

Abstract
Burkholderia cenocepacia and Burkholderia multivorans are opportunistic drug-resistant pathogens that account for the majority of Burkholderia cepacia complex infections in cystic fibrosis patients and also infect other immunocompromised individuals. While they share similar genetic compositions, B. cenocepacia and B. multivorans exhibit important differences in pathogenesis. We have developed reconciled genome-scale metabolic network reconstructions of B. cenocepacia J2315 and B. multivorans ATCC 17616 in parallel (designated iPY1537 and iJB1411, respectively) to compare metabolic abilities and contextualize genetic differences between species. The reconstructions capture the metabolic functions of the two species and give insight into similarities and differences in their virulence and growth capabilities. The two reconstructions have 1,437 reactions in common, and iPY1537 and iJB1411 have 67 and 36 metabolic reactions unique to each, respectively. After curating the extensive reservoir of metabolic genes in Burkholderia, we identified 6 genes essential to growth that are unique to iPY1513 and 13 genes uniquely essential to iJB1411. The reconstructions were refined and validated by comparing in silico growth predictions to in vitro growth capabilities of B. cenocepacia J2315, B. cenocepacia K56-2, and B. multivorans ATCC 17616 on 104 carbon sources. Overall, we identified functional pathways that indicate B. cenocepacia can produce a wider array of virulence factors compared to B. multivorans, which supports the clinical observation that B. cenocepacia is more virulent than B. multivorans. The reconciled reconstructions provide a framework for generating and testing hypotheses on the metabolic and virulence capabilities of these two related emerging pathogens.
INTRODUCTION
Multidrug-resistant pathogens are a severe health concern and can cause chronic infections in a variety of patient populations with limited recourse for treatment. Here, we investigate two multidrug-resistant species, Burkholderia cenocepacia and Burkholderia multivorans, of the Burkholderia cepacia complex (BCC) which are considered dangerous and difficult to treat in patients with cystic fibrosis (CF), chronic granulomatous disease, or compromised immune systems (1). With larger genomes (8.06 Mbp and 7.01 Mbp, respectively) than many other multidrug-resistant pathogens, they also contain an expanded reservoir of genes that may assist their ability to avoid clinical eradication (2–4). Because they are nosocomial, transmissible between patients, and also routinely acquired (and reacquired) from the environment, B. cenocepacia and B. multivorans are the two BCC species most commonly isolated from the sputum of CF patients (5–8). In patients with CF, pulmonary infection with BCC can contribute to the rapid deterioration of lung function known as cepacia syndrome, a necrotizing pneumonia that can lead to bacteremia, septicemia, and increased mortality (9, 10). A combination of high antibiotic resistance and decreased immune function in patients makes B. cenocepacia and B. multivorans extremely difficult to eradicate once lung colonization is established (11, 12). The potential to cause chronic infection as well as rapid decline in health makes BCC spp. clinically important emerging human pathogens.
Further complicating treatment is the variability in infection course and severity in patients infected with B. cenocepacia or B. multivorans (or occasionally both species). Compared to patients infected with Pseudomonas aeruginosa, the most common CF lung pathogen in adults, patients infected with BCC can have a substantially worse prognosis (13, 14). Cepacia syndrome has historically been associated with B. cenocepacia, but infections with B. multivorans have on occasion also induced this syndrome (8). B. cenocepacia causes greater mortality than B. multivorans and has also been seen to replace B. multivorans infections (15). However, many centers in multiple countries are showing a shift within BCC infection incidence from predominantly B. cenocepacia to B. multivorans infections that is not fully understood (8). These differences between B. cenocepacia and B. multivorans provide motivation for a comparative systems analysis of these two BCC species. Comparing and contrasting the metabolic functions of B. cenocepacia and B. multivorans can help elucidate their metabolic adaptability, mechanisms of pathogenicity, and other underlying contributors to differing clinical outcomes, such as cepacia syndrome.
Genome-scale metabolic reconstructions of multiple organisms can be used to study and predict phenotypic differences between related species or strains based on their genetic content (16). For example, a recent study used the framework of two previously published reconstructions of Staphylococcus aureus N315, the ERGO bioinformatics suite and the KEGG pathway suite, to assemble a consensus species-level reconstruction of S. aureus. Strain-specific enzyme annotations were then incorporated to develop 13 reconciled models of S. aureus strains that identified differences in common essential enzymes (17). Meaningful biological comparison becomes more complex when using species-level reconstructions. These reconstructions must be intensively curated in such a way that all artifacts from the model-building process are reconciled, as performed in the reconstruction-based comparison of two species of Pseudomonas (18). Artifacts that must be reconciled include differences in naming conventions, reaction stoichiometries, and, importantly, annotations of gene functions and their implementation in gene-protein-reaction (GPR) associations. Proper reconciliation of genome-scale metabolic reconstructions is crucial for studying the genetic and phenotypic differences between the organisms.
Here, we present reconciled genome-scale metabolic reconstructions of B. cenocepacia and B. multivorans, two of the largest bacterial reconstructions built to date in terms of the numbers of genes and reactions incorporated, correlating with the large genomes of the pathogens. A previously published reconstruction of B. cenocepacia J2315, iKF1028, using the ToBiN reconstruction platform, accounted for 859 reactions, 834 metabolites, and 1,028 genes and was used as a reference for our curation efforts (19). However, we chose to begin with draft reconstructions that were both built using the Model SEED tool (20), currently the most widely used publicly available reconstruction platform, to aid our comparative analyses. Our B. cenocepacia reconstruction is substantially larger than iKF1028, while including much of the knowledge gained from this prior study. The reconstructions for B. cenocepacia J2315 and B. multivorans ATCC 17616 were developed and manually curated in parallel to ensure consistency in all aspects of the model-building process. In particular, the two reconstructions were reconciled in the annotation and assignment of orthologous gene functions, which span 1,437 common metabolic reactions.
The reconstructions of B. cenocepacia J2315 and B. multivorans ATCC 17616 are denoted formally as iPY1537 and iJB1411, respectively, following established naming conventions (21). However, for the sake of clarity, iPY1537 and iJB1411 will be referred to as iBC and iBM throughout this article for “in silico B. cenocepacia” and “in silico B. multivorans,” respectively. The reconstructions were validated by comparing growth predictions with substrate utilization experiments using B. cenocepacia J2315, B. cenocepacia K56-2, and B. multivorans ATCC 17616. Through the reconstruction and reconciliation process, we reannotated the functions of a collection of genes. Predictions of genes essential for growth were made by simulating in silico growth of iBC and iBM, which has relevance in drug target identification. Pathogenic characteristics of the two species were compared by predicting the production capacity of an array of metabolites involved in virulence. This study provides a framework for investigating the metabolic architectures of two clinically important BCC species, emphasizing metabolic connections to virulence and pathogenicity, as well as a guide for parallel reconstruction and comparison of genome-scale metabolic networks of related organisms.
MATERIALS AND METHODS
Metabolic reconstructions of B. cenocepacia and B. multivorans.
The complete sequenced genomes of B. cenocepacia J2315 and B. multivorans ATCC 17616 were used as the starting points for the metabolic network reconstruction process shown in Fig. 1. The J2315 strain of B. cenocepacia was isolated from a cystic fibrosis patient (22), and the ATCC 17616 strain of B. multivorans was an environmental isolate obtained from the soil (23). The DNA genomic sequences of B. cenocepacia J2315 (GenBank accession no. NC_011000 to NC_011003) and B. multivorans ATCC 17616 (GenBank accession no. NC_010084, NC_010086, NC_010087, and NC_010070) were downloaded from the NCBI GenBank database.
FIG 1.
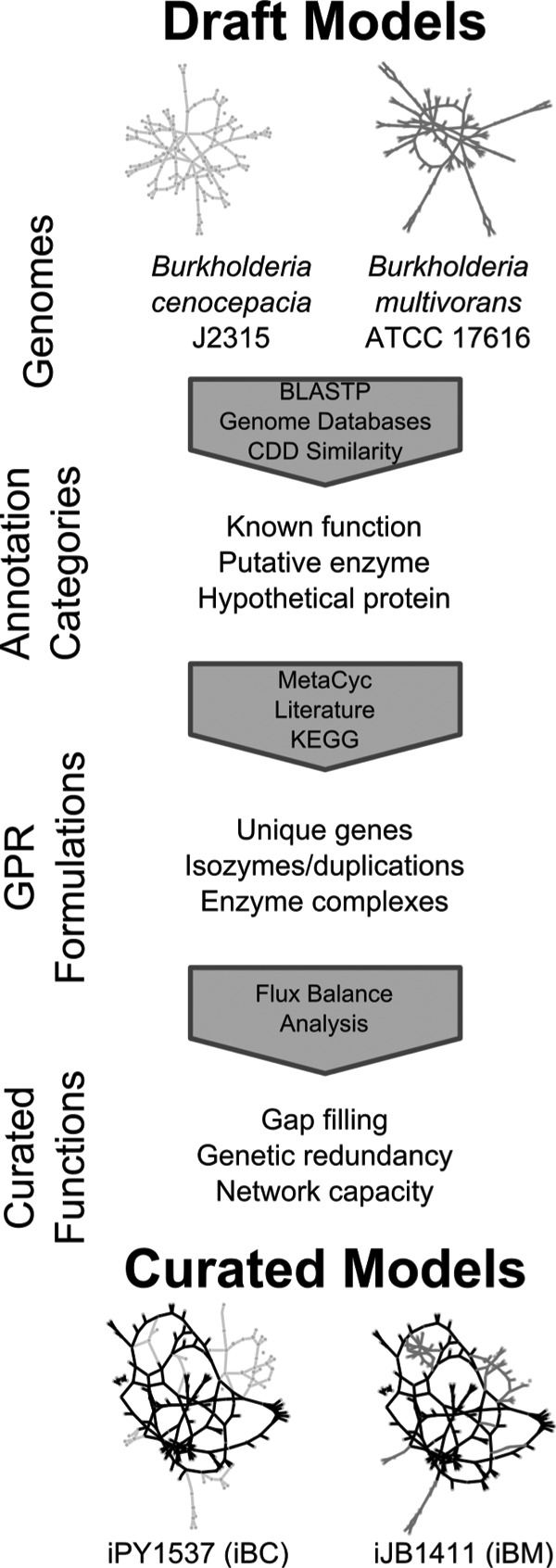
Overview of the reconstruction and reconciliation process. The annotated genomes of B. cenocepacia J2315 and B. multivorans ATCC 17616 were used to generate automated draft metabolic reconstructions using Model SEED (20, 70). The gene-protein-reaction associations of the draft reconstructions were then further manually curated and annotated using information from online databases and literature references. Curation of the two reconstructions was done in parallel to ensure the proper assignment of homologous and orthologous genes. The reconstructions were then functionally curated through iterative validations of in silico and experimental data. The final reconciled models for B. cenocepacia and B. multivorans are designated iPY1537 and iJB1411, respectively, but are referred to throughout as iBC and iBM for clarity. Abbreviations: GPR, gene-protein-reaction; BLASTP, Protein Basic Local Alignment Search Tool; CDD, Conserved Domain Database; KEGG, Kyoto Encyclopedia of Genes and Genomes.
The Model SEED was used to generate draft reconstructions of B. cenocepacia and B. multivorans (20). The Model SEED requires annotated genomes as input in order to produce draft reconstructions. While the finished annotation for B. cenocepacia had already been uploaded from NCBI and converted to the SEED format, the genome of B. multivorans was not included in the SEED database. The genome of B. multivorans was downloaded from the NCBI Genome database and was submitted to the RAST server for annotation (24), and then the Model SEED was used to produce draft reconstructions of both species. Differences in annotation quality between these submission methods were evident in the resulting draft reconstructions and addressed during the manual curation process. We allowed SEED to gap-fill needed reactions to enable growth on SEED's complex medium formulation (all exchange reactions open) and default biomass formulation in the draft construction process.
We then used biological databases such as KEGG (25), Metacyc (26), and the Burkholderia Genome Database (BGD) (27) to evaluate the draft reconstructions. Specifically, we used the gene and reaction annotations in KEGG and compared them with those of the draft reconstructions. Protein BLAST was used to determine genes that likely code for isozymes, both within each genome as well as between the two genomes. This process expanded the size of the models, as well as reduced the number of non-gene-associated reactions (gap-filled by SEED) needed in rich media to 15 reactions (11 unique enzymes), as shown in Table S1 in the supplemental material. Ten more reactions were used in gap-filling virulence-related pathways and growth under particular minimal medium conditions. Thirty-seven reactions and 39 reactions were removed from the original SEED drafts of iBC and iBM, respectively, due to low annotation evidence, low likelihood of enzyme activity, or infeasible loops formed in the model. The final curated models iPY1537 and iJB1411 are available as spreadsheets in the supplemental material and as SBML files on the Papin lab website (http://www.bme.virginia.edu/csbl/downloads-burkholderia-cepacia.php).
FBA of iBC and iBM.
Flux balance analysis (FBA) was used to assess in silico growth of iBC and iBM (28). Mathematically, the FBA problem is formulated as follows: maximize vbiomass such that Sv = 0 and vmin ≤ v ≤ vmax.
In the FBA framework, the reactions of a metabolic network are represented using a stoichiometric matrix, S, such that the rows of S correspond to the metabolites and the columns correspond to the reactions. The elements of the matrix are the stoichiometric coefficients such that Sij is the coefficient for metabolite i in reaction j. At steady state, we have the relation Sv = 0, where the vector v represents the fluxes of each of the reactions. The fluxes are conventionally in units of mmol/gDW/h (where gDW is g dry cell weight), with the exception of vbiomass, which has units of h−1 (representative of the growth rate). Because Sv = 0 is underdetermined, FBA uses linear programming to optimize the flux through an objective reaction (typically, the biomass reaction, denoted as vbiomass). The biomass reaction includes the relative weights of the metabolites required to form biomass and details of its formulation are described below and in the supplemental material. vmin and vmax are vectors of the upper and lower bounds of the reaction fluxes, respectively. Consequently, medium conditions are imposed by constraining the bounds of the exchange reactions, which represent the uptake and secretion of metabolites in the model (e.g., vmin < 0 for a metabolite that is imported and vmax > 0 for a metabolite that is secreted). The formulations of the different medium conditions are described in the supplemental material. All FBA simulations were conducted using the COBRA Toolbox in MATLAB (29).
Biomass reaction formulation.
Burkholderia-specific biomass reactions were formulated for iBC and iBM. To evaluate the capability of growth in a variety of environments and genetic backgrounds, the biomass reactions are often set as the objective functions in FBA, and the numerical value of the flux through the biomass reactions corresponds to the theoretical maximal growth rates under a given medium condition (30). The constituents of the biomass reactions represent the molecular components that are required for growth. These molecular components are grouped according to the main macromolecular components, which include protein, DNA, RNA, lipopolysaccharide (LPS), peptidoglycan, glycogen, lipids, and polyamine pools. Burkholderia species exhibit unique fatty acid and lipid compositions, such as the presence of fatty acids containing cyclopropane rings, and these were accounted for in the formulation of the composition of the biomass reaction (31). The biochemical reactions needed to synthesize many of these unique lipids and fatty acids have been included in the reconstructions based on work done previously (19). The stoichiometric coefficients for constituents of the biomass reactions of iBC and iBM were based on experimental evidence for Burkholderia and related organisms where possible. Otherwise, values from the well-annotated biomass reaction of Escherichia coli were used (32). Values specific to the two species were used when possible (e.g., RNA, DNA, and protein). Otherwise, for constituents not specific to either species, the same value was used for both reconstructions (e.g., lipids, fatty acids, cell wall, and ATP maintenance). The supplemental material describes the full process of formulating the Burkholderia-specific biomass reactions based on established methods (33).
Predicting essential genes.
Essential genes are defined as genes for which there is no in silico growth under a given medium condition (maximum flux through vbiomass = 0) when the reaction or reactions associated with that gene are removed from the network. To simulate the loss of a gene in the model, gene-protein-reaction (GPR) associations are evaluated to determine which reactions are not allowed to carry flux when the gene is removed. GPRs link genetic information, such as isozymes, gene duplications, and enzyme complexes, to the reactions in the reconstructions using Boolean logic. Perturbations of the GPRs enable the prediction of phenotypic changes as a function of genetic changes. For example, the fluxes for the reactions affected by a gene deletion are constrained by setting the corresponding vmax and vmin to 0, and the model is then optimized for growth to evaluate the effect of the missing gene.
In silico medium conditions.
Medium conditions were formulated for lysogeny broth (LB), M9 minimal medium, and synthetic cystic fibrosis medium (SCFM) as in previous publications (34, 35). The medium conditions were defined by setting the lower and upper bounds of the exchange reactions to specify metabolites that are available or unavailable. Full descriptions of the medium conditions are presented in the supplemental material. All medium conditions allow for the exchange of typical salts and water. LB contains amino acids, glucose, and salts. SCFM contains lactate in addition to similar carbon sources to LB; in addition, the uptake rates of all nutrients present in SCFM are constrained to 10 mmol/gDW/h to represent the reduced nutrient availability in the cystic fibrosis lung environment, as done elsewhere (36). In M9 minimal medium, a single carbon source is enabled for uptake at 10 mmol/gDW/h to represent tested compounds in in vitro Biolog growth screening (34).
Prediction of virulence factor production capacity.
Virulence factor production capacity was assessed by adding a demand reaction to the model for each virulence-related compound. Maximizing the flux through this reaction allows prediction of the maximum theoretical production level under a given medium condition, such as with each of the individual carbon substrates in SCFM. The trade-off between growth and virulence factor production was assessed by maximizing the flux through a demand reaction for each factor while constraining the biomass flux to various percentages of its maximum value when simulated under SCFM conditions.
Network visualization.
MetDraw was used to generate visual representations of the metabolic networks (http://www.metdraw.com; P. A. Jensen and J. A. Papin, submitted for publication). SBML versions of the models were used as inputs to MetDraw, and the resulting network visualizations were output as SVG files.
Growth phenotype screening.
We conducted growth screening using Biolog PM1 and PM2A microplates (Biolog, Inc., Hayward, CA). Instead of adding redox dye and measuring oxidation of each carbon source included in the screen, we evaluated growth in each well using optical density measurements at 700 nm (OD700) to avoid error due to pigment production. Bacteria were scraped from an LB plate and resuspended in Biolog inoculating fluid PM IF-0a GN/GP to reach an OD of 0.07. Biolog microplate wells were inoculated with 100 μl of this suspension, and plates were then incubated at 37°C for 48 h. OD700 was measured at time zero and every 12 h thereafter. Experiments with B. cenocepacia J2315 were carried out using two biological replicates each for PM1 and PM2A, while experiments with B. cenocepacia K56-2 and B. multivorans ATCC 17616 were performed using three biological replicates. Growth was evaluated based on the resulting growth curves and maximum change in OD for each carbon source compared to the maximum change in OD of inoculated control wells with no carbon source. Additionally, we evaluated the ability of each organism to catabolize cysteine and tryptophan, which are amino acids present in cystic fibrosis lung sputum but not included in the Biolog plates (35). These amino acids were each added at concentrations of 20 mM to M9 salts to create cysteine and tryptophan minimal media. Colonies of each Burkholderia strain were scraped from LB plates and resuspended to reach an OD of 0.07 in each respective minimal medium. Three replicates of 200 μl of each inoculated strain-medium combination were plated in a 96-well microplate and grown under conditions replicating the Biolog screen.
An alternate strain-specific network reconstruction of B. cenocepacia K56-2 was created by conducting a genome-scale reciprocal BLASTP with an E value cutoff of 0.01 with no low-complexity filter using CLC Main Workbench (CLC bio, Aarhus, Denmark). The genome sequence of B. cenocepacia K56-2, a strain isolated from a cystic fibrosis patient, has recently been published (Genbank accession no. ALJA00000000). Protein sequences were derived from annotated open reading frames (ORFs) in B. cenocepacia J2315 and B. cenocepacia K56-2 (83). Results of this BLASTP comparison are included in Table S2 in the supplemental material. Nearly all genes incorporated in iBC matched genes present in B. cenocepacia K56-2, and the 7 genes that did not have significant matches (E value of >10−30) did not affect predictions in this study.
RESULTS
Metabolic network reconstructions.
Here, we present the metabolic networks of B. cenocepacia and B. multivorans as valuable tools that we use as a framework to perform our comparative analyses (Fig. 1). Figure 2A provides a quantitative comparison of the reconstructions in contrast to the previously published B. cenocepacia J2315 reconstruction (iKF1028) and the reconstruction of the closely related pathogen Pseudomonas aeruginosa PAO1 (iMO1086). It also highlights the substantial increase in scope of the reconciled network reconstructions. iBC accounts for the functions of 1,537 genes, 1,506 reactions, and 1,280 metabolites. iBM accounts for the functions of 1,411 genes, 1,473 reactions, and 1,245 metabolites. Most of the reactions for each reconstruction are broadly concentrated in lipid, amino acid, and carbohydrate metabolism (Fig. 2B), but reactions in over 60 canonical metabolic pathways (as classified by KEGG) are included in the reconstructions. Figure 2C shows a map of the reconstructions that contrasts the shared and unique metabolic reactions of each species. (A high-resolution version can be found in Fig. S1 in the supplemental material.) iBC has nearly double the number of unique reactions compared to iBM as well as a substantially higher number of incorporated genes (Fig. 2A). There are more unique reactions in iBC in the amino acid and carbohydrate subsystems; however, iBM has more unique reactions in the lipid subsystem (Fig. 2B). The reconstructions enable functional comparative analysis of these differences through examination of gene essentiality, growth capacity, and virulence factor production capabilities.
FIG 2.

Statistics of metabolic network reconstructions. (A) Statistics of iBC and iBM are shown. For reference, statistics for the previously reconstructed models iMO1086 (P. aeruginosa PAO1) and iKF1028 (B. cenocepacia J2315) are listed. (B) Distribution of the reactions in iBC and iBM by metabolic pathways are presented. The light gray, dark gray, and black stacked bars show the reactions that are shared, unique to iBC, and unique to iBM, respectively. Numbers next to the dark gray and black bars indicate their numerical values. (C) A visualization of iBC and iBM shows the reactions unique to iBC, reactions unique to iBM, and reactions common to both reconstructions as red, blue, and gray lines, respectively.
Model curation and validation.
Substantial manual curation of the draft Model SEED reconstructions was performed in order to build an up-to-date knowledgebase of genetic and metabolic information for B. cenocepacia and B. multivorans and resulted in the addition of a substantial number of reactions. For example, we added 13 reactions to iBC involved in tryptophan catabolism and 17 reactions to iBM involved in phenylalanine catabolism that were missing from the original Model SEED draft reconstructions; species-specific functionality of the associated pathways had been investigated in Burkholderia cenocepacia J2315 in the literature (37, 38). After databases and literature were canvassed as explained in Materials and Methods, B. cenocepacia J2315 and B. multivorans ATCC 17616 were experimentally tested for growth on Biolog phenotype microplates to generate in vitro data to guide further curation decisions and enable validation of substrate utilization predictions.
The Biolog screen of B. cenocepacia J2315 showed markedly limited growth under many of the in vitro minimal medium conditions, ranging from no apparent growth to growth at a substantially lower rate than B. multivorans ATCC 17616 on many substrates. To further investigate this outcome, we also performed identical Biolog screens on B. cenocepacia K56-2 after noting nearly identical genomic content with respect to the metabolic genes incorporated in iBC, as shown in Table S2 in the supplemental material. Given the high sequence similarity between the B. cenocepacia strains, we used iBC to predict B. cenocepacia growth, which we compared with equivalent B. cenocepacia K56-2 in vitro data (Fig. 3) and B. cenocepacia J2315 in vitro data. The same analysis was performed with iBM and B. multivorans ATCC 17616. Complete in vitro data and in silico growth comparisons for all assessed strains are included in Table S3 in the supplemental material.
FIG 3.

Validation of in silico growth predictions. Gray and white boxes indicate the growth ability of bacteria provided a single carbon source. “D” denotes discrepancies between experimental and in silico results.
When comparing growth between the two B. cenocepacia strains on the 192 carbon sources, 53 substrates enabled quantifiable growth of both J2315 and K56-2. Twenty-eight substrates did not enable quantifiable growth of J2315, in contrast to successful utilization by K56-2, while only 4 substrates enabled quantifiable growth of J2315 but no quantifiable growth of K56-2. Biolog plates introduce temporal limitations to assessing growth because of potential effects of evaporation after 48 h of incubation that complicate the evaluation of a slow-growing strain like J2315. The limited growth time frame as well as potential unaccounted for regulatory differences between strains may in combination explain the difference between in vitro results and in silico predictions that reflect the collected genetic evidence for the catabolic pathways in iBC. At minimum, this is an opportunity for comparative study in the future, as we found no metabolic evidence for the growth defects of J2315 compared to K56-2 within the context of our expansive models. Perhaps certain enzymes integral to basic metabolic processes are operating at low efficiency due to point mutations, or there are alterations in transcriptional control that could be addressed via a future integration of regulatory and metabolic reconstructions. In light of this, we chose to use B. cenocepacia K56-2 in vitro data when assessing the accuracy of our growth predictions as growth versus no growth on a given substrate was possible to assess confidently with the Biolog plates.
Of the 190 Biolog carbon source substrates experimentally tested, 102 substrates were accounted for in iBC and iBM and could be tested for in silico growth using FBA. In addition, growth on the two amino acids cysteine and tryptophan was separately evaluated given their presence in the CF lung, and discrepancies were identified during the curation process. After further curation efforts, iBC and iBM correctly matched the experimental growth phenotypes of 53 substrates for which transporters were incorporated in the reconstructions (44 correct growth and 9 correct no-growth phenotypes each). For the other 31 substrates for which transporters were incorporated, iBC, iBM, or both failed to match experimental growth phenotypes. B. cenocepacia and B. multivorans did not exhibit growth in vitro on 14 substrates, and because iBC and iBM lack transporters for these substrates, they predicted correct no-growth phenotypes. Finally, 6 substrates did not have transporters in iBC and iBM; however, B. cenocepacia, B. multivorans, or both grew on the substrates in vitro. Overall, iBC and iBM predicted the correct growth phenotype with 75% and 76% accuracy (78 and 79 out of 104 substrates), respectively (see Table S3 in the supplemental material). Sixty-seven substrates enabled the same accurate in silico and in vitro phenotypes in both models, 13 substrates showed the same type of discrepancy between in silico and in vitro phenotypes in both models, and the remaining 24 substrates showed discrepancies in phenotype between B. cenocepacia and B. multivorans. Although the remaining 88 carbon sources were not incorporated in the reconstructions and thus not analyzed in silico, B. cenocepacia and B. multivorans were capable of growing on 24 and 32 additional substrates in vitro, respectively. These data are provided in Table S3.
With curation efforts guided by the in vitro data, iBC and iBM both underpredict growth on lysine, and iBM underpredicts growth on tryptophan (Fig. 3). Genomic evidence to support the addition of reactions that would fix these inconsistencies could not be found. For example, no evidence for several key reactions in lysine degradation could be found in iBC (glutarate-coenzyme A [CoA] ligase [EC 6.2.1.6], 3-hydroxyacyl-CoA dehydrogenase [EC 1.1.1.35]). The difference in branched-chain amino acid catabolism between reconstructions is due to the identification of three B. cenocepacia genes that appear to have high sequence similarity with the bkd operon of other bacteria, which enables catabolism of isoleucine, leucine, and valine (39). These genes were not present in B. multivorans. However, neither species is capable of in vitro growth on valine or leucine, and B. multivorans is unexpectedly capable of in vitro growth on isoleucine (Fig. 3).
These differences in experimental and in silico growth are potentially due to incomplete and/or incorrect annotations of genes in the databases. For example, while we explored homology of current hypothetical proteins with known enzymes in lysine catabolism, the homology may be below the thresholds we used, or novel lysine degradation reactions may exist. Another possible reason for the discrepancy is control by regulatory systems not accounted for in our current metabolic network reconstruction. These differences present opportunities for further study of metabolism of the BCC.
Through our curation of the models from the SEED drafts to the presented final versions, iBC gained 269 reactions and lost 116 reactions, and GPRs were substantively altered in a further 324 reactions. iBM gained 297 reactions, lost 106 reactions, and had a further 352 reactions subjected to GPR alterations. In summary, more than a third of each model's individual content is different from the original draft reconstructions due to major additions and improvements to both reactions and GPR formulations during the curation and reconciliation process, resulting in the identification of 1,437 reactions common to both iBC and iBM. The scope of these changes emphasizes the value of manual curation after automated network reconstruction, and the functional implications of these differences are presented below.
Refinement of genome annotation.
Manual curation of iBC and iBM aided in the identification of many proposed genome annotation refinements of B. cenocepacia and B. multivorans. A subset of our changes and additions to GPRs were based on the assignment of novel function to genes previously annotated as coding for hypothetical or putative proteins. Many of the annotation refinements were the identification of potential isozymes and duplicated genes for a given reaction, but refinements also included genes that fill in gaps in model pathways and putative functional assignment of 10 hypothetical proteins. Table 1 summarizes these annotation refinements and a selection of other high-confidence refinements of interest. A table of moderate confidence annotations and list of reactions that are missing gene associations are included in Table S1 in the supplemental material. Examples of genome annotation refinements are presented below.
TABLE 1.
Proposed reannotations of gene functions resulting from the curation and reconciliation process
|
B. cenocepacia |
B. multivorans |
Proposed annotation | Gene name | Protein ID | Evidence | ||
|---|---|---|---|---|---|---|---|
| Locus tag | Current annotation | Locus tag | Current annotation | ||||
| BCAS0031 | Hypothetical protein | Bmul_6140 | Hypothetical protein | l-2,4-Diaminobutyric acid transaminase | doeD | EC 2.6.1.- | E values of 1E−154 and 1E−155, respectively, to Helo_3661 in H. elongata (40) |
| BCAM1003 | Putative epimerase | Bmul_4613 | NAD-dependent epimerase/dehydratase | GDP-d-mannose reductase | bceM | EC 1.1.1.187 | E values of 1E−141 and 1E−129, respectively, to Bcep1808_4471 in B. vietnamiensis (41) |
| BCAS0105 | Hypothetical protein | — | — | dTDP-4-dehydrorhamnose reductase | rfbD | EC 1.1.1.133 | E value of 1E−139 to Bmul_6045, which is annotated as a dTDP-4-dehydrorhamnose reductase |
| BCAM2148 | Hypothetical protein | Bmul_3704 | Hypothetical protein | Malate dehydrogenase | mdh | EC 1.1.1.37 | E values of 7E−30 and 3E−27 to BCAM1263 and Bmul_4451, respectively, which are annotated as malate/l-lactate dehydrogenases |
| BCAS0285 | Hypothetical protein | — | — | Argininosuccinate lyase | argH | EC 4.3.2.1 | CDD E value of 3E−27 to argininosuccinate lyase; E value of 9E-17 to Bcep1808_5167, which is annotated as an argininosuccinate lyase in B. vietnamiensis |
| —a | — | Bmul_6038 | Hypothetical protein | 4-Carboxy-4-hydroxy-2-oxoadipate (CHA) aldolase | galC | EC 4.1.3.17 | CDD E value of 1E−95 to CHA aldolase; E value of 2E−80 and 95% coverage to PP_2514 in Pseudomonas putida (81); no ortholog in B. cenocepacia |
| BCAM0581 | Enoyl-CoA hydratase | Bmul_5121 | Enoyl-CoA hydratase | 3-Hydroxydodecanoyl-ACP hydratase/cis-2-dodecenoyl-ACP thioesterase | In vitro characterization of BCAM0581 showed bifunctional product (40); Bmul_5121 has E value of 1E−162 to BCAM0581 | ||
| — | — | Bmul_3969 | Autoinducer synthesis protein | N-Acylhomoserine lactone synthase | cepI | EC 2.3.1.184 | E value of 8E−81 to BCAM1870, which is annotated as cepI |
| BCAL2418 | Hypothetical protein | Bmul_0955 | 6-Phosphogluconolactonase | Gluconolactonase | ppgL | EC 3.1.1.17 | E values of 2E−84 and 2E−83 to PA4204 in P. aeruginosa, which codes for a characterized periplasmic gluconolactonase (43) |
| BCAL1668 | Putative amino acid transport system | Bmul_1621 | Extracellular solute-binding protein | Cysteine ABC transporter | cjaA | CjaA | E values of 1E−25 and 1E−25 to Cj0982 in Campylobacter jejuni, which was shown to be a high-affinity cysteine transporter (44) |
| BCAS0145, BCAL1224, BCAS0735, BCAM1221, BCAM0361 | Metallo peptidase, allantoate amidohydrolase | Bmul_5349, Bmul_4490, Bmul_2063, Bmul_5235 | Allantoate amidohydrolase | β-Ureidopropionase | EC 3.5.1.6 | All genes have E values of <1E−100 to PP_0614 in P. putida, which was shown to be a β-ureidopropionase (82) | |
| BCAL1428 | Putative amine catabolism-related protein | Bmul_1894 | Xylose isomerase domain-containing protein | myo-Inosose-2 dehydratase | iolE | EC 4.2.1.44 | E values of 2E−56 and 4E−58 to SMc00433 in Sinorhizobium meliloti (46) |
| BCAL1429 | Putative TPP-binding acetolactate synthase | Bmul_1893 | Thiamine pyrophosphate protein | 3d-(3,5/4)-Trihydroxycyclohexane-1,2-dione hydrolase | iolD | E values of 1E−168 and 1E−166 to SMc01166 in S. meliloti (46) | |
| BCAL1427 | myo-Inositol catabolism protein | Bmul_1895 | myo-Inositol catabolism IolB domain-containing protein | 5-Deoxy-d-glucuronate isomerase | iolB | EC 5.3.1.- | E values of 3E−71 and 2E−71 to SMc00432 in S. meliloti (46) |
| BCAL1430 | Putative carbohydrate kinase | Bmul_1892 | Ribokinase-like domain-containing protein | 5-Dehydro-2-deoxygluconokinase | iolC | EC 2.7.1.92 | E values of 1E−147 and 1E−143 to SMc01165 in S. meliloti (46) |
| BCAL2435 | Na+/solute symporter family protein | Bmul_0940 | Na+/solute symporter | Monocarboxylate permease | mctP | MctP | E values of 1E−161 and 1E−155 to mctP in Rhizobium leguminosarum, which was shown to code for a pyruvate transporter (47) |
Dashes indicate the absence of a corresponding reannotation in one of the genomes.
(i) Ornibactin synthesis.
During our refinement process, we added pathways enabling the synthesis of ornibactin, a key Burkholderia-specific siderophore that the literature indicates is associated with increased virulence of some clinical isolates (48). A model of ornibactin synthesis has been proposed based on a study of B. cenocepacia 715j transposon mutants identifying nonribosomal peptide synthases and associated enzymes grouped into an operon (the orb operon) that enables ornibactin production (49). This operon of 14 genes involved in ornibactin synthesis, export, and uptake was annotated in the BGD for B. cenocepacia J2315. However, despite identification of a likely orb operon induced by iron-responsive regulator Fur in B. multivorans ATCC 17616 (42), most of the B. multivorans genes are annotated as putative enzymes or hypothetical proteins in the BGD. We have incorporated the proposed synthesis pathways in both reconstructions and include the associated annotation updates in Table S1 in the supplemental material.
(ii) Ectoine degradation.
As another example of a refinement that involves novel assignment of gene function, Fig. 4A shows our proposed annotation of orthologous hypothetical proteins encoded by BCAS0031 and Bmul_6140 as an essential enzyme in ectoine degradation. Ectoine, produced by many prokaryotes as a solute that assists in maintaining an osmotic equilibrium, can be used as a growth substrate by environmental bacteria, including some halophilic and soil bacteria (40). Ectoine production has been shown to be upregulated under stress conditions and, interestingly, in mutant strains of soil bacteria that are resistant to antibiotics targeting the cell envelope (e.g., Streptomyces coelicolor) and protein synthesis (e.g., Nocardiopsis sp. strain FU40) (50, 51). These studies theorize that ectoine upregulation may be part of a broad secondary metabolism response to changes in regulation due to resistance-related mutations. Through literature mining and similarity searches via BLASTP, we determined that BCAS0031 and Bmul_6140 have high similarities to Helo_3661 in Halomonas elongata DSM 2581 (E values of 10−154 and 10−155, respectively) (40). Through mutational studies, Helo_3661 was found to be an l-2,4-diaminobutyric acid transaminase (DoeD) and BCAS0031 and Bmul_6140 and the associated reactions in iBC and iBM were subsequently included. These genes are also part of apparent operons that also contain doeABC, lending credence to this reannotation (40). A search for sequences similar to BCAS0031 and Bmul_6140 across all bacteria via BLASTP showed that many of the orthologous genes were annotated as encoding hypothetical proteins or generic class III aminotransferases. This refinement enabled a fully functional ectoine degradation pathway in the models that had not been previously identified in B. cenocepacia and B. multivorans genome databases or the literature.
FIG 4.

Examples of network refinements and reannotations. (A) Both B. cenocepacia (BC) and B. multivorans (BM) genomes include genes that allow for the degradation of ectoine. BCAS0031 and Bmul_6140, the products of which are currently annotated as hypothetical proteins, were identified as orthologs of Helo_3661 in H. elongata, which encodes DoeD. (B) Curation of the tryptophan degradation pathway identified a gap for the essential reaction catalyzed by Kmo, which led to the addition of Kmo to both iBC and iBM without an assigned gene. The gene for HaaO was present only in B. cenocepacia and allows for the synthesis of quinolinate. (C) B. cenocepacia has the rhlABC genes necessary for rhamnolipid synthesis, while B. multivorans does not. Abbreviations: DoeA, ectoine hydrolase; DoeB, N-α-acetyl-l-2,4-diaminobutyric acid deacetylase; DoeC, aspartate-semialdehyde dehydrogenase; DoeD, diaminobutyric acid transaminase; KynA, tryptophan 2,3-dioxygenase (EC 1.13.11.11); KynB, kynurenine formamidase (EC 3.5.1.9); KynU, kynureninase (EC 3.7.1.3); Kmo, kynurenine 3-monooxygenase (EC 1.14.13.9); HaaO, 3-hydroxyanthranilate-3,4-dioxygenase (EC 1.13.11.6); RhlA, rhamnosyltransferase chain A; RhlB, rhamnosyltransferase chain B; RhlC, rhamnosyltransferase chain 2.
(iii) Cepacian synthesis.
Another type of model refinement is the gap-filling of metabolic pathways that are known to be present in Burkholderia. For example, as part of our effort to include pathways for the biosynthesis of virulence-related compounds, reactions required for the production of the Burkholderia exopolysaccharide cepacian were added. Literature evidence suggests that the production of GDP-d-rhamnose, one of the sugar residues of cepacian, is catalyzed by a GDP-d-mannose reductase (RMD) (41). In Burkholderia vietnamiensis G4, the RMD protein is encoded by Bcep1808_4471, also known as bceM (52). A similarity search of Bcep1808_4471 via BLASTP shows that bceM in B. cenocepacia and B. multivorans is carried by BCAM1003 and Bmul_4613 (E values of 10−141 and 10−129, respectively). Although the BCAM1003 and Bmul_4613 products are currently listed as a putative epimerase and an NAD-dependent epimerase/dehydratase in the BGD, respectively, they have been assigned as RMDs in iBC and iBM, and we suggest a consistent update to the database annotations.
(iv) Tryptophan catabolism.
The in vitro carbon source growth screens further aided in identifying gaps in iBC and iBM. In our growth screens, B. cenocepacia and B. multivorans were observed to grow on M9 minimal medium supplemented with tryptophan as the sole carbon source. However, the draft reconstructions initially could not grow in silico under these conditions. Literature mining revealed that kynurenine-3-monooxygenase (Kmo) catalyzes an essential reaction in the tryptophan degradation pathway (Fig. 4B). A study identified orthologs of a cluster of tryptophan catabolic genes similar to those in Bacillus cereus 10897 in B. cenocepacia J2315, with the exception of the gene encoding Kmo, which suggests the existence of a nonorthologous form of Kmo in Burkholderia (37). The reaction catalyzed by Kmo was then added to iBC and iBM without a gene assignment. This model refinement identifies a current gap in our genomic knowledge of Burkholderia, even though there is evidence for the enzymatic reaction (Fig. 4B) and the experimental screen provides evidence of its assumed function.
Further curation of the tryptophan degradation pathway led to refinements that showed species-specific differences. Literature evidence has shown that hydroxyanthranilate-3,4-dioxygenase (HaaO) also catalyzes an essential step required for the degradation of tryptophan in B. cenocepacia (37). BCAM2130 encodes HaaO in B. cenocepacia; however, an ortholog could not be found via a BLASTP search in B. multivorans (Fig. 4B). In this case, the reaction was included in iBC but not in iBM. Lack of HaaO and its reaction contributes to the inability of iBM to grow in silico with tryptophan as the sole carbon source. However, artificial inclusion of the reaction in iBM still did not allow for in silico growth, suggesting that other reactions needed for the catabolism of tryptophan are missing in iBM. This was one of the inconsistencies between the in vitro and in silico growth screens that we were unable to rectify through model refinement. This inconsistency demonstrates how the capability of the entire metabolic network is assessed when testing for growth; network gaps are not always direct or trivial and provide hypotheses for further experimentation.
(v) Rhamnolipid synthesis.
Species-specific differences are also exemplified within the rhamnolipid synthesis pathway. Rhamnolipid production has been linked to enhanced P. aeruginosa virulence in the cystic fibrosis lung by enabling invasion of host epithelial cells and affecting biofilm assembly, structural maintenance, and dispersion (53, 54). We located genes in B. cenocepacia that were previously unidentified as the rhamnolipid synthesis operon rhlABC but could not find genes encoding enzymes in this operon in B. multivorans (Fig. 4C). The rhlABC genes in B. cenocepacia are BCAM2340, BCAM2338, and BCAM2336, which have BLASTP E values of 7 × 10−63, 5 × 10−86, and 4 × 10−63 to the orthologous genes in P. aeruginosa PAO1 (PA3479, PA3478, and PA1130, respectively). Our additions resulted in a functional rhamnolipid synthesis pathway in iBC, while iBM was unable to produce rhamnolipids. Currently we are unaware of literature supporting rhamnolipid production in B. cenocepacia or B. multivorans, but nonpathogenic Burkholderia thailandensis and Burkholderia plantarii as well as pathogenic Burkholderia pseudomallei can produce rhamnolipids under specific conditions (55–57). However, a transcriptomic screening study by Sass et al. showed significant upregulation of the genes we identified as the rhl operon in B. cenocepacia during stationary-phase growth in minimal medium (58). These studies support the likelihood of the ability of B. cenocepacia to produce rhamnolipids, which may contribute to the enhanced virulence of B. cenocepacia over B. multivorans during chronic lung infections.
Comparison of gene content.
Our model curation efforts resulted in a high number of genes associated with each reaction in the reconstructions compared to other large, well-curated reconstructions. We counted the unique genes in the GPR of each reaction in each model, finding an average of 3.24 genes per reaction in iBC, 3.01 genes per reaction in iBM, and 1.9 genes per reaction in the P. aeruginosa reconstruction iMO1086. This increase in genes per reaction in the BCC reconstructions appears to be due to higher numbers of isozymes and gene duplications as implemented in GPRs and may be evidenced by the larger genomes of the BCC species compared to P. aeruginosa. By dividing the total number of genes associated with the reactions in each subsystem by the number of reactions in each subsystem, the average number of genes per reaction per subsystem was calculated (Fig. 5). For context, the average numbers of genes associated with each reaction grouped by subsystems in iBC, iBM, and iMO1086 are compared.
FIG 5.

Distribution of the number of genes per reaction in iBC and iBM by subsystem. The average number of genes per reaction per subsystem was calculated by dividing the total number of genes in the GPRs of the reactions (rxns) in each subsystem by the number of reactions in each subsystem. Solid diamonds and X's show the average number of genes per reaction for iBC and iBM, respectively. For reference, the average number of genes per reaction for iMO1086 (P. aeruginosa PAO1) is shown as open circles. For each subsystem, the three values in brackets denote the number of reactions for iBC, iBM, and iMO1086.
While iMO1086 shows a higher number of average genes per reaction in a subset of amino acid metabolism pathways and a few other subsystems, overall, iBC has the most genes per reaction across all subsystems, followed by iBM and iMO1086. The most notable increases in the average number of genes associated with reactions in iBC over the other reconstructions are incorporated in lipid metabolism. However, iBM may have higher metabolic capacity in certain energy and carbohydrate metabolic pathways as it has a higher gene-per-reaction average in these pathways. Pathways where iBC and iBM have similar numbers of genes associated with a reaction include metabolism of amino acids as well as terpenoid and polyketide metabolism. The reconstructions offer a method of probing specific subsystems to identify gene duplications or isozymes that may indicate concentrated genetic redundancy.
Gene essentiality.
An important consequence of the presence of increased genes per reaction in iBC and iBM compared to reconstructions of other bacteria was the reduction in the number of genes predicted to be essential for growth. During in silico growth in LB medium, our models predicted 66 essential genes in iBC and 73 essential genes in iBM (Table 2). Sixty of these genes were orthologs between iBC and iBM, 6 genes were uniquely essential in iBC, and 13 genes were uniquely essential in iBM. In comparison, iMO1086 required 150 genes to grow in silico on LB (with an accuracy of 83.9%) (18). Because iBM and iBC are not currently reconciled with iMO1086, we compared our predicted essential genes with a list of potentially essential P. aeruginosa PAO1 genes identified experimentally; these PAO1 genes had no recorded transposon mutants as identified in the Pseudomonas Genome Database based on genome-scale transposon mutagenesis libraries (59). As shown in Table S4 in the supplemental material, 35 out of 78 predicted essential Burkholderia genes matched P. aeruginosa PAO1 probable essential genes using a BLAST comparison. Another 12 predicted essential Burkholderia genes matched PAO1 genes with likely isozymes or duplications. Two predicted essential Burkholderia genes had no match to any PAO1 gene locus. The low number of potential essential Burkholderia genes compared to other species corresponds with a recent study that created promoter-based conditional mutants to identify essential Burkholderia cenocepacia K56-2 genes (60). However, the authors saw unexpectedly low essential operon hit rates during their mutant library screening.
TABLE 2.
Comparison of essential gene predictions
| Gene category | Locus tag: |
Gene abbreviation(s) | Enzyme name | EC no. | |
|---|---|---|---|---|---|
| B. cenocepacia | B. multivorans | ||||
| iBC unique essential genes | BCAL0800 | prs | Ribose-phosphate pyrophosphokinase | 2.7.6.1 | |
| BCAL0902 | d,d-Heptose 1,7-bisphosphate phosphatase | ||||
| BCAL2356 | Phosphatidylserine decarboxylase | 4.1.1.65 | |||
| BCAL3389 | tktA | Transketolase | 2.2.1.1 | ||
| BCAL3428 | nrdB | Ribonucleotide-diphosphate reductase subunit β | 1.17.4.1 | ||
| BCAL3429 | Ribonucleotide-diphosphate reductase subunit α | 1.17.4.1 | |||
| Common essential genes | BCAL0162 | Bmul_0198 | gmhA | Phosphoheptose isomerase | 5.3.1.28 |
| BCAL0508 | Bmul_3079 | Lipid A biosynthesis lauroyl acyltransferase | 2.3.1.- | ||
| BCAL0509 | Bmul_3078 | metK | S-Adenosylmethionine synthetase | 2.5.1.6 | |
| BCAL0612 | Bmul_2976 | glmU | UDP-N-acetylglucosamine pyrophosphorylase | 2.7.7.23 | |
| BCAL0743 | Bmul_0450 | gpsA | NAD(P)H-dependent glycerol-3-phosphate dehydrogenase | 1.1.1.94 | |
| BCAL0817 | Bmul_0528 | kdsC | Putative 3-deoxy-d-manno-octulosonate 8-phosphate phosphatase | 3.1.3.45 | |
| BCAL0818 | Bmul_0529 | Putative arabinose 5-phosphate isomerase | 5.3.1.13 | ||
| BCAL1269 | Bmul_2021 | glmM | Phosphoglucosamine mutase | 5.4.2.10 | |
| BCAL1281 | Bmul_1997 | Hypothetical protein (ornithine-acyl-ACP N-acyltransferase in Burkholderia sp. strain 383) | |||
| BCAL1556 | Bmul_1683 | rpiA | Ribose-5-phosphate isomerase A | 5.3.1.6 | |
| BCAL1887 | Bmul_1460 | ndk | Nucleoside diphosphate kinase | 2.7.4.6 | |
| BCAL1987 | Bmul_1360 | purL | Phosphoribosylformylglycinamidine synthase | 6.3.5.3 | |
| BCAL2061 | Bmul_1287 | guaA | GMP synthase | 6.3.5.2 | |
| BCAL2078 | Bmul_1270 | lpxB | Lipid A disaccharide synthase | 2.4.1.182 | |
| BCAL2079 | Bmul_1269 | lpxA | UDP-N-acetylglucosamine acyltransferase | 2.3.1.129 | |
| BCAL2080 | Bmul_1268 | fabZ | (3R)-Hydroxymyristoyl-ACP dehydratase | 4.2.1.- | |
| BCAL2081 | Bmul_1267 | lpxD | UDP-3-O-(3-hydroxymyristoyl) glucosamine N-acyltransferase | 2.3.1.191 | |
| BCAL2089 | Bmul_1259 | pyrH | Uridylate kinase | 2.7.4.- | |
| BCAL2101 | Bmul_1247 | dapD | 2,3,4,5-Tetrahydropyridine-2,6-carboxylate N-succinyltransferase | 2.3.1.117 | |
| BCAL2103 | Bmul_1245 | dapE | Succinyl-diaminopimelate desuccinylase | 3.5.1.18 | |
| BCAL2146 | Bmul_1207 | ask | Aspartate kinase | 2.7.2.4 | |
| BCAL2154 | Bmul_1199 | UDP-2,3-diacylglucosamine hydrolase | 3.6.1.54 | ||
| BCAL2180 | Bmul_1162 | 2-Dehydro-3-deoxyphosphooctonate aldolase | 2.5.1.55 | ||
| BCAL2181 | Bmul_1161 | pyrG | CTP synthetase | 6.3.4.2 | |
| BCAL2289 | Bmul_1080 | Glutamate racemase | |||
| BCAL2355 | Bmul_1017 | Putative phosphatidyltransferase | 2.7.8.8 | ||
| BCAL2388 | Bmul_0984 | Hypothetical protein (glucose-1-phosphate adenylyltransferase in iKF1028) | |||
| BCAL2389 | Bmul_0983 | purD | Phosphoribosylamine-glycine ligase | 6.3.4.13 | |
| BCAL2403 | Bmul_0965 | Putative LPS core biosynthesis protein | 2.4.-.- | ||
| BCAL2759 | Bmul_0751 | Tetraacyldisaccharide 4′-kinase | 2.7.1.130 | ||
| BCAL2761 | Bmul_0749 | kdsB | 3-Deoxy-manno-octulosonate cytidylyltransferase | 2.7.7.38 | |
| BCAL2770 | Bmul_0740 | Putative glycerol-3-phosphate acyltransferase PlsY | 2.3.1.15 | ||
| BCAL2836 | Bmul_0675 | purK | Phosphoribosylaminoimidazole carboxylase ATPase subunit | 4.1.1.21 | |
| BCAL2837 | Bmul_0674 | purE | Phosphoribosylaminoimidazole carboxylase catalytic subunit | 4.1.1.21 | |
| BCAL2838 | Bmul_0673 | purC | Phosphoribosylaminoimidazole-succinocarboxamide synthase | 6.3.2.6 | |
| BCAL2912 | Bmul_2218 | thyA | Thymidylate synthase | 2.1.1.45 | |
| BCAL2944 | Bmul_2251 | hldD | ADP-l-glycero-d-manno-heptose-6-epimerase | 5.1.3.20 | |
| BCAL2951 | Bmul_2258 | cmk | Cytidylate kinase | 2.7.4.14 | |
| BCAL3012 | Bmul_2401 | gmk | Guanylate kinase | 2.7.4.8 | |
| BCAL3110 | Bmul_2494 | waaA | 3-Deoxy-d-manno-octulosonic-acid transferase | 2.4.99.12 | |
| BCAL3113 | Bmul_2497 | manB | Phosphomannomutase | 5.4.2.2 | |
| BCAL3133 | Bmul_2598 | rmlC | dTDP-4-keto-6-deoxy-d-glucose 3,5-epimerase | 5.1.3.13 | |
| BCAL3134 | Bmul_2597 | rmlA | Glucose-1-phosphate thymidylyltransferase | 2.7.7.24 | |
| BCAL3239 | Bmul_4484 | Glucosyltransferase | |||
| BCAL3261 | Bmul_2624 | purM | Phosphoribosylaminoimidazole synthetase | 6.3.3.1 | |
| BCAL3336 | Bmul_2693 | purH | Phosphoribosylaminoimidazolecarboxamide formyltransferase | 2.1.2.3 | |
| BCAL3361 | Bmul_2717 | purB | Adenylosuccinate lyase | 4.3.2.2 | |
| BCAL3397 | Bmul_2756 | Putative phosphatidylglycerophosphatase | 3.1.3.27 | ||
| BCAL3460 | Bmul_2834 | ddl | d-Alanine-d-alanine ligase | 6.3.2.4 | |
| BCAL3461 | Bmul_2835 | murC | UDP-N-acetylmuramate-l-alanine ligase | 6.3.2.8 | |
| BCAL3462 | Bmul_2836 | murG | Undecaprenyldiphospho-muramoylpentapeptide β-N-acetylglucosaminyltransferase | 2.4.1.227 | |
| BCAL3464 | Bmul_2838 | murD | UDP-N-acetylmuramoyl-l-alanyl-d-glutamate synthetase | 6.3.2.9 | |
| BCAL3465 | Bmul_2839 | mraY | Phospho-N-acetylmuramoyl-pentapeptide-transferase | 2.7.8.13 | |
| BCAL3466 | Bmul_2840 | murF | UDP-N-acetylmuramoylalanyl-d-glutamyl-2,6-diaminopimelate-d-alanyl-d-alanyl ligase | 6.3.2.10 | |
| BCAL3467 | Bmul_2841 | murE | UDP-N-acetylmuramoylalanyl-d-glutamate-2,6-diaminopimelate ligase | 6.3.2.13 | |
| BCAM0683 | Bmul_3371 | ceoR | LysR family regulatory protein | ||
| BCAM0986 | Bmul_4626 | asd | Aspartate-semialdehyde dehydrogenase | 1.2.1.11 | |
| BCAM0998 | Bmul_4615 | purF | Amidophosphoribosyltransferase | 2.4.2.14 | |
| BCAM1337 | Bmul_4402 | Glycosyltransferase | |||
| BCAM2044 | Bmul_3751 | Putative asparagine synthetase | 6.3.5.4 | ||
| iBM unique essential genes | Bmul_0742 | murB | UDP-N-acetylenolpyruvoylglucosamine reductase | 1.1.1.158 | |
| Bmul_1262 | Phosphatidate cytidylyltransferase | 2.7.7.41 | |||
| Bmul_2179 | 3-Oxoacyl-(acyl carrier protein) synthase II | 2.3.1.179 | |||
| Bmul_2182 | fabD1, fabD2 | UDP-3-O-(3-hydroxymyristoyl) N-acetylglucosamine deacetylase | 3.5.1.108 | ||
| Bmul_2183 | fabH1, fabH2 | 3-Oxoacyl-ACP synthase | 2.3.1.41 | ||
| Bmul_2184 | plsX | Putative glycerol-3-phosphate acyltransferase | 2.3.1.15 | ||
| Bmul_2221 | Dihydrofolate reductase | 1.5.1.3 | |||
| Bmul_2735 | dapB | Dihydrodipicolinate reductase | 1.3.1.26 | ||
| Bmul_2829 | lpxC | UDP-3-O-(3-hydroxymyristoyl) N-acetylglucosamine deacetylase | 3.5.1.108 | ||
| Bmul_3080 | dapF | Diaminopimelate epimerase | 5.1.1.7 | ||
| Bmul_3287 | prpB | 2-Methylisocitrate lyase | 4.1.3.30 | ||
| Bmul_3288 | Methylcitrate synthase | 2.3.3.5 | |||
| Bmul_4654 | prpD | 2-Methylcitrate dehydratase | 4.2.1.79 | ||
The difference between the overall number of essential genes in iBC and iBM compared to P. aeruginosa is likely due to the high number of isozymes and duplicate genes included in our GPRs as well as the comprehensive GPR curation, which added robustness to the performance of particular functions. Many of the predicted essential genes were located in expected pathways, such as nucleotide metabolism, energy metabolism, and lipid metabolism. The common essential genes represent opportunities to target both species with the same treatment. The high number of unique essential genes predicted for each species offers new hypotheses regarding species-specific targets in developing novel treatments for B. cenocepacia versus B. multivorans infections.
When the unique essential genes predicted for each model were compared, a gene associated with dihydrofolate reductase (DHFR) was only present in iBM. DHFR, coded for by Bmul_2221, is the target of trimethoprim, an antibiotic commonly effective against pathogens such as E. coli and Haemophilus influenzae (61). However, trimethoprim has shown variable effectiveness against different strains of the BCC (62), and our essentiality analysis indicates no apparent matching essential gene in iBC. Instead, two genes were associated with this reaction in iBC by the Model SEED tool. One gene, BCAL2915, is annotated as dfrA, a trimethoprim-resistant variant of the DHFR gene (63), and has high similarity to Bmul_2221. The other gene, BCAL1859, is annotated on the BGD as a hypothetical protein. When conducting a BLASTP search with BCAL1859, the most similar functionally annotated matches were to DHFR genes in Deinococcus and Aeromonas species (E values of ∼10−50 and ∼10−40, respectively) with no other highly similar matches. Interestingly, a study of in vitro antimicrobial susceptibility determined that B. multivorans ATCC 17616 is notably more susceptible to trimethoprim than B. cenocepacia J2315, with a MIC of 2 mg/liter compared to 64 mg/liter (62). Determination of whether this apparently rare variant of DHFR (BCAL1859) is connected with the elevated trimethoprim resistance of B. cenocepacia J2315 would require further experimental study, but the identification of a previously unannotated DHFR isozyme in the more virulent of our two BCC species of interest is an important consequence of our comparative study of essential genes.
Virulence factor production capacity.
Our models enable in silico investigation of how the nutritional environment of the cystic fibrosis lung contributes to production of important factors in initial colonization and chronic infection by Burkholderia. iBC and iBM were used to predict the ability of each species to produce an array of virulence-related molecules while under varied growth demands or when presented with limited nutritional resources. We include prototypical virulence factors, such as biofilm-related exopolysaccharide cepacian, immune response triggering, phagocytosis-resistant lipopolysaccharide (LPS), and quorum sensing signals that enable communication with other bacteria (64). We also include the production of rhamnolipids due to their role in biofilm formation and regulation, as previously mentioned. Intracellular signaling molecules such as the polyamines putrescine and spermidine are not essential for growth in many environments, but their loss induces significant phenotypic changes in various bacteria and they are considered important in the regulation of biofilm production and other virulence pathways (65). Ornibactin enables iron acquisition, and salicylate potentially acts as a siderophore and is also required for production of other siderophores; both compounds have been connected with enhanced virulence both clinically and in animal models (48). Homogentisate is a precursor of the melanin-like pigment produced by some Burkholderia species that provides protection from reactive oxygen and nitrogen species (66, 67).
We evaluated the in silico production capacities of each of these compounds under different growth and medium constraints, as explained in Materials and Methods (Fig. 6). We analyzed the trade-off between virulence factors and biomass by predicting the maximum production of each virulence factor under a given requirement for biomass flux in SCFM (Fig. 6A). For the majority of the virulence factors that iBC and iBM can produce, production capacity decreases approximately linearly as the percentage of maximum biomass flux constraint is increased (Fig. 6A). However, both models predict that ornibactin and homogentisate can be produced at a sustained level over a wide range of growth rates until constrained by the demand for resources necessary for near-optimal levels of biomass production. Also, both models predict that maximum cepacian production occurs at non-zero biomass production requirements. This dependency between cepacian production and growth is a result of complex interrelationships between cofactors and by-products upstream of the cepacian biosynthesis pathway. Additionally, while iBC is capable of de novo synthesis of all of the tested virulence factors, iBM contains incomplete biosynthetic pathways for the production of spermidine, salicylate, and di-rhamnolipid, resulting in the inability to produce these molecules. The models enable the identification of both the missing genes that entirely prevent production and altered upstream pathways that limit final production levels of a given virulence factor in iBM.
FIG 6.

iBC and iBM virulence factor production capacities. (A) Trade-off between virulence factor production and growth (biomass production) was predicted by optimizing the production of an array of virulence factors while constraining the flux of the biomass reaction to various percentages of its maximum value under SCFM conditions. Presented values were normalized from 0 to 100% for each virulence factor across all biomass flux constraint conditions. While iBC is capable of producing all tested virulence factors, iBM contains incomplete biosynthetic pathways for the production of spermidine, salicylate, and di-rhamnolipid. (B) The contribution of the carbon source components of SCFM to the production of the virulence factors was predicted. For each virulence factor, production was maximized under minimal medium conditions supplemented with each SCFM carbon source individually. Presented values were normalized from 0 to 100% for each virulence factor across all single-substrate conditions. (C) Each column of production capacities was averaged along all virulence factors to calculate an average production capacity on each substrate for each model, as shown in the heat map via text and shading. Capacity averages below 50 are in white text for contrast. Colors are consistent with the color bars of panels A and B. For both iBC and iBM, on average, l-tyrosine supports the highest average production capacity for both models.
The relationship between changing in vivo nutrient resources, colonization, and infection has been highlighted as an understudied potential target of novel therapies in an array of host-pathogen relationships (68). To better understand the role of the individual components of SCFM in the production of the various virulence factors, we assessed in silico virulence factor production capacities on each carbon source present in SCFM without any biomass flux constraints (Fig. 6B). The production capacities for each virulence factor were normalized by the maximum possible value across the different individual carbon sources for that molecule. For both models, on average, tyrosine and glucose were the carbon sources that contributed the most toward the production of the virulence factors. An example of an exception is that tyrosine contributes greatly to cepacian production in iBC but not in iBM (Fig. 6B). Homogentisate is produced at high levels on l-phenylalanine for both iBC and iBM, but other factors are only produced at average levels on the same substrate. The models also predict that each virulence factor can be produced at some level on any single carbon source that also enables growth in that model, as shown in Fig. 3. These results suggest variabilities in virulence factor production given changing substrate availability and predict potential pathways to target for therapeutic virulence inhibition.
To evaluate the potential production advantage imparted by each analyzed substrate to the full set of virulence factors, we averaged the virulence factor production capacity for all compounds by each substrate in Fig. 6C. The results suggest that general virulence factor production is well supported by catabolism of l-tyrosine, d-glucose, l-arginine, and l-ornithine and poorly supported by glycine, d-lactate, and l-serine. B. cenocepacia has higher-capacity averages on all substrates capable of supporting virulence compound production. The difference in averages of substrates supporting any virulence factor production in both models highlights a potential species-specific substrate preference for l-proline by B. cenocepacia. Ultimately, these analyses offers an interesting set of hypotheses regarding substrates that may be important for maximal virulence activity during infection of cystic fibrosis patients, identify potential variations in substrate preferences between each Burkholderia species, and emphasize the enhanced pathogenic abilities of B. cenocepacia in comparison to B. multivorans.
DISCUSSION
This study involved the parallel generation and comparison of two new genome-scale metabolic reconstructions for the multidrug-resistant pathogens B. cenocepacia and B. multivorans. The reconstructions share 1,437 reactions, with 68 additional reactions unique to iBC and 36 additional reactions unique to iBM. iBC accounts for the function of 1,537 genes, while iBM accounts for the function of 1,411 genes. The reconstructions incorporated pathways necessary for growth on an array of substrates, species-specific biomass formulations, and virulence factor synthesis pathways common to related bacteria as well as specific to BCC pathogens. Models were validated using experimental growth screens on over 100 carbon sources, including substrates abundant in the cystic fibrosis lung environment. This process enabled us to refine the genome annotations of each species by reannotating an array of hypothetical and putative proteins, evaluate the consequences of the large genomes of B. cenocepacia and B. multivorans via GPR comparisons, make predictions regarding unique and shared essential genes for each pathogen, and evaluate the enhanced virulence factor production capacity of B. cenocepacia in comparison to B. multivorans.
The reconstructions provide a framework for contextualizing the genes of B. cenocepacia and B. multivorans in relation to growth and virulence factor production capacities. iBC and iBM are new tools that can enable the interrogation of interdependent functions of the large BCC genomes. The reconstruction process guided our evaluation of the current genome annotations and in several cases led us to propose annotation refinements. Additionally, our comparison of in silico predictions with in vitro growth screening identified pathways that were clearly functional in vitro, such as lysine degradation and tryptophan degradation, that we could not make function in silico by addition of any known enzymes/genes. Thus, this analysis highlighted potentially uncharacterized enzymes that enable the function of certain pathways in Burkholderia and may also be active in other organisms. Our genome annotation refinements also generated model-driven hypotheses that can be followed up experimentally, such as the species-specific production of rhamnolipids and spermidine and the degradation of ectoine.
Tools for semiautomatic generation of genome-scale metabolic models have proved useful for expediting the reconstruction process (69–71). In this study, using the Model SEED pipeline to concurrently generate the two reconstructions aided the manual curation and reconciliation phases since the differences in content and function between the two reconstructions could more easily be tracked and evaluated in parallel. The parallel curation process also ensured that the two reconstructions were of higher detail and quality than if the two reconstructions were developed in isolation, since genetic evidence for reactions were exhaustively cross-checked between the two species. It is unlikely that our reconstructions have incorporated every metabolic function of each species, and further curation will be required as the BCC knowledgebase grows. Growth prediction discrepancies on lysine, tryptophan, and branched-chain amino acids have highlighted areas to be investigated further experimentally.
The size and scope of the reconstructions in addition to high average gene count per reaction likely contributed to the reduced number of predicted essential genes in comparison to other reconstructions. To date, while there are emerging computational efforts to predict essential genes (72), no complete transposon mutagenesis library of B. cenocepacia or B. multivorans is available, and our network-driven in silico gene essentiality predictions serve as hypotheses for future work. The essential genes predicted to be unique to each species also offer interesting opportunities for targeted therapeutics. Given the general similarity of the network architectures in terms of the common incorporated reactions, the unique essential gene sets are a novel outcome of our comprehensive formulation and comparison of the GPR associations.
Our comparative analysis enabled system-wide evaluations as well as the evaluation of a single reaction or gene functionality that could be tied to a particular model prediction, which is a strength of genome-scale metabolic modeling. Modeling reaction activity using the reconstructions and constraint-based analysis allowed us to evaluate altered production capacity between models due to specific reaction differences in upstream pathways that limited final production levels. These analyses provide specific, testable hypotheses regarding important metabolic functions that are less intuitive than hypotheses generated through gene essentiality analysis alone.
Ultimately, each comparative analysis performed in this study supports the conclusion that B. cenocepacia has enhanced metabolic capacity over B. multivorans. The predicted growth rate on most in silico media of iBC was equal to or higher than that of iBM and notably higher on SCFM, a medium designed to replicate the environment of the cystic fibrosis lung (see Table S3 in the supplemental material). iBC also showed higher capacity for virulence factor production over iBM on a range of substrates found in CF lung sputum. Our reconstruction process enabled the organization and functional evaluation of the increased genetic redundancy (e.g., isozymes and gene duplications) across a range of pathways that were available to potentially supplement any disrupted gene function in iBC compared to iBM. This added genetic redundancy resulted in both metabolic robustness and increased opportunity for the evolutionary divergence of duplicated genes to perform new functions (73). We have shown that these reconciled metabolic reconstructions offer a way to develop and investigate intriguing hypotheses for the enhanced virulence of B. cenocepacia—a higher capacity for genetic adaptation in the CF lung in addition to the flexibility provided by a large number of distinct catabolic pathways.
In summary, our reconciled metabolic models of the emerging multidrug-resistant pathogens B. cenocepacia and B. multivorans are valuable tools that can aid in comparative analysis of metabolic capacity, identification of novel therapeutic targets and strategies, and prediction of key phenotypes of pathogenesis. Here, we have thoroughly characterized the metabolism of the type strains of B. cenocepacia and B. multivorans. Our network reconstructions can be used to contextualize high-throughput transcriptomic and proteomic data (58, 74–77) to provide further insight into gene regulation and downstream phenotypes. Additionally, our reconstructions can be used as established starting points to analyze pathogenesis and physiology of the growing list of sequenced Burkholderia strains, including both clinical and environmental isolates (78). The other members of the BCC are also human pathogens, while related species Burkholderia mallei and Burkholderia pseudomallei are dangerous bioterror agents that would be ideal candidates for in silico study. Other nonpathogenic Burkholderia species are of great interest to metabolic engineering projects in bioremediation and agricultural biotechnology as soil pathogens that excel in catabolism of certain pollutants as well as the production of antimicrobials that protect plant health (79, 80). Given their notably large genomes and capacity for metabolic adaptation as a key factor in pathogenesis, the reconstructions of B. cenocepacia and B. multivorans offer tremendous potential for future development of treatment strategies to combat chronic infections and a model for the metabolic analysis and comparison of similar dangerous pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by NIH RO1 GM088244 and Cystic Fibrosis Foundation Grant 1060 to J.A.P., NIH T32 GM008715 to P.Y., and Cystic Fibrosis Foundation Grant GOLDB12P0 and NIH R21 AI103653 to J.B.G.
We thank Kevin M. D'Auria and other members of the Papin laboratory for helpful comments on the manuscript.
Footnotes
Published ahead of print 25 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00997-13.
REFERENCES
- 1.Mahenthiralingam E, Baldwin A, Vandamme P. 2002. Burkholderia cepacia complex infection in patients with cystic fibrosis. J. Med. Microbiol. 51:533–538 [DOI] [PubMed] [Google Scholar]
- 2.Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081. 10.1086/533452 [DOI] [PubMed] [Google Scholar]
- 3.Slama TG. 2008. Gram-negative antibiotic resistance: there is a price to pay. Crit. Care 12(Suppl 4):S4. 10.1186/cc6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12. 10.1086/595011 [DOI] [PubMed] [Google Scholar]
- 5.De Boeck K, Malfroot A, Van Schil L, Lebecque P, Knoop C, Govan JRW, Doherty C, Laevens S, Vandamme P. 2004. Epidemiology of Burkholderia cepacia complex colonisation in cystic fibrosis patients. Eur. Respir. J. 23:851–856. 10.1183/09031936.04.00118804 [DOI] [PubMed] [Google Scholar]
- 6.Jones AM, Dodd ME, Govan JRW, Barcus V, Doherty CJ, Morris J, Webb AK. 2004. Burkholderia cenocepacia and Burkholderia multivorans: influence on survival in cystic fibrosis. Thorax 59:948–951. 10.1136/thx.2003.017210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coenye T, Spilker T, Van Schoor A, LiPuma J, Vandamme P. 2004. Recovery of Burkholderia cenocepacia strain PHDC from cystic fibrosis patients in Europe. Thorax 59:952–954. 10.1136/thx.2003.019810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horsley A, Webb K, Bright-Thomas R, Govan J, Jones A. 2011. Can. early Burkholderia cepacia complex infection in cystic fibrosis be eradicated with antibiotic therapy? Front. Cell. Infect. Microbiol. 1:18. 10.3389/fcimb.2011.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isles A, Maclusky I, Corey M, Gold R, Prober C, Fleming P, Levison H. 1984. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J. Pediatr. 104:206–210. 10.1016/S0022-3476(84)80993-2 [DOI] [PubMed] [Google Scholar]
- 10.Glass S, Govan J. 1986. Pseudomonas cepacia—fatal pulmonary infection in a patient with cystic fibrosis. J. Infect. 13:157–158. 10.1016/S0163-4453(86)92953-1 [DOI] [PubMed] [Google Scholar]
- 11.Aaron SD, Ferris W, Henry DA, Speert DP, Macdonald NE. 2000. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am. J. Respir. Crit. Care Med. 161:1206–1212. 10.1164/ajrccm.161.4.9907147 [DOI] [PubMed] [Google Scholar]
- 12.Hancock RE. 1998. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis. 27:S93–S99. 10.1086/514909 [DOI] [PubMed] [Google Scholar]
- 13.Snell G, de Hoyos A, Krajden M, Winton T, Maurer J. 1993. Pseudomonas cepacia in lung transplant recipients with cystic fibrosis. Chest 103:466–471. 10.1378/chest.103.2.466 [DOI] [PubMed] [Google Scholar]
- 14.Marolda C, Hauröder B, John M, Michel R, Valvano M. 1999. Intracellular survival and saprophytic growth of isolates from the Burkholderia cepacia complex in free-living amoebae. Microbiology 145:1509–1517. 10.1099/13500872-145-7-1509 [DOI] [PubMed] [Google Scholar]
- 15.Mahenthiralingam E, Vandamme P, Campbell M, Henry D, Gravelle A, Wong L, Davidson A, Wilcox P, Nakielna B, Speert D. 2001. Infection with Burkholderia cepacia complex genomovars in patients with cystic fibrosis: virulent transmissible strains of genomovar III can replace Burkholderia multivorans. Clin. Infect. Dis. 33:1469–1475. 10.1086/322684 [DOI] [PubMed] [Google Scholar]
- 16.Oberhardt MA, Palsson BØ, Papin JA. 2009. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 5:320. 10.1038/msb.2009.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee D-S, Burd H, Liu J, Almaas E, Wiest O, Barabási A-L, Oltvai ZN, Kapatral V. 2009. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 191:4015–4024. 10.1128/JB.01743-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oberhardt MA, Puchałka J, Martins dos Santos VAP, Papin JA. 2011. Reconciliation of genome-scale metabolic reconstructions for comparative systems analysis. PLoS Comput. Biol. 7:e1001116. 10.1371/journal.pcbi.1001116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang K, Zhao H, Sun C, Lam CMC, Chang S, Zhang K, Panda G, Godinho M, Martins dos Santos VAP, Wang J. 2011. Exploring the metabolic network of the epidemic pathogen Burkholderia cenocepacia J2315 via genome-scale reconstruction. BMC Syst. Biol. 5:83. 10.1186/1752-0509-5-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. 2010. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28:977–982. 10.1038/nbt.1672 [DOI] [PubMed] [Google Scholar]
- 21.Reed JL, Vo TD, Schilling CH, Palsson BØ. 2003. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 4:R54. 10.1186/gb-2003-4-9-r54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holden MTG, Seth-Smith HMB, Crossman LC, Sebaihia M, Bentley SD, Cerdeño-Tárraga AM, Thomson NR, Bason N, Quail MA, Sharp S, Cherevach I, Churcher C, Goodhead I, Hauser H, Holroyd N, Mungall K, Scott P, Walker D, White B, Rose H, Iversen P, Mil-Homens D, Rocha EPC, Fialho AM, Baldwin A, Dowson C, Barrell BG, Govan JR, Vandamme P, Hart CA, Mahenthiralingam E, Parkhill J. 2009. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 191:261–277. 10.1128/JB.01230-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanier R, Palleroni N, Duodoroff M. 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43:159–271. 10.1099/00221287-43-2-159 [DOI] [PubMed] [Google Scholar]
- 24.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28:27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, Walk TC, Zhang P, Karp PD. 2008. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 36(Suppl 1):D623–D631. 10.1093/nar/gkm900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winsor GL, Khaira B, Van Rossum T, Lo R, Whiteside MD, Brinkman FS. 2008. The Burkholderia Genome Database: facilitating flexible queries and comparative analyses. Bioinformatics 24:2803–2804. 10.1093/bioinformatics/btn524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orth JD, Thiele I, Palsson BØ. 2010. What is flux balance analysis? Nat. Biotechnol. 28:245–248. 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schellenberger J, Que R, Fleming RMT, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BØ. 2011. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6:1290–1307. 10.1038/nprot.2011.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feist AM, Palsson BO. 2010. The biomass objective function. Curr. Opin. Microbiol. 13:344–349. 10.1016/j.mib.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor CJ, Anderson AJ, Wilkinson SG. 1998. Phenotypic variation of lipid composition in Burkholderia cepacia: a response to increased growth temperature is a greater content of 2-hydroxy acids in phosphatidylethanolamine and ornithine amide lipid. Microbiology 144:1737–1745. 10.1099/00221287-144-7-1737 [DOI] [PubMed] [Google Scholar]
- 32.Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BØ. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3:121. 10.1038/msb4100155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chavali AK, Whittemore JD, Eddy JA, Williams KT, Papin JA. 2008. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol. Syst. Biol. 4:177. 10.1038/msb.2008.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oberhardt MA, Puchałka J, Fryer KE, Martins dos Santos VAP, Papin JA. 2008. Genome-scale metabolic network analysis of the opportunistic pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 190:2790–2803. 10.1128/JB.01583-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer KL, Aye LM, Whiteley M. 2007. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189:8079–8087. 10.1128/JB.01138-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oberhardt MA, Goldberg JB, Hogardt M, Papin JA. 2010. Metabolic network analysis of Pseudomonas aeruginosa during chronic cystic fibrosis lung infection. J. Bacteriol. 192:5534–5548. 10.1128/JB.00900-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colabroy K, Begley T. 2005. Tryptophan catabolism: identification and characterization of a new degradative pathway. J. Bacteriol. 187:7866–7869. 10.1128/JB.187.22.7866-7869.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yudistira H, McClarty L, Bloodworth RAM, Hammond SA, Butcher H, Mark BL, Cardona ST. 2011. Phenylalanine induces Burkholderia cenocepacia phenylacetic acid catabolism through degradation to phenylacetyl-CoA in synthetic cystic fibrosis sputum medium. Microb. Pathog. 51:186–193. 10.1016/j.micpath.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 39.Madhusudhan KT, Luo J, Sokatch JR. 1999. In vitro transcriptional studies of the bkd operon of Pseudomonas putida: L-branched-chain amino acids and D-leucine are the inducers. J. Bacteriol. 181:2889–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwibbert K, Marin-Sanguino A, Bagyan I, Heidrich G, Lentzen G, Seitz H, Rampp M, Schuster SC, Klenk H-P, Pfeiffer F, Oesterhelt D, Kunte HJ. 2011. A blueprint of ectoine metabolism from the genome of the industrial producer Halomonas elongata DSM 2581T. Environ. Microbiol. 13:1973–1994. 10.1111/j.1462-2920.2010.02336.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sousa SA, Feliciano JR, Pinheiro PF, Leitão JH. 2013. Biochemical and functional studies on the Burkholderia cepacia complex bceN gene, encoding a GDP-D-mannose 4,6-dehydratase. PLoS One 8:e56902. 10.1371/journal.pone.0056902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuhara S, Komatsu H, Goto H, Ohtsubo Y, Nagata Y, Tsuda M. 2008. Pleiotropic roles of iron-responsive transcriptional regulator Fur in Burkholderia multivorans. Microbiology 154:1763–1774. 10.1099/mic.0.2007/015537-0 [DOI] [PubMed] [Google Scholar]
- 43.Tarighi S, Wei Q, Cámara M, Williams P, Fletcher MP, Kajander T, Cornelis P. 2008. The PA4204 gene encodes a periplasmic gluconolactonase (PpgL) which is important for fitness of Pseudomonas aeruginosa. Microbiology (Reading, England) 154:2979–2990. 10.1099/mic.0.2008/018465-0 [DOI] [PubMed] [Google Scholar]
- 44.Müller A, Thomas GH, Horler R, Brannigan JA, Blagova E, Levdikov VM, Fogg MJ, Wilson KS, Wilkinson AJ. 2005. An ATP-binding cassette-type cysteine transporter in Campylobacter jejuni inferred from the structure of an extracytoplasmic solute receptor protein. Mol. Microbiol. 57:143–155. 10.1111/j.1365-2958.2005.04691.x [DOI] [PubMed] [Google Scholar]
- 45. Reference deleted.
- 46.Kohler PRA, Zheng JY, Schoffers E, Rossbach S. 2010. Inositol catabolism, a key pathway in Sinorhizobium meliloti for competitive host nodulation. Appl. Environ. Microbiol. 76:7972–7980. 10.1128/AEM.01972-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hosie A, Allaway D, Poole P. 2002. A monocarboxylate permease of Rhizobium leguminosarum is the first member of a new subfamily of transporters. J. Bacteriol. 184:5436–5448. 10.1128/JB.184.19.5436-5448.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darling P, Chan M, Cox AD, Pamela A. 1998. Siderophore production by cystic fibrosis isolates of Burkholderia cepacia siderophore production by cystic fibrosis isolates of Burkholderia cepacia. Infect. Immun. 66:874–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agnoli K, Schwager S, Uehlinger S, Vergunst A, Viteri DF, Nguyen DT, Sokol PA, Carlier A, Eberl L. 2012. Exposing the third chromosome of Burkholderia cepacia complex strains as a virulence plasmid. Mol. Microbiol. 83:362–378. 10.1111/j.1365-2958.2011.07937.x [DOI] [PubMed] [Google Scholar]
- 50.Hesketh A, Hill C, Mokhtar J, Novotna G, Tran N, Bibb M, Hong H-J. 2011. Genome-wide dynamics of a bacterial response to antibiotics that target the cell envelope. BMC Genomics 12:226. 10.1186/1471-2164-12-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derewacz DK, Goodwin CR, Mcnees CR, Mclean JA, Bachmann BO. 2013. Antimicrobial drug resistance affects broad changes in metabolomic phenotype in addition to secondary metabolism. Proc. Natl. Acad. Sci. U. S. A. 110:2336–2341. 10.1073/pnas.1218524110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferreira AS, Leitão JH, Silva IN, Pinheiro PF, Sousa SA, Ramos CG, Moreira LM. 2010. Distribution of cepacian biosynthesis genes among environmental and clinical Burkholderia strains and role of cepacian exopolysaccharide in resistance to stress conditions. Appl. Environ. Microbiol. 76:441–450. 10.1128/AEM.01828-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zulianello L, Canard C, Köhler T, Caille D, Lacroix J-S, Meda P. 2006. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect. Immun. 74:3134–3147. 10.1128/IAI.01772-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pamp SJ, Tolker-Nielsen T. 2007. Multiple roles of biosurfactants in structural biofilm development by Pseudomonas aeruginosa. J. Bacteriol. 189:2531–2539. 10.1128/JB.01515-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dubeau D, Déziel E, Woods DE, Lépine F. 2009. Burkholderia thailandensis harbors two identical rhl gene clusters responsible for the biosynthesis of rhamnolipids. BMC Microbiol. 9:263. 10.1186/1471-2180-9-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andrä J, Rademann J, Howe J, Koch MHJ, Heine H, Zähringer U, Brandenburg K. 2006. Endotoxin-like properties of a rhamnolipid exotoxin from Burkholderia (Pseudomonas) plantarii: immune cell stimulation and biophysical characterization. Biol. Chem. 387:301–310. 10.1515/BC.2006.040 [DOI] [PubMed] [Google Scholar]
- 57.Häußler S, Nimtz M, Domke T, Wray V, Steinmetz I. 1998. Purification and characterization of a cytotoxic exolipid of Burkholderia pseudomallei. Infect. Immun. 66:1588–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sass AM, Schmerk C, Agnoli K, Norville PJ, Eberl L, Valvano MA, Mahenthiralingam E. 2013. The unexpected discovery of a novel low-oxygen-activated locus for the anoxic persistence of Burkholderia cenocepacia. ISME J. 7:1568–1581. 10.1038/ismej.2013.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winsor GL, Lam DKW, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock REW, Brinkman FSL. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600. 10.1093/nar/gkq869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bloodworth RAM, Gislason AS, Cardona ST. 2013. Burkholderia cenocepacia conditional growth mutant library created by random promoter replacement of essential genes. Microbiol. Open 2:243–258. 10.1002/mbo3.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith C, Powell K. 2000. Review of the sulfonamides and trimethoprim. Pediatr. Rev. 21:368–371 [PubMed] [Google Scholar]
- 62.Nzula S, Vandamme P, Govan JR. 2002. Influence of taxonomic status on the in vitro antimicrobial susceptibility of the Burkholderia cepacia complex. J. Antimicrob. Chemother. 50:265–269. 10.1093/jac/dkf137 [DOI] [PubMed] [Google Scholar]
- 63.Kehrenberg C, Schwarz S. 2005. dfrA20, a novel trimethoprim resistance gene from Pasteurella multocida. Antimicrob. Agents Chemother. 49:414–417. 10.1128/AAC.49.1.414-417.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loutet SA, Valvano MA. 2010. A decade of Burkholderia cenocepacia virulence determinant research. Infect. Immun. 78:4088–4100. 10.1128/IAI.00212-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams BJ, Du R-H, Calcutt MW, Abdolrasulnia R, Christman BW, Blackwell TS. 2010. Discovery of an operon that participates in agmatine metabolism and regulates biofilm formation in Pseudomonas aeruginosa. Mol. Microbiol. 76:104–119. 10.1111/j.1365-2958.2010.07083.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu GY, Nizet V. 2009. Color me bad: microbial pigments as virulence factors. Trends Microbiol. 17:406–413. 10.1016/j.tim.2009.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keith KE, Killip L, He P, Moran GR, Valvano MA. 2007. Burkholderia cenocepacia C5424 produces a pigment with antioxidant properties using a homogentisate intermediate. J. Bacteriol. 189:9057–9065. 10.1128/JB.00436-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown SA, Palmer KL, Whiteley M. 2008. Revisiting the host as a growth medium. Nat. Rev. Microbiol. 6:657–666. 10.1038/nrmicro1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, Nielsen J. 2013. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 9:e1002980. 10.1371/journal.pcbi.1002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Devoid S, Overbeek R, Dejongh M, Vonstein V, Best AA, Henry C. 2013. Automated genome annotation and metabolic model reconstruction in the SEED and Model SEED. Methods Mol. Biol. 985:17–45. 10.1007/978-1-62703-299-5_2 [DOI] [PubMed] [Google Scholar]
- 71.Arakawa K, Yamada Y, Shinoda K, Nakayama Y, Tomita M. 2006. GEM System: automatic prototyping of cell-wide metabolic pathway models from genomes. BMC Bioinformatics 7:168. 10.1186/1471-2105-7-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juhas M, Stark M, von Mering C, Lumjiaktase P, Crook DW, Valvano MA, Eberl L. 2012. High confidence prediction of essential genes in Burkholderia cenocepacia. PLoS One 7:e40064. 10.1371/journal.pone.0040064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11:97–108. 10.1038/nrg2689 [DOI] [PubMed] [Google Scholar]
- 74.Blazier AS, Papin JA. 2012. Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 3:299. 10.3389/fphys.2012.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sass A, Marchbank A, Tullis E, Lipuma JJ, Mahenthiralingam E. 2011. Spontaneous and evolutionary changes in the antibiotic resistance of Burkholderia cenocepacia observed by global gene expression analysis. BMC Genomics 12:373. 10.1186/1471-2164-12-373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoder-Himes DR, Konstantinidis KT, Tiedje JM. 2010. Identification of potential therapeutic targets for Burkholderia cenocepacia by comparative transcriptomics. PLoS One 5:e8724. 10.1371/journal.pone.0008724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zlosnik JEA, Speert DP. 2010. The role of mucoidy in virulence of bacteria from the Burkholderia cepacia complex: a systematic proteomic and transcriptomic analysis. J. Infect. Dis. 202:770–781. 10.1086/655663 [DOI] [PubMed] [Google Scholar]
- 78.Mukhopadhyay S, Thomason MK, Lentz S, Nolan N, Willner K, Gee JE, Glass MB, Inglis TJJ, Merritt A, Levy A, Sozhamannan S, Mateczun A, Read TD. 2010. High-redundancy draft sequencing of 15 clinical and environmental Burkholderia strains. J. Bacteriol. 192:6313–6314. 10.1128/JB.00991-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mahenthiralingam E, Urban TA, Goldberg JB. 2005. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3:144–156. 10.1038/nrmicro1085 [DOI] [PubMed] [Google Scholar]
- 80.Suárez-Moreno ZR, Caballero-Mellado J, Coutinho BG, Mendonça-Previato L, James EK, Venturi V. 2012. Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb. Ecol. 63:249–266. 10.1007/s00248-011-9929-1 [DOI] [PubMed] [Google Scholar]
- 81.Nogales J, Canales A, Jiménez-Barbero J, Serra B, Pingarrón JM, García JL, Díaz E. 2011. Unravelling the gallic acid degradation pathway in bacteria: the gal cluster from Pseudomonas putida. Mol. Microbiol. 79:359–374. 10.1111/j.1365-2958.2010.07448.x [DOI] [PubMed] [Google Scholar]
- 82.Ogawa J, Shimizu S. 1994. Beta-ureidopropionase with N-carbamoyl-alpha-L-amino acid amidohydrolase activity from an aerobic bacterium, Pseudomonas putida IFO 12996. Eur. J. Biochem. 223:625–630. 10.1111/j.1432-1033.1994.tb19034.x [DOI] [PubMed] [Google Scholar]
- 83.Varga JJ, Losada L, Zelazny AM, Kim M, McCorrison J, Brinkac L, Sampaio EP, Greenberg DE, Singh I, Heiner C, Ashby M, Nierman WC, Holland SM, Goldberg JB. 2013. Draft genome sequences of Burkholderia cenocepacia ET12 lineage strains K56-2 and BC7. Genome Announc. 1(5):e00841-13. 10.1128/genomeA.00841-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.