

Abstract
We compared the classification accuracy of two sections of the fungal internal transcribed spacer (ITS) region, individually and combined, and the 5′ section (about 600 bp) of the large-subunit rRNA (LSU), using a naive Bayesian classifier and BLASTN. A hand-curated ITS-LSU training set of 1,091 sequences and a larger training set of 8,967 ITS region sequences were used. Of the factors evaluated, database composition and quality had the largest effect on classification accuracy, followed by fragment size and use of a bootstrap cutoff to improve classification confidence. The naive Bayesian classifier and BLASTN gave similar results at higher taxonomic levels, but the classifier was faster and more accurate at the genus level when a bootstrap cutoff was used. All of the ITS and LSU sections performed well (>97.7% accuracy) at higher taxonomic ranks from kingdom to family, and differences between them were small at the genus level (within 0.66 to 1.23%). When full-length sequence sections were used, the LSU outperformed the ITS1 and ITS2 fragments at the genus level, but the ITS1 and ITS2 showed higher accuracy when smaller fragment sizes of the same length and a 50% bootstrap cutoff were used. In a comparison using the larger ITS training set, ITS1 and ITS2 had very similar accuracy classification for fragments between 100 and 200 bp. Collectively, the results show that any of the ITS or LSU sections we tested provided comparable classification accuracy to the genus level and underscore the need for larger and more diverse classification training sets.
INTRODUCTION
Fungi are one of the most diverse groups of eukaryotic organisms on Earth, with estimates that range from 1.5 to 5.1 million species (1, 2). The use of next-generation sequencing (NGS) is playing a major role in the discovery of new species and ecological studies of fungi. Large molecular data sets are being generated at an extraordinary rate (3–6), but diversity estimations and taxonomic identification at all taxonomic levels are constrained by the lack of accurate, comprehensive taxonomic databases and information on the accuracy of classification tools for comparison of environmental survey data. The detection of emergent fungal diseases, the determination of biogeographical patterns, and definition of strategies for conservation of fungi are just a few examples of research areas that are challenged by the lack of reliable databases and tools (7, 8). The large number of sequences generated from platforms of high-throughput sequencing also demand fast and accurate algorithms for sequence analysis and taxonomic classification of fungi.
The entire internal transcribed spacer (ITS) rRNA region (approximately 600 bp in length) is composed of two hypervariable regions (ITS1 and ITS2) with the highly conserved 5.8S rRNA gene between them (Fig. 1). The ITS region has been used for many years for diversity estimations and taxonomic identification of fungal isolates and uncultured taxa (9–11) and was adapted as the barcode region for Fungi by the Consortium for the Barcode of Life (12). The large-subunit rRNA (LSU) region, located immediately downstream of the ITS, has also been widely used for phylogenetic assignment of cultures (13–15) and for environmental surveys (6, 16). A 5′ section of the gene, 635 to 651 bp in length, contains two hypervariable regions (D2 and D3) that discriminate among most fungal genera (17).
FIG 1.
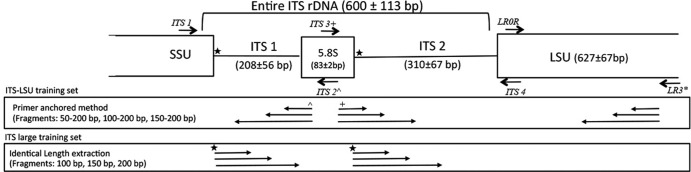
Primer locations in the ITS region, showing the variable ITS1 and ITS2 regions and sequence length in the ITS-LSU training set. Two fragment extraction methods were utilized. Fragment sizes are shown with arrows.
The use of the rRNA gene regions for fungal identification presents several advantages over the use of functional genes (protein-coding genes): high sequence variability for identification of the large majority of fungi at the species level; a high number of copies per cell, which provides a direct advantage when little DNA is available (e.g., for herbarium specimens or certain environmental samples); conserved primer sites not subject to variable third-codon positions; and a growing number of sequences represented by curated fungal cultures and environmental samples in public databases (12, 18–22). The ITS and LSU regions each have strengths and weaknesses as molecular signatures for fungal identification and environmental surveys. Drawbacks associated with use of the ITS region are limitations of low taxonomic resolution for some species delimitations (23, 24), difficulty in fungus-specific PCR primer design in this region, and high variability that precludes the use of alignments and tree-based methods for analysis of environmental sequences (18, 21). The LSU region is generally considered less variable than the ITS region, which can limit taxonomic resolution at the species levels and diversity analysis. However, it is amenable to sequence alignment and phylogenetic identification of new clades. While both regions represent information-rich sequences for fungal identification (19), they have not been directly compared for use with classifier approaches.
Little information is available on the classification accuracy of commonly used sections of the ITS (i.e., ITS1, ITS2, or combined sections) (Fig. 1) or the LSU, with different tools of analysis, such as the naive Bayesian classifier of the Ribosomal Database Project (RDP) or BLASTN (12, 25, 26). With the availability of public analysis tools for ITS and LSU sequence analysis (17, 27–30), this information is fundamental to determine the optimal sections and the criteria required to optimize primer design and selection of sequencing platforms and to improve analysis at the genus and species levels.
The naive Bayesian classifier has been available for analysis of bacterial and archaeal sequences for many years (31). Recently, Liu et al. (17) created a hand-curated fungal database of the large-subunit rRNA (LSU) gene and demonstrated that the naive Bayesian classifier can be used for accurate classification of fungal sequences using this LSU database. Recent surveys of soil fungal communities have successfully used this database to track fungal composition in different locations and responses to environmental perturbations (6, 53).
We evaluated two new hand-curated sequence training sets, one that contains sequences spanning the ITS and LSU regions (modified from reference 12) and a larger database containing only ITS region sequences. We compared the performance of the naive Bayesian classifier for classification of ITS1, ITS2, the entire ITS region (ITS1 plus 5.8S rRNA plus ITS2), and the 5′ section of the LSU. We then compared the performance of the naive Bayesian classifier to BLASTN using fragment sizes ranging from 50 to ∼600 bp and employing a 50% bootstrap cutoff to improve taxonomic classification. The naive Bayesian classifier and supporting databases for ITS and LSU regions are available through the RDP website, http://rdp.cme.msu.edu/classifier/classifier.jsp.
MATERIALS AND METHODS
Fungal ITS-LSU gene training set.
To compare classification accuracy among sections of the ITS and LSU, we needed a training set that contained sequences spanning both the ITS and LSU regions for each fungal isolate. We downloaded 1,125 sequences, published by the fungal barcode of life consortium (12), from the NCBI nucleotide and taxonomy database using NCBI Entrez in batch mode. After removal of duplicate sequences and sequences with unclear taxonomy, a total of 1,091 fungal ITS gene sequences spanning 20 classes and 6 phyla were recovered. The taxonomic composition of this ITS-LSU training set is shown in Table S1 in the supplemental material. Four genera that were each represented by only a single sequence (here termed singletons) were excluded from leave-one-out cross-validation (LOOCV) analyses for classification accuracy to avoid incorrect taxonomic representation due to the lack of representative sequences, as reported by Liu et al. (17). The resulting data set used for LOOCV classification accuracy included 1,087 sequences. Every sequence (excluding singletons) was evaluated as a query against the data set to determine taxonomic classification accuracy and bootstrap support using in-house Perl scripts (17). Classification accuracy was evaluated from phylum to genus to determine differences between LSU and ITS. We did not perform analysis at the species level because when using a LOOCV approach, the species level accuracy is determined by the number of different strains (the same species) present in the data set, and the current data sets that include both ITS and LSU regions (see, e.g., reference 12) do not have adequate coverage at the species level to conduct rigorous or informative species-level comparisons. The availability of more accurate databases will facilitate in the future comparisons among different taxonomic ranks (e.g., Ascomycota versus Basidiomycota or species-level analysis). With the current state of the public databases, analyses comparing species or even phyla will be highly affected by the lack of enough species representation and/or the taxonomic uncertainty that is more prevalent for certain taxonomic ranks.
Large fungal ITS gene training set.
A set of 9,838 fungal ITS GenBank sequences were manually checked and downloaded from the NCBI database. Sequences were selected from published phylogenies in the peer-reviewed literature or in NCBI using the search tool. Sequences were also obtained from other sources, including AFTOL publications (http://aftol.org/), MycoBank (http://www.mycobank.org), CBS type cultures (http://www.cbs.knaw.nl/Collections/ ), and mor (32). Curation of the database was intended to ensure that (i) the taxonomic placement was consistent across the database, (ii) all taxa had some information associated with each taxonomic rank (at least Fungi incertae sedis as a minimum for fungi with uncertain taxonomic placement), and (iii) each sequence contained no errors (blank spaces, a family name instead of an order name, or spelling errors). In addition, we reduced duplications and normalized taxonomy assignments; there were many incidences where family names/order names were inconsistent or different for taxa with the same name. We used Index Fungorum (http://www.indexfungorum.org) as a guideline to reflect currently accepted taxonomic placement. Using NCBI Entrez in batch mode and an in-house Perl script, we extracted the sequence and taxonomy information from the NCBI nucleotide and taxonomy database (17). The ITS gene sequences were aligned using the program MUSCLE (33) using the 5.8S rRNA gene and PCR primer sequences as guidelines to determine the correct orientation and location of the ITS1 and ITS2 regions. Alignments were trimmed using MEGA (34), and sequences that had only a small portion of the ITS region (50 to 100 bp) or poor quality (a large number of undefined nucleotides) were excluded. The final hand-curated database contained 8,967 fungal ITS gene sequences, and this final data set was designated the “large ITS training set” for LOOCV analyses using the naive Bayesian classifier and BLASTN. The database contains 36 classes, including 118 orders, 332 families, and 1,110 genera (Table 1).
TABLE 1.
Taxonomic composition of the large ITS training set used for LOOCV comparisons of ITS1 and ITS2
| Kingdom (n = 1) | Domain (n = 4) | Phylum (n = 12) | Class (n = 36) | Order (n = 118) | No. of: |
||
|---|---|---|---|---|---|---|---|
| Families (n = 332) | Genera (n = 1,110) | Sequences (n = 8,967) | |||||
| Eukaryota | Fungi | Ascomycota | Lecanoromycetes | Acarosporales | 1 | 2 | 2 |
| Lecanorales | 8 | 26 | 126 | ||||
| Pertusariales | 4 | 4 | 7 | ||||
| Ostropales | 2 | 6 | 8 | ||||
| Lecanoromycetes incertae sedis | 1 | 1 | 1 | ||||
| Peltigerales | 1 | 1 | 1 | ||||
| Dothideomycetes | Jahnulales | 1 | 2 | 2 | |||
| Dothideomycetes incertae sedis[O] | 1 | 1 | 76 | ||||
| Botryosphaeriales | 1 | 21 | 101 | ||||
| Capnodiales | 13 | 54 | 507 | ||||
| Pleosporales | 12 | 44 | 294 | ||||
| Dothideales | 3 | 11 | 81 | ||||
| Myriangiales | 1 | 2 | 22 | ||||
| Hysteriales | 1 | 2 | 2 | ||||
| Trypetheliales | 1 | 1 | 1 | ||||
| Sordariomycetes | Xylariales | 5 | 26 | 83 | |||
| Sordariomycetes incertae sedis[O] | 1 | 1 | 25 | ||||
| Hypocreales | 7 | 46 | 1,596 | ||||
| Calosphaeriales | 1 | 7 | 26 | ||||
| Microascales | 4 | 7 | 32 | ||||
| Sordariales | 3 | 6 | 56 | ||||
| Chaetosphaeriales | 2 | 2 | 9 | ||||
| Coniochaetales | 1 | 1 | 3 | ||||
| Diaporthales | 7 | 26 | 501 | ||||
| Glomerellales | 1 | 3 | 127 | ||||
| Lulworthiales | 1 | 1 | 1 | ||||
| Magnaporthales | 1 | 2 | 23 | ||||
| Ophiostomatales | 1 | 4 | 72 | ||||
| Trichosphaeriales | 2 | 2 | 3 | ||||
| Pezizomycetes | Pezizales | 9 | 28 | 178 | |||
| Ascomycota incertae sedis[C] | Ascomycota incertae sedis[O] | 1 | 1 | 67 | |||
| Eurotiomycetes | Chaetothyriales | 4 | 16 | 54 | |||
| Eurotiomycetes incertae sedis | 2 | 2 | 3 | ||||
| Eurotiales | 2 | 9 | 185 | ||||
| Pyrenulales | 2 | 3 | 3 | ||||
| Mycocaliciales | 1 | 1 | 1 | ||||
| Onygenales | 2 | 3 | 13 | ||||
| Verrucariales | 1 | 9 | 19 | ||||
| Saccharomycetes | Saccharomycetales | 7 | 17 | 96 | |||
| Leotiomycetes | Helotiales | 7 | 47 | 215 | |||
| Erysiphales | 1 | 3 | 23 | ||||
| Leotiomycetes incertae sedis | 5 | 12 | 73 | ||||
| Geoglossomycetes | Geoglossales | 1 | 2 | 2 | |||
| Lichinomycetes | Lichinales | 2 | 2 | 3 | |||
| Neolectomycetes | Neolectales | 1 | 1 | 1 | |||
| Orbiliomycetes | Orbiliales | 1 | 2 | 6 | |||
| Taphrinomycetes | Taphrinales | 2 | 2 | 3 | |||
| Arthoniomycetes | Arthoniales | 1 | 3 | 3 | |||
| Basidiomycota | Agaricomycetes | Agaricales | 29 | 212 | 1,980 | ||
| Agaricomycetes incertae sedis | 1 | 1 | 1 | ||||
| Russulales | 11 | 32 | 263 | ||||
| Amylocorticiales | 1 | 1 | 1 | ||||
| Boletales | 20 | 45 | 231 | ||||
| Atheliales | 1 | 7 | 28 | ||||
| Auriculariales | 3 | 6 | 22 | ||||
| Polyporales | 14 | 74 | 366 | ||||
| Thelephorales | 3 | 14 | 106 | ||||
| Cantharellales | 7 | 18 | 190 | ||||
| Corticiales | 1 | 24 | 114 | ||||
| Gomphales | 2 | 7 | 99 | ||||
| Geastrales | 2 | 2 | 9 | ||||
| Gloeophyllales | 1 | 3 | 5 | ||||
| Hymenochaetales | 4 | 16 | 56 | ||||
| Hysterangiales | 1 | 1 | 3 | ||||
| Phallales | 1 | 3 | 7 | ||||
| Sebacinales | 1 | 6 | 25 | ||||
| Trechisporales | 1 | 2 | 3 | ||||
| Agaricostilbomycetes | Agaricostilbales | 3 | 7 | 7 | |||
| Microbotryomycetes | Sporidiobolales | 4 | 7 | 27 | |||
| Heterogastridiales | 1 | 1 | 1 | ||||
| Leucosporidiales | 1 | 3 | 5 | ||||
| Microbotryales | 1 | 2 | 61 | ||||
| Microbotryomycetes incertae sedis[O] | 1 | 1 | 1 | ||||
| Ustilaginomycetes | Ustilaginales | 2 | 8 | 23 | |||
| Urocystales | 3 | 3 | 3 | ||||
| Pucciniomycetes | Pucciniales | 7 | 12 | 86 | |||
| Platygloeales | 1 | 1 | 1 | ||||
| Septobasidiales | 1 | 1 | 1 | ||||
| Tremellomycetes | Cystofilobasidiales | 2 | 7 | 30 | |||
| Filobasidiales | 1 | 1 | 4 | ||||
| Tremellales | 3 | 10 | 126 | ||||
| Tremellomycetes incertae sedis[O] | 1 | 1 | 66 | ||||
| Dacrymycetes | Dacrymycetales | 1 | 4 | 4 | |||
| Entorrhizomycetes | Entorrhizales | 1 | 1 | 1 | |||
| Exobasidiomycetes | Entylomatales | 1 | 1 | 2 | |||
| Exobasidiales | 1 | 1 | 2 | ||||
| Exobasidiomycetes incertae sedis[O] | 1 | 1 | 4 | ||||
| Malasseziales | 1 | 1 | 2 | ||||
| Microstromatales | 3 | 4 | 9 | ||||
| Doassansiales | 1 | 1 | 1 | ||||
| Tilletiales | 1 | 2 | 4 | ||||
| Georgefischeriales | 1 | 1 | 1 | ||||
| Cystobasidiomycetes | Erythrobasidiales | 1 | 3 | 3 | |||
| Mixiomycetes | Mixiales | 1 | 1 | 1 | |||
| Wallemiomycetes | Wallemiales | 1 | 1 | 1 | |||
| Blastocladiomycota | Blastocladiomycetes | Blastocladiales | 2 | 5 | 22 | ||
| Chytridiomycota | Chytridiomycetes | Chytridiales | 5 | 11 | 15 | ||
| Cladochytriales | 1 | 2 | 2 | ||||
| Spizellomycetales | 4 | 10 | 24 | ||||
| Rhizophydiales | 3 | 3 | 11 | ||||
| Lobulomycetales | 1 | 1 | 1 | ||||
| Chytridiomycetes incertae sedis[O] | 1 | 1 | 1 | ||||
| Chytridiomycota | Monoblepharidomycetes | Monoblepharidales | 2 | 2 | 3 | ||
| Fungi incertae sedis[P] | Fungi incertae sedis[C] | Entomophthorales | 3 | 5 | 26 | ||
| Mucorales | 8 | 12 | 52 | ||||
| Harpellales | 1 | 3 | 18 | ||||
| Mortierellales | 1 | 2 | 38 | ||||
| Zoopagales | 1 | 2 | 2 | ||||
| Glomeromycota | Glomeromycetes | Diversisporales | 2 | 2 | 2 | ||
| Glomerales | 1 | 1 | 3 | ||||
| Paraglomerales | 1 | 1 | 3 | ||||
| Neocallimastigomycota | Neocallimastigomycetes | Neocallimastigales | 1 | 4 | 14 | ||
| Zygomycota | Zygomycota incertae sedis | Dimargaritales | 1 | 1 | 1 | ||
| Endogonales | 1 | 1 | 1 | ||||
| Kickxellales | 1 | 2 | 3 | ||||
| Protozoa | Protozoa incertae sedis | Ichthyosporea | Ichthyophonida | 1 | 1 | 1 | |
| Viridiplantae | Streptophyta | Streptophyta incertae sedis | Asterales | 1 | 1 | 1 | |
| Stramenopiles | Xanthophyceae | Xanthophyceae incertae sedis | Vaucheriales | 1 | 1 | 1 | |
Genetic region representation.
All conserved sections of the ITS region (a small portion of the 3′ end of the small-subunit rRNA [SSU] gene, the 5.8S rRNA gene between ITS1 and ITS2, and the 5′ end of the LSU gene) were aligned using MUSCLE to generate the master alignment. The actual ITS1 and ITS2 sections are so highly variable they cannot be aligned with confidence, and the alignments using surrounding conserved regions were conducted to determine that all sequences contained ITS1, ITS2, and the 5.8S rRNA gene and were oriented in the 5′-to-3′ direction (Fig. 1). The location of the 5.8S rRNA gene was determined following positions as described by Bell et al. (35). The quality of the alignments was determined by visual evaluation of the conserved regions and identification of primer sites for primers ITS1, ITS4, ITS2, and ITS3 as a guideline (http://nature.berkeley.edu/brunslab/tour/primers.html). For LSU alignments, sequences were trimmed at the LR0R and LR3 primer sites (http://biology.duke.edu/fungi/mycolab/primers.htm).
Extraction of different test fragments for comparisons of the ITS and LSU regions.
Classification accuracy was evaluated for ITS1, ITS2, and LSU for different fragment lengths. The first comparison was based on full-length regions for each section (ITS1, 208 ± 57 bp; ITS2, 310 ± 67 bp; entire ITS, 599 ± 113 bp; and LSU, 627 ± 51 bp), allowing the variation in sequence length between the sections. Two methods were then used to extract fragments of different sizes to enable comparisons that were not biased by sequence length. The first method, termed “primer anchored,” created length range extractions for fragment sizes from 50 to 200 bp, 100 to 200 bp, and 150 to 200 bp based on PCR primer positions commonly used for the ITS1, ITS2, and LSU regions and was used with the ITS-LSU training set (Fig. 1). For LSU, test sequences were anchored at the 3′ end of the LR3 primer in the reverse direction. For ITS1, the fragments were anchored to the 3′ end of the ITS2 primer in reverse direction, and the ITS2 fragments were anchored at the 3′ end of the ITS3 primer in the forward direction. LR3 was used for the LSU analysis because Liu et al. (17) showed that the D2 region upstream of the LR3 priming site is the most informative for LSU taxonomic classification. A minimum length cutoff of 50 bp was chosen because the naive Bayesian classifier does not perform well with sequences smaller than 50 bases (data not shown), and the majority of current high-throughput platforms produce fragments larger than 50 bp. This poor performance with short fragments is likely due to insufficient information to conduct accurate taxonomic classification.
The sequence length extractions described above represent sequences that are often obtained using current PCR and sequencing platforms. Due to primer location, the fragments will contain portions of the 5.8S rRNA gene region and 28S rRNA gene in addition to the actual ITS sequences. This results in 31 to 33 additional nucleotides that lie in the adjacent conserved genes for the ITS1 and ITS2 regions (Fig. 1). To facilitate the comparison of the ITS1 and ITS2 regions without these adjacent conserved regions, a second method, termed “identical-length extraction,” was used, which created fragments of identical size for the ITS1 and ITS2 sections in the large ITS training set. Sequences of 100 bp, 150 bp, and 200 bp were created for LOOCV testing based on the start positions of the ITS1 and ITS2 regions using the alignment of the SSU, 5.8S rRNA, and LSU sections to identify the specific start sites (Fig. 1).
Naive Bayesian classifier and bootstrap analysis.
The Java tool of the naive Bayesian classifier was obtained from RDP's sourceforge page (http://sourceforge.net/projects/rdp-classifier/) and installed locally. The naive Bayesian classifier provides rapid taxonomic assignment of rRNA sequences, with reference to a training set of sequences of known taxonomy (17). For each query sequence, a subset was chosen from all 8-base overlapping subsequences (words) in the query. The joint probability of observing the selected words was calculated for each taxon and the sequence assigned to the taxa with the highest probability based on the naive Bayesian assumption that the probabilities for each word are independent (31). The sampling is repeated for 100 bootstrap trials to provide an estimate of confidence in the assignment. The naive Bayesian classifier assigns query sequences at each taxonomic rank from phylum to genus and provides a bootstrap confidence estimate at each rank. The naive Bayesian classifier is available in web-based and stand-alone formats (31).
For each naive Bayesian classifier-based LOOCV test, a single random sequence was removed from the training set as a query sequence to test its taxonomic placement against the remaining training sequence set. The process was repeated for all sequences in the training set. In-house Perl scripts were then used to parse the taxonomy assignment.
Evaluation of BLASTN classification.
We used BLASTN as a tool to compare the accuracy of the naive Bayesian classifier. BLASTN was installed locally from http://www.ncbi.nlm.nih.gov/. Both short sequences with a maximum of 200 bp (50 to 200 bp) and full-length sequences with an average range from 200 to 600 bp were tested in this study. BLASTN parameters were set to a word size of 7 for short sequences and the default 11 for full-length sequences and an E value threshold of 1,000. For each BLASTN-based LOOCV test, a single sequence was reserved from the ITS and LSU gene training set sequences as the test sequence, and the remaining sequences in the database comprised a reformatted BLASTN database. The process was repeated for all sequences in the training set. An in-house Perl script was used to obtain BLASTN information for the top hits. Analysis was conducted using a Mac OS X (10.5.8) server with a 2.66-GHz Quad-Core Intel Xeon processor and 3 GB of 1,066-MHz DDR3 memory. Bootstrap and BLASTN analyses were conducted for the ITS-LSU training set (1,087 sequences) and for the large ITS training data set (8,967 sequences).
Assessment of taxonomic assignment consistency.
To assess classification accuracy, we used Matthew's correlation coefficient (MCC) as a statistical measure of the quality of the classification (36). This statistic is used in machine learning control theory by measuring four metrics to decompose classification accuracies acquired from different classification tools, and it facilitates the identification of false positives and negatives. For a set of query read fragments, we counted the assignment combinations that could be considered consistent positives, consistent negatives, divergent positives, and divergent negatives. A set of assignments was considered a consistent positive if the query read fragment was assigned to the same and correct taxonomy by each classification tool. A consistent negative was a set of queries that were assigned to the same but incorrect taxonomy. A divergent positive denoted that at least one classification tool's assignment was correct. A divergent negative was a query read fragment that was assigned to different taxonomies by each classification tool and none of them were correctly assigned. There are numerous methods to weigh these four values. To evenly balance the terms, we used Matthew's correlation coefficient (MCC). This coefficient can vary between −1 and +1 and represents a value for measuring diversity between taxonomy classification results (e.g., different classifier, primers, or data sets). A coefficient of +1 represents a perfect prediction, 0 represents no better than random prediction, and −1 indicates total disagreement between prediction and observation (36).
RESULTS
LSU versus ITS classification accuracy using the ITS-LSU training set.
Accuracy in the classification of full-length ITS and LSU regions was compared for the naive Bayesian classifier and BLASTN (Fig. 2). With the exception of the ITS1 region, the accuracy levels obtained by the naive Bayesian classifier and BLASTN were very similar for ITS2, the entire ITS, and LSU. At the genus level, classification accuracy was 94 to 95% when either the entire ITS or LSU region was used, 93% for the ITS2 section, and 89 to 91% for ITS1 section (Fig. 2). Similar performance was obtained at the family level, with 98 to 99% classification accuracy for the entire ITS, the LSU region, or the ITS2 section. When BLAST and the naive Bayesian classifier were compared, BLASTN showed the lowest performance at all taxonomic levels for ITS1, with 89% accuracy at the genus level and 93% at the family level. The naive Bayesian classifier showed higher accuracy for ITS1 in comparison with BLASTN, with 91% accuracy at the genus level and 96% accuracy at the family level.
FIG 2.

Classification accuracy at each taxonomic level using different rRNA gene regions for LOOCV testing with the naive Bayesian classifier (NBC) and BLASTN approaches. Numbers are percentages of correctly classified query sequences from the ITS-LSU database. The naive Bayesian classifier was trained by full-length sequences without a bootstrap cutoff.
When full-length sequence regions were compared, the LSU region slightly outperformed the individual ITS1 and ITS2 regions but not the entire ITS region (Fig. 2). With the naive Bayesian classifier, the average accuracies for the ITS1, ITS2, entire ITS, and LSU regions at the genus level were 90.81% (ITS1), 93.18% (ITS2), 94.60% (entire ITS), and 94.60% (LSU). With BLASTN, the average accuracies at the genus level were about 88.72% (ITS1), 93.09% (ITS2), 94.79% (entire ITS), and 93.16% (LSU). The modest improvement in accuracy observed for the LSU region versus ITS1 or ITS2 is likely due to its length advantage, since the average length of LSU for this data set was 626 bp, versus 208 bp and 309 bp for the ITS1 and ITS2 regions, respectively (Fig. 1). The accuracy of the LSU region was very similar in comparison with that of the entire ITS region (ITS1 plus 5.8S rRNA plus ITS2) (599 bp), and the entire ITS had a higher accuracy than the individual ITS1 or ITS2 sections (Fig. 2). This observation suggests that longer sequences provide a higher discriminatory power than shorter sequences, for either the ITS or LSU region, regardless of the classification method.
These results were confirmed using Matthew's correlation coefficient (MCC) as a statistical measure of the quality of the classification using different regions or fragment sizes (see Table S2 in the supplemental material). A higher number of consistent positives was observed for longer sequences. For example, for the LSU we obtained 91% of consistent positive assignments, versus 87.2% for ITS1 and 90.33% for ITS2 (see Table S2 in the supplemental material). The MCC values between BLASTN and naive Bayesian classifier results were 51%, 36%, and 46% for LSU, ITS1, and ITS2, respectively.
To correct for a possible bias due to sequence length, we conducted a comparison using three sequence length ranges for the ITS and LSU regions. We also evaluated the effect of bootstrap support at a 50% cutoff. The accuracy of BLASTN and the naive Bayesian classification varied by only 1% to 2% for three minimum-length cutoffs that were obtained using the primer-anchored method (50 to 200 bp, 100 to 200 bp, and 150 to 200 bp) (data not shown). Therefore, we applied a global cutoff of 50- to 200-bp fragments (1,054 nonsingleton sequences out of 1,089 sequences longer than 50 bp) (see Fig. S1 in the supplemental material). The classification accuracies for the naive Bayesian classifier and BLASTN were very similar, with a slightly better performance for BLASTN (by 1 to 2%). The major effect was observed when a bootstrap cutoff was used for the naive Bayesian classifier. For all cases, the use of a 50% bootstrap cutoff improved the classification accuracy in comparison with BLASTN (1 to 2%) (see Fig. S1 in the supplemental material).
Classification accuracy for ITS1, ITS2, and LSU varied between 91 to 92% without the use of a bootstrap cutoff. The ITS1 and ITS2 sections had 1 to 2% higher accuracy at the genus level (94 to 95%) with respect to LSU (93%) when a 50% bootstrap cutoff was used with the naive Bayesian classifier (Fig. 3). With the use of a 50% bootstrap cutoff, the classification accuracy improved for all three sections tested, and ITS1 showed the highest improvement. The ITS1 section improved from 91% to 95% at the genus level and from 96% to 99% at the family level for each of the fragment sizes (Fig. 3). The classification accuracy using the ITS2 region improved from 92% to 94%, and the accuracy for the LSU fragments improved from 91% to 93%. The minimum-length cutoff effect was not obvious, although slightly different numbers of sequences (1,054, 1,043, and 910, respectively) were retained after applying 50- to 200-bp, 100- to 200-bp, and 150- to 200-bp cutoffs (Fig. 3).
FIG 3.

Classification accuracy comparison for the ITS1, ITS2, and LSU regions using the naive Bayesian classifier without a bootstrap or with a 50% bootstrap cutoff. The y axis shows the percentages of LOOCV sequences that were accurately classified. (A) ITS1 region; (B) ITS2 region; (C) LSU region. Query sequences of three lengths were extracted using the primer-anchored method.
The impact of using a bootstrap cutoff on sequence retention was also evaluated. About 5.5%, 2.8%, and 2.4% of the sequences in our training set were not retained at genus level assignment for the ITS1, ITS2, and LSU regions, respectively (Fig. 4), and smaller fragments were eliminated at a higher percentage for all the regions when a bootstrap cutoff was applied (Fig. 4). ITS1 showed the lowest sequence retention regardless of minimum-length cutoff. Considering the trade-off between higher accuracy but reduced number of reliable sequences remaining in the training set and the variable MCC results obtained for ITS1 and ITS2 depending of the fragment size (see Table S2 in the supplemental material), the performance of ITS1 against ITS2 did not differ when the ITS-LSU training data set was used.
FIG 4.

Percentage of sequences removed from the ITS-LSU training set after applying a 50% bootstrap cutoff with the naive Bayesian classifier. The y axis shows the percentage of sequence removed. Query sequences of three sizes were extracted using the primer-anchored method.
Comparisons between the ITS1 and ITS2 sections using the large ITS training set.
A larger, more comprehensive fungal sequence training data set was created to test differences in performance between the ITS1 and ITS2 sections. The larger data set consisted of 8,966 sequences (8,494 nonsingletons), representing 1,110 genera, 335 families, and 118 orders of fungi (Table 1). In general, the accuracy achieved using the classifier was 4 to 11% lower with this large training set than with the smaller, less comprehensive ITS-LSU training set used to compare the ITS and LSU regions (see Table S3 in the supplemental material). These results were expected because the smaller training set contained only ITS and LSU highly curated barcode sequences, and this larger training set is likely to contain more taxonomy conflicts due to the diversity of the resources that were used to create the training data set.
BLASTN showed higher accuracy than the naive Bayesian classifier without the use of a bootstrap cutoff. At the genus level, ITS1 and ITS2 had the same accuracy (84%), with an improvement of 1% when the entire ITS region was evaluated (Fig. 5). For the naive Bayesian classifier, ITS1 showed the lowest accuracy (80% accuracy) without the use of bootstrap, and the entire ITS had the highest level of accuracy (83%). The family-level accuracy of full-length sequences ranged from 90 to 95% accuracy. We observed a slightly better performance of the naive Bayesian classifier than of BLASTN with the use of a 50% bootstrap cutoff on full-length ITS1 or ITS2 sequences (Fig. 5). The entire ITS (average length, 588 bp), ITS1 (211 bp), and ITS2 (298 bp) had the same level of accuracy with the classifier when the 50% bootstrap cutoff was used, with 87% accuracy at the genus level. The corresponding accuracy using BLASTN for the different regions was 84 to 85%.
FIG 5.

Comparison of the full lengths of ITS1, ITS2, and entire ITS sections using the naive Bayesian classifier (NBC) or BLASTN. The classifier was used with and without a 50% bootstrap cutoff.
To eliminate any sequence length bias, the ITS1 and ITS2 sections were also tested using specific fragment sizes of 100 bp, 150 bp, and 200 bp with and without a 50% bootstrap cutoff. ITS1 and ITS2 showed very similar performance with the 200-bp and 150-bp fragments (Fig. 6). Classification accuracy at the genus level was 91 to 92% for ITS1 and ITS2 for the 200-bp and 150-bp fragments with the 50% bootstrap cutoff. Classification accuracy was higher for the ITS1 section than for the ITS2 section when a shorter (100-bp) sequence was used as long as a 50% bootstrap cutoff was employed. Classification accuracy at the family and order levels ranged from 92 to 98% for the different fragment sizes. In general, 10 to 20% of the sequences were removed from the analysis with the use of a 50% bootstrap cutoff at the genus level.
FIG 6.

Accuracy comparison using the ITS1 and ITS2 sections with the naive Bayesian classifier at its default setting and a 50% bootstrap cutoff for exact-length sequences of 100 bp (A), 150 bp (B), and 200 bp (C). The y axes show percentages of accuracy (lines) and of sequences removed (bars).
DISCUSSION
A number of factors that can influence classification accuracy of the fungal rRNA operon sequences were evaluated in this study. These included two databases with different compositions and sequence numbers for the ITS region, the use of a naive Bayesian classifier and BLASTN approaches for classification, the use of three ITS sections (full ITS, ITS1, and ITS2) in comparison with a 5′ region of the LSU gene, different sequence lengths likely achieved by current sequencing technologies, and the impact of employing a bootstrap cutoff calculation to improve classification accuracy. Overall, we found that the underlying database had the highest impact on classification accuracy using a LOOCV analysis, followed by the use of a bootstrap cutoff that increased accuracy at the expense of removing unreliable sequences. All of the gene regions provided excellent classification at higher taxonomic intervals, and differences at the genus level were small (1 to 2%) across the regions used (ITS1, ITS2, entire ITS, and LSU) or across sequence lengths from 100 to 200 bp. The choice of gene region and classification approach/stringency can be optimized within the framework of information provided here, as needed for different studies that may focus on different fungal clades or data sets. Clearly, improvements in the taxonomic scope and depth of publicly available databases will increase classification accuracy for all applications (17, 27, 37).
Through our comparisons, we provide two publicly available databases upon which the scientific community may build: an 8,967-member, hand-curated ITS database and an ITS-LSU training set modified from that described in reference 12. First, by focusing on a parallel ITS-LSU training set, where each taxon was represented by the ITS and LSU regions, we were able to provide direct comparisons of rRNA gene regions. Second, the larger ITS database was created to broadly survey sequences to represent all major phyla as much as possible instead of focusing deeply on particular families, genera, and species. For environmental survey applications, a broad database with good representation at higher-level taxa is most important because many environmental sequences are not closely related to known genera or families (e.g., in recent soil surveys [6, 38–40]). Future efforts will require incorporation of taxa represented only by environmental sequences from NCBI and the incorporation of databases that incorporated community curation efforts by experts in the field such as those conducted by UNITE and the Fungal Consortium for the Barcode of Life (12, 21, 22, 27).
Database composition.
Our study and prior studies have shown that the quality and success of automated fungal classification were greatly influenced by reliable reference sequences, sequence length, and the algorithms used in the analysis (19, 25, 41). The two databases used in this study provided different levels of classification accuracy, with the ITS-LSU training achieving 4 to 11% higher accuracy in LOOCV comparisons than the large ITS training set (see Table S3 in the supplemental material). The large ITS training set is more comprehensive but has lower coverage in many less-characterized lineages. Many environmental surveys show a high percentage of “novel” or inadequately classified taxa, so it is important to create the mechanisms to include novel clades in curated databases. This result underscores the importance of generating and maintaining accurate and comprehensive databases for use by the scientific community. Common problems that impact the quality of fungal classification include the presence of misclassified sequences (approximately 20% of the sequences in NCBI) (37), the high number of polyphyletic groups common in fungal taxonomy, reflecting the need for more phylogenetic studies, and sequences with uncertain or unknown taxonomic placement (incertae sedis) at all taxonomic levels, such as those frequently found in common mitosporic Ascomycota.
Potential reasons why the accuracy dropped when the larger training data set was applied included the presence of many less-well-studied fungal sequences in the larger ITS data set, including taxa with unknown taxonomic placement at different classification levels. Another likely reason could be due to the biased data set itself. If a reference data set contains many sequences derived from some environments and few associated with others, these could lead to a substantial variation in classification quality. In addition, the use of LOOCV at the sequence level (31, 36, 42–45) where a single sequence with a known annotation is held out from a reference database and classified using the remainder might not be perfect. Although not reported yet from fungal community study, it is known that natural environments contain “microdiverse” clusters of closely related bacterial strains (44). If the distribution from the fungal community had a similar pattern, then it would lead to a biased training set with a greater extent of diversity within certain phyla only. Therefore, it is essential to continue efforts to enlarge curated and more diverged fungal databases to cover a greater number of taxonomic classes.
Naive Bayesian classifier versus BLASTN and effect of bootstrap cutoff.
At higher taxonomic levels (phylum, class, and order), the classification accuracies of the naive Bayesian classifier and BLASTN were quite similar. Porter and Golding (25) compared different classification methods and showed that BLASTN was consistently better than MEGAN (29) and SAP (Statistical Assignment Program) (46) for the entire ITS and partial ITS sequences. In the current study with the use of a 50% bootstrap cutoff, the naive Bayesian classifier showed the highest classification accuracy at the genus level, with 1 to 5% higher accuracy than BLASTN. This was especially true for the ITS1 region, with an improvement of up to 5% in classification accuracy. The naive Bayesian classifier provided multiple other advantages over BLASTN, including an alignment-independent algorithm, the availability of classification accuracy values as a measure of certainty, and computational speed. The higher speed of the naive Bayesian classifier than of GreenGenes, BLAST, MEGAN, and other classification tools is well documented (17, 25, 30, 43). The naive Bayesian classifier is therefore ideal for regions such as the ITS that cannot be aligned with confidence and are commonly use in environmental studies.
LSU versus ITS.
In addition to the generation of a high-quality ITS reference database, our analyses showed that a fragment length of more than 300 bp could influence the quality of sequence classification. The longer queries for the LSU region and the entire ITS outperformed the individual ITS1 and ITS2 sections at the genus level (Fig. 2), showing that sequence length needs to be taken into consideration when selecting genetic markers and sequencing platforms (25, 26). However, short sequences from 50 to 200 bp can still yield high-accuracy classification for both the ITS and LSU regions, especially if a bootstrap cutoff is used with the naive Bayesian classifier as described by Liu et al. (17) and in this study. At higher taxonomic levels, the performances of the LSU and ITS regions were very similar. At lower levels (genus), the individual ITS1 and ITS2 showed 2 to 3% higher accuracy using the naive Bayesian classifier with a 50% bootstrap cutoff than the LSU region. Sequence length affected classification accuracy with each rRNA region. With identical sequence lengths, accuracy using ITS1, ITS2, or the LSU region was within 0 to 1%, illustrating that the database and the use of a bootstrap cutoff have greater influence on classification accuracy than the choice of rRNA gene region. Even if smaller fragments have lower resolution for fungal phylogenetics (47), they still provide a very valuable resource for taxonomic identification if reliable database sequences are available.
ITS1 versus ITS2.
Using 50- to 200-bp sequence lengths, the performances of ITS1 and ITS2 were very similar (<1% difference) with either the ITS-LSU training set or the large ITS training set. Several studies have shown little difference between ITS1 and ITS2 when the regions are used to determine community composition, but in general ITS1 seems to show higher variability (20, 48). ITS2 might have better overall performance if we considered that with the ITS-LSU training set more sequences were retained after applying a 50% bootstrap cutoff and similar numbers were retained for the 150-bp and 200-bp fragments for the larger training database. In addition, IT2 could be more variable than ITS1 for some taxa (49) and has a secondary structure signal that could be used for fungal phylogenetics (50, 51). The availability of multiple universal primers in the 5.8S rRNA gene provides additional advantages for the ITS2 region. The longer fragments lengths now available with the different NGS platforms will also target part of the LSU region, and this region can be aligned for more detailed phylogenetic analysis of novel fungal taxa (12).
With new sequencing technologies, the largest portion of the diversity of fungi will soon be represented by sequence data, bringing new challenges to automated fungal classification (21, 52). To effectively classify sequences obtained from PCR primer-based environmental and metagenomic studies, new guidelines and methods to annotate and name operational taxonomic units (OTUs) that are represented only by sequenced data need to be defined (22). In addition, curation of available sequences for specimens deposited in collections will require organization of the community to improve collaborations among taxonomists, systematists, ecologists, and bioinformatics specialists (19, 52).
Supplementary Material
ACKNOWLEDGMENTS
We thank Rebecca Mueller, Cedar N. Hesse, and the reviewers for their valuable suggestions on the manuscript and Zachary Gossage, Katrina Sandona, and Terri Tobias for their assistance with the development of the training data set.
This study was supported by the U.S. Department of Energy, Office of Biological and Environmental Research, through a Science Focus Area grant (2009LANLF260) (C.R.K., A.P.-A., and K.-L.L.). Additional support was provided by the NSF (grant 0919510) (A.P.-A.), Western Illinois University, and the National Science Council in Taiwan (NSC97-2917-I-006-111) (K.-L.L.).
Footnotes
Published ahead of print 15 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02894-13.
REFERENCES
- 1.Blackwell M. 2011. The fungi: 1, 2, 3. 5.1 million species? Am. J. Bot. 98:426–438. 10.3732/ajb.1000298 [DOI] [PubMed] [Google Scholar]
- 2.O'Brien H, Parrent J, Jackson J, Moncalvo J-M, Vilgalys R. 2005. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microbiol. 71:5544–5550. 10.1128/AEM.71.9.5544-5550.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jumpponen A, Jones K. 2009. Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere. New Phytol. 184:438–448. 10.1111/j.1469-8137.2009.02990.x [DOI] [PubMed] [Google Scholar]
- 4.Blaalid R, Carlsen T, Kumar S, Halvorsen R, Ugland KI, Fontana G, Kauserud H. 2012. Changes in the root-associated fungal communities along a primary succession gradient analysed by 454 pyrosequencing. Mol. Ecol. 21:1897–1908. 10.1111/j.1365-294X.2011.05214.x [DOI] [PubMed] [Google Scholar]
- 5.Tedersoo L, Nilsson RH, Abarenkov K, Jairus T, Sadam A, Saar I, Bahram M, Bechem E, Chuyong G, Kõljalg U. 2010. 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytol. 188:291–301. 10.1111/j.1469-8137.2010.03373.x [DOI] [PubMed] [Google Scholar]
- 6.Weber CF, Vilgalys R, Kuske CR. 2013. Changes in fungal community composition in response to elevated atmospheric CO2 and nitrogen fertilization varies with soil horizon. Front. Microbiol. 4:78. 10.3389/fmicb.2013.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hibbett DS, Ohman A, Kirk PM. 2009. Fungal ecology catches fire. New Phytol. 184:279–282. 10.1111/j.1469-8137.2009.03042.x [DOI] [PubMed] [Google Scholar]
- 8.Aime MC, Brearley FQ. 2012. Tropical fungal diversity: closing the gap between species estimates and species discovery. Biodivers. Conserv. 21:2177–2180. 10.1007/s10531-012-0338-7 [DOI] [Google Scholar]
- 9.Kelly LJ, Hollingsworth PM, Coppins BJ, Ellis CJ, Harrold P, Tosh J, Yahr R. 2011. DNA barcoding of lichenized fungi demonstrates high identification success in a floristic context. New Phytol. 191:288–300. 10.1111/j.1469-8137.2011.03677.x [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Nilsson RH, Lopez-Giraldez F, Zhuang W, Dai Y, Johnston PR, Townsend JP. 2011. Tasting soil fungal diversity with earth tongues: phylogenetic test of SATé alignments for environmental ITS data. PLoS One 6:e19039. 10.1371/journal.pone.0019039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landeweert R, Leeflang P, Kuyper TW, Hoffland E, Rosling A, Wernars K, Smit E. 2003. Molecular identification of ectomycorrhizal mycelium in soil horizons. Appl. Environ. Microbiol. 69:327–333. 10.1128/AEM.69.1.327-333.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W. 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U. S. A. 109:6241–6246. 10.1073/pnas.1117018109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnold AE, Miadlikowska J, Higgins KL, Sarvate SD, Gugger P, Way A, Hofstetter V, Kauff F, Lutzoni F. 2009. A phylogenetic estimation of trophic transition networks for ascomycetous fungi: are lichens cradles of symbiotrophic fungal diversification? Syst. Biol. 58:283–297. 10.1093/sysbio/syp001 [DOI] [PubMed] [Google Scholar]
- 14.James TY, Letcher PM, Longcore JE, Mozley-Standridge SE, Porter D, Powell MJ, Griffith GW, Vilgalys R. 2006. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98:860–871. 10.3852/mycologia.98.6.860 [DOI] [PubMed] [Google Scholar]
- 15.James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, Celio G, Gueidan C, Fraker E, Miadlikowska J, Lumbsch HT, Rauhut A, Reeb V, Arnold AE, Amtoft A, Stajich JE, Hosaka K, Sung G-H, Johnson D, O'Rourke B, Crockett M, Binder M, Curtis JM, Slot JC, Wang Z, Wilson AW, Schüssler A, Longcore JE, O'Donnell K, Mozley-Standridge S, Porter D, Letcher PM, Powell MJ, Taylor JW, White MM, Griffith GW, Davies DR, Humber RA, Morton JB, Sugiyama J, Rossman AY, Rogers JD, Pfister DH, Hewitt D, Hansen K, Hambleton S, Shoemaker RA, Kohlmeyer J, Volkmann-Kohlmeyer B, Spotts RA, et al. 2006. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443:818–822. 10.1038/nature05110 [DOI] [PubMed] [Google Scholar]
- 16.Lekberg Y, Schnoor T, Kjøller R, Gibbons SM, Hansen LH, Al-Soud WA, Sørensen SJ, Rosendahl S. 2012. 454-sequencing reveals stochastic local reassembly and high disturbance tolerance within arbuscular mycorrhizal fungal communities. J. Ecol. 100:151–160. 10.1111/j.1365-2745.2011.01894.x [DOI] [Google Scholar]
- 17.Liu K-L, Porras-Alfaro A, Kuske CR, Eichorst SA, Xie G. 2012. Accurate, rapid taxonomic classification of fungal large-subunit rRNA genes. Appl. Environ. Microbiol. 78:1523–1533. 10.1128/AEM.06826-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vrålstad T. 2011. ITS, OTUs and beyond—fungal hyperdiversity calls for supplementary solutions. Mol. Ecol. 20:2873–2875. 10.1111/j.1365-294X.2011.05149.x [DOI] [PubMed] [Google Scholar]
- 19.Begerow D, Nilsson H, Unterseher M, Maier W. 2010. Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 87:99–108. 10.1007/s00253-010-2585-4 [DOI] [PubMed] [Google Scholar]
- 20.Blaalid R, Kumar S, Nilsson RH, Abarenkov K, Kirk PM, Kauserud H. 2013. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 13:218–224. 10.1111/1755-0998.12065 [DOI] [PubMed] [Google Scholar]
- 21.Bates ST, Ahrendt S, Bik HM, Bruns TD, Caporaso JG, Cole J, Dwan M, Fierer N, Gu D, Houston S, Knight R, Leff J, Lewis C, Maestre JP, McDonald D, Nilsson RH, Porras-Alfaro A, Robert V, Schoch C, Scott J, Taylor DL, Parfrey LW, Stajich JE. 2013. Meeting report: Fungal ITS Workshop (October 2012). Stand. Genomic Sci. 8:118–123. 10.4056/sigs.3737409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüßler A, Scott JA, Senés C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson K-H. 2013. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22:5271–5277. 10.1111/mec.12481 [DOI] [PubMed] [Google Scholar]
- 23.Gazis R, Rehner S, Chaverri P. 2011. Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Mol. Ecol. 20:3001–3013. 10.1111/j.1365-294X.2011.05110.x [DOI] [PubMed] [Google Scholar]
- 24.Lindner DL, Gargas A, Lorch JM, Banik MT, Glaeser J, Kunz TH, Blehert DS. 2010. DNA-based detection of the fungal pathogen Geomyces destructans in soils from bat hibernacula. Mycologia 103:241–246. 10.3852/10-262 [DOI] [PubMed] [Google Scholar]
- 25.Porter TM, Golding GB. 2011. Are similarity- or phylogeny-based methods more appropriate for classifying internal transcribed spacer (ITS) metagenomic amplicons? New Phytol. 192:775–782. 10.1111/j.1469-8137.2011.03838.x [DOI] [PubMed] [Google Scholar]
- 26.Porter TM, Golding GB. 2012. Factors that affect large subunit ribosomal DNA amplicon sequencing studies of fungal communities: classification method, primer choice, and error. PLoS One 7:e35749. 10.1371/journal.pone.0035749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kõljalg U, Larsson K-H, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Vrålstad T, Ursing BM. 2005. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166:1063–1068. 10.1111/j.1469-8137.2005.01376.x [DOI] [PubMed] [Google Scholar]
- 28.Taylor DL, Houston S. 2011. Fungal genomics, p 141–155 In Xu J-R, Bluhm BH. (ed), Fungal genomics: methods and protocols. Humana Press, Totowa, NJ [Google Scholar]
- 29.Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386. 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caporaso JG. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:334. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hibbett DS, Nilsson RH, Snyder M, Fonseca M, Costanzo J, Shonfeld M. 2005. Automated phylogenetic taxonomy: an example in the homobasidiomycetes (mushroom-forming fungi). Syst. Biol. 54:660–668. 10.1080/10635150590947104 [DOI] [PubMed] [Google Scholar]
- 33.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599. 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- 35.Bell G, DeGennard L, Gelfand D, Bishop R, Valenzuela P, Rutter W. 1977. RNA genes of Saccharomyces cerevisiae. J. Biol. Chem. 252:8118–8125 [PubMed] [Google Scholar]
- 36.Baldi P, Soren B. 2001. Bioinformatics: the machine learning approach, 2nd ed. MIT Press, Cambridge, MA [Google Scholar]
- 37.Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson K-H, Kõljalg U. 2006. Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS One 1:e59. 10.1371/journal.pone.0000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eichorst SA, Kuske CR. 2012. Identification of cellulose-responsive bacterial and fungal communities in geographically and edaphically different soils by using stable isotope probing. Appl. Environ. Microbiol. 78:2316–2327. 10.1128/AEM.07313-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porras-Alfaro A, Herrera J, Sinsabaugh RL, Odenbach KJ, Lowrey T, Natvig DO. 2008. Novel root fungal consortium associated with a dominant desert grass. Appl. Environ. Microbiol. 74:2805–2813. 10.1128/AEM.02769-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porras-Alfaro A, Herrera J, Natvig DO, Lipinski K, Sinsabaugh RL. 2011. Diversity and distribution of soil fungal communities in a semiarid grassland. Mycologia 103:10–21. 10.3852/09-297 [DOI] [PubMed] [Google Scholar]
- 41.Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, Angenent LT, Knight R, Ley RE. 2012. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 6:94–103. 10.1038/ismej.2011.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu CH, Lee SM, Vanlare JM, Kasper DL, Mazmanian SK. 2008. Regulation of surface architecture by symbiotic bacteria mediates host colonization. Proc. Natl. Acad. Sci. U. S. A. 105:3951–3956. 10.1073/pnas.0709266105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu D, Hartman A, Ward N, Eisen JA. 2008. An automated phylogenetic tree-based small subunit rRNA taxonomy and alignment pipeline (STAP). PLoS One 3:e2566. 10.1371/journal.pone.0002566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, Polz MF. 2004. Fine-scale phylogenetic architecture of a complex bacterial community. Nature 430:551–554. 10.1038/nature02649 [DOI] [PubMed] [Google Scholar]
- 45.Sundquist A, Bigdeli S, Jalili R, Druzin ML, Waller S, Pullen KM, El-Sayed YY, Taslimi MM, Batzoglou S, Ronaghi M. 2007. Bacterial flora-typing with targeted, chip-based pyrosequencing. BMC Microbiol. 7:108. 10.1186/1471-2180-7-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munch K, Boomsma W, Huelsenbeck JP, Willerslev E, Nielsen R. 2008. Statistical assignment of DNA sequences using Bayesian phylogenetics. Syst. Biol. 57:750–757. 10.1080/10635150802422316 [DOI] [PubMed] [Google Scholar]
- 47.Min XJ, Hickey DA. 2007. Assessing the effect of varying sequence length on DNA barcoding of fungi. Mol. Ecol. Notes 7:365–373. 10.1111/j.1471-8286.2007.01698.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bazzicalup OAL, Bálint M, Schmitt I. 2013. Comparison of ITS1 and ITS2 rDNA in 454 sequencing of hyperdiverse fungal communities. Fungal Ecol. 6:102–109. 10.1016/j.funeco.2012.09.003 [DOI] [Google Scholar]
- 49.Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H. 2008. Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. Online 4:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coleman AW. 2007. Pan-eukaryote ITS2 homologies revealed by RNA secondary structure. Nucleic Acids Res. 35:3322–3329. 10.1093/nar/gkm233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krüger D, Gargas A. 2008. Secondary structure of ITS2 rRNA provides taxonomic characters for systematic studies—a case in Lycoperdaceae (Basidiomycota). Mycol. Res. 112:316–330. 10.1016/j.mycres.2007.10.019 [DOI] [PubMed] [Google Scholar]
- 52.Hibbett DS, Ohman A, Glotzer D, Nuhn M, Kirk P, Nilsson RH. 2011. Progress in molecular and morphological taxon discovery in Fungi and options for formal classification of environmental sequences. Fungal Biol. Rev. 25:38–47. 10.1016/j.fbr.2011.01.001 [DOI] [Google Scholar]
- 53.Lothamer K, Brown SP, Mattox JD, Jumpponen A. 2013. Comparison of root-associated communities of native and non-native ectomycorrhizal hosts in an urban landscape. Mycorrhiza. 10.1007/s00572-013-0539-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.