

Abstract
Pseudomonas aeruginosa is distinguished by its broad metabolic diversity and its remarkable capability for adaptation, which relies on a large collection of transcriptional regulators and alternative sigma (σ) factors. The largest group of alternative σ factors is that of the extracytoplasmic function (ECF) σ factors, which control key transduction pathways for maintenance of envelope homeostasis in response to external stress and cell growth. In addition, there are specific roles of alternative σ factors in regulating the expression of virulence and virulence-associated genes. Here, we analyzed a deletion mutant of the ECF σ factor SigX and applied mRNA profiling to define the SigX-dependent regulon in P. aeruginosa in response to low-osmolarity-medium conditions. Furthermore, the combination of transcriptional data with chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq) led to the identification of the DNA binding motif of SigX. Genome-wide mapping of SigX-binding regions revealed enrichment of downstream genes involved in fatty acid biosynthesis, type III secretion, swarming and cyclic di-GMP (c-di-GMP) signaling. In accordance, a sigX deletion mutant exhibited altered fatty acid composition of the cell membrane, reduced cytotoxicity, impaired swarming activity, elevated c-di-GMP levels, and increased biofilm formation. In conclusion, a combination of ChIP-seq with transcriptional profiling and bioinformatic approaches to define consensus DNA binding sequences proved to be effective for the elucidation of the regulon of the alternative σ factor SigX, revealing its role in complex virulence-associated phenotypes in P. aeruginosa.
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic bacterial pathogen that can be distinguished by its exceptional high capability to adapt and survive in various and challenging habitats and hosts, including animals, plants, and the human host. The necessary means for bacterial adaptation processes critically rely on sensing and quickly responding to the specific extracellular conditions encountered. One common way to achieve rapid activation of genes in response to fluctuating environmental conditions is the use of extracytoplasmic function (ECF) sigma (σ) factors that are especially abundant in P. aeruginosa (1, 2). ECF σ factors serve as important regulators, and they are increasingly recognized as factors regulating expression of virulence genes and virulence-associated genes (3–5). The activity of most of the ECF σ factors are modulated by inner membrane sensor proteins that act as anti-sigma factors. An off-switch of the anti-sigma factor in response to specific environmental changes thereby presumably leads to the release of the cognate σ factor and thus allows recruitment of the RNA polymerase to initiate expression of the specific σ factor-dependent gene regulon (6). So far, cell envelope stress, iron limitation, and oxidative stress have been demonstrated to play a pivotal role during host infection and were described to activate ECF σ factors (7, 8). In addition to the best-studied P. aeruginosa ECF σ factors AlgU and PvdS, SigX has been investigated in recent studies in the context of transcriptional regulation of the outer membrane protein OprF (9, 10). P. aeruginosa SigX shares 49% sequence similarity to σw of Bacillus subtilis, which is induced by osmotic stress, phage infection, or interruption of cell wall biosynthesis following antibiotic treatment (2, 11). In P. aeruginosa deletion of sigX led to impaired growth under low-salt concentrations and reduced oprF expression (9). Later, Bouffartigues and colleagues confirmed these data and presented a link between lowered sodium chloride concentrations and the transcription of oprF due to the activation of the sigX promoter (10). As the ECF σ factor SigX was shown to be essential for survival under low-osmolarity-medium conditions and seems to be involved in responses to osmotic and cell wall stresses (9, 10), it was suggested that the SigX regulon might be larger than anticipated.
In this study, we constructed a sigX deletion mutant in P. aeruginosa PA14 and used mRNA profiling and chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq) to identify the binding motif and the respective global ECF σ factor SigX-dependent regulon in response to low-osmolarity-medium conditions. The combination of ChIP-seq with transcriptional profiling and bioinformatic approaches to define consensus DNA binding sequences is an increasingly important approach for elucidating transcriptional regulatory mechanisms in prokaryotes and will enable the dissection of even very complex gene regulatory networks.
MATERIALS AND METHODS
Strains and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Unless otherwise stated, all P. aeruginosa strains were cultivated in lysogeny broth (LB) at 37°C with shaking at 180 rpm. LB contained none, 8 mM, 80 mM, 120 mM, 154 mM, 200 mM, 428 mM, or 500 mM sodium chloride (NaCl) or 240 mM sucrose (corresponding to 120 mM NaCl) as an alternative osmolyte. When required (e.g., for plasmid maintenance or induction of gene expression), 30 μg ml−1 gentamicin and 0.5% l-arabinose (Sigma) were added.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant feature(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | Strain used for all standard cloning experiments | 55 |
| E. coli S17-1 | Mobilizing strain for RP4 Mob-containing plasmids | 56 |
| PA14 | Wild-type reference strain | 57 |
| PA14 ΔsigX::Gmr | sigX::Gmr mutant of PA14 wild-type | This study |
| PA14 ΔsigX | sigX mutant of PA14 wild-type | This study |
| Plasmids | ||
| pJN105 | Broad-host-range low-copy-number vector pBBR1-MCS5 harboring araC-PBAD cassette from pBAD18, Gmr | 12 |
| pJN105-RBS-sigX | sigX ORF with optimized start and stop codon and preceding RBS cloned into pJN105 using EcoRI and XbaI sites, Gmr | This study |
| pJN105-RBS-sigX-his8 | sigX ORF with optimized start and stop codon, preceding RBS and C-terminal 8×His coding sequence cloned into pJN105 using EcoRI and XbaI sites, Gmr | This study |
| pBBR1-MCS5-TT-RBS-lux | Broad-host-range low-copy-number vector pBBR1-MCS5 harboring luxCDABE and terminators lambda T0 rrnB1 T1 for plasmid-based transcriptional fusions, Gmr | 13 |
| pBBR1-MCS5-TT-PsigX-RBS-lux | −492 to +1 fragment upstream of sigX ORF cloned into pBBR1-MCS5-TT-RBS-lux using BamHI and XmaI sites, Gmr | This study |
| pBBR1-MCS5-TT-PaccB-RBS-lux | −254 to −144 fragment upstream of the accB ORF cloned into pBBR1-MCS5-TT-RBS-lux using BamHI and EcoRI sites, Gmr | This study |
| pBBR1-MCS5-TT-PcheY2-RBS-lux | −209 to +1 fragment upstream of cheY2 ORF cloned into pBBR1-MCS5-TT-RBS-lux using SpeI and HindIII sites, Gmr | This study |
| pUC18-mini-Tn7T-Gm-lacZ | pUC18 derivative encoding mini-Tn7 element with transcriptional terminators and lacZ fusion, Gmr | 17 |
| pEX18TAp | Gene replacement vector with MCS from pUC18, oriT+ sacB+ Apr | 58 |
| pEX18TAp2 | pEX18TAp derivative, 845-bp fragment containing 5S rRNA and lacZ-alpha genes and MCS removed by inverse PCR, novel MCS generated with unique restriction sites for XhoI, PstI, SmaI/XmaI, XbaI, SacI, HindIII, NheI, NotI, MluI, KpnI, BamHI, EcoRI, Apr | This study |
| pEX18TAp2-up-sigX-do-sigX | pEX18TAp2 harboring a 485-bp upstream and 381-bp downstream region of sigX ORF with a junction sequence encoding a KpnI-site and three shifted stop codons, Apr | This study |
| pEX18TAp2-up-sigX-Gm-do-sigX | pEX18TAp2-up-sigX-do-sigX containing an FRT-flanked Gmr cassette amplified from pUC18-mini-Tn7T-Gm-lacZ and introduced at a KpnI site, Apr Gmr | This study |
| pFLP3 | FLP expression vector, sacB+ oriT+ Apr Tetr | 59 |
ORF, open reading frame; RBS, ribosome binding site; MCS, multiple cloning site; FRT, Flp recognition target.
Plasmid and strain construction.
Primers used are listed in Table S1 in the supplemental material. Cloning was performed in Escherichia coli DH5α using standard molecular biology techniques. For sigX overexpression in P. aeruginosa, the sigX gene was amplified by PCR using a forward primer harboring a ribosomal binding site and an optimized start codon (TTG→ATG) and a reverse primer with an optimized stop codon (TAG→TGA). PCR products were digested with EcoRI and XbaI and introduced into the corresponding sites of the broad-host-range plasmid pJN105 under the control of PBAD resulting in pJN105-RBS-sigX (12). For ChIP-seq experiments, pJN105-RBS-sigX-his8 was constructed using a reverse primer additionally encoding eight copies of a His tag (8×His). Reporter plasmids for bioluminescence assays were produced by ligation of promoter regions into pBBR1-MCS5-TT-RBS-lux (13). The promoter regions selected for transcriptional luxCDABE fusions upstream of the sigX, accB, and cheY2 gene (PA14_02660) and the restriction sites are indicated in Table 1. Plasmids were transferred into P. aeruginosa by electroporation as previously described (14).
The PA14 ΔsigX::Gmr deletion mutant was constructed according to a modified protocol using overlap extension PCR as described previously (15). The gene replacement vector pEX18TAp was modified by inverse PCR to remove the coding sequence for 5S rRNA. In addition, the resulting vector pEX18TAp2 encompasses a novel multiple cloning site (MCS) established by primer extension. Regions up- and downstream of sigX were amplified by PCR as indicated in Table S1 in the supplemental material. The primers Mut-sigX-up-RV and Mut-sigX-down-FW harbored complementary sequences coding for three shifted stop codons and a KpnI restriction site. The two PCR products were fused in a second PCR, and the obtained fragment was introduced in pEX18TAp2 at BamHI and NotI restriction sites, resulting in pEX18TAp2-up-sigX-do-sigX. Finally, pEX18TAp2-up-sigX-Gm-do-sigX was produced by ligation of an FLP-excisable gentamicin cassette amplified from pUC18-mini-Tn7T-Gm-lacZ into pEX18TAp2-up-sigX-do-sigX. This construct was transformed in E. coli S17-1 and transferred into the PA14 wild-type strain by conjugation. Single crossovers were selected on gentamicin. Counterselection in BM2 medium (16) supplemented with sucrose was used to force plasmid resolution, resulting in PA14 ΔsigX::Gmr. The gentamicin cassette was excised using the FLP expression vector pFLP3 as described elsewhere (17) to obtain PA14 ΔsigX. All vectors as well as PA14 ΔsigX::Gmr were confirmed by DNA sequencing.
Growth curves.
Growth of bacterial strains was recorded by spectrophotometric measurement of the optical density at 600 nm (OD600) of 10-ml cultures incubated in 50-ml flasks at 37°C at 180 rpm in LB supplemented with different sodium chloride concentrations as indicated in Fig. 1 and Fig. S1 in the supplemental material. OD600 values were recorded in triplicates every hour for a time period of 24 h.
FIG 1.

Growth phenotypes of the ΔsigX deletion mutant. (A) Colony morphologies of the PA14 wild-type (WT) strain and the ΔsigX mutant on blood agar plates after 16 h of incubation at 37°C. (B) Colony morphologies on Congo red agar after incubation for 7 days at room temperature. (C) Growth curves of the wild-type PA14 strain and the ΔsigX mutant cultivated at 37°C in LB with different concentrations of sodium chloride (NaCl). One representative of three replicates is shown. t, time.
Congo red plate assay.
Congo red agar plates were prepared as described previously by Friedman and Kolter (18). Briefly, LB medium without NaCl was solidified with 1.6% agar and supplemented with 40 μg ml−1 Congo red dye. Five microliters of bacterial cell cultures at an OD600 of 0.025 was spotted on the agar and incubated at room temperature for 7 days.
Bioluminescence assays.
The sodium chloride-dependent activity of SigX was analyzed via bioluminescence-based reporter assays by measuring the activity of the sigX promoter (PsigX) itself and of its target promoter PaccB. To confirm the specificity of the assay, the background luminescence from reporter strains harboring the promoterless or the RpoS-dependent promoter PcheY2 construct was determined in parallel. For each reporter strain two individual cultures were grown to exponential growth stage in 1:5 diluted LB. Next, 5 × 109 bacteria (OD600 of 5.0) were harvested and resuspended in 1 ml of inoculation fluid IF-0 (Biolog). Cell suspensions were diluted with inoculation fluid IF-10 (Biolog) to an initial OD600 of 0.05 and supplemented with the following sodium chloride concentrations: 0, 8, 80, 120, 154, 200, 428, and 500 mM. A black 96-well microtiter plate with a transparent and flat bottom was inoculated with 100 μl of bacterial suspension and incubated for 6 h at 37°C without shaking. Bioluminescence as well as the OD600 was measured in intervals of 30 min. Promoter activities are given as the relative luminescence divided by the OD600 (relative light units [RLU] OD600−1) over time. The values were corrected by subtracting the corresponding RLU OD600−1 of promoterless vector controls.
mRNA profiling.
For mRNA profiling, two independent experiments were performed, and each experiment included pooling of three individual main cultures. RNA was prepared from PA14 wild-type, PA14 ΔsigX::Gmr, PA14(pJN105), and PA14(pJN105-RBS-sigX) strains growing in 10 ml of LB containing 8 mM sodium chloride at 37°C with shaking. To induce sigX overexpression, l-arabinose was added to a final concentration of 0.5% to PA14(pJN105-RBS-sigX) and the corresponding control PA14(pJN105) at an OD600 of 0.5 for 45 min. RNA extraction, cDNA library preparation, and deep sequencing were performed as previously described (19). In brief, cells were harvested after addition of RNA Protect Buffer (Qiagen), and RNA was isolated from cell pellets using an RNeasy Plus Kit (Qiagen). mRNA enrichment was performed using a MicrobExpress Kit (Ambion). RNA was fragmented and ligated to specific RNA adapters containing a hexameric barcode sequence for multiplexing. The resulting RNA libraries were reverse transcribed and amplified, resulting in cDNA libraries ready for sequencing. All samples were sequenced on an Illumina Genome Analyzer II-x in the single end mode with 36 cycles.
Quantification of gene expression.
Sequence reads were separated according to their barcodes, and barcode sequences were removed. The short reads (36 nucleotides [nt]) that were obtained for the PA14 samples were used without any trimming. Sequences were mapped to the genome sequence of the reference strain wild-type P. aeruginosa PA14 using Stampy (20) with default settings.
The R package DESeq (version 1.10.1) (21) was used for differential gene expression analysis. Briefly, the reads-per-gene data were prefiltered to get rid of rRNA and tRNA genes and then normalized for variation in library size/sequencing depth by using the estimateSizeFactor function of DESeq. Differentially expressed genes were identified using the nbinomTest function based on the negative binomial model after prefiltering by overall variance. In whole-transcriptome approaches, detection power is reduced due to the large number of genes that are tested for differential expression. Prefiltering has been demonstrated to address this issue by removing those genes that are unlikely to be differentially expressed and so reduce the overall number of statistical tests that have to be performed (22). Therefore, we used gene-by-gene filtering by overall variance of the variance stabilization-transformed data with the 40th percentile boundary as the threshold for prefiltering. The Benjamini-Hochberg correction was used to control the false-discovery rate (FDR) at 5% to determine the list of regulated genes.
Data quality assessment and quality control were performed on the variance-stabilizing data that were generated by reestimating the dispersion using the method “blind” in DESeq to ensure that the variance-stabilizing transformation is not informed about the design and not biased toward a result supporting the design (21). We used three different methods to check the data quality: a matrix of scatter plots of the variance stabilization-transformed data of all overexpressed/deletion mutant replicates against each other (see Fig. S2 in the supplemental material), a principal component analysis of the samples using the 100 most variable genes (see Fig. S3), and hierarchical cluster analysis of samples using the 50 most variable genes displayed in a heat map that also shows clustering of the expression values of these genes (see Fig. S4). All three approaches provide evidence that the corresponding replicates form clusters which are significantly different from the cluster of other samples and confirm the quality of our results. Overexpressed and deletion mutant samples were used in duplicate; if the quality control failed on one sample, the high-throughput sequencing of the RNA transcripts (RNA-seq) was repeated, and the samples were reevaluated. Genes were identified as differentially expressed if they fulfilled the following criteria: (i) their logarithmic fold change was higher than 1 or lower than −1 in a comparison of the sigX mutant with the wild-type strain or of the sigX-overexpressing strain with the empty vector control strain and (ii) the Benjamini-Hochberg-corrected P value was smaller than 5%.
Chromatin immunoprecipitation followed by deep sequencing.
For each ChIP-seq approach, two independent experiments were performed, and each experiment included pooling of two individual cultures of 50 ml. ChIP-seq was applied to four 20-ml cultures of PA14(pJN105-RBS-sigX-his8) and PA14(pJN105) as a control strain under the same culture conditions as described in the section on mRNA profiling. Individual cultures were combined and incubated with a final concentration of 0.5% formaldehyde for 5 min at room temperature with gentle agitation to conserve DNA-protein interactions. The reaction was quenched by addition of glycine to a final concentration of 137 mM for 2 min at room temperature and gentle agitation. Cells were harvested at 2,500 × g for 20 min at 4°C and washed first with 10 ml of chilled phosphate-buffered saline (PBS) and then with 10 ml of chilled Tris-buffered saline (TBS). Finally, cells were washed with 1 ml of chilled TBS and transferred to 1.5-ml tubes, and cell pellets were stored at −70°C. Cell pellets were resuspended in 0.5 ml of lysis buffer (10 mM Tris-HCl, pH 8, 20% [wt/vol] sucrose, 50 mM NaCl, 10 mM EDTA) and lysozyme (20 mg ml−1) was added to a final concentration of 4 mg ml−1. The reaction mix was incubated at 37°C for 30 min. Cell suspensions were combined and transferred into a 15-ml tube containing 1.5 ml of immunoprecipitation (IP) buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% [vol/vol] Triton X-100, 0.1% [wt/vol] sodium deoxycholate) supplemented with 0.1% SDS and proteases inhibitors (Roche) and incubated on ice. Under constant cooling, DNA was fragmented to an average size of 200 to 250 bp by sonication for 5 cycles at 45 s at level 4 with a 90% duty cycle (Branson Sonicator S250 Analogue), and aliquots were stored at −70°C. Next, cell debris was removed from cell extracts by centrifugation, and for each experiment, in total 3 ml of cell extract was subjected to chromatin immunoprecipitation with 15 μl of anti-6×His tag antibody (ab9108; Abcam) overnight at 4°C and with rotation. DNA-σ factor-antibody complexes were captured with Dynabeads protein G (100.04D; Invitrogen) for 1 h at room temperature with rotation and isolated using a magnetic stand (Qiagen). Beads were washed three times with IP buffer and eluted in two steps with 100 μl and 50 μl of elution buffer (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 1% [wt/vol] SDS) for 15 min at 65°C on a rocking platform. Eluate (100 μl) was incubated with 1 μl of RNase A (100 mg ml−1) for 30 min at 65°C, and 5 μl of proteinase K (20 mg ml−1) was added and incubated for 1 h at 50°C and then for 6 h at 65°C, followed by another incubation with 5 μl of proteinase K (20 mg ml−1) for 1 h at 50°C. Immunoprecipitated DNA was recovered using a QIAquick PCR Purification kit (Qiagen) and subjected to a modified linear DNA amplification (LinDA) protocol described recently (23). Major changes included additional DNA purification after every reaction step until in vitro transcription, use of modified LinDA primer (see Table S1 in the supplemental material), and RNA isolation with an RNeasy Plus Kit (Qiagen) instead of phenol-chloroform extraction followed by ethanol precipitation. For next-generation sequencing, up to 50 ng of DNA was used to prepare libraries using a TruSeq DNA sample preparation kit according to the low-throughput protocol (Illumina), which encompasses in our case the following steps: DNA end repair, adenylation of 3′ ends, ligation of adapters, and purification, enrichment, and validation of processed DNA fragments. Finally, prepared DNA was subjected to Illumina sequencing platforms.
Bioinformatic analysis of ChIP-seq data.
Adapter sequences were removed using the fastq-mcf script that is part of the EA-utils package (24). During the same step, reads were trimmed allowing for minimal quality of 10 at their ends. We used the Bowtie aligner (25) to map the reads against the PA14 reference sequence. The observed genomic read coverage was more than 100 times in both experiments. Model-based analysis of ChIP-seq (MACS) (26) was applied for peak detection using a P value cutoff of 5% and shift size 30 for the peak modeling, making use of the relevant control samples. Promoter hits were considered significant when they were detected in both ChIP-seq approaches with an enrichment factor (EF) of at least 3 and a P value of ≤0.01. Statistical analysis of the obtained 624 candidates revealed less than one false positive (6,014 × 0.012) with a P value of 0 according to a hypergeometric test in R using the Phyper command on a basis of 6,014 possible candidates (see Fig. S5 in the supplemental material).
Definition and functional profiling of the SigX regulon.
A SigX motif search was applied using the MEME suite (27) on promoter regions whose respective genes (i) showed both a SigX-dependent downregulation in PA14 ΔsigX::Gmr and upregulation in PA14(pJN105-RBS-sigX) and (ii) were either identified in ChIP-seq experiments and/or were defined to be the first gene of a transcriptional unit according to Wurtzel et al. (28). These criteria were met by 20 candidates. Promoter regions were defined as sequences 500 bp upstream of the respective start codon. The parameters occurrence (0 or 1 per sequence), width (minimum, 30 nucleotides; maximum, 40 nucleotides), and number of sites (minimum, 10) were specified, and the DNA option “search given strand only” was activated. Furthermore, a background Markov model was supplied. Next, the obtained motif was submitted to FIMO (29) to identify putative SigX binding sites in all promoter regions of the PA14 genome. Promoter hits with a P value of ≤5 × 10−6 were regarded as significant, resulting in 1,578 candidates with a reidentification of 17 of 20 promoters selected for motif search. To define the primary SigX regulon, genes were selected which fulfilled at least two of the following three criteria: (i) exhibited SigX-dependent regulation of expression, (ii) had a promoter that was enriched in ChIP-seq experiments, and (iii) had a promoter that contained a SigX binding site. Finally, statistical significance of these candidate genes was checked by performing a hypergeometric test on the intersections ChIP-seq/RNA-seq, RNA-seq/motif search, and ChIP-seq/motif search. Only groups of genes whose P values were less than 0.05 as well as genes which were hit in all three approaches were considered to be part of the primary SigX regulon. This final set of 267 genes was functionally characterized using the PseudoCAP annotation (30). To improve this profiling, the PseudoCAP PA14 annotation was updated by adding the PseudoCAP classes of PAO1 homologs to corresponding PA14 genes. Over- or underrepresentation was calculated by comparing normalized PseudoCAP classes experimentally detected and normalized PseudoCAP classes annotated using the following equation: x = (number of specific PseudoCAP classes detected/number of all PseudoCAP classes detected)/(number of specific PseudoCAP classes annotated/number of all PseudoCAP classes annotated), where an x of ≥1.5 is defined as overrepresentation and an x of ≤0.66 is defined as underrepresentation.
Fatty acid composition analysis.
Bacteria were harvested after 20 h of planktonic growth in 10 ml of LB supplemented with 8 mM sodium chloride (NaCl), 120 mM NaCl, 500 mM NaCl, or 240 mM sucrose. Cellular fatty acid extraction was performed as described elsewhere with 2 g of wet cells from the cell pellet to extract the total lipids and to prepare the fatty acids for separation by gas chromatography (31, 32). The fatty acid composition is provided as the area percentage of the total fatty acids. All measurements were performed in triplicate.
Cytotoxicity assay.
Cytotoxicity of wild-type P. aeruginosa PA14 and the ΔsigX mutant was assessed by infecting A549-Gluc cells as described previously (33). A549-Gluc cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U ml−1 of penicillin, 100 g ml−1 of streptomycin, and 10% fetal calf serum (DMEM complete). Cell cultures were grown at 37°C with 5% CO2. A549-Gluc cells were generated from A549 cells by lentiviral gene transfer as described previously (34, 35). A549-Gluc cells were grown in 96-well plates to 90 to 100% confluence. Cells were washed once with phosphate-buffered saline (PBS) prior to infection with P. aeruginosa strains, which were cultivated in LB with either 8 mM, 120 mM, or 500 mM sodium chloride to an OD600 of 0.1 to 0.5. Cells were infected at a multiplicity of infection (MOI) of 10. Plates were centrifuged for 5 min at 500 × g to facilitate contact between bacterial and epithelial cells. Cell culture supernatants were collected after 3 h of incubation at 37°C with 5% CO2 following a centrifugation step to pellet out bacteria and cell debris. Gaussia luciferase activity was measured for 0.1 s using an LB 960 Centro XS3 plate luminometer (Berthold Technologies) after the addition of 60 μl of 10 μM coelenterazine (PJK GmbH).
Biofilm formation assays.
The ability of bacteria to adhere to polyvinyl chloride (PVC) was determined by the use of a modified previously described protocol (36). Overnight cultures were diluted to an OD600 of 0.02 in LB supplemented with 8 mM, 120 mM, or 500 mM NaCl or 240 mM sucrose, and 100 μl of this bacterial suspension was inoculated in 96-well PVC plates (Costar). The plates were sealed with air-permeable membranes (Greiner Bio-One) and incubated under static conditions at 37°C in a humid atmosphere. After 24 h, planktonic bacteria were removed, and the wells were washed with water before being stained with 0.1% (wt/vol) crystal violet at room temperature. The staining solution was removed after 30 min; the wells were washed with water and air dried before the retained crystal violet was destained with 95% ethanol for 30 min at room temperature. For quantification, 125 μl of the solution was transferred to fresh polystyrene microtiter plates (Nunc), and the absorbance was measured at 550 nm. Three biological replicates with eight technical replicates were used to calculate mean values and standard deviations.
Quantification of c-di-GMP.
For the quantification of the intracellular cyclic di-GMP (c-di-GMP) levels, 5-ml bacterial suspensions were harvested after 20 h of cultivation in LB supplemented with 8 mM sodium chloride (NaCl), 120 mM NaCl, 500 mM NaCl, or 240 mM sucrose. The c-di-GMP extraction and quantification by high-performance liquid chromatography (HPLC)-coupled tandem mass spectrometry was performed as described previously with isotope-labeled [13C15N]-c-di-GMP as an internal standard (37). The collected supernatants of the c-di-GMP extraction were stored overnight at −20°C to allow complete protein precipitation. The c-di-GMP concentrations are given as pmol of c-di-GMP per mg of protein as mean values from three biological and two technical replicates each. The protein concentration was determined with Roti Nanoquant reagent (Roth) according to the manufacturer's instructions.
Motility assays.
Swarming and swimming motility assays were performed as described elsewhere (16, 38). Briefly, swarming was evaluated on BM2 glucose plates (62 mM potassium phosphate buffer [pH 7], 2 mM MgSO4, 10 μM FeSO4, 0.4% [wt/vol] glucose) containing 0.5% agar supplemented with 0.1% Casamino Acids, and swimming capability was determined on BM2 glucose plates solidified with 0.3% agar; 8 mM, 120 mM, or 500 mM NaCl or 240 mM sucrose was added to the motility plates. Plates were inoculated with 2 μl of a preculture at an OD600 of >1 and incubated for 16 h at 37°C. Swimming motility was evaluated by measuring the diameter of the swimming zone from three biological replicates, and diameters were recorded as mean values with standard deviations. Twitching assays were performed on LB plates solidified with 1.5% agar and supplemented with 8 mM, 120 mM, or 500 mM NaCl or 240 mM sucrose. After 48 h of incubation at 37°C, the agar was carefully removed, and the diameter of the twitching zone at the plastic-agar surface was measured.
Gene expression data accession number.
RNA-seq and ChIP-seq raw as well as processed data have been submitted to the Gene Expression Omnibus (GEO) database under accession number GSE50937.
RESULTS
Growth defect of a PA14 ΔsigX mutant is enhanced under low-salt-medium conditions.
When constructing a ΔsigX mutant in the P. aeruginosa PA14 strain, we observed variant colony morphology and growth behavior of the mutant in comparison to its respective wild type. As shown in Fig. 1A the ΔsigX mutant exhibited smaller and less smooth colonies on Columbia blood agar plates. The variant colony morphology became even more apparent when the bacteria were cultured on Congo red agar plates without salt at room temperature (Fig. 1B). A growth defect of a ΔsigX mutant in the PAO1 strain has been described previously by Brinkman et al. (9), and an involvement of SigX in the regulation and adaptation to osmotic stress and, in particular, to low osmolarity has been suggested (9, 10). In line with these previous observations, the PA14 ΔsigX mutant exhibited delayed growth with an extended lag phase in liquid LB cultures, which became most apparent under low-sodium-chloride-medium (8 mM NaCl) conditions (Fig. 1C).
SigX promoter activity is induced by low sodium chloride concentrations.
To define the role of SigX in adaptation to osmotic stress, we performed bioluminescence reporter assays to measure the sigX promoter activity in response to different salt concentrations. A PsigX-luxCDABE transcriptional fusion was constructed for real-time examination of in vivo sigX expression kinetics and promoter responses to altered osmolarity. Indeed, we found that decreasing the osmolarity resulted in increased sigX promoter activity in the PA14 wild-type strain (Fig. 2).
FIG 2.

Promoter activity of PsigX. Bioluminescence-based reporter assays were used to measure the promoter activity of PsigX in the PA14 wild-type strain cultivated at 37°C in LB with different NaCl concentrations. One representative of three replicates is shown. t, time.
Transcriptional profile upon sigX overexpression and sigX deletion.
With the aim to identify the global impact of SigX on the PA14 transcriptome, we performed mRNA sequencing and recorded the transcriptome of the ΔsigX mutant compared to its respective PA14 wild-type strain upon growth under low-salt-medium (8 mM NaCl) conditions. A total of 255 genes were downregulated in the ΔsigX mutant relative to levels in PA14, whereas 247 genes were upregulated at least 2-fold. We also overexpressed sigX in the PA14 wild-type strain and again recorded the transcriptional profile. As observed in the ΔsigX mutant, sigX overexpression resulted in considerable growth inhibition (see Fig. S1 in the supplemental material). We found 191 genes downregulated when sigX was overexpressed in trans, whereas 374 genes were found to be upregulated compared to levels in the PA14 wild-type strain carrying the empty vector. Genes which were found to be downregulated in the ΔsigX mutant or upregulated upon sigX overexpression are listed in Table S2 in the supplemental material. They represent the global effect of SigX on the transcriptional profile of PA14 when it is cultivated under low-salt conditions and reflect not only primary but also secondary and tertiary effects. Only 26 of these genes were downregulated in the ΔsigX mutant and were at the same time regulated in opposite direction upon sigX overexpression. These 26 genes are listed in Table 2 and most likely are directly regulated by the alternative ECF σ factor SigX. As expected, sigX itself was among these 26 genes, as well as three hypothetical genes, eight genes involved in fatty acid metabolism (fabBGD, accAC, acpP, PA14_00060, and PA14_68360), six genes involved in type III secretion (popD, pscCF, and exsBDC), and oprM, mexA, lpxB, cmpX, and yegE. The YegE protein was predicted to be involved in c-di-GMP metabolism as it contains a GGDEF domain often found in diguanylate cyclases and a modified EAL (ELL) motif characteristic for phosphodiesterases (39).
TABLE 2.
Genes downregulated in PA14 ΔsigX::Gmr and upregulated in a sigX-overexpressing strain
| PA14 locus no. | Gene namea | Fold change in gene expression in: |
Product namec | |
|---|---|---|---|---|
| PA14 ΔsigX::Gmr vs WT PA14b | PA14(pJN105sigX) vs PA14(pJN105) | |||
| PA14_00060 | −2.5 | 8.8 | Putative acyltransferase | |
| PA14_00480 | −2.3 | 15.0 | Conserved hypothetical protein | |
| PA14_05530 | mexA | −2.1 | 4.1 | RND multidrug efflux membrane fusion protein MexA precursor |
| PA14_05550 | oprM | −2.1 | 3.4 | Major intrinsic multiple antibiotic resistance efflux outer membrane protein OprM precursor |
| PA14_08560 | tyrZ | −2.3 | 4.5 | Tyrosyl-tRNA synthetase 2 |
| PA14_09400 | phzS | −13.3 | 3.0 | Flavin-containing monooxygenase |
| PA14_17220 | lpxB | −2.2 | 3.1 | Lipid A disaccharide synthase |
| PA14_17270 | accA | −2.9 | 5.8 | Acetyl-CoA carboxylase, carboxyl transferase, alpha subunit |
| PA14_20750 | −2.2 | 2.1 | Putative chemotaxis signal transduction protein | |
| PA14_25650 | fabD | −6.0 | 13.0 | Malonyl- CoA-[acyl carrier protein] transacylase |
| PA14_25660 | fabG | −3.1 | 8.7 | 3-Oxoacyl-[acyl carrier protein] reductase |
| PA14_25670 | acpP | −2.1 | 4.8 | Acyl carrier protein |
| PA14_25900 | -3.4 | 14.4 | Putative short-chain alcohol dehydrogenase | |
| PA14_40560 | −8.2 | 19.2 | Conserved hypothetical protein | |
| PA14_41575 | sigX | −50.7 | 19.0 | ECF sigma factor SigX |
| PA14_41590 | cmpX* | −4.2 | 5.2 | Putative cytoplasmic membrane protein |
| PA14_42310 | pscF | −5.3 | 6.7 | Type III export protein PscF |
| PA14_42350 | pscC | −5.3 | 10.2 | Type III secretion outer membrane protein PscC precursor |
| PA14_42380 | exsD* | −3.8 | 5.7 | Conserved hypothetical protein |
| PA14_42400 | exsB | −11.9 | 10.1 | Exoenzyme S synthesis protein B |
| PA14_42430 | exsC | −4.0 | 6.6 | Exoenzyme S synthesis protein C precursor |
| PA14_42440 | popD | −7.2 | 2.2 | Translocator outer membrane protein PopD precursor |
| PA14_43690 | fabB | −3.1 | 8.6 | Beta-ketoacyl-ACP synthase I |
| PA14_49160 | yegE* | −4.0 | 12.8 | Conserved hypothetical protein |
| PA14_64110 | accC | −3.0 | 7.3 | Biotin carboxylase |
| PA14_68360 | −7.2 | 6.0 | Putative beta-ketoacyl synthase | |
An alternative gene name was provided (indicated by the asterisk) when no gene name was annotated.
WT, wild-type.
CoA, coenzyme A.
Genome-wide mapping of SigX-binding regions.
To define the primary regulon of SigX, we complemented our transcriptome data with chromatin immunoprecipitation (ChIP-seq) experiments. We constructed a variant of SigX containing an 8×His tag, introduced this into the PA14 wild-type strain, and upon l-arabinose-induced sigX overexpression and growth under low-salt conditions, we identified SigX-bound genomic DNA to determine the σ factor binding sites of SigX on a genome-wide level. As many as 329 genomic regions showed at least 3-fold enrichment in both ChIP-seq approaches (log2 ≥ 1.58; P value of ≤0.01). Of these regions, 94% were located within 500 bp upstream of the putative translation start sites (ATG) of a total of 624 genes as some of the enriched regions could be assigned to two adjacent genes with opposite orientations. We next applied a motif search using the MEME suite (27) on the promoter regions whose respective genes exhibited SigX-dependent regulation of expression, as listed in Table 2, and were either identified in the ChIP-seq experiments to bind SigX and/or were defined as the first gene within a transcriptional unit. Figure 3 displays the sequence logo with the SigX consensus sequence exhibiting relatively low sequence conservation. A bipartite motif with two boxes separated by a spacer of 20 nucleotides can be recognized, which is characteristic for σ factors (40).
FIG 3.
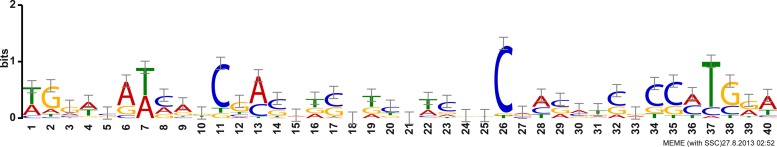
Logo of the SigX binding motif. A SigX motif search was performed with the MEME suite (27). The displayed motif is based on 20 of 20 submitted promoter regions with an E value of 6.2 × 10−11. Small-scale correction is indicated by gray error bars.
Defining the primary SigX regulon.
To further characterize the primary regulon of SigX, we identified the genes that (i) exhibited SigX-dependent regulation of expression (603 genes) (see Table S2 in the supplemental material), (ii) were identified in the ChIP-seq experiments to bind SigX in the promoter region (624 genes), and (iii) displayed a SigX motif in their promoter regions (Fig. 4) (41). The 267 genes that fulfilled at least two of these three criteria and were confirmed to be of statistical significance (P value of ≤0.05) are listed in Table S3. Twenty-two out of 26 genes found to be upregulated upon sigX overexpression and at the same time downregulated in the ΔsigX mutant (Table 2) were among them. To functionally profile genes of the primary SigX regulon, we used the PseudoCAP annotation (30). Nine functional groups were enriched in the group of SigX-dependent genes (Fig. 5). In line with the finding that SigX transcriptionally impacted fatty acid biosynthesis genes as well as genes involved in type III secretion, the PseudoCAP categories fatty acid/phospholipid metabolism as well as protein secretion/export apparatus and secreted factors were clearly enriched. Moreover, genes involved in chemotaxis and motility/attachment were significantly affected by SigX. In addition, we found enrichment of genes linked to stress conditions as indicated by the overrepresentation of PseudoCAP classes such as adaptation/protection, chaperone/heat shock proteins, translation, posttranslational modification, and degradation as well as genes related to phage, transposon, and plasmid. Interestingly, genes linked to basic processes such as replication, transcription, and cell division were underrepresented in the primary SigX regulon (Fig. 5).
FIG 4.
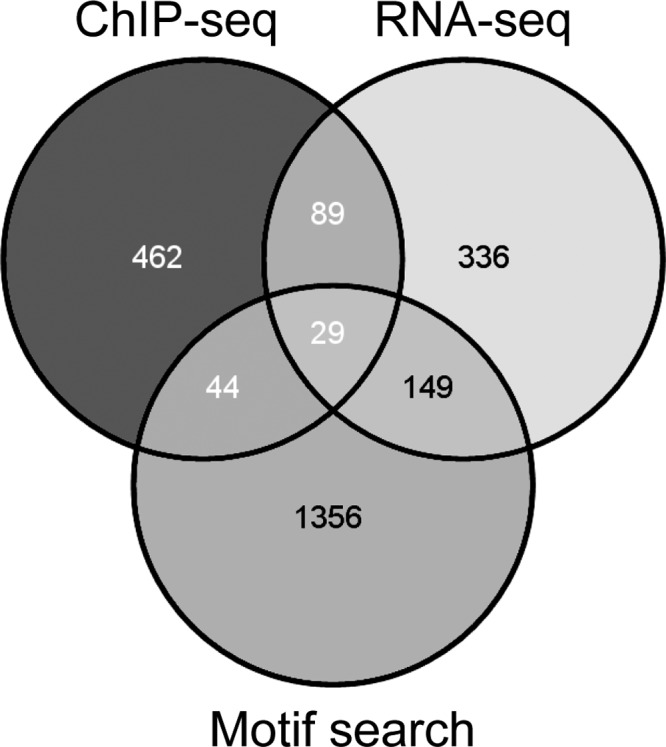
Quantitative analysis of the SigX regulon. The Venn diagram (41) shows genes whose promoter regions were captured in ChIP experiments (enrichment factor of ≥3; P ≤ 0.01), genes which were downregulated in ΔsigX compared to PA14 wild-type levels and/or upregulated in a sigX-overexpressing strain compared to a PA14 wild-type strain carrying the empty vector (fold change of ≥2; FDR of ≤0.05) according to RNA-seq, and genes whose promoters harbor a SigX binding site (P value of ≤5 × 10−6). Genes in all intersections except ChIP-seq/motif search were defined as the primary SigX regulon.
FIG 5.

Functional profiling of the primary SigX regulon. The PseudoCAP annotation (30) was used to categorize the members of the primary SigX regulon, and enrichment of specific classes of genes relative to their distribution in PA14 is displayed as the enrichment factor. Overrepresented classes (values of ≥1.5) are highlighted in black, while underrepresented classes (values of ≤0.66) are shown in white. Gray bars indicate no significant impact of SigX on the corresponding PseudoCAP function.
SigX-dependent promoter activity is induced by low sodium chloride concentrations.
To confirm our genome-wide SigX mapping data, we performed bioluminescence-based reporter assays and as an example measured the promoter activity of one of the SigX target genes, namely, accB, involved in the regulation of fatty acid biosynthesis, in response to different salt concentrations. Clearly, time-resolved studies showed increased activation for the accB promoter fused to the reporter gene complex luxCDABE under low and medium NaCl concentrations when it was introduced into the PA14 wild-type strain (Fig. 6). In contrast, the promoter PcheY2, which is controlled by RpoS (42), did not show any promoter activity for the tested NaCl concentrations (data not shown).
FIG 6.

Promoter activity of PaccB. Bioluminescence-based reporter assays were used to measure promoter activity of PaccB in the PA14 wild-type strain cultivated at 37°C in LB with different NaCl concentrations. One representative of three replicates is shown. t, time.
The ΔsigX mutant exhibits altered fatty acid composition.
Since many genes involved in fatty acid metabolism were downregulated in the ΔsigX mutant, we measured the overall composition of the fatty acids in the bacterial cell membrane by using gas chromatography. As depicted in Fig. 7, the fatty acid composition of the PA14 wild-type strain, irrespective of the medium osmolarity, consisted mainly of saturated palmitic acid (C16:0) and cis-vaccenic acid (C18:1ω7), which comprised more than 85% of the total fatty acids. In contrast, the spectrum of fatty acids was more diverse for the ΔsigX mutant, which was severely impaired in the production of C16:0 under all medium conditions and displayed reduced C18:1ω7 under low osmolarity. This phenotype of reduced amounts of the main fatty acid components in the cell membrane was especially apparent under low-salt-medium conditions and could be at least in part complemented by growing the bacteria under higher salt concentrations.
FIG 7.

Fatty acid composition analysis. Total fatty acid composition analysis for the PA14 wild-type (WT) strain and the ΔsigX mutant as the percentage of the area of total fatty acids from extracts cultivated under different osmolarities. Data represent the mean values of three biological replicates, with error bars indicating the standard deviations. Levels of statistical significance were calculated using a two-tailed unpaired t test (***, P < 0.001; **, P < 0.01).
The ΔsigX mutant exhibits reduced cytotoxicity.
The SigX regulon comprised not only genes involved in fatty acid biosynthesis but also several involved in type III secretion. In our transcriptomic data (see Table S2 in the supplemental material), a total of 27 genes were downregulated in the sigX mutant and/or upregulated in the sigX-overexpressing strain, including important needle, translocator, and regulatory proteins. We hypothesized that reduced expression of the type III secretion machinery should lead to decreased virulence behavior of the sigX mutant in comparison to the wild type. Thus, we performed a Gaussia luciferase assay with A549 cells to determine the cytotoxicity of the ΔsigX mutant compared to the PA14 wild-type strain after growth under different salt concentrations. As depicted in Fig. 8, infection with the wild-type PA14 reduced the viability of A459 cells up to 40%. In contrast, incubation with the ΔsigX mutant did not impact the viability of A459 cells. Varying the salt concentrations did not influence the cytotoxic effect of the PA14 wild-type strain, whereas the ΔsigX mutant was more cytotoxic in LB supplemented with 120 mM NaCl than in very low or high NaCl concentrations (Fig. 8).
FIG 8.

Cytotoxicity determined by a Gaussia luciferase assay with A549 cells. The viability of A549 cells was determined after 3 h of infection (MOI of 10) with the PA14 wild-type (WT) strain and the ΔsigX mutant. Prior to infection bacteria were cultivated with different concentrations of sodium chloride (NaCl). The viability of A549 cells is given as the percentage relative to uninfected medium controls. Data represent the mean values of six replicates, with error bars indicating the standard deviations. Levels of statistical significance were calculated using a two-tailed unpaired t test (***, P < 0.001; **, P < 0.01).
Biofilm formation is altered in the ΔsigX mutant.
Our primary SigX regulon (see Table S3 in the supplemental material) includes four genes (PA14_37690, PA14_49160, PA14_63210, and PA14_64050) that encode a GGDEF and/or EAL or HD-GYP domain protein which were previously associated with c-di-GMP turnover (43). The second messenger c-di-GMP is involved in the regulation of biofilm formation in P. aeruginosa (44, 45). Thus, we aimed at testing whether SigX impacts biofilm formation in PA14. We therefore analyzed the capability of the bacteria to attach to surfaces by the use of the crystal violet assay and stained pellicles formed in static cultures of 96-well plates at the air-liquid interface. Figure 9 clearly shows increased attachment of the ΔsigX mutant in LB supplemented with 120 mM NaCl or 240 mM sucrose, whereas this difference could not be observed under high- or low-salt conditions. The reduced growth rate of the ΔsigX mutant in low-osmolarity medium might explain the diminished attachment phenotype and underlines the importance of SigX to adapt to low osmolarity.
FIG 9.

Biofilm phenotype of the ΔsigX deletion mutant. Quantification of crystal violet-stained biofilms after 24 h of static growth in LB with different sodium chloride (NaCl) concentrations or with 240 mM sucrose. Data represent the mean values of three biological replicates, with error bars indicating the standard deviations. Levels of statistical significance were calculated using a two-tailed unpaired t test (***, P < 0.001).
c-di-GMP concentrations are elevated in the ΔsigX mutant.
We next sought to determine whether the increased attachment of the ΔsigX mutant is accompanied by elevated levels of c-di-GMP. Indeed, under conditions of physiological osmolarity (LB with 120 mM NaCl or 240 mM sucrose), we found elevated levels of c-di-GMP in the ΔsigX mutant after 20 h of growth (Fig. 10). As in the attachment assay, this difference was not observed under high- or low-salt conditions. Again, the failure to adapt to low salt concentrations of the ΔsigX mutant might explain the diminished differences in c-di-GMP levels under low-salt-medium conditions.
FIG 10.

Intracellular c-di-GMP concentrations. Intracellular c-di-GMP concentrations for the PA14 wild-type (WT) strain and the ΔsigX mutant were determined after 20 h of growth in LB with different sodium chloride (NaCl) concentrations or with 240 mM sucrose at 37°C. Data represent the mean values of three biological replicates, with error bars indicating the standard deviations. Levels of statistical significance were calculated using a two-tailed unpaired t test (**, P < 0.01; ns, not significant).
Deletion of sigX impacts motility.
We also analyzed the impact of sigX deletion on the PA14 motility phenotype. Clearly, swimming motility in the PA14 wild-type strain was dependent on the medium salt concentrations. Both high and low osmolarity reduced the swimming activity of PA14 (Fig. 11A). Although the ΔsigX mutant exhibited a reduced swimming zone compared to its respective PA14 wild type, the ratio of reduced swimming was constant over a wide range of NaCl concentrations (Fig. 11A). This indicates that the impaired swimming motility of the ΔsigX mutant is independent of the osmolarity. The ΔsigX mutant did not swarm under any medium condition tested (Fig. 11B), whereas the PA14 wild-type strain exhibited efficient swarming motility and was strongly inhibited only by 500 mM NaCl. The twitching motility was not affected in the ΔsigX mutant. However, again, high osmolarity (500 mM NaCl) reduced the twitching ability in the PA14 wild type and the ΔsigX mutant (Fig. 11C).
FIG 11.

Motility phenotypes of ΔsigX. (A) Swimming assay for the PA14 wild-type (WT) strain and the ΔsigX mutant. The swimming agar was supplemented with different concentrations of sodium chloride (NaCl) or sucrose. Data represent the mean values of three biological replicates, with error bars indicating the standard deviations. Statistical analysis using a two-tailed unpaired t test revealed that all differences between PA14 wild-type and ΔsigX mutant were significant (P < 0.01). (B) Swarming assay for the PA14 wild-type (WT) strain and the ΔsigX mutant. The swarming agar was supplemented with different concentrations of NaCl or sucrose. (C) Twitching motility of the PA14 wild-type (WT) strain and the ΔsigX mutant on LB agar plates supplemented with different concentrations of NaCl or sucrose after 48 h of growth at 37°C. Data represent the mean values of three biological replicates, with error bars indicating the standard deviations. Levels of statistical significance were calculated using a two-tailed unpaired t test (***, P < 0.001; **, P < 0.01).
DISCUSSION
To date 24 σ factors have been described in P. aeruginosa (2). Among them, the ECF σ factors play a crucial role in the transmission of extracellular conditions to the cytoplasm and the initiation of a timely response to the specific extracellular conditions encountered by the bacterium. Despite their crucial role in bacterial adaptation processes, the ECF σ factors, with the exception of AlgU and PvdS, have not been the subject of intense research in P. aeruginosa.
Since the first use of ChIP-chip to study protein-DNA interactions in a bacterium (46), the regulons of various transcription factors have been successfully identified on a global scale in several bacterial pathogens (47–49). In the present study, we applied transcriptional profiling and ChIP-seq under conditions shown to activate the alternative ECF σ factor SigX in order to define the DNA binding consensus sequence of SigX and the primary SigX regulon in P. aeruginosa. The transcriptional data provided in Table S2 reflect the transcriptional profile upon sigX deletion or sigX overexpression in PA14 and comprise also secondary effects, e.g., due to altered expression of other transcription factors. To gain more information about direct targets of the alternative σ factor SigX, we additionally applied the ChIP-seq technique to measure the association of SigX with transcribed regions of DNA in vivo. Categorization of the 624 SigX target promoters as identified by ChIP-seq led to the identification of a large subset of promoter sites upstream of genes that were also differentially regulated in a ΔsigX mutant or sigX-overexpressing strain (18.9%), and more than 24% of those harbored a SigX sequence motif. Since transcriptional profiling measures the consequences of the binding of a protein rather than its actual binding, the transcriptional profile also reflects secondary and tertiary effects on gene expression. This may account for the large number of genes detected to be differentially regulated in a ΔsigX mutant or sigX-overexpressing strain (603 genes, of which 178 also exhibited a SigX motif in the respective promoter sequence) but that did not show SigX binding in vivo. On the other hand, for a large fraction of the in vivo SigX targets (535, of which 73 harbored a SigX motif), no detectable SigX-dependent effect on the transcription of the neighboring genes was observed. This phenomenon of unexpected protein-DNA interactions that could not be identified using transcriptional profiling has been observed before (46, 50, 51) and might be attributed to the fact that the transcriptome profile might have been determined under not fully activated conditions, whereas ChIP-seq involves overexpression of SigX as the DNA binding protein. In addition, ChIP-seq is not a strand-specific method, and one peak might be assigned to two genes. This phenomenon in combination with low motif conservation also provides an explanation for the poor statistical significance of genes within the ChIP-seq/motif search intersection. Despite the fact that the global regulons as determined by ChIP-seq and transcriptional profiling do not fully overlap and that not all of the SigX-regulated genes and those found to bind SigX in vivo also exhibit a consensus DNA binding sequence, we demonstrate that the combination of the techniques plus rigorous statistical testing served well to describe the SigX regulon in P. aeruginosa. It appears that SigX is a master regulator of bacterial adaptation to osmotic stress that impacts more than 250 genes. Of note, we identified not only several genes involved in adaptation/protection, heat shock response, chemotaxis, motility/attachment, and modulation of fatty acid and phospholipid metabolism to belong to the SigX regulon but also virulence and virulence-associated genes linked to the protein secretion/export apparatus or secreted factors. We show that deletion of sigX leads to increased biofilm formation and reduced swarming motility, which is in line with previous studies demonstrating an inverse regulation of swarming and biofilm formation (52–54). Our finding of reduced cytotoxicity and elevated c-di-GMP levels in the sigX deletion mutant suggests that in P. aeruginosa expression of the type III secretion system is enhanced and that c-di-GMP production is repressed in a SigX-dependent manner under low-osmolarity-medium conditions. In a previous study, Bouffartigues and colleagues demonstrated that in the P. aeruginosa strain PAO1, expression of the outer membrane porin OprF is dependent on SigX when the bacteria is cultivated in LB medium. The oprF gene is located 108 bp downstream of the sigX gene and was suggested before to be transcribed both monocistronically and bicistronically (10). Although we observed a slight downregulation of oprF in our PA14 sigX mutant (log2 fold change of −0.726; P = 0.075), oprF is not listed in our transcriptomic data (see Table S2 in the supplemental material) due to the stringent filter criteria. However, both the sigX and oprF promoter regions were successfully immunoprecipitated in our ChIP-seq approach, indicating a direct influence of SigX on oprF expression. In addition, our data show that the gene located upstream of sigX, cmpX, which encodes a thus far uncharacterized membrane protein, is also a member of the SigX regulon. Thereby, SigX seems to directly regulate cmpX expression as the cmpX gene was not only differentially expressed in the sigX mutant as well as in the sigX overexpressing strain but also contains a SigX consensus sequence in its promoter region and was identified in our ChIP-seq experiments.
Overall, much remains to be learned about the physiology of the SigX-mediated response and how the ability of the bacteria to integrate information from multiple sources will be reflected by the pattern of connections among the σ factor regulon and other systems that influence the development of complex phenotypes such as swarming, biofilm formation, cytotoxicity, and fatty acid biosynthesis.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Annette Garbe for perfect technical assistance by performing the c-di-GMP quantification measurements, Esther Surges for fatty acid determinations, Monique Duwe for performing bioluminescence assays, and the Genome Analytics group of the Helmholtz Center for Infection Research for establishing the ChIP-seq libraries and providing Sanger and deep-sequencing service. The A459-Gluc cells were kindly provided by Thomas Pietschmann, Twincore GmbH.
This work was supported by an ERC starter grant (RESISTOME 260276) and by the German Research Foundation (DFG SFB 900) and the Federal Ministry of Education and Research. A.B. was supported by the International Research Training Group 1273 funded by the German Research Foundation, and S.S. was supported by the Helmholtz International Graduate School for Infection Research, grant number VH-GS-202.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01034-13.
REFERENCES
- 1.Brooks BE, Buchanan SK. 2008. Signaling mechanisms for activation of extracytoplasmic function (ECF) sigma factors. Biochim. Biophys. Acta 1778:1930–1945. 10.1016/j.bbamem.2007.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Potvin E, Sanschagrin F, Levesque RC. 2008. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 32:38–55. 10.1111/j.1574-6976.2007.00092.x [DOI] [PubMed] [Google Scholar]
- 3.Hendrickson EL, Plotnikova J, Mahajan-Miklos S, Rahme LG, Ausubel FM. 2001. Differential roles of the Pseudomonas aeruginosa PA14 rpoN gene in pathogenicity in plants, nematodes, insects, and mice. J. Bacteriol. 183:7126–7134. 10.1128/JB.183.24.7126-7134.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazmierczak MJ, Wiedmann M, Boor KJ. 2005. Alternative sigma factors and their roles in bacterial virulence. Microbiol. Mol. Biol. Rev. 69:527–543. 10.1128/MMBR.69.4.527-543.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaturongakul S, Raengpradub S, Wiedmann M, Boor KJ. 2008. Modulation of stress and virulence in Listeria monocytogenes. Trends Microbiol. 16:388–396. 10.1016/j.tim.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho TD, Ellermeier CD. 2012. Extra cytoplasmic function σ factor activation. Curr. Opin. Microbiol. 15:182–188. 10.1016/j.mib.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helmann JD. 2002. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 46:47–110. 10.1016/S0065-2911(02)46002-X [DOI] [PubMed] [Google Scholar]
- 8.Bashyam MD, Hasnain SE. 2004. The extracytoplasmic function sigma factors: role in bacterial pathogenesis. Infect. Genet. Evol. 4:301–308. 10.1016/j.meegid.2004.04.003 [DOI] [PubMed] [Google Scholar]
- 9.Brinkman FS, Schoofs G, Hancock RE, De Mot R. 1999. Influence of a putative ECF sigma factor on expression of the major outer membrane protein, OprF, in Pseudomonas aeruginosa and Pseudomonas fluorescens. J. Bacteriol. 181:4746–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouffartigues E, Gicquel G, Bazire A, Bains M, Maillot O, Vieillard J, Feuilloley MGJ, Orange N, Hancock REW, Dufour A, Chevalier S. 2012. Transcription of the oprF Gene of Pseudomonas aeruginosa is dependent mainly on the SigX sigma factor and is sucrose induced. J. Bacteriol. 194:4301–4311. 10.1128/JB.00509-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schöbel S, Zellmeier S, Schumann W, Wiegert T. 2004. The Bacillus subtilis σW anti-sigma factor RsiW is degraded by intramembrane proteolysis through YluC. Mol. Microbiol. 52:1091–1105. 10.1111/j.1365-2958.2004.04031.x [DOI] [PubMed] [Google Scholar]
- 12.Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203. 10.1016/S0378-1119(98)00601-5 [DOI] [PubMed] [Google Scholar]
- 13.Gödeke J, Heun M, Bubendorfer S, Paul K, Thormann KM. 2011. Roles of two Shewanella oneidensis MR-1 extracellular endonucleases. Appl. Environ. Microbiol. 77:5342–5351. 10.1128/AEM.00643-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi K-H, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391–397. 10.1016/j.mimet.2005.06.001 [DOI] [PubMed] [Google Scholar]
- 15.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 16.Overhage J, Bains M, Brazas MD, Hancock REW. 2008. Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J. Bacteriol. 190:2671–2679. 10.1128/JB.01659-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi K-H, Schweizer HP. 2006. Mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 1:153–161. 10.1038/nprot.2006.24 [DOI] [PubMed] [Google Scholar]
- 18.Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675–690. 10.1046/j.1365-2958.2003.03877.x [DOI] [PubMed] [Google Scholar]
- 19.Dötsch A, Eckweiler D, Schniederjans M, Zimmermann A, Jensen V, Scharfe M, Geffers R, Häussler S. 2012. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS One 7:e31092. 10.1371/journal.pone.0031092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lunter G, Goodson M. 2011. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21:936–939. 10.1101/gr.111120.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourgon R, Gentleman R, Huber W. 2010. Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. U. S. A. 107:9546–9551. 10.1073/pnas.0914005107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shankaranarayanan P, Mendoza-Parra M-A, van Gool W, Trindade LM, Gronemeyer H. 2012. Single-tube linear DNA amplification for genome-wide studies using a few thousand cells. Nat. Protoc. 7:328–338. 10.1038/nprot.2011.447 [DOI] [PubMed] [Google Scholar]
- 24.Aronesty E. 2011. ea-utils: Command-line tools for processing biological sequencing data. http://code.google.com/p/ea-utils
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, Sorek R, Lory S. 2012. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 8:e1002945. 10.1371/journal.ppat.1002945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winsor GL, Lo R, Ho Sui SJ, Ung KS, Huang S, Cheng D, Ching WK, Hancock RE, Brinkman FS. 2005. Pseudomonas aeruginosa Genome Database and PseudoCAP: facilitating community-based, continually updated, genome annotation. Nucleic Acids Res. 33:D338–D343. 10.1093/nar/gki047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abraham W-R, Meyer H, Lindholst S, Vancanneyt M, Smit J. 1997. Phospho- and sulfolipids as biomarkers of Caulobacter sensu lato , Brevundimonas and Hyphomonas. Syst. Appl. Microbiol. 20:522–539. 10.1016/S0723-2020(97)80022-7 [DOI] [Google Scholar]
- 32.Abraham WR, Strömpl C, Bennasar A, Vancanneyt M, Snauwaert C, Swings J, Smit J, Moore ER. 2002. Phylogeny of Maricaulis Abraham et al. 1999 and proposal of Maricaulis virginensis sp. nov., M. parjimensis sp. nov., M. washingtonensis sp. nov. and M. salignorans sp. nov. Int. J. Syst. Evol. Microbiol. 52:2191–2201. 10.1099/ijs.0.02248-0 [DOI] [PubMed] [Google Scholar]
- 33.Gödeke J, Pustelny C, Häussler S. 2013. Recycling of peptidyl-tRNAs by peptidyl-tRNA hydrolase counteracts azithromycin-mediated effects on Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57:1617–1624. 10.1128/AAC.02582-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haid S, Windisch MP, Bartenschlager R, Pietschmann T. 2010. Mouse-specific residues of claudin-1 limit hepatitis C virus genotype 2a infection in a human hepatocyte cell line. J. Virol. 84:964–975. 10.1128/JVI.01504-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gentzsch J, Hinkelmann B, Kaderali L, Irschik H, Jansen R, Sasse F, Frank R, Pietschmann T. 2011. Hepatitis C virus complete life cycle screen for identification of small molecules with pro- or antiviral activity. Antiviral Res. 89:136–148. 10.1016/j.antiviral.2010.12.005 [DOI] [PubMed] [Google Scholar]
- 36.O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461. 10.1046/j.1365-2958.1998.00797.x [DOI] [PubMed] [Google Scholar]
- 37.Spangler C, Böhm A, Jenal U, Seifert R, Kaever V. 2010. A liquid chromatography-coupled tandem mass spectrometry method for quantitation of cyclic di-guanosine monophosphate. J. Microbiol. Methods 81:226–231. 10.1016/j.mimet.2010.03.020 [DOI] [PubMed] [Google Scholar]
- 38.Yeung AT, Torfs EC, Jamshidi F, Bains M, Wiegand I, Hancock RE, Overhage J. 2009. Swarming of Pseudomonas aeruginosa is controlled by a broad spectrum of transcriptional regulators, including MetR. J. Bacteriol. 191:5592–5602. 10.1128/JB.00157-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee DG, Neely AN, Hyodo M, Hayakawa Y, Ausubel FM, Lory S. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. U. S. A. 103:2839–2844. 10.1073/pnas.0511090103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paget MS, Helmann JD. 2003. The σ70 family of sigma factors. Genome Biol. 4:203. 10.1186/gb-2003-4-1-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliveros JC. 2007. VENNY. An interactive tool for comparing lists with Venn diagrams. Spanish Center for Biotechnology, Madrid, Spain: http://bioinfogp.cnb.csic.es/tools/venny/index.html [Google Scholar]
- 42.Schuster M, Hawkins AC, Harwood CS, Greenberg EP. 2004. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol. Microbiol. 51:973–985. 10.1046/j.1365-2958.2003.03886.x [DOI] [PubMed] [Google Scholar]
- 43.Galperin MY, Nikolskaya AN, Koonin EV. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203:11–21. 10.1111/j.1574-6968.2001.tb10814.x [DOI] [PubMed] [Google Scholar]
- 44.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7:263–273. 10.1038/nrmicro2109 [DOI] [PubMed] [Google Scholar]
- 45.Sondermann H, Shikuma NJ, Yildiz FH. 2012. You've come a long way: c-di-GMP signaling. Curr. Opin. Microbiol. 15:140–146. 10.1016/j.mib.2011.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laub MT, Chen SL, Shapiro L, McAdams HH. 2002. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc. Natl. Acad. Sci. U. S. A. 99:4632–4637. 10.1073/pnas.062065699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M. 2009. Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol. Microbiol. 73:1072–1085. 10.1111/j.1365-2958.2009.06832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perkins TT, Davies MR, Klemm EJ, Rowley G, Wileman T, James K, Keane T, Maskell D, Hinton JCD, Dougan G, Kingsley RA. 2013. ChIP-seq and transcriptome analysis of the OmpR regulon of Salmonella enterica serovars Typhi and Typhimurium reveals accessory genes implicated in host colonization. Mol. Microbiol. 87:526–538. 10.1111/mmi.12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, Gomes A, Rustad T, Dolganov G, Glotova I, Abeel T, Mahwinney C, Kennedy AD, Allard R, Brabant W, Krueger A, Jaini S, Honda B, Yu W-H, Hickey MJ, Zucker J, Garay C, Weiner B, Sisk P, Stolte C, Winkler JK, Van de Peer Y, Iazzetti P, Camacho D, Dreyfuss J, Liu Y, Dorhoi A, Mollenkopf H-J, Drogaris P, Lamontagne J, Zhou Y, Piquenot J, Park ST, Raman S, Kaufmann SHE, Mohney RP, Chelsky D, Moody DB, Sherman DR, Schoolnik GK. 2013. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499:178–183. 10.1038/nature12337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grainger DC, Aiba H, Hurd D, Browning DF, Busby SJW. 2007. Transcription factor distribution in Escherichia coli: studies with FNR protein. Nucleic Acids Res. 35:269–278. 10.1093/nar/gkl1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wade JT, Struhl K, Busby SJW, Grainger DC. 2007. Genomic analysis of protein-DNA interactions in bacteria: insights into transcription and chromosome organization. Mol. Microbiol. 65:21–26. 10.1111/j.1365-2958.2007.05781.x [DOI] [PubMed] [Google Scholar]
- 52.Kuchma SL, Brothers KM, Merritt JH, Liberati NT, Ausubel FM, O'Toole GA. 2007. BifA, a cyclic-di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J. Bacteriol. 189:8165–8178. 10.1128/JB.00586-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caiazza NC, Merritt JH, Brothers KM, O'Toole GA. 2007. Inverse regulation of biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J. Bacteriol. 189:3603–3612. 10.1128/JB.01685-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Ditmarsch D, Boyle KE, Sakhtah H, Oyler JE, Nadell CD, Déziel É Dietrich LEP, Xavier JB. 2013. Convergent evolution of hyperswarming leads to impaired biofilm formation in pathogenic bacteria. Cell Rep. 4:697–708. 10.1016/j.celrep.2013.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith SS, Michael MZ, Graham MW. 1989. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 17:3469–3478. 10.1093/nar/17.9.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 57.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U. S. A. 103:2833–2838. 10.1073/pnas.0511100103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. 10.1016/S0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
- 59.Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443–448. 10.1038/nmeth765 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.