

Abstract
Flavobacterium hibernum, isolated from larval habitats of the eastern tree hole mosquito, A. triseriatus, remained suspended in the larval feeding zone much longer (8 days) than other bacteria. Autofluorescent protein markers were developed for the labeling of F. hibernum with a strong flavobacterial expression system. Green fluorescent protein (GFP)-tagged F. hibernum cells were quickly consumed by larval mosquitoes at an ingestion rate of 9.5 × 104/larva/h. The ingested F. hibernum cells were observed mostly in the foregut and midgut and rarely in the hindgut, suggesting that cells were digested and did not pass the gut viably. The NanoLuc luciferase reporter system was validated for quantitative larval ingestion rate and bacterial fate analyses. Larvae digested 1.87 × 105 cells/larva/h, and few F. hibernum cells were excreted intact. Expression of the GFP::Cry11A fusion protein with the P20 chaperone protein from Bacillus thuringiensis H-14 was successfully achieved in F. hibernum. Whole-cell bioassays of recombinant F. hibernum exhibited high larvicidal activity against A. triseriatus in microplates and in microcosms simulating tree holes. F. hibernum cells persisted in microcosms at 100, 59, 30, and 10% of the initial densities at days 1, 2, 3, and 6, respectively, when larvae were absent, while larvae consumed nearly all of the F. hibernum cells within 3 days of their addition to microcosms.
INTRODUCTION
Quantitative studies of the ingestion and digestion of microorganisms by insects can inform the significance of microorganisms in nutrition and symbioses (1). The interaction between bacteria and their associated hosts or predators has been investigated by culture-dependent (viable plating) and/or culture-independent methods (such as rRNA gene sequencing, quantitative PCR, or terminal restriction fragment length polymorphism) (2–4). Our previous studies using some of the procedures described above showed that tree hole larval habitats of Aedes triseriatus mosquitoes contain diverse microbial components and that flavobacteria are a prominent bacterial group. Bacterial community structure was greatly shaped by mosquito larval feeding, and flavobacteria were much more abundant in the water column of natural tree holes after A. triseriatus larvae were removed (5). This finding indicates that the bacteria were a major food source for mosquito larvae in these habitats (4). Previous observations also indicate that flavobacteria are associated with detrital surfaces and therefore may transform the allochthonous organic matter that drives the trophic relationships within the tree hole ecosystem (4, 5). Recent studies using pyrosequencing techniques showed that the flavobacteria Elizabethkingia and Chryseobacterium were associated with mosquitoes at various developmental stages as their symbionts (2, 6). Extracts from Elizabethkingia revealed antibacterial, antifungal, and also antiparasitic activities, indicating that some flavobacteria may have a significant impact on mosquito health (2). Thus, further study of interactions between flavobacteria and mosquitoes and/or other microorganisms will help us understand how these important bacteria contribute to mosquito production, nutrition, and development (7). However, the above-described approaches have limited application in systems biology because they lack the ability to directly observe bacteria in situ.
The development of sensitive reporters for flavobacteria can facilitate more sophisticated quantitative study of the interactions between such bacteria and their associated predators or hosts. Autofluorescent protein (AFP)-labeled bacteria such as Escherichia coli (9), Mycobacterium ulcerans (10), Pseudomonas aeruginosa (11), and many others have been used to study their interaction with mosquitoes. For example, Geavgaard et al. engineered P. aeruginosa with a green fluorescent protein (GFP) reporter to study the feeding behavior of larval mosquitoes (Culex quinquefasciatus) (11). Both planktonic and biofilm-forming P. aeruginosa bacteria were acquired by larval C. quinquefasciatus and were concentrated in the midgut after 24 h of feeding. The existence of fluorescent P. aeruginosa in larval mosquitoes was confirmed by PCR assays, indicating that it was appropriate to use the GFP reporter-tagged bacterial strain to study the interaction with larval mosquitoes (11). However, quantitative studies of the feeding behavior of larval mosquitoes (uptake or digestion rate and biocontrol potential) were not reported. They pointed out that utilization of GFP-labeled strains was more limited in the analysis of pupae and adults because of a high level of background autofluorescence in them (11). Recently, various bioluminescent enzymes have been used to develop more sensitive reporters (12). NanoLuc, derived from the luminescent sea shrimp Oplophorus gracilirostris, is a novel, ATP-independent, and brighter luciferase (13). It is regarded as the “next generation reporter” because it is one of the most sensitive and versatile reporters and has been successfully used to analyze metabolic pathways in mammalian cells (13). However, its utilization in bacterial cells has not been studied.
Tree hole- or container-breeding mosquitoes such as A. triseriatus obtain nutrients primarily by consuming available microorganisms on the surface of detritus or suspended in the water column (14). Studies in our lab have shown that about half of their feeding time is spent at or near the surface of the water column (14). Exploring the potential to utilize flavobacteria as expression vehicles for larvicidal toxin genes is a logical extension of our studies of larval feeding ecology in these habitats (15, 16). Novel recombinant bioinsecticides have been developed by expressing larvicidal toxin genes from Bacillus thuringiensis subsp. israelensis and/or B. sphaericus in Gram-negative bacteria, including Enterobacter amnigenus (17), Caulobacter crescentus (18), Ancylobacter aquaticus (19), Asticcacaulis excentricus (20), and Pseudomonas putida (21). The above-described strategy partially ameliorated the problems associated with applications of formulations of B. thuringiensis subsp. israelensis and B. sphaericus that sediment out of the larval feeding zone rapidly and have shortened periods of efficacy because of UV inactivation (22). Nevertheless, most of the hosts for attempted larvicidal gene expression are laboratory strains or their derivatives. Unfortunately, it was difficult to obtain measurable larvicidal activity when these genetically engineered microorganisms (GEMs) were tested in microcosms (Y. Xu, data not shown). There are few reports on tracking of the fate of GEMs in mosquitoes or microcosms simulating natural mosquito habitats. It is possible that the lab strains studied were originally isolated from soil and persisted poorly in the aquatic microcosms into which they were introduced. The environmental flavobacteria isolated from larval mosquitoes' feeding zone may overcome this deficiency. However, the genetic manipulation of environmental flavobacteria is extremely difficult; the available molecular tools, such as selectable markers, replicable plasmids, and expression systems from proteobacteria, do not function in flavobacteria (23). Flavobacteria have unique transcriptional and translational mechanisms that contrast markedly with those in proteobacteria (24). Recently, flavobacterial expression based on a strong promoter of outer membrane protein A (PompA) was established and has been proved to be functional in several Flavobacterium species (8, 15), Cytophaga hutchinsonii (25), and Capnocytophaga canimorsus (26), providing the opportunity to conduct comprehensive genetic studies and overexpress functional products in flavobacteria. However, the use of this expression system in environmental flavobacteria has not been reported.
Given the above-described background, the objectives of the present study were to (i) develop molecular tools for the genetic study of Flavobacterium hibernum, a fast-growing bacterium isolated from native mosquito habitats; (ii) investigate if A. triseriatus larvae ingest and digest F. hibernum efficiently; and (iii) test whether or not F. hibernum expresses larvicidal toxins sufficiently to kill mosquito larvae.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
E. coli JM109 was used for cloning. E. coli S17 (λ pir) was used for conjugation. E. coli strains were routinely grown in Luria-Bertani (LB) broth. Casitone-yeast extract (CYE) medium was routinely used for Flavobacterium culture. Liquid cultures were grown with shaking (ca. 200 rpm) at either 25°C (Flavobacterium) or 37°C (E. coli). For solid LB medium, Bacto agar (Difco, Detroit, MI) was added to a final concentration of 20 g/liter. Ampicillin (100 μg/ml) was added for plasmid selection in E. coli, and erythromycin (Em; 100 μg/ml) was added for plasmid selection in Flavobacterium.
Recombinant DNA methods.
The translational fusion of GFP and Cry11A was constructed as follows. The cry11A+p20 operon was amplified with primers Walker25 (TCCCCCGGGATGGAAGATAGTTCTTTAGATAC) and Walker26 (GCATGCTTAAGTTAAATAAGTCATTGTTAC) with pMMB731 (Table 1) as the template according to standard procedures (28). The PCR product (cry11A+p20) was first inserted into a T Easy vector (pSCH539) (Table 1). Expression of the p20 gene was included at a position 283 bp downstream of cry11A because it is a molecular chaperone of Cry11A (21). The insert was released from pSCH539 by SmaI and SphI digestion and inserted into the same sites of plasmid pSCH529, which harbored the gfp gene (15), creating the translation fusion cry11A operon construct (gfp::cry11A+p20) on T Easy (pSCH551). Plasmid pSCH551 was digested with restriction enzymes BamHI and SphI and transferred into the same sites of Fj29 (15), replacing gfp, leading to toxin expression plasmid pSCH550 (Table 1). Plasmid pSCH550 was introduced into F. hibernum by conjugation, and transformants with Em resistance were screened under fluorescence microscopy by the protocol described in reference 23. Colonies emitting bright green fluorescence were subcultured and selected by fluorometry and Western blot analysis (see below). SCH616 was chosen for further studies because it showed the highest GFP::Cry11A expression based on GFP fluorescence determination. Plasmid pSCH144 without the expression cassette was introduced into F. hibernum as a control (strain SCH734) (Table 1). Plasmids pSCH342 (yellow fluorescent protein [YFP]), pFj29 (GFP), pSCH443 (mStrawberry fluorescent protein [mStraw]), and pSCH445 (mOrange fluorescent protein [mOrange]) (15) were introduced into F. hibernum separately in this study, creating reporter strains SCH391 (YFP), SCH376 (GFP), SCH449 (mStraw), and SCH447 (mOrange) (Table 1).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) and/or plasmid constructiona | Reference or source |
|---|---|---|
| E. coli | ||
| S17-1 | hsdR17 (rK− mK−) recA RP4-2 (Tc::Mu-Km::Tn7 Strr) | 27 |
| JM109 | F′ [traD36 proAB+ lacIq lacZΔM15]/recA1 supE44 endA1 hsdR17 gyrA96 relA1 thi-1 mcrA Δ(lac-proAB) | Promega |
| F. hibernum | ||
| WT | 23 | |
| SCH376 | Strain carrying plasmid pFj29 (PompA+gfp) | This study |
| SCH391 | Strain carrying plasmid pSCH342 (PompA+yfp) | This study |
| SCH447 | Strain carrying plasmid pSCH445 (PompA+mOrange) | This study |
| SCH449 | Strain carrying plasmid pSCH443 (PompA+mStrawberry) | This study |
| SCH616 | Strain carrying expression cassette PompA+gfp::cry11A+p20 | This study |
| SCH733 | Strain carrying plasmid pSCH722 (PompA+nluc) | This study |
| SCH734 | Strain carrying plasmid pSCH144 | This study |
| Plasmids | ||
| pGEM-T Easy | Cloning vector; Ampr | Promega |
| pMMB731 | cry11A+p20 expression vector for Pseudomonas, Ampr | 21 |
| pFj29 | PompA+gfp on pCP29; Emr (Ampr) | 15 |
| pNL1.1 | nluc gene template; Ampr | Promega |
| pSCH144 | Plasmid derived from pCP29 | 15 |
| pSCH342 | PompA+yfp on pCP29; Emr (Ampr) | 15 |
| pSCH443 | PompA+mStrawberry on pCP29; Emr (Ampr) | This study |
| pSCH445 | PompA+mOrange on pCP29; Emr (Ampr) | This study |
| pSCH529 | GFP::P50 on T Easy vector; Ampr | 15 |
| pSCH539 | cry11A+p20 operon on T Easy vector, Ampr | This study |
| pSCH550 | PompA+gfp::cry11A+p20 on pFj29; Emr (Ampr) | This study |
| pSCH551 | gfp::cry11A+p20 on T Easy vector, Ampr | This study |
| pSCH703 | nluc gene fragment on T Easy vector; Ampr | This study |
| pSCH722 | PompA+nluc on pCP29; Emr (Ampr) | This study |
Antibiotic resistance phenotype: Ampr, ampicillin resistance; Emr, erythromycin resistance. Unless indicated otherwise, antibiotic resistance phenotypes are those expressed in E. coli. Antibiotic resistance phenotypes in parentheses are those expressed in Flavobacterium strains but not in E. coli.
The nluc gene encoding the NanoLuc reporter was amplified by using plasmid pNL1.1 (Table 1) with forward primer Walker118 (GGATCCAATATTCTATAATAAAAAACAGTATGGTCTTCACACTCGAAGATTTCG) and reverse primer Walker100 (GCATGCTTACGCCAGAATGCGTTCGCAC) and cloned into the T Easy vector (pSCH703). nluc was next released from pSCH703 with BamHI and SphI and inserted into the same sites of pFj29, leading to NanoLuc expression plasmid pSCH722. Plasmid pSCH722 was introduced into F. hibernum by conjugation, leading to NanoLuc reporter strain SCH733 (Table 1).
Western blotting.
Cells were collected by centrifugation, washed with 0.1 M phosphate-buffered saline (PBS, pH 7.4), and diluted in Laemmli sample buffer (Bio-Rad) supplemented with 2-mercaptoethanol (5%). Proteins were separated by 4 to 20% SDS-PAGE (Bio-Rad), transferred onto polyvinylidene difluoride membrane (EMD Millipore), and reacted with anti-Cry11A antiserum generated as previously described (21). Peroxidase-conjugated goat anti-rabbit immunoglobulin G was used as the secondary antibody, and the reaction was developed with SuperSignal (Pierce, Rockford, IL).
Epifluorescence microscopy.
Cells tagged with AFPs were visualized with an Olympus Provis AX70 microscope, equipped with appropriate filters, a mercury lamp, and a DP-50 digital camera linked to an external PC. Cells were washed with PBS twice and then resuspended in PBS. Second-instar mosquito larvae were fed with various AFP-tagged cells at room temperature for 2 h, killed with 100% ethanol, transferred onto a thin layer of 0.1% agarose on a microscope slide, and observed with the appropriate filter (15).
Determination of AFP and NanoLuc reporter activity.
F. hibernum cells harboring constructs with confirmed sequences were cultured in CYE medium. Quantitative analysis of AFP or NanoLuc production was performed with a SpectraMax M5 spectrophotometer (Molecular Devices, Sunnyvale, CA). Aliquots (200 μl) of cultures were centrifuged, washed with 0.1 M phosphate-buffered saline (PBS, pH 7.4), diluted in the same buffer to an optical density at 600 nm (OD600) of 0.4, and subjected to fluorescence determination in a 96-well microtiter plate (black and clear bottom; Costar, Corning, NY). GFP fluorescence was determined at an excitation wavelength of 490 nm, an emission wavelength of 530 nm, and a cutoff of 515 nm. Strain SCH734 without any AFP genes was used as the blank for the calculation of relative fluorescence units (15).
For determination of NanoLuc reporter activity, the cells were sampled, diluted, and immediately added to an equal volume of NanoLuc assay buffer (Promega, Madison, WI), and light intensity was quantified in 96-well microtiter plates with a plate reader according to the manufacturer's protocol. If necessary, flavobacterial cells were first lysed with Passive Lysis Buffer (Promega) and lysozyme and then the lysate were mixed with an equal volume of NanoLuc assay buffer as described above.
Suspension test.
Overnight bacterial cultures of F. hibernum W22, F. johnsoniae, B. thuringiensis, E. coli, and P. putida KT2440 were centrifuged, washed, and adjusted to an OD600 of 1.0 with water. A 5-ml sample of each cell suspension was placed in a sterile glass tube (12 by 75 mm). The tube was kept at room temperature and photographed every 2 days.
Ingestion study.
A. triseriatus larvae (third instar; Michigan State University lab strain) were starved for ca. 4 h in Milli-Q (purified distilled) water prior to mixing with F. hibernum. The bacterial cells were grown overnight, collected by centrifugation, washed twice with PBS, shaken in PBS for 1 h to remove excessive nutrients from the cell surface, and fed to larvae for specified times (see Results). For the preliminary ingestion study with the GFP reporter strain (SCH376), it was diluted to a concentration of 1 × 106 cells/ml and used as the feeding solution. Five alive or dead larvae were transferred to 2 ml of the feeding solution (n = 5). Cells in the feeding solution were sampled and counted immediately with a Petroff-Hausser counting chamber by epifluorescence microscopy.
For the ingestion study with a NanoLuc reporter strain (SCH733), third-instar larvae (6 days past hatching), fourth-instar larvae (9 days past hatching), and pupae (12 days past hatching) were used. First- and second-instar larvae were not used because it was difficult to estimate their ingestion rate because of their small size. SCH733 was extensively washed in PBS and adjusted to ca. 7.3 × 105 cells/ml as the feeding solution. Six larvae were transferred into 3 ml of feeding solution (n = 3). Cells remaining in the feeding solution were sampled at the times specified, and NanoLuc reporter activity was assayed according to standard assay procedures.
Digestion study.
Third-instar A. triseriatus larvae were starved for 2 h in sterile water before incubation with reporter strain SCH733 for 4 h (ca. 2.8 × 109 cells/ml). Mosquito larvae were extensively rinsed and transferred to a six-well plate (2 ml of sterile H2O in each well; n = 3). Bacteria inside the larvae and the incubation solution were sampled at each time point (0, 0.5, 1, 1.5, 2, 2.5, 3, or 4 h). To sample the internal bacteria, the three larvae were pooled, homogenized with a sterile pestle, resuspended in H2O, and subjected to NanoLuc reporter analysis immediately.
Microcosm study.
Artificial larval mosquito habitats (microcosms) were constructed in 50-ml sterile plastic tubes (Corning) containing 50 mg of senescent American beech leaf detritus and distilled water. One milliliter of a tree hole water inoculum (to allow the formation of microcosms that simulate tree holes) was added to the microcosms. Microcosms were incubated at room temperature for 2 days to condition the leaf detritus. The final volume of microcosms was adjusted to ∼25 ml. After incubation for another 24 h, six second-instar larvae were transferred into the microcosms (n = 3). Experimental control 1 was the same as that described above but without the addition of mosquito larvae. Experimental control 2 was similar to control 1, but the leaf detritus and water were sterilized by autoclaving. SCH733 was added to the above-described microcosms, and its final concentration was adjusted to be 4.7 × 105 cells/ml. Cells in microcosms were gently mixed before sampling, washed with PBS, and resuspended in the same volume of PBS. Fifty microliters was subjected to the standard luciferase activity assay. Mosquito larvae were periodically inspected and replaced as necessary (with larvae from extra replicates under the same conditions) to maintain a constant population.
Larvicidal bioassays.
Overnight cultures of F. hibernum cells were harvested by centrifugation and washed with sterile water three times. A series of dilutions was made in sterile distilled water for the range of cell concentrations to be tested. For the bioassay in water, four early-third-instar larvae of A. triseriatus were added to six-well plates. Each well held a 3-ml cell suspension (concentrations ranged from 1.8 × 105 to 1.8 × 109 cells/ml). Tests were performed with six replicates of each dilution. The surviving larvae were counted after 24 h of incubation at room temperature. For bioassays of larvicidal activity in microcosms, six early-third-instar larvae of A. triseriatus (reared in microcosms) were added to microcosms prepared as described above (concentrations ranged from 1.6 × 106 to 6.5 × 109 cells/ml). Tests were performed with three replicates of each dilution. The surviving larvae were counted after 48 h of incubation at room temperature. The 50% lethal concentrations (LC50s) and their 95% confidence limits were calculated with PROC PROBIT (SAS 9.3) (29).
RESULTS
Suspension characteristics of selected Gram-positive and Gram-negative bacteria.
The ability to stay in suspension varied dramatically between types of bacteria. (Fig. 1). B. thuringiensis sedimented quickly after 6 h, and the broth completely cleared in 2 days (Fig. 1). Lab strains of E. coli, P. putida, and F. johnsoniae also began to sediment after 2 days, and nearly half of them sedimented in 6 days, while F. hibernum cells generally remained suspended longer (Fig. 1). Therefore, the suspension potentials of the selected strains were qualitatively ranked as follows: F. hibernum > F. johnsoniae > E. coli > P. putida > B. thuringiensis.
FIG 1.
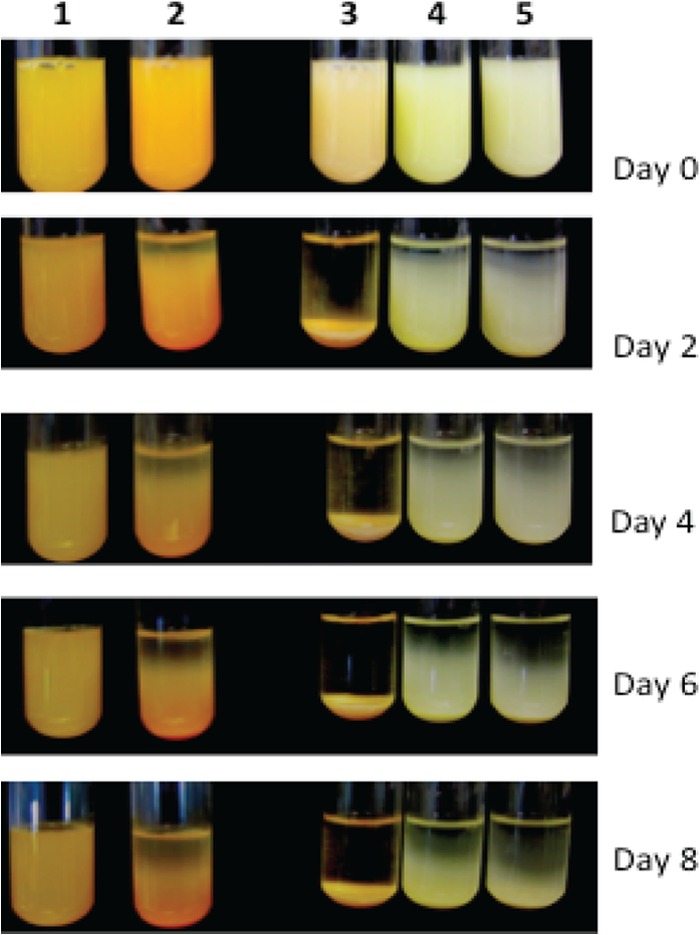
Demonstration of suspensions of selected bacteria. The representative cultures used were F. hibernum (column 1), F. johnsoniae (column 2), B. thuringiensis (column 3), E. coli (column 4), and P. putida (column 5). These bacteria were larvicidal toxin producers or were used as hosts for toxin expression. Five-milliliter volumes of cells grown overnight were adjusted to an OD600 of 1.0, aliquoted into test tubes (12 by 75 mm), and incubated while stationary at room temperature. Pictures were taken every 2 days.
Flavobacterial ingestibility by mosquito larvae.
Four different constructs carrying the genes for the AFPs GFP, YFP, mStrawberry, and mOrange were individually introduced into F. hibernum cells. Transformants carrying reporter constructs emitted bright fluorescence under epifluorescence microscopy. As shown in Fig. 2, AFP fluorescence was readily detected in the larval gut, indicating that mosquito larvae efficiently ingested AFP-tagged bacteria. Fluorescence was not observed in larvae fed control cells (SCH743) (Fig. 2). Fluorescing bacteria were concentrated mostly in the foregut and midgut. Very few fluorescent bacteria were observed in the hindgut (Fig. 2). Rates of bacterial ingestion by larvae were initially studied with GFP-labeled F. hibernum cells (Fig. 3). Approximately 50% of the initial F. hibernum cells remained in the incubation solution after 2 h (Fig. 3). The total number of fluorescent bacteria in the control samples containing dead larvae did not decrease. The disappearance of flavobacterial cells from the feeding solution was due mostly to ingestion by mosquito larvae because the larval gut was dominated by bacterial cells emitting green fluorescence while only a negligible amount of fluorescent cells could be detected on the larval surface (Fig. 2). The rate of ingestion of bacteria by mosquito larvae was estimated to be 9.5 × 104 cells/larva/h.
FIG 2.
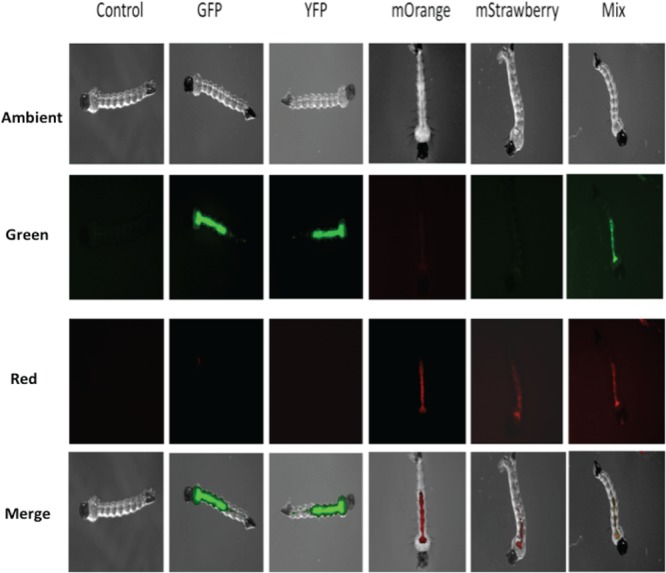
Locations of ingested F. hibernum cells in mosquito larvae. The genes for the reporters GFP, YFP, mOrange, and mStrawberry were expressed in F. hibernum W22 with the flavobacterial expression system. Mosquito larvae were fed cultures carrying AFP reporters for ∼2 h, processed as described in Materials and Methods, and observed by epifluorescence microscopy. The control was SCH743 without AFP. The mixed reporter strains used were those tagged with GFP (SCH376) or mOrange (SCH447).
FIG 3.
Ingestion of F. hibernum cells tagged with a GFP reporter by mosquito larvae. Cells tagged with GFP were incubated with third-instar mosquito larvae for 2 h; the control was dead mosquito larvae. The cells were counted by epifluorescence microscopy with a Petroff-Hausser counting chamber. Values are means ± standard deviations; n = 5.
As a first step in using the NanoLuc reporter gene (nluc) in F. hibernum, we determined the relationship between the bacterial cell number and luminescence for this construct. As shown in Fig. S1 in the supplemental material, there was a strong linear relationship between bioluminescence values and bacterial concentrations over a range of 7 × 102 to 7 × 107 cells/ml. Thus, the NanoLuc marker had sufficient sensitivity to allow the detection of as few as 700 cells/ml (see Fig. S1). In the feeding studies, cell numbers determined by NanoLuc in controls with no larvae did not vary much within the test period (3 h), showing that NanoLuc in flavobacteria is stable as a reporter (Fig. 4). Also, pupae did not acquire bacterial cells, as expected. In contrast, third- or fourth-instar larval mosquitoes ingested SCH733 quickly (Fig. 4). “Fast” (within the first hour) and “slow” (between the first and third hours) ingestion phases were identified within the test period. The ingestion rates of fourth- and third-instar larvae were 1.6 × 105 and 1.0 × 105 cells/larva/h, respectively, in the fast ingestion stage. The ingestion rates of fourth- and third-instar larvae were estimated to be 3.4 × 104 and 5.8 × 104 cells/larva/h, respectively, in the slow ingestion phase.
FIG 4.
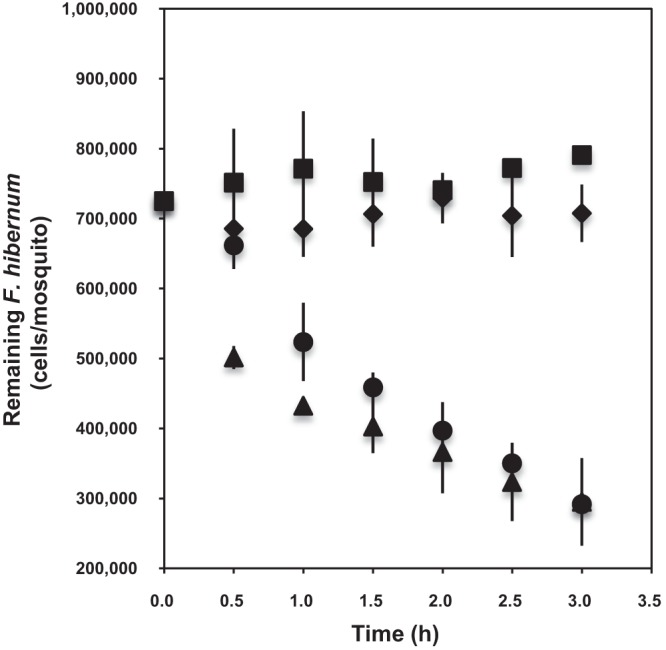
Studies of ingestion of F. hibernum with the NanoLuc reporter by mosquito larvae. Third-instar mosquito larvae (▲, 6 days old), fourth-instar larvae (●, 9 days old), and pupae (◆, 12 days old) were used for digestion analysis; the control had no larvae added (◾). Larvae were starved for 2 h in water before incubation with reporter strain SCH733 (PompA+nluc). Cells were adjusted to a concentration of ca. 7.3 × 105/ml. Bacterial cells were sampled and subjected to the standard NanoLuc reporter assay as described in Materials and Methods. Values are means ± standard deviations; n = 3.
Ingestion of F. hibernum cells by larvae in artificial microcosms was further investigated for 6 days (Fig. 5). Up to 30% of the F. hibernum bacteria were found to be ingested by larval mosquitoes within the initial 24 h, while there was no change in the control samples absent of larvae with or without tree hole water (tree hole water was used to introduce more “native” microbes or protozoans). In the presence of larvae, the F. hibernum level further decreased to only 10% of the initial level after 48 h. When larvae were absent from the controls with a tree hole water inoculum under the same conditions, F. hibernum cells retained up to 60% of the initial level after 48 h. The F. hibernum cell concentration was close to the lower detection limit in the presence of larval mosquitoes after 72 h. However, up to 31 and 10% of the cells remained in the control samples (tree hole water, no larvae) under the same conditions after 72 and 144 h. There was no strong evidence that other microorganisms or protozoans significantly affected F. hibernum cell concentrations under our testing conditions because the numbers of F. hibernum cells in the two controls (with and without the addition of tree hole water) were similar.
FIG 5.
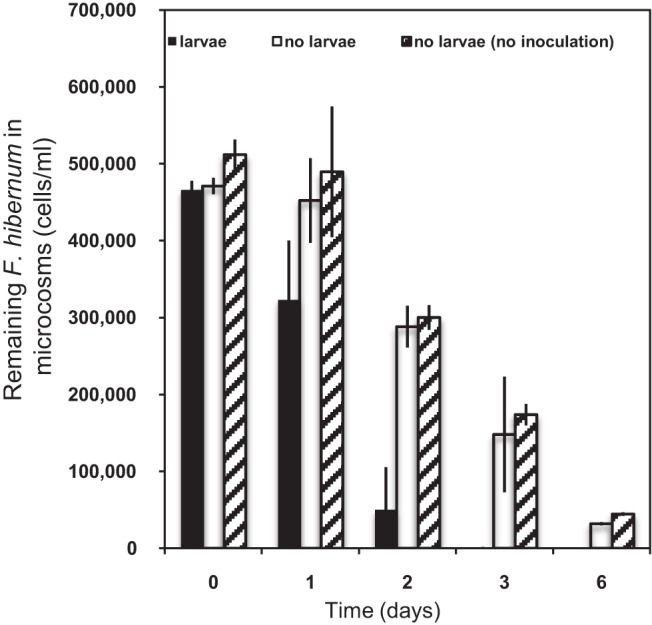
Uptake of F. hibernum by larval mosquitoes in microcosms. Microcosms were prepared as described in Materials and Methods. Second-instar mosquito larvae were added to a microcosms (20 ml) consisting of an inoculum of tree hole water and beech leaf detritus (black bars). The controls are a tree hole water inoculum with no larvae (blank bars) and an inoculum with no larvae or tree hole water (line-filled bars). SCH733 (PompA+nluc) cells were diluted into the above-described vials at a concentration of ca. 4.7 × 105/ml. Bacterial cells were sampled and subjected to the standard NanoLuc reporter assay as described in Materials and Methods. Values are means ± standard deviations; n = 6.
Flavobacterial digestibility by larval mosquitoes.
The digestibility of SCH733 by mosquito larvae was tested at specific time points (Fig. 6). A single larval gut contained up to 4.3 × 105 cells after feeding for 4 h (time zero, initial time for digestion). Forty-nine percent of the initial F. hibernum cells were detected in the larvae when the fed larvae were transferred to fresh buffer for 1 h. We detected 22.2% of the F. hibernum cells in the gut if no other source of food was provided for 2 h (Fig. 6). Only negligible levels of SCH733 cells were found to be released into the incubation solution compared to those in the larvae, indicating that F. hibernum cells were digested quickly (Fig. 6). The rate of bacterial cell digestion was roughly estimated to be 1.87 × 105/larva/h within the first 2 h. However, it decreased to 0.58 × 105/larva/h between 1 and 3 h. SCH733 cell numbers in larval mosquitoes did not change between 3 and 4 h.
FIG 6.
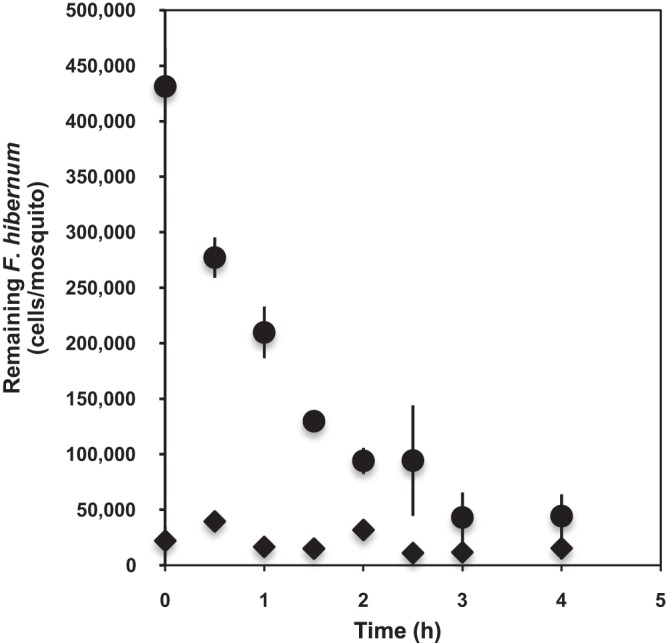
Analysis of the digestibility of F. hibernum with a NanoLuc reporter by mosquito larvae. Third-instar larvae (4 days old) were starved for 2 h in sterile water before incubation with reporter strain SCH733 (ca. 2.8 × 109 cells/ml). The fed larvae were next rinsed and transferred into fresh sterile H2O for a digestion study. Larvae and the incubation solution were sampled at 0, 0.5, 1, 1.5, 2, 2.5, 3, and 4 h and subjected to the standard NanoLuc reporter assay as described in Materials and Methods. ◆, SCH733 cells in incubation solution; ●, SCH733 cells that remained in mosquito larvae.
Expression of larvicidal cry11A+p20 genes in Flavobacterium.
We attempted to express the cry11A+p20 operon in F. johnsoniae but could not obtain stable transformants (15). In this study, we reengineered the expression construct by inserting a gfp gene into the N terminus of cry11A (gfp::cry11A). This method was successfully used to tag binary toxin component P51 or P42 (15). It is a high-throughput approach to obtain transformants with good expression based on the GFP fluorescence if the fused component does not affect GFP folding in vivo (15). The introduction of pSCH558 (gfp::cry11A+p20) into F. hibernum resulted in some Em-resistant colonies. Fifteen fluorescent colonies were first selected from ∼200 recombinants under UV, and their relative GFP fluorescence emission was quantified by fluorescence spectrometry (Fig. 7A). GFP::Cry11A production in the selected colonies was confirmed by Western blotting with Cry11A antiserum (Fig. 7B). A predominant band with a molecular mass of 98 kDa was close to the theoretical value of fused GFP::Cry11A, indicating that the expression of fused GFP::Cry11A was successful and GFP fluorescence may be used to represent the level of Cry11A production (Fig. 7A and B). One of the colonies, Gcry6 (later named SCH616), showing the highest fluorescence, was further chosen for Cry11A production and larvicidal activity assays. Compared to the cell growth of the control, SCH616 cell growth inhibition was not obvious until the bacteria entered the mid-log phase (ca. 9 h) (Fig. 8A). However, ∼21% inhibition was detected after 24 h, indicating that overexpression of Cry11A was possibly toxic to flavobacteria in the stationary phase to some extent (Fig. 8A). The relative GFP fluorescence in SCH616 increased with cell growth in the construct carrying gfp::cry11A but not in wild-type (WT) cells without the larvicidal genes (Fig. 8B). The maximum GFP production was detected in construct SCH616 after a 24-h incubation, indicating that higher Cry11A production occurred in the stationary phase (Fig. 8B). Western blot results confirmed that the higher GFP fluorescence (9 versus 24 h) correlated with increasing Cry11A production (Fig. 8C).
FIG 7.
Screening of F. hibernum transformants with a high level of gfp:cry11A expression based on GFP fluorescence determination. The screening construct (PompA+gfp::cry11A+p20) was created by translational fusion of gfp and cry11A. Transformants with green fluorescence were screened under epifluorescence microscopy. (A) Fifteen selected transformants were cultured in CYE medium overnight and quantified by fluorescence spectrometry as described in Materials and Methods. The control was F. hibernum W22 transformed with plasmid without the larvicidal construct. (B) Western blot assay of the selected transformants with higher fluorescence. The cell number was adjusted to an OD600 of 0.1, and ca. 20 μl of cells was subjected to SDS-PAGE.
FIG 8.

Growth and Cry11A production of recombinant strain SCH616 (GFP::Cry11A+P20). (A) Comparison of the growth of SCH616 (▲) and the WT (■). (B) Comparison of the green fluorescence emitted by SCH616 (■) and the WT (♦) grown in CYE medium supplemented with Em by fluorescence spectrometry (see Materials and Methods). The relative green fluorescence was normalized with the OD600. (C) Western blot analysis of GFP::Cry11A production by SCH616 and the WT. Cultures were sampled at 9 and 24 h, respectively. The cell number was adjusted to an OD600 of 0.1, and ca. 20 μl of cells was subjected to SDS-PAGE. Western blot analysis was performed as described in Materials and Methods. The control was F. hibernum WT strain W22. (D) Bioassay of SCH616 (■) and WT (∙) conducted with A. triseriatus larvae in water. Results were recorded after 24 h. Values are means ± standard deviations; n = 4. (E) Bioassay of SCH616 (■) and WT (∙) conducted with A. triseriatus larvae in a microcosm system. Results were recorded after 48 h. Values are means ± standard deviations; n = 6.
SCH616 cells (GFP::Cry11A+P20) were tested for larvicidal activity against A. triseriatus (early third instar) in distilled water and also in microcosms (Fig. 8D and E). Survival of mosquito larvae in the negative-control samples without the larvicidal gene was 100% in water (Fig. 8D). It was nontoxic when the dose of SCH616 was lower than 1.8 × 105 cells/ml. The LC50 was estimated to be 8.2 × 106 cells/ml by the standard assay (Fig. 8D). SCH616 also showed toxicity against A. triseriatus in microcosms with an estimated LC50 of 4.7 × 107 cells/ml (Fig. 8E), indicating that SCH616 has great potential for use in field studies.
DISCUSSION
In our study system, one of the most efficient ways to control mosquito larvae would be to use flavobacteria as expression hosts to produce larvicidal proteins from B. thuringiensis subsp. israelensis or B. sphaericus if the selected bacteria are ingested and digested efficiently and if these bacteria persist in the natural environment. F. hibernum isolated from native larval mosquito habitats did not show sedimentation in water for up to 8 days, indicating that it is a good candidate for development as a bioinsecticide for larval mosquito control. We then examined if F. hibernum cells were ingested by mosquito larvae efficiently or not. To do this, four different AFP genes were expressed in F. hibernum with the strong flavobacterial expression system (15). This technique offers a good opportunity to study bacterium-bacterium or bacterium-insect interaction in a different environment such as in the larval or adult mosquito gut (2, 6, 7). Furthermore, fluorescent bacteria allowed us not only to qualitatively investigate the location of ingested flavobacteria (Fig. 2) but also to test their ingestion efficiency (Fig. 3). Quantitative assays of the feeding behavior of larval mosquitoes is difficult and challenging even with an AFP-tagging method. Determination of fluorescent flavobacteria by fluorimetry or fluorescence-activated cell sorting methods is problematic because of the problem of separating bacterial cells from mosquito gut or tissues. GFP fluorescence can bleach when a long UV exposure time is needed. Also, it is hard to automate this method when a large amount of samples is processed.
Because of these limitations, we explored a more sensitive reporter for a quantitative study of the interaction between larval mosquitoes and bacteria. NanoLuc showed excellent reporter characteristics in F. hibernum; e.g., it provided highly sensitive quantitation with broad linearity (see Fig. S1 in the supplemental material). Furthermore, the relative NanoLuc reporter activity in F. hibernum varied only slightly among the different growth phases (see Fig. S2); it was stable within 24 h when used in a water or microcosm study (Fig. 4 and 5). Collectively, the effects of environmental factors and growth phase on nluc expression were minor in vivo. The reporter strain with NanoLuc allowed us to investigate larval ingestion rates in a detailed and reproducible manner. Fourth-instar larvae ingested bacteria around 50% faster than third-instar larvae did within the first hour (“fast phase”). The larger size of fourth-instar larvae was obviously a factor in their higher initial ingestion rates (the head capsule of fourth-instar larvae was at least 30% larger than that of third-instar larvae). Ingestion rates likely declined faster in the older larvae than in third-instar larvae simply because fourth-instar larvae engorge faster. External bacterial cell concentrations would be diminishing more rapidly in the fourth-instar feeding medium, and this could also influence ingestion rates (30). Very few studies have reported the feeding rate with native bacteria as a model in larval mosquitoes. In previous studies, the most common approach to estimation of the ingestion rate used solid particulates, which likely differ from native bacteria in size and feeding stimuli (31, 32). Larval mosquito feeding behavior can be greatly affected by the food source. Thiery et al. investigated the ingestion of cyanobacterial strains by Culex pipiens and Anopheles gambiae mosquitoes by a culture-dependent method and found that the numbers of cells ingested were dependent on the cyanobacterial strains and varied with the developmental stage and species of mosquitoes (33). Late (fourth)-instar larvae ingested more cyanobacterial cells than early (second)-instar larvae when fed on the same cell density, which agrees with what we found in this study.
The survival and fate of bacteria, including GEMs or WT bacteria, introduced into an aquatic ecosystem could be affected by many factors, including biotic factors, trophic conditions, water chemistry, bacterial strain differences, etc. For example, the E. coli cells added survived well in sterile water while their numbers declined dramatically in natural water (no cells were detected after 3 days), indicating that biotic factors were responsible for the decline of the introduced microbes (34). In our study, only 10% of the added F. hibernum cells were retained in the presence of larval mosquitoes but at least 60% of them were detected in controls lacking larvae after 48 h, indicating that mosquito larvae acquired F. hibernum efficiently in microcosms but that a residual population persisted (Fig. 5). F. hibernum cells remained in microcosms at 30% (1.5 × 105 cells/ml) and 10% (3.2 × 104 cells/ml) of the initial levels after 72 and 144 h, respectively. Iwasaki et al. reported that genetically engineered P. putida did not persist well in microcosms (lake water) because only less than 1 and 0.01% of the initial GEMs were detected within 3 and 7 days, respectively (35). In our initial studies, a relatively simple artificial microcosm consisting of distilled water, beech leaf detritus, and tree hole water allowed some persistence of F. hibernum (Fig. 5). Although our inoculum included predators of bacteria such as ciliated protozoans, we did not quantify their abundance, and high initial numbers of F. hibernum bacteria may have allowed their persistence and adaptation to bacterivores even in the absence of mosquito larvae (36). However, because the persistence of F. hibernum was similar in microcosms that were autoclaved, this study suggests that the bacterial strain was not particularly susceptible to protozoan predation, nor was it necessarily affected by competing bacterial species. A more detailed investigation should be done to further determine how the input F. hibernum interacts with indigenous microbes and protozoans in natural tree hole systems, and the NanoLuc reporter provides an excellent method by which to track the fate of flavobacteria under these complex conditions.
Toxins such as Cry11A will not function well unless ingested bacteria can be efficiently digested by larval mosquitoes. Our results showed that third-instar larvae consumed F. hibernum very rapidly and digested up to 80% of the ingested bacteria. The digestibility of bacteria acquired by aquatic insects such as the mayflies (Ephemera danica) and larval mosquitoes (C. pipiens and A. gambiae) has been investigated (33, 37). Aeromonas hydrophila and Citrobacter freundii cells were digested efficiently in the mayfly gut, but Acinetobacter and Flavobacterium sp. cells were not, indicating that the digestion of bacteria is selective and dependent on the species or strain involved (37). The same bacteria may even be differently digestible by different larval mosquitoes (9, 33). Therefore, the digestibility of bacteria should be carefully considered if they are used as hosts to produce larvicidal proteins that cannot be secreted.
The cry11A+p20 operon was expressed in Flavobacterium strains here to test its potential for the biocontrol of mosquitoes. In one of our previous studies, the expression of the cry11A+p20 cluster was difficult while the expression of the helper protein p20 gene was successful in F. johnsoniae (15). The failure of cry11A+p20 expression previously may have been due to the cytotoxicity of overexpressed proteins. However, the expression of the genes for individual binary toxins (gfp::p42 and gfp::p50) in F. johnsoniae strains proved to be no problem (15). Furthermore, the combined binary toxins were larvicidal to C. quinquefasciatus, A. gambiae, and A. triseriatus mosquitoes. Therefore, the expression of GFP had no influence on the function of the larvicidal protein P42 or P51 (15). In this study, GFP::Cry11A in F. hibernum improved the efficiency of screening for the target protein Cry11A based on fluorescence emission and also allowed us simultaneously to track the cells' location and fate in microcosms. More importantly, the recombinant Cry11A in F. hibernum was toxic to the tree hole mosquito A. triseriatus in microcosms (Fig. 8), which is very promising for the biocontrol of A. triseriatus and related species. However, there is room for further improvement despite the high toxicity of the F. hibernum clone (SCH616) described here. For instance, the expression of additional larvicidal genes synergistic with others, such as those encoding chitinase, cyt1A, or binary toxins from B. sphaericus, will help improve toxicity and expand the effective range to additional mosquito species and larval habitats. Flavobacteria are good chitinase producers (30). Our future studies will investigate if overexpression of flavobacterial chitinases will help enhance the overall larvicidal toxicity of strain SCH616. Also, an inducible expression system is warranted to alleviate the overexpression of larvicidal proteins that are toxic to host cells (S. Chen, unpublished data). Collectively, F. hibernum and other members of the family Flavobacteriaceae isolated from tree holes show great potential as desirable recombinant insecticides because they dwell in the larval feeding zone, can be efficiently ingested and digested by mosquito larvae, and appear to persist in simulated larval habitats.
Conclusion.
We successfully tagged F. hibernum, an environmental isolate from native mosquito larval habitats, with various sensitive reporters (AFPs and NanoLuc). The reporter system developed in this study allowed us to accurately study its interaction with the tree hole mosquito A. triseriatus, and the results showed that larvae were ingested and digested by F. hibernum, which provided direct evidence that some of the flavobacteria in the natural tree holes function as a food source for larval mosquitoes. Our studies further demonstrated that overexpression of larvicidal genes from B. thuringiensis subsp. israelensis (cry11A+p20) in F. hibernum was toxic to third-instar larvae in microcosms. This study shows that the genetic engineering of bacterial isolates from natural larval mosquito habitats continues to show promise for future mosquito biocontrol. The reporter system developed in this study can also aid in elucidating the pathogenic mechanisms of flavobacteria, given the fact that many flavobacteria are animal pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark McBride (University of Wisconsin—Milwaukee) and Michael Bagdasarian (Michigan State University) for generously providing E. coli-Flavobacterium shuttle plasmids and several bacterial strains.
This project was funded by NIH grant R37AI21884.
Footnotes
Published ahead of print 2 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03319-13.
REFERENCES
- 1.Merritt RW, Dadd RH, Walker ED. 1992. Feeding behavior, natural food, and nutritional relationships of larval mosquitoes. Annu. Rev. Entomol. 37:349–374. 10.1146/annurev.en.37.010192.002025 [DOI] [PubMed] [Google Scholar]
- 2.Ngwa CJ, Glöckner V, Abdelmohsen UR, Scheuermayer M, Fischer R, Hentschel U, Pradel G. 2013. 16S rRNA gene-based identification of Elizabethkingia meningoseptica (Flavobacteriales: Flavobacteriaceae) as a dominant midgut bacterium of the Asian malaria vector Anopheles stephensi (Dipteria: Culicidae) with antimicrobial activities. J. Med. Entomol. 50:404–412. 10.1603/ME12180 [DOI] [PubMed] [Google Scholar]
- 3.Pumpuni CB, Demaio J, Kent M, Davis JR, Beier JC. 1996. Bacterial population dynamics in three Anopheline species: the impact on Plasmodium sporogonic development. Am. J. Trop. Med. Hyg. 54:214–218 [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Chen S, Kaufman MG, Maknojia S, Bagdasarian M, Walker ED. 2008. Bacterial community structure in tree hole habitats of Ochlerotatus triseriatus: influences of larval feeding. J. Am. Mosq. Control Assoc. 24:219–227. 10.2987/5666.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman MG, Chen S, Walker ED. 2008. Leaf-associated bacterial and fungal taxa shifts in response to larvae of the tree hole mosquito, Ochlerotatus triseriatus. Microb. Ecol. 55:673–684. 10.1007/s00248-007-9310-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Gilbreath TM, Pukula K, Yan G, Xu J. 2011. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6:e24767. 10.1371/journal.pone.0024767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engel P, Moran NA. 2013. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol. Rev. 37:699–735. 10.1111/1574-6976.12025 [DOI] [PubMed] [Google Scholar]
- 8.McBride MJ, Kempf MJ. 1996. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 178:583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavshin A, Oshaghi M, Vatandoost H, Yakhchali B, Raeisi A, Zarenejad F. 2013. Escherichia coli expressing a green fluorescent protein (GFP) in Anopheles stephensi: a preliminary model for paratransgenesis. Symbiosis 60:17–24. 10.1007/s13199-013-0231-5 [DOI] [Google Scholar]
- 10.Wallace JR, Gordon MC, Hartsell L, Mosi L, Benbow ME, Merritt RW, Small PLC. 2010. Interaction of Mycobacterium ulcerans with mosquito species: implications for transmission and trophic relationships. Appl. Environ. Microbiol. 76:6215–6222. 10.1128/AEM.00340-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geavgaard JC, Turnbull MW, McNealy TL. 2010. A novel technique for feeding and confirming uptake of bacteria in larvae of the southern house mosquito, Culex quinquefasciatus (Diptera: Culicidae). Fla. Entomol. 93:577–583. 10.1653/024.093.0416 [DOI] [Google Scholar]
- 12.Ozawa T, Yoshimura H, Kim SB. 2013. Advances in fluorescence and bioluminescence imaging. Anal. Chem. 85:590–609. 10.1021/ac3031724 [DOI] [PubMed] [Google Scholar]
- 13.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. 2012. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7:1848–1857. 10.1021/cb3002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker ED, Lawson DL, Merritt RW, Morgan WT, Klug MJ. 1991. Nutrient dynamics, bacterial populations, and mosquito productivity in tree hole ecosystems and microcosms. Ecology 72:1529–1546. 10.2307/1940953 [DOI] [Google Scholar]
- 15.Chen S, Kaufman MG, Bagdasarian M, Bates AK, Walker ED. 2010. Development of an efficient expression system for Flavobacterium strains. Gene 458:1–10. 10.1016/j.gene.2010.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Federici BA, Park HW, Bideshi DK, Wirth MC, Johnson JJ. 2003. Recombinant bacteria for mosquito control. J. Exp. Biol. 206:3877–3885. 10.1242/jeb.00643 [DOI] [PubMed] [Google Scholar]
- 17.Khampang P, Luxananil P, Tanapongpipat S, Chungjatupornchai W, Panyim S. 2001. Recombinant Enterobacter amnigenus highly toxic to Anopheles dirus mosquito larvae. Curr. Microbiol. 43:448–451. 10.1007/s002840010337 [DOI] [PubMed] [Google Scholar]
- 18.Thanabalu T, Hindley J, Brenner S, Oei C, Berry C. 1992. Expression of the mosquitocidal toxins of Bacillus sphaericus and Bacillus thuringiensis subsp. israelensis by recombinant Caulobacter crescentus, a vehicle for biological control of aquatic insect larvae. Appl. Environ. Microbiol. 58:905–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yap W, Thanabalu HT, Porter AG. 1994. Expression of mosquitocidal toxin genes in a gas-vacuolated strain of Ancylobacter aquaticus. Appl. Environ. Microbiol. 60:4199–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JW, Yap WH, Thanabalu T, Porter AG. 1996. Efficient synthesis of mosquitocidal toxins in Asticcacaulis excentricus demonstrates potential of Gram-negative bacteria in mosquito control. Nat. Biotechnol. 14:343–347. 10.1038/nbt0396-343 [DOI] [PubMed] [Google Scholar]
- 21.Xu Y, Nagai M, Bagdasarian M, Smith TW, Walker ED. 2001. Expression of the p20 gene from Bacillus thuringiensis H-14 increases Cry11A toxin production and enhances mosquito larvicidal activity in recombinant Gram-negative bacteria. Appl. Environ. Microbiol. 67:3010–3015. 10.1128/AEM.67.7.3010-3015.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griego VM, Spence KD. 1978. Inactivation of Bacillus thuringiensis spores by ultraviolet and visible light. Appl. Environ. Microbiol. 35:906–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Bagdasarian M, Kaufman MG, Walker ED. 2007. Characterization of strong promoters from an environmental Flavobacterium hibernum strain by using a green fluorescent protein-based reporter system. Appl. Environ. Microbiol. 73:1089–1100. 10.1128/AEM.01577-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Bagdasarian M, Kaufman MG, Bates AK, Walker ED. 2007. Mutational analysis of the ompA promoter from Flavobacterium johnsoniae. J. Bacteriol. 189:5108–5118. 10.1128/JB.00401-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Ji X, Chen N, Li P, Liu W, Lu X. 2012. Development of replicative oriC plasmids and their versatile use in genetic manipulation of Cytophaga hutchinsonii. Appl. Microbiol. Biotechnol. 93:697–705. 10.1007/s00253-011-3572-0 [DOI] [PubMed] [Google Scholar]
- 26.Mally M, Cornelis GR. 2008. Genetic tools for studying Capnocytophaga canimorsus. Appl. Environ. Microbiol. 74:6369–6377. 10.1128/AEM.01218-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 28.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 29.SAS Institute, Inc 2011. SAS/STAT 9.3 user's guide. SAS Institute, Inc., Cary, NC [Google Scholar]
- 30.McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, Goltsman E, Wang W, Xu J, Hunnicutt DW, Staroscik AM, Hoover TR, Cheng YQ, Stein JL. 2009. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75:6864–6875. 10.1128/AEM.01495-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aly C, Mulla S. 1986. Orientation and ingestion rates of larval Anopheles albimanus in response to floating particles. Entomol. Exp. Appl. 42:83–90. 10.1111/j.1570-7458.1986.tb02191.x [DOI] [Google Scholar]
- 32.Ben-Dov E, Saxena D, Wang Q, Manasherob R, Boussiba S, Zaritsky A. 2003. Ingested particles reduce susceptibility of insect larvae to Bacillus thuringiensis. J. Appl. Entomol. 127:146–152. 10.1046/j.1439-0418.2003.00732.x [DOI] [Google Scholar]
- 33.Thiery I, Nicolas L, Rippka R, Marsac NT. 1991. Selection of cyanobacteria isolated from mosquito breeding sites as a potential food source for mosquito larvae. Appl. Environ. Microbiol. 57:1354–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao W, Feng R. 1990. Survival of genetically engineered Escherichia coli in natural soil and river water. J. Appl. Bacteriol. 68:319–325. 10.1111/j.1365-2672.1990.tb02881.x [DOI] [PubMed] [Google Scholar]
- 35.Iwasaki K, Uchiyama H, Yagi O. 1993. Survival and impact of genetically engineered Pseudomonas putida harboring mercury resistance gene in aquatic microcosms. Biosci. Biotechnol. Biochem. 57:1264–1269. 10.1271/bbb.57.1264 [DOI] [PubMed] [Google Scholar]
- 36.Walker E, Kaufman M, Merritt R. 2010. An acute trophic cascade among microorganisms in the tree hole ecosystem following removal of omnivorous mosquito larvae. Community Ecol. 11:171–178. 10.1556/ComEc.11.2010.2.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Austin DA, Baker JH. 1988. Fate of bacteria ingested by larvae of the freshwater mayfly, Ephemera danica. Microb. Ecol. 15:323–332. 10.1007/BF02012645 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.