggest that the differential abundances resulted primarily from the lack of TpIc3 and not from a difference in growth modes. In the case of fermenting cells, the differential protein abundances were in remarkable agreement with the transcription changes (see Tables S3 and S4 in the supplemental material). In parallel with the transcription data, the proteins CooS and CooC, needed for CO dehydrogenase formation (Dde_3028 and Dde_3029), were also significantly more abundant in I2, regardless of culturing conditions, although the accumulation of CO as an end product was not markedly higher in I2 (Table 2).
Proteins that appeared to be decreased in abundance in I2 during sulfate respiration were two of the proteins encoded in the nine-gene region from Dde_1258 to Dde_1250 that includes genes for the enzymes for succinate production from pyruvate (Table 3). All nine proteins were identified in G20, but only five were found in I2, presumably because the abundance of the remaining four was below the detection limit. Although ratios were not meaningful for the four proteins not observed in I2, these four were apparently less abundant in the I2 extracts than in the G20 extracts. Similarly, in fermenting cells, the proteins encoded by the genes in this region were observed at lower frequencies in I2. The overall decrease in protein abundance for this metabolic pathway was in excellent congruence with the downregulation of the transcripts from these genes (Table 3) and the lack of succinate as an excreted metabolic end product (Table 2).
Two hydrogenases, the [NiFeSe] and the [NiFe] isozyme 1 hydrogenases (Dde_2134 to Dde_2138), were more abundant in fermenting cells of I2 than in fermenting cells of G20 (Table 3). Changes in transcription of other hydrogenases (Dde_0081-0082, Dde_2280-2281, Dde_3755-3756) were not detected, and only one [Fe] hydrogenase subunit was identified.
To summarize, Fig. 3 shows the normalized relative protein abundances detected for enzymes in the major pathways of pyruvate metabolism in cultures of G20 and I2 growing on lactate-sulfate or pyruvate medium. For the enzymatic steps illustrated, the isozymes putatively most abundant were chosen for the proteomic comparison; however, the abundances of other isozymes showed similar trends or no significant differences (see Table S4 in the supplemental material). Those enzymes already discussed as being increased in I2 were those for formate or CO metabolism, while those for succinate production from pyruvate were all strongly decreased, regardless of the growth substrates. Clearly, the isozymes with predicted activity for lactate or oxaloacetate metabolism were not found to be differentially accumulated in the mutant. Interestingly the enzymes needed for substrate-level phosphorylation from pyruvate (pyruvate ferredoxin oxidoreductase, phosphotransacetylase, and acetyl kinase) were not affected by the lack of TpIc3. However, the enzymes needed for this pathway in the fermenting cells, surprisingly, appeared to be about 2-fold decreased relative to those in cells respiring sulfate.
FIG 3.

Comparisons of protein abundances in the primary redox reactions possible for carbon metabolism during lactate-supported sulfate respiration and pyruvate fermentation by D. alaskensis G20 and CycA mutant I2. Protein abundances are relative counts from the most abundant isozyme of each enzyme denoted A through J. Abundance values given to the left of an arrow in the enzyme pathway are from G20, and those values to the right of arrow, in bold, are from I2. Multiple values for some enzymes reflect the abundance of each putative subunit of the particular isozyme. Sulfate respiration was achieved in lactate-sulfate (60 mM/30 mM) medium, and pyruvate fermentation used 60 mM pyruvate medium. Proteomic data obtained for all annotated isozymes for the enzymes listed (A to J) are given in Table 4. ND, no protein was detected.
Finally, we explored the accurate mass tag and time protein data for any evidence supporting compensatory c-type cytochromes that might serve for electron storage or transfer in the absence of TpIc3. As expected, this c3 cytochrome (Dde_3182) was more abundant in G20 (∼12-fold) than in I2, where peptides were at the limit of detection, possibly due to rare suppression of the CycA mutation. At least 14 c-type cytochromes were annotated in the genome and confirmed by predicted heme binding motifs in the protein sequences (data not shown). Only the level of the decaheme cytochrome (Dde_0580) was increased >3-fold in I2 compared with its level in G20 during sulfate respiration but not during fermentation. A putative cytochrome c554 (Dde_2858) showed a significant increase of 4.4-fold in I2 fermenting cells; however, there was no increase in sulfate-respiring cells. The roles of these cytochromes in the electron circuitry of I2 remain to be determined.
DISCUSSION
The tetraheme cytochrome TpIc3 found in Desulfovibrio strains has been predicted to be an electron donor or recipient for a large number of redox-active proteins. The periplasmic localization of c-type cytochromes (46) limits the functional interactions of TpIc3 with colocalized proteins and those transmembrane complexes that are exposed to the periplasm. The isolation of D. alaskensis G20 mutants lacking TpIc3 by plasmid or transposon interruption of the gene encoding TpIc3, cycA (22, 26), established that this cytochrome is required for H2- or formate-supported growth with sulfate as the electron acceptor but is not essential for the reduction of sulfate when lactate is the source of electrons. The oxidation of lactate by Desulfovibrio strains produces pyruvate, which proceeds to be oxidatively decarboxylated, with acetate ultimately being excreted. In our culture of the CycA mutant I2, about 88% of the electrons from the metabolism of lactate to acetate were accounted for in the sulfide measured (Table 2). Therefore, electrons from the intermediate pyruvate oxidation step appeared in sulfide. The observation that the electrons from pyruvate did not support sulfate reduction with pyruvate as the source of carbon and electrons for I2 was unexpected. Proteomics analyses indicated that the proteins needed for sulfate reduction to sulfite, i.e., sulfate adenylyltransferase (Dde_2265), APS reductase (Dde_1109 and Dde_1110), and the associated Qmo transmembrane complex (Dde_1111 to Dde_1113), were abundant in both wild-type G20 and the mutant lacking TpIc3, even when fermenting pyruvate (Table 3). Thus, the lack of sulfate reduction with electrons from pyruvate by I2 did not result from the absence of the enzymes for the process. Also, proteins for bisulfite reduction were present and not differentially affected by the cycA mutation, and sulfite could be reduced with pyruvate as the electron donor (data not shown). We suggest that some of the electrons from pyruvate oxidation may travel to the periplasm, where they enter the cytochrome c3 pool before returning through transmembrane complexes for sulfate reduction (Fig. 4).
FIG 4.
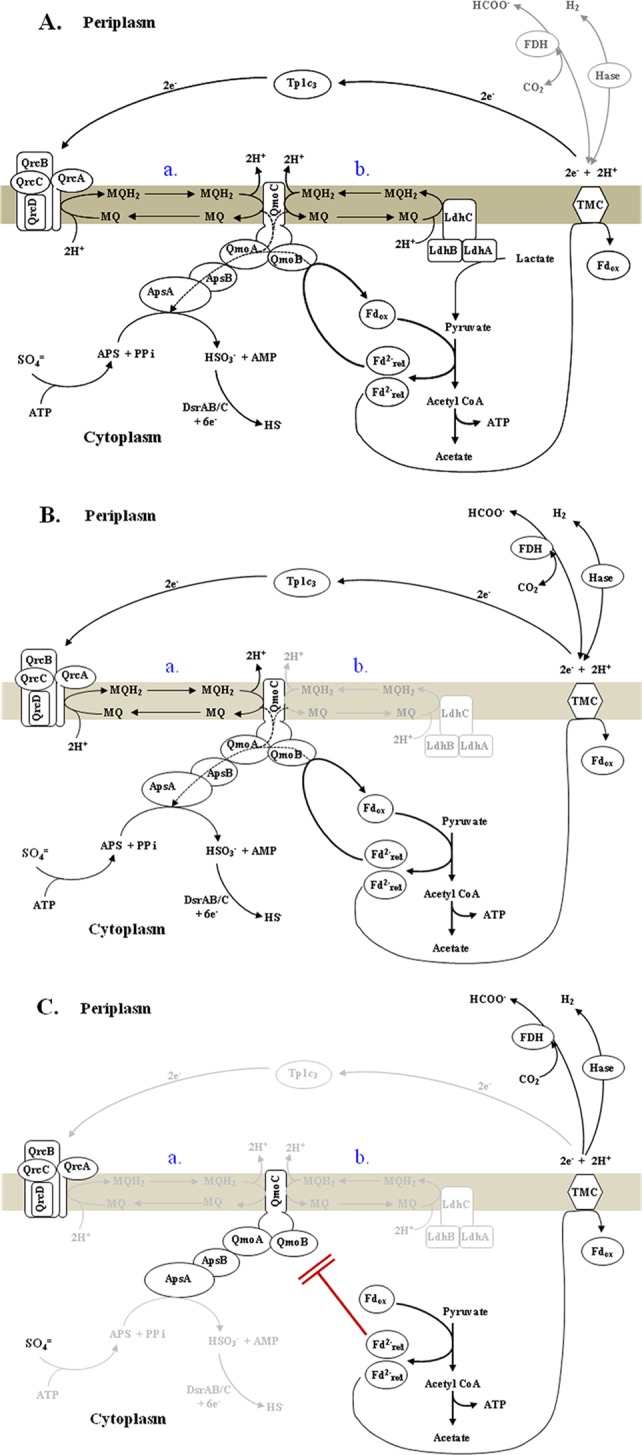
Proposed model of electron flow from lactate by confurcation to APS for sulfite production in D. alaskensis G20 in exponential phase of growth. (A) Wild-type cultures with lactate-sulfate medium where small amounts of reductant appear in hydrogen (8%) and formate (not detected) (Table 2). Menaquinol is produced by either route a or b or both route a and b (indicated in blue) in the model. (B) Wild-type cultures with pyruvate-sulfate medium where small amounts of reductant are produced as hydrogen (9%) and formate (9%). Menaquinol is produced by electrons delivered to the Qrc complex by the TpIc3 by route a. (C) CycA mutant cells with pyruvate sulfate medium unable to generate menaquinol by either route a or b and, therefore, not able to use reduced ferredoxin for reduction of APS. Reductant appears in hydrogen (88%) and formate (12%). In this model, the abbreviation of TMC is used as a generic term for an unidentified transmembrane complex. FDH, formate dehydrogenase; Hase, hydrogenase; MQ, menaquinone; Fd, ferredoxin; PPi, inorganic pyrophosphate.
The question then becomes whether there are two routes for the electrons from pyruvate in D. alaskensis G20. We would suggest that the growth properties of this mutant that we observed are consistent with this possibility (Fig. 4). The recent proposal that the transmembrane complex QmoABC participates in a confurcation of electrons to APS reductase for bisulfite production (45, 47, 48) provides a feasible explanation of the phenotype of the I2 mutant. In that scheme, reduced menaquinone (E0′ menaquinol = −75 mV) in partnership with an electron donor of lower potential would be needed to overcome the membrane potential and provide electrons to APS (E0′ APS/HSO3− = −60 mV). We propose that the electron donor partner is reduced ferredoxin (E0′, ca. −500 mV) produced from pyruvate:ferredoxin 2-oxidoreductase, the primary enzyme for pyruvate oxidation.
When lactate (E0′ = −190 mV) is the growth substrate for G20 (Fig. 4A), lactate dehydrogenase (12), the membrane-bound flavoprotein, may deliver electrons directly to menaquinone, producing the menaquinol needed for the confurcating QmoABC complex to bring about APS reduction. The pyruvate resulting from lactate oxidation produces reduced ferredoxin, which may provide the second source of electrons to the QmoABC complex. From the metabolites accumulated in lactate-sulfate cultures (Table 2), reduced ferredoxin could be oxidized at a transmembrane conduit where the electrons are provided to periplasmic c-type cytochromes, hydrogenases, and/or formate dehydrogenases. The cytochrome pool, hydrogen, and/or formate could act as a temporary sink of reductant and the source of electrons for the Qrc complex that is an alternative entry of electrons to the menaquinone pool.
When wild-type G20 is given pyruvate as the sole source of carbon and electrons with sulfate as the terminal acceptor (Fig. 4B), lactate dehydrogenase no longer supplies menaquinol for the confurcating QmoABC complex for sulfate reduction. We propose that a portion of the electrons from pyruvate must enter the periplasm and the cytochrome c pool, especially targeting TpIc3. That cytochrome then delivers electrons to the QrcABCD transmembrane complex, which has been shown to be capable of reducing menaquinone and is necessary for sulfate reduction (21, 22). The menaquinol produced by QrcABCD reduces the QmoABC complex. The remaining reduced ferredoxin could be directly oxidized by the QmoABC complex as the second source of electrons during confurcation for delivery of reductant to APS reductase accomplishing sulfate reduction.
Finally, when I2 (lacking TpIc3) is provided with pyruvate and sulfate as growth substrates (Fig. 4C), menaquinol is not available from either the lactate dehydrogenase or the QrcABCD complex; therefore, QmoABC is unable to carry out confurcation. Reduced ferredoxin would be produced but could not be oxidized by the QmoABC complex without the second source of electrons. The reduced ferredoxin would need to be reoxidized by an unidentified transmembrane complex releasing electrons to the periplasm, where they are quantitatively used for the production of hydrogen and formate as the pyruvate is consumed.
It must be remembered that Desulfovibrio strains differ widely in the array of electron carriers and their energy conversion pathways (14). As a result, the phenotypes resulting from mutations of a component of electron transfer in G20 may not extrapolate to other Desulfovibrio species.
In addition to sulfate respiration capacity, G20 is among several Desulfovibrio species which are able to disproportionate fumarate (49). The observations that I2 could not grow by fumarate disproportionation or produce detectable succinate when oxidizing lactate or pyruvate (Table 2) were not anticipated. Dde_1258 through Dde_1253 (as transcribed) are proposed to form a single operon encoding fumarate reductase, fumarate hydratase, and an isozyme of malic enzyme sufficient for succinate production from pyruvate. The inability of I2 to metabolize fumarate appears to be a direct result of the decrease in both transcripts and proteins from these genes (Fig. 3; Tables 3 and 4). The intriguing question then becomes how the elimination of TpIc3 causes a decrease in expression of these genes.
Regulation of transcription of the genes for fumarate reduction in other bacterial systems has been shown to be mediated by the FNR protein, a global regulator mediating gene expression for adaptation to oxygen-limiting conditions (50). The G20 strain has four paralogs of CRP/FNR proteins (51), yet none of these paralogs has an amino acid sequence necessary for formation of a 4Fe-4S center, the cofactor of FNR sensing oxidative changes (50). The paralog in D. alaskensis G20 that has the highest homology to FNR of E. coli (27% identity and 49% similarity over 92% of the protein), Dde_2644, has no cysteine residues, leaving the involvement of this protein doubtful. In an attempt to identify another possible regulator, a search of open reading frames flanking the genes for fumarate reduction in G20 revealed genes encoding a putative histidine kinase sensor and a response regulator (Dde_1261 and Dde_1260, respectively). Preliminary evidence from a transposon mutant with a mutation in the histidine kinase gene supported a role for this two-component regulator in controlling expression of fumarate reductase, but the signal for expression changes remains to be determined.
Finally, the data presented here are consistent with the hydrogen cycling model for energy metabolism of G20 growing with pyruvate but not lactate (15, 16). A portion of the electrons released from pyruvate oxidation likely travels from the cytoplasm to the abundant periplasmic TpIc3 by a transmembrane complex or complexes linked to proton pumping, effectively moving hydrogen equivalents to the periplasm. With sulfate available as the terminal electron acceptor, reduced TpIc3 delivers electrons to the transmembrane QrcABCD complex (23). Depending on the rate of oxidation by the Qrc complex, reduced TpIc3 could be used to form small amounts of hydrogen or formate. The inner membrane QrcD subunit reduces the menaquinone pool, producing menaquinol that can be oxidized by the Qmo complex. The latter has been shown to be required by all electron donors for transfer of electrons to APS reductase, the enzyme responsible for the first two-electron reduction step of sulfate (20). The fact that the mutants of G20 lacking TpIc3 or QrcA cannot grow on pyruvate, formate, or H2 with sulfate as the terminal electron acceptor supports the flow of electrons suggested. The observations made here, however, do not provide direct evidence that the electrons move from the cytoplasm as molecular H2 during this process.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by U.S. Department of Energy (DOE) Office of Basic Energy Sciences grants DE-FG02-87ER13713 (to B.J.R.-G. and J.D.W.) and the Office of Biological and Environmental Research (BER) program on BioHydrogen Production and BioEthanol grant DE-FG02-083464691 (to K.L.K., B.J.R.-G., and J.D.W.). S.D.B. and I.P. were supported by DOE BER through DE-FG02-083464691 at the Oak Ridge National Laboratory, managed by the University of Tennessee-Battelle LLC for the DOE under contract DE-AC05-00OR22725. K.L.K., E.S.S., S.D.B., and J.D.W. are participants in ENIGMA, Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under contract DE-AC02-05CH11231. The proteomic portion of this research was performed using EMSL, a national scientific user facility sponsored by the U.S. Department of Energy's Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory.
K.L.K., B.J.R.-G., I.P., S.D.B., and J.D.W. were responsible for the experimental design; B.J.R.-G. performed the metabolic analysis; B.J.R.-G. and E.S.S. performed growth curves; I.P. and S.D.B. performed the biomass production for omic analysis and carried out the transcriptomics; EMSL performed the proteomics analysis; and K.L.K. performed the proteomic data interpretation. K.L.K., B.J.R.-G., S.D.B., and J.D.W. wrote the manuscript.
Footnotes
Published ahead of print 15 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02963-13.
REFERENCES
- 1.Hamilton WA. 2003. Microbially influenced corrosion as a model system for the study of metal microbe interactions: a unifying electron transfer hypothesis. Biofouling 19:65–76. 10.1080/0892701021000041078 [DOI] [PubMed] [Google Scholar]
- 2.Lee W, Lewandowski Z, Okabe S, Characklis WG, Avci R. 1993. Corrosion of mild steel underneath aerobic biofilms containing sulfate-reducing bacteria. Part I. At low dissolved oxygen concentration. Biofouling 7:197–216. 10.1080/08927019309386254 [DOI] [Google Scholar]
- 3.Lovley DR, Phillips EJP, Gorby YA, Landa ER. 1991. Microbial reduction of uranium. Nature 350:413–416. 10.1038/350413a0 [DOI] [Google Scholar]
- 4.Wall JD, Krumholz LR. 2006. Uranium reduction. Annu. Rev. Microbiol. 60:149–166. 10.1146/annurev.micro.59.030804.121357 [DOI] [PubMed] [Google Scholar]
- 5.Thauer RK, Stackebrandt E, Hamilton WA. 2007. Energy metabolism and phylogenetic diversity of sulphate-reducing bacteria, p 1–37 In Barton LL, Hamilton WA. (ed), Sulphate-reducing bacteria: environmental and engineered systems. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 6.Thauer RK, Jungermann K, Decker K. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41:100–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powell B, Mergeay M, Christofi N. 1989. Transfer of broad host-range plasmids to sulphate-reducing bacteria. FEMS Microbiol. Lett. 59:269–274. 10.1111/j.1574-6968.1989.tb03123.x [DOI] [Google Scholar]
- 8.Van den Berg WAM, Stokkermans JPWG, van Dongen WMAM. 1989. Development of a plasmid transfer system for the anaerobic sulphate reducer, Desulfovibrio vulgaris. J. Biotechnol. 12:173–184. 10.1016/0168-1656(89)90014-X [DOI] [Google Scholar]
- 9.Keller KL, Wall JD. 2011. Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front. Microbiol. 2:135. 10.3389/fmicb.2011.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pankhania IP, Spormann AM, Hamilton WA, Thauer RK. 1988. Lactate conversion to acetate, CO2 and H2 in cell suspensions of Desulfovibrio vulgaris (Marburg): indications for the involvement of an energy driven reaction. Arch. Microbiol. 150:26–31. 10.1007/BF00409713 [DOI] [Google Scholar]
- 11.Postgate JR. 1984. The sulphate reducing bacteria, 2nd ed. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 12.Stams AJM, Hansen TA. 1982. Oxygen-labile L(+) lactate dehydrogenase activity in Desulfovibrio desulfuricans. FEMS Microbiol. Lett. 13:389–394 [Google Scholar]
- 13.Pereira IAC, Haveman SA, Voordouw G. 2007. Biochemical and genetic and genomic characterization of anaerobic electron transport pathways in sulphate-reducing delta proteobacteria. In Barton LL, Hamilton WA. (ed), Sulphate-reducing bacteria: environmental and engineered systems. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 14.Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. 2011. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2:69. 10.3389/fmicb.2011.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odom JM, Peck HD. 1981. Localization of dehydrogenases, reductases and electron transfer components in the sulfate-reducing bacterium Desulfovibrio gigas. J. Bacteriol. 147:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peck HD, Jr, LeGall J, Lespinat PA, Berlier Y, Fauque G. 1987. A direct demonstration of hydrogen cycling by Desulfovibrio vulgaris employing membrane-inlet mass spectrometry. FEMS Microbiol. Lett. 40:295–299. 10.1111/j.1574-6968.1987.tb02042.x [DOI] [Google Scholar]
- 17.Hauser LJ, Land ML, Brown SD, Larimer F, Keller KL, Rapp-Giles BJ, Price MN, Lin M, Bruce DC, Detter JC, Tapia R, Han CS, Goodwin LA, Cheng J-F, Pitluck S, Copeland A, Lucas S, Nolan M, Lapidus ALA, Palumbo V, Wall JD. 2011. Complete genome sequence and updated annotation of Desulfovibrio alaskensis G20. J. Bacteriol. 193:4268–4269. 10.1128/JB.05400-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, Eisen JA, Ward N, Methe B, Brinkac LM, Daugherty SC, Deboy RT, Dodson RJ, Durkin AS, Madupu R, Nelson WC, Sullivan SA, Fouts D, Haft DH, Selengut J, Peterson JD, Davidsen TM, Zafar N, Zhou L, Radune D, Dimitrov G, Hance M, Tran K, Khouri H, Gill J, Utterback TR, Feldblyum TV, Wall JD, Voordouw G, Fraser CM. 2004. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22:554–559. 10.1038/nbt959 [DOI] [PubMed] [Google Scholar]
- 19.Matias PM, Pereira IAC, Soares CM, Carrondo MA. 2005. Sulphate respiration from hydrogen in Desulfovibrio bacteria: a structural biology overview. Prog. Biophys. Mol. Biol. 89:292–329. 10.1016/j.pbiomolbio.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 20.Zane GM, Yen H-C, Wall JD. 2010. Effect of the deletion of qmoABC and the promoter-distal gene encoding a hypothetical protein on sulfate reduction in Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 76:5500–5509. 10.1128/AEM.00691-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venceslau SS, Lino RR, Pereira IAC. 2010. The QRC membrane complex, related to the alternative complex III, is a menaquinone reductase involved in sulfate respiration. J. Biol. Chem. 285:22774–22783. 10.1074/jbc.M110.124305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Luo Q, Wofford NQ, Keller KL, McInerney MI, Wall JD, Krumholz LR. 2009. A molybdopterin oxidoreductase is involved in H2 oxidation in Desulfovibrio desulfuricans G20. J. Bacteriol. 191:2675–2682. 10.1128/JB.01814-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venceslau SS, Mantos D, Pereira IAC. 2011. EPR characterization of the new QRC complex from sulfate reducing bacteria and its ability to form a supercomplex with hydrogenase and TpIc. FEBS Lett. 585:2177–2181. 10.1016/j.febslet.2011.05.054 [DOI] [PubMed] [Google Scholar]
- 24.Madej MG, Nasiri HR, Hilgendorff NS, Schwalbe H, Lancaster CR. 2006. Evidence for transmembrane proton transfer in a dihaem-containing membrane protein complex. EMBO J. 25:4963–4970. 10.1038/sj.emboj.7601361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groh JL, Luo Q, Ballard JD, Krumholz LR. 2005. A method adapting microarray technology for signature-mutagenesis of Desulfovibrio desulfuricans G20 and Shewanella oneidensis MR-1 in anaerobic sediment survival experiments. Appl. Environ. Microbiol. 71:7064–7074. 10.1128/AEM.71.11.7064-7074.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapp-Giles BJ, Casalot L, English RS, Ringbauer JA, Jr, Dolla A, Wall JD. 2000. Cytochrome c3 mutants of Desulfovibrio desulfuricans. Appl. Environ. Microbiol. 66:671–677. 10.1128/AEM.66.2.671-677.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruschi M. 1994. Cytochrome c3 (Mr 26,000) isolated from sulfate-reducing bacteria and its relationships to other polyhemic cytochromes from Desulfovibrio. Methods Enzymol. 243:140–155 [DOI] [PubMed] [Google Scholar]
- 28.Weimer PJ, Van Kavelaar MJ, Michel CB, Ng TK. 1988. Effect of phosphate on the corrosion of carbon steel and on the composition of corrosion products in the two-stage continuous cultures of Desulfovibrio desulfuricans. Appl. Environ. Microbiol. 54:386–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rousset M, Casalot L, Rapp-Giles BJ, Dermoun Z, de Philip P, Belaich JP, Wall JD. 1998. New shuttle vectors for the introduction of cloned DNA in Desulfovibrio. Plasmid 39:114–122. 10.1006/plas.1997.1321 [DOI] [PubMed] [Google Scholar]
- 30.Wall JD, Rapp-Giles BJ, Rousset M. 1993. Characterization of a small plasmid from Desulfovibrio desulfuricans and its use for shuttle vector construction. J. Bacteriol. 175:4121–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh J, Fund E, Price MN, Dehal PS, Davis RW, Giaever G, Nislow C, Arkin AP, Deutschbauer A. 2010. A universal TagModule collection for parallel genetic analysis of microorganisms. Nucleic Acids Res. 38:1–14. 10.1093/nar/gkp829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 33.Li MZ, Elledge SJ. 2007. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4:251–256. 10.1038/nmeth1010 [DOI] [PubMed] [Google Scholar]
- 34.Cord-Ruwisch R. 1985. A quick method for the determination of dissolved and precipitated sulfides in cultures of sulfate-reducing bacteria. J. Microbiol. Methods 4:33–36. 10.1016/0167-7012(85)90005-3 [DOI] [Google Scholar]
- 35.Chhabra SR, He Q, Huang KH, Gaucher SP, Alm EJ, He Z, Hadi MZ, Hazen TC, Wall JD, Zhou J, Arkin AP, Singh AK. 2006. Global analysis of heat shock response in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 188:1817–1828. 10.1128/JB.188.5.1817-1828.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, He Z, Zhou J. 2005. Selection of optimal oligonucleotide probes for microarrays using multiple criteria, global alignment and parameter estimation. Nucleic Acids Res. 33:6114–6123. 10.1093/nar/gki914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang S, Tschaplinski TJ, Engle NL, Carroll SL, Martin SL, Davison BH, Palumbo AV, Rodriguez MJ, Brown SD. 2009. Transcriptomic and metabolomic profiling of Zymomonas mobilis during aerobic and anaerobic fermentation. BMC Genomics 10:34. 10.1186/1471-2164-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norbeck AD, Monroe ME, Adkins JN, Anderson KK, Daly DS, Smith RD. 2005. The utility of accurate mass and LC elution time information in the analysis of complex proteomes. Am. Soc. Mass Spectrom. 16:1239–1249. 10.1016/j.jasms.2005.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo Q, Hixson KK, Callister SJ, Lipton MS, Morris BEL, Krumholz LR. 2007. Proteome analysis of Desulfovibrio desulfuricans G20 mutants using the accurate mass and time (AMT) tag approach. J. Proteome Res. 6:3042–3053. 10.1021/pr070127o [DOI] [PubMed] [Google Scholar]
- 40.Elias DA, Monroe ME, Marshall MJ, Romine MF, Beliaev AS, Fredrickson JK. 2005. Global detection and characterization of hypothetical proteins in Shewanella oneidensis MR-1 using LC-MS based proteomics. Proteomics 5:3120–3130. 10.1002/pmic.200401140 [DOI] [PubMed] [Google Scholar]
- 41.Shen Y, Tolic N, Zhao R, Pasa-Tolic L, Li L, Berger SJ, Harkewicz R, Anderson GA, Belov ME, Smith RD. 2001. High-throughput proteomics using high-efficiency multiple-capillary liquid chromatography with on-line high-performance ESI FTICR mass spectrometry. Anal. Chem. 73:3011–3021. 10.1021/ac001393n [DOI] [PubMed] [Google Scholar]
- 42.Eng JK, McCormack AL, Yates JR., III 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989. 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- 43.Grant WM. 1947. Colorimetric determination of sulfur dioxide. Anal. Chem. 19:345–346. 10.1021/ac60005a023 [DOI] [Google Scholar]
- 44.Larsen RA, Wilson MM, Guss AM, Metcalf WW. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178:193–201. 10.1007/s00203-002-0442-2 [DOI] [PubMed] [Google Scholar]
- 45.Buckel W, Thauer RK. 2013. Energy conservation via electron bifurcating ferrodoxin reduction and proton/Na+ translocating ferrodoxin oxidation. Biochim. Biophys. Acta 1827:94–113. 10.1016/j.bbabio.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 46.Kranz RG, Richard-Fogal C, Taylor JS, Frawley ER. 2009. Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 73:510–528. 10.1128/MMBR.00001-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grein F, Ramos AR, Venceslau SS, Pereira IAC. 2013. Unifying concepts in anaerobic respiration: insights from dissimilatory sulfur metabolism. Biochim. Biophys. Acta 1827:145–160. 10.1016/j.bbabio.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 48.Ramos AR, Keller KL, Wall JD, Pereira IAC. 2012. The membrane QmoABC complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Front. Microbiol. 3:137. 10.3389/fmicb.2012.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaunmüller T, Kelly DJ, Glöckner FO, Uden G. 2006. Succinate dehydrogenases functioning by a reverse redox loop mechanism and fumarate reductase in sulphate-reducing bacteria. Microbiology 152:2443–2453. 10.1099/mic.0.28849-0 [DOI] [PubMed] [Google Scholar]
- 50.Kiley PJ, Beinert H. 1998. Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. FEMS Microbiol. Rev. 22:341–352. 10.1111/j.1574-6976.1998.tb00375.x [DOI] [PubMed] [Google Scholar]
- 51.Zhou A, Chen YI, Zane GM, He Z, Hemme CL, Joachimiak MP, Baumohl JK, He Q, Fields MW, Arkin AP, Wall JD, Hazen TC, Zhou J. 2012. Functional characterization of Crp/Fnr-type global transcriptional regulators in Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 78:1168–1177. 10.1128/AEM.05666-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller KL, Bender KS, Wall JD. 2009. Development of a markerless genetic exchange system in Desulfovibrio vulgaris Hildenborough and its use in generating a strain with increased transformation efficiency. Appl. Environ. Microbiol. 75:7682–7691. 10.1128/AEM.01839-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller JDA, Wakerley DS. 1966. Growth of sulphate-reducing bacteria by fumarate dismutation. J. Gen. Microbiol. 43:101–107. 10.1099/00221287-43-1-101 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.