

Abstract
The xylem-limited bacterium Xylella fastidiosa is the causal agent of several plant diseases, most notably Pierce's disease of grape and citrus variegated chlorosis. We report the isolation and characterization of the first virulent phages for X. fastidiosa, siphophages Sano and Salvo and podophages Prado and Paz, with a host range that includes Xanthomonas spp. Phages propagated on homologous hosts had observed adsorption rate constants of ∼4 × 10−12 ml cell−1 min−1 for X. fastidiosa strain Temecula 1 and ∼5 × 10−10 to 7 × 10−10 ml cell−1 min−1 for Xanthomonas strain EC-12. Sano and Salvo exhibit >80% nucleotide identity to each other in aligned regions and are syntenic to phage BcepNazgul. We propose that phage BcepNazgul is the founding member of a novel phage type, to which Sano and Salvo belong. The lysis genes of the Nazgul-like phage type include a gene that encodes an outer membrane lipoprotein endolysin and also spanin gene families that provide insight into the evolution of the lysis pathway for phages of Gram-negative hosts. Prado and Paz, although exhibiting no significant DNA homology to each other, are new members of the phiKMV-like phage type, based on the position of the single-subunit RNA polymerase gene. The four phages are type IV pilus dependent for infection of both X. fastidiosa and Xanthomonas. The phages may be useful as agents for an effective and environmentally responsible strategy for the control of diseases caused by X. fastidiosa.
INTRODUCTION
The plant-pathogenic bacterium Xylella fastidiosa is the causative agent of a number of economically important diseases, including Pierce's disease of grape, phony peach disease, periwinkle wilt, citrus variegated chlorosis, almond leaf scorch, oleander leaf scorch, and coffee leaf scorch (1). Existing disease control methods are often only partially successful and, in the case of systemic insecticides used to control vector populations, may be potentially harmful to the environment (2, 3). Recently, there has been renewed interest in the application of bacteriophages (phage) as an environmentally acceptable mode for the control of bacterial plant disease (4).
To this end, our group isolated and characterized the first temperate (lysogenic) phage of X. fastidiosa, Xfas53 (5). However, temperate phage should not be used as biocontrol agents because of their capacity for lysogenic conversion, superinfection immunity, and risk of generalized transduction. Instead, virulent (lytic) phages are needed for an effective and sustainable phage-based control strategy (6). Here we report the isolation and characterization of the first virulent siphophages and podophages for X. fastidiosa that are members of the Nazgul-like phage and the phiKMV-like phage types, respectively.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. X. fastidiosa strains were cultured at 28°C in PW-M broth (PW-MB) (7) or on PW-M agar (PW-MA) plates (PW-MB with 20 g/liter plant cell culture-tested agar [Sigma]) (5). PW-M soft agar (PW-MSA) (PW-MB with 7.5 g/liter plant cell culture-tested agar) was used for overlays, with 5- to 7-day-old plate cultures used to make indicator suspensions in PW-M broth (optical density at 600 nm [OD600] = ∼0.5). PD3 agar (20 g/liter) (8) was used for the preparation of electrocompetent cells and transformations. For X. fastidiosa cultures carrying antibiotic resistance cassettes or plasmids, the medium was supplemented with chloramphenicol (Cm) (5 μg/ml) or kanamycin (Km) (5 μg/ml). Xanthomonas strains were cultured at 28°C in tryptone nutrient broth (TNB) (9) or tryptone nutrient agar (TNA) (TNB lacking KNO3 with 20 g/liter agar). TNA soft agar (TNSA) (TNB lacking KNO3 with 7.5 g/liter agar) was used for overlays, with 18-h plate cultures being used to make indicator suspensions in TNB (OD600 = ∼0.5). For Xanthomonas cultures harboring plasmids, the medium was supplemented with Km (50 μg/ml). Yeast tryptone broth (YTB) and yeast tryptone sucrose agar (YTSA) (YTB with 20 g/liter agar and sucrose at a final concentration of 15% [wt/vol]) were used for the resolution of mutants (10). Plasmid constructs were generated by using Escherichia coli DH5α MCR as the host. E. coli strains were cultured at 37°C in LB broth or LB agar (11). For E. coli cultures harboring plasmids, medium was supplemented with Cm (50 μg/ml), Km (50 μg/ml), or ampicillin (Amp) (100 μg/ml), as appropriate. Plant extracts were plated onto PW-MA or TNA supplemented with cycloheximide (40 μg/ml), designated PW-MC and TNAC, respectively.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype and/or relevant feature(s)a | Reference or source |
|---|---|---|
| Strains | ||
| X. fastidiosa | ||
| Temecula 1 | PD strain (X. fastidiosa subsp. fastidiosa) (ATCC 700964) | 79 |
| Ann-1 | Oleander isolate (X. fastidiosa subsp. sandyi) (ATCC 700598) | 80 |
| Dixon | Almond isolate (X. fastidiosa subsp. multiplex) (ATCC 700965) | 80 |
| XF53 | Grape isolate (X. fastidiosa subsp. fastidiosa) | 5 |
| XF54 | Grape isolate (X. fastidiosa subsp. fastidiosa) | 5 |
| XF95 | Oleander isolate (X. fastidiosa subsp. sandyi) | 5 |
| XF134 | Grape isolate (X. fastidiosa subsp. fastidiosa) | This study |
| XF136 | Grape isolate (X. fastidiosa subsp. fastidiosa) | This study |
| XF140 | Grape isolate (X. fastidiosa subsp. fastidiosa) | This study |
| XF141 | Grape isolate (X. fastidiosa subsp. fastidiosa) | This study |
| XF15-1 | Temecula 1; ΔpilA::Kmr | This study |
| XF15-1-1 | XF15-1; NS1::Cmr pilA | This study |
| TM1 | Temecula 1; PD1693::Tn5 | 26 |
| tonB1 | Temecula 1; PD0843::Tn5 | 52 |
| Xanthomonas | ||
| EC-12 | Xanthomonas sp., rice isolate (ATCC PTA-13101) | This study |
| North 40 | X. axonopodis pv. citri, sweet orange isolate | N. Wang, University of Florida |
| Ft. Basinger | X. axonopodis pv. citri, sweet orange isolate | N. Wang, University of Florida |
| Block 22 | X. axonopodis pv. citri, sweet orange isolate | N. Wang, University of Florida |
| Jal-4 | X. euvesicatoria, jalapeno isolate | Laboratory stock |
| Presidio-4 | Xanthomonas sp., rice isolate | Laboratory stock |
| EC-12-1 | EC-12; unmarked deletion of pilA | This study |
| EC-12-1-1 | EC-12-1; pMo168::pilA | This study |
| E. coli | ||
| DH5αMCR | F− ϕ80lacZΔM15 Δ(lacZYA-argF) recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relAl deoR U169 | Life Technologies, Inc. (Gaithersburg, MD) |
| Plasmids | ||
| pUC19 | pMB1 replicon; cloning vector; Apr | 81 |
| pXF004 | RSF1010::Kmr; broad-host-range vector | 25 |
| pSJA101 | Isogenic to pUC19; Temecula 1 pilA downstream-flanking region cloned into XmaI and XbaI sites | This study |
| pSJA102 | Isogenic to pSJA101; Kmr cassette cloned into KpnI and XmaI sites | This study |
| pSJA103 | Isogenic to pSJA102; Temecula 1 pilA upstream-flanking region cloned into SacI and KpnI sites | This study |
| pAX1Cm | Cmr cassette and multiple-cloning site on pAX1 backbone | 7 |
| pSJA104 | Isogenic to pAX1Cm; Temecula 1 pilA cloned into XbaI and XhoI sites | This study |
| pMo130 | Suicide vector for allelic exchange; ColE1 ori RK2 oriT xylE sacB Kmr | 10 |
| pSJA105 | Isogenic to pMo130; EC-12 pilA upstream-flanking region cloned into NheI and BglII sites | This study |
| pSJA106 | pSJA105 with EC-12 pilA downstream-flanking region cloned into BglII and HindIII sites | This study |
| pMo168 | Replicative vector; ori pBBR1 mob+ xylE Kmr | 10 |
| pSJA107 | pMo168::EC-12 pilA | This study |
Ap, ampicillin; Cm, chloramphenicol; Km, kanamycin.
Bacteriophage isolation and purification.
Plant and sewage samples were assayed for the presence of phage able to form plaques on X. fastidiosa strain Temecula 1. Plant extracts were prepared by macerating 10 g of plant tissue (Oryzae sativa or weeds obtained from rice fields in Jefferson County and Wharton County, TX) in 50 ml of P-buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 8 mM MgSO4) using a mortar and pestle, strained through a sterile double layer of cheese cloth to remove large particles, and filter sterilized. Sewage influent collected from a municipal wastewater treatment plant in Brazos County, TX, was centrifuged twice and filter sterilized. Plant and sewage filtrates were directly screened for phage activity using the spot test and soft agar overlay method (5). Soft agar overlays containing 100 μl of X. fastidiosa strain Temecula 1 (∼108 CFU/ml) as an indicator host were allowed to solidify, spotted with 10-μl drops of serially diluted filtrates in P-buffer, and assayed after incubation at 28°C for 5 to 7 days. Filtrates producing either plaques or cleared zones were serially diluted in P-buffer and assayed by using the overlay method, where 100 μl of each dilution was mixed directly with the host suspension in tempered soft agar before overlaying. Individual plaques formed on overlays of X. fastidiosa strain Temecula 1 were excised, suspended in P-buffer, and filter sterilized, and titers were determined. This procedure was repeated three times to obtain single plaque isolates. Additionally, filtrates were screened for phage activity by using Xanthomonas strain EC-12 (isolated from rice [see below]), as described above. Plaques were purified three times by using Xanthomonas strain EC-12, and purified phages were tested for activity on X. fastidiosa strain Temecula 1.
High-titer phage plate lysates (>1 × 1010 PFU/ml) were prepared by harvesting overlay plates of X. fastidiosa strain Temecula 1 or Xanthomonas strain EC-12 exhibiting confluent lysis. After being flooded with 5 ml of P-buffer, the soft agar overlay was macerated, clarified by centrifugation, and filter sterilized. The resulting lysates were stored at 4°C. High-titer phage lysates were further purified by isopycnic CsCl centrifugation, as previously described (5).
Bacterial isolation, purification, and identification.
Samples were tested for the presence of X. fastidiosa by using quantitative real-time PCR (qRT-PCR) with X. fastidiosa-specific primers (INF2 and INR1), as previously described (12). Additionally, nonfiltered plant extracts and sewage samples were plated onto PW-MC or TNAC, incubated at 28°C, and evaluated for growth (3 days on TNAC and 10 days on PW-MC). Representative single colonies were picked from plates and streak purified three times to obtain stocks on appropriate media. Xanthomonas isolates were identified by sequencing of the 16S-23S internal transcribed spacer (ITS) region (primers used are listed in Table S1 in the supplemental material). One Xanthomonas isolate, EC-12, was selected for further use in this study.
Phage DNA isolation and genome sequencing.
DNA was isolated from CsCl-purified phage suspensions as previously described (13). Phages Sano, Salvo, Prado, and Paz were sequenced as part of two pools of phage genomes by using the 454 pyrosequencing method (Emory GRA Genomics Core). Phage genomic DNA was prepared from phage isolates as described above and mixed in equimolar amounts to a final concentration of ca. 100 ng/μl. The pooled DNA was sheared, ligated with a multiplex identifier (MID) tag specific for each of the four pools, and sequenced by pyrosequencing using a full-plate reaction on a Roche FLX Titanium sequencer according to the manufacturer's protocols. The pooled phage DNA was present in two sequencing reactions. The trimmed FLX Titanium flowgram outputs corresponding to each of the four pools were assembled individually by using Newbler Assembler version 2.5.3 (454 Life Sciences), by adjusting settings to include only reads containing a single MID per assembly. The identity of individual contigs was determined by PCR using primers generated against contig sequences and individual phage genomic DNA preparations as the template. The coverages, calculated as assembled reads (in nucleotides) divided by the size of the genome, were 41-, 62-, 140-, and 46-fold for phages Sano, Salvo, Prado, and Paz, respectively. Sequencher 4.8 (Gene Codes Corporation) was used for sequence assembly and editing. Phage chromosomal end structures were determined experimentally. Cohesive (cos) ends for phages Sano and Salvo were determined by sequencing off the ends of the phage genome and sequencing the PCR products derived by amplification through the ligated junction of circularized genomic DNA, as previously described (13). Direct terminal repeats for phages Prado and Paz were determined by sequencing off the ends of the phage genome with primers designed within the first and last identified genes. Protein-coding regions were initially predicted by using GeneMark.hmm (14), refined by manual analysis in Artemis (15), and analyzed by using BLAST (E value cutoff of 0.005) (16). Proteins of particular interest were additionally analyzed by InterProScan (17), HHpred searches against the pdb70_29jun13 database (18), the NCBI Conserved Domain Database (19), LipoP (20), SignalP 4.1 (21), and TMHMM (22). The sequence of phage Salvo gene 36 was confirmed by PCR and sequencing of the resulting PCR product.
Transmission electron microscopy.
Electron microscopy of CsCl-purified bacteriophage (∼1 × 1011 PFU/ml) was performed by diluting stock with P-buffer. Phages were applied onto thin 400-mesh carbon-coated Formvar grids, stained with 2% (wt/vol) uranyl acetate, and air dried. Specimens were observed on a JEOL 1200EX transmission electron microscope operating at an acceleration voltage of 100 kV. Five virions of each phage were measured to calculate mean values and standard deviations for dimensions of capsid and tail, where appropriate.
Host range and efficiency of plating.
Host ranges of purified phages (propagated on X. fastidiosa strain Temecula 1) were determined by the serial dilution spot test method, as described above, using a panel of X. fastidiosa and Xanthomonas isolates as indicator hosts. Phage sensitivities of type IV pilus mutants and complements were tested similarly. Efficiency of plating (EOP) was determined by calculating the ratio of the phage plaque titer obtained with a heterologous (nonpropagating) host to that obtained with a homologous (propagating) host. All experiments were done in triplicate.
One-step growth curve.
One-step growth curves were used to determine the burst size and latent period of the phages (23). Liquid cultures of logarithmically growing Xanthomonas strain EC-12 were infected with individual phages at a multiplicity of infection (MOI) of ∼3 and allowed to adsorb at 28°C for 5 min. To stop further phage adsorption, cultures were diluted 1,000-fold in TNB. Infected centers were incubated at 28°C with constant shaking (150 rpm). Samples were taken at 3-min intervals, immediately filter sterilized, and plated in soft agar lawns of Xanthomonas strain EC-12. All experiments were done in triplicate.
Bacteriophage adsorption.
The kinetics of phage adsorption was determined as previously described (5). Liquid cultures of logarithmically growing cells (X. fastidiosa strain Temecula 1 or Xanthomonas strain EC-12) were infected with individual phages (propagated on homologous hosts) at an MOI of ∼0.1. The mixture was incubated at room temperature with shaking (150 rpm). Samples were taken (2-h intervals for X. fastidiosa strain Temecula 1 and 2-min intervals for Xanthomonas strain EC-12) and immediately filter sterilized, and titers were determined. The rate of phage particle disappearance is defined as dP/dt = −kBP, where B is the concentration of bacteria, P is the concentration of free phage at any time (t), and k is the adsorption rate constant in ml cell−1 min−1 (24). All experiments were done in triplicate.
Construction of the X. fastidiosa strain Temecula 1 pilA deletion mutant and complementation in trans.
The X. fastidiosa pilA (GenBank accession number NP_780105.1) open reading frame (ORF) from nucleotides (nt) 2256755 to 2257201 (X. fastidiosa Temecula 1 [accession number NC_004556.1]) was removed and replaced with a Kmr cassette by site-directed gene disruption, as described previously by Matsumoto et al. (25), except that pUC19 served as the cloning vector and the downstream pilA, Kmr cassette (amplified from pXF004), and upstream pilA regions were cloned sequentially. Primers used are listed in Table S1 in the supplemental material. The deletion of pilA and replacement with the Kmr cassette were confirmed by PCR.
Complementation of X. fastidiosa strain Temecula 1 pilA deletion mutant strain XF15-1 was accomplished by introducing a wild-type copy of pilA using a chromosome-based complementation system (25). Insertion of pilA and the Cmr cassette into the neutral site (NS1) of putative complements was confirmed by PCR amplification using primer pair NS1-f and NS1-r (25) and sequencing of the product.
Construction of the Xanthomonas strain EC-12 pilA deletion mutant and complementation in trans.
Deletion of pilA in Xanthomonas strain EC-12 was performed as described previously by Hamad et al. (10), except that cells were transformed using electroporation. Primers used are listed in Table S1 in the supplemental material. PCR primers were designed based on the annotated pilA sequence of the Xanthomonas strain EC-12 draft genome. The deletion of pilA was confirmed by PCR. To complement Xanthomonas strain EC-12-1 (ΔpilA), wild-type pilA was introduced in trans (10). The presence of the wild-type pilA was confirmed by PCR and sequencing for confirmation.
Twitching motility assay.
Twitching motility was examined as previously described (26). Briefly, late-log-phase liquid cultures of X. fastidiosa strain Temecula 1 (5 days), Xanthomonas strain EC-12 (18 h), and their derivatives were centrifuged and resuspended to 200 μl in liquid medium as appropriate (1 × 109 CFU/ml). Five-microliter cell suspensions were spotted onto appropriate solid medium modified with 1.2% agar and incubated at 28°C (X. fastidiosa [5 days] and Xanthomonas [18 h]). Pseudomonas aeruginosa strain PAO1, grown in a similar manner at 37°C for 24 h, was used as a positive control. Following incubation, colony edge morphology was visualized by using an Olympus SZ-PT microscope equipped with a QImaging Go-21 CMOS camera system. Colonies with a peripheral fringe were designated twitch positive.
Lysogen formation assay.
To assay for phage lysogen formation, survivors of phage infection were tested for the presence of prophages. For each phage, bacteria were infected at an input MOI of ∼3 and plated in a soft agar overlay. Plates were monitored for colony growth (10 to 15 days for X. fastidiosa strain Temecula 1 and 2 to 3 days for Xanthomonas strain EC-12). Individual colonies that emerged were picked, purified (three times), and retested for phage sensitivity by spotting dilutions of the same phage in a soft agar overlay. Primer pairs specific to Sano and Salvo primase or Prado and Paz helicase genes (see Table S1 in the supplemental material) were then used to test for the presence of prophage sequences in the phage-insensitive isolates. Wild-type bacterial DNA was used as the negative control, and wild-type bacterial DNA spiked with phage DNA served as the positive control.
To test whether evidence for abortive lysogeny (i.e., the establishment of repression) could be found, we followed a procedure described previously by Gill et al. (27), except that reversibly bound phages were removed by three successive washes. Liquid cultures of logarithmically growing Xanthomonas strain EC-12 were cultured to an OD600 of ∼0.3 to 0.5. One-milliliter aliquots were pelleted by centrifugation and resuspended in 0.20 ml of phage lysate (harvested in TNB) or sterile TNB. After a 30-min incubation at 25°C, cell-phage mixtures were centrifuged, the supernatant was removed, and the titer of adsorbed phage was determined. In preliminary experiments, it was determined that phages were reversibly bound, which affected the MOIactual (28) calculation. To circumvent this problem and to obtain an accurate MOIactual, cells were resuspended in sterile TNB, allowed to incubate for 5 min at 25°C, and centrifuged, and supernatants were removed. This procedure was repeated three times to remove unbound phage. The titer of each supernatant was determined to determine PFU. Cell pellets were resuspended in 0.20 ml of sterile TNB, serially diluted, and plated to enumerate the bacterial survivors remaining following phage exposure. From these data, the MOIactual, i.e., the ratio of the number of adsorbed phage to the number of CFU in the phage-free controls, was calculated. These MOIactual values were used to calculate the predicted proportion of uninfected cells by using the Poisson distribution. This experiment was replicated three times, using both Sano and Prado.
Nucleotide sequence accession numbers.
In accordance with Texas A&M Center for Phage Technology policy, novel phages were given names for mnemonic purposes. Names were checked for uniqueness by literature searching and were prefixed with Xfa, which is the ReBase species acronym for X. fastidiosa (http://rebase.neb.com/). Completed phage genomes were deposited in GenBank under the following accession numbers: KF626665 for XfaSano, KF626668 for XfaSalvo, KF626667 for XfaPrado, and KF626666 for XfaPaz. For simplicity, the phage names are used without the Xfa prefix. Phages Sano, Salvo, Prado, and Paz were deposited in the ATCC under accession numbers ATCC PTA-13096, ATCC PTA-13095, ATCC PTA-13099, and ATCC PTA-13100, respectively.
RESULTS
Phage isolation and characterization.
Our initial efforts focused on the screening of rice and weed extracts for phages that formed plaques on lawns of X. fastidiosa strain Temecula 1. Extracts yielded 10 to 105 PFU/g of tissue on lawns of X.fastidiosa. X.fastidiosa was not detected in extracts (by direct plating or qRT-PCR to limits of detection). However, Xanthomonas hosts were isolated from extracts, including EC-12. Subsequently, samples from both plant tissues and sewage were routinely screened against both X. fastidiosa strain Temecula 1 and Xanthomonas strain EC-12. Of the phages isolated, Sano, Salvo, Prado, and Paz are representative.
The physical and host range properties of the four representative phages are summarized in Tables 2 and 3 and Fig. 1. Phages Sano and Salvo exhibit siphophage morphology, with isometric heads and noncontractile tails (Fig. 1A and B, respectively), whereas phages Prado and Paz exhibit podophage morphology, with isometric heads and short stubby tails (Fig. 1C and D, respectively). All four phages formed plaques on both X. fastidiosa and Xanthomonas hosts, with differences in host range (Table 3). The siphophages formed small clear plaques, whereas the podophages formed large clear plaques.
TABLE 2.
General, physiological, and structural characteristics of phages Sano, Salvo, Prado, and Paz
| Feature | Value for phaged |
|||
|---|---|---|---|---|
| Sano | Salvo | Prado | Paz | |
| Host strain | Temecula 1 | EC-12 | Temecula 1 | Temecula 1 |
| Mean capsid width (nm) (SD)a | 64 (±2.1) | 64 (±0.8) | 69 (±1.4) | 68 (±1.1) |
| Mean tail length (nm) (SD)a | 204 (±2.3) | 207 (±4.2) | NAd | NA |
| Mean k (ml cell−1 min−1) ± SDb | ||||
| Temecula 1 | (4.33 ± 0.28) × 10−12 | (4.33 ± 0.28) × 10−12 | (4.33 ± 0.28) × 10−12 | (4.33 ± 0.28) × 10−12 |
| EC-12 | (5.48 ± 0.23) × 10−10 | (4.58 ± 0.30) × 10−10 | (7.26 ± 0.42) × 10−10 | (5.98 ± 0.37) × 10−10 |
| Mean burst size (PFU/cell) (SD)c | 100 (±10.1) | 112 (±8.3) | 99 (±13.2) | 104 (±12.5) |
Phage physical dimensions are the means of measurements of five virions, and values in parentheses indicate standard deviations.
Phage adsorption rate constants, k, are the means of three independent experiments, and values in parentheses indicate standard deviations. Phage stocks were propagated on a homologous host.
Phage bursts are the means of three independent experiments, and values in parentheses indicate standard deviations. Xanthomonas strain EC-12 was used as the host.
NA, not applicable.
TABLE 3.
Phage host range
| Strain | Presence of phage |
|||
|---|---|---|---|---|
| Sano | Salvo | Prado | Paz | |
| X. fastidiosa | ||||
| Temecula 1 | +a | + | + | + |
| Ann-1 | + | + | + | + |
| Dixon | + | + | + | + |
| XF53 | + | + | + | + |
| XF54 | + | + | + | + |
| XF95 | + | + | + | + |
| XF134 | − | + | + | + |
| XF136 | − | + | + | − |
| XF140 | + | + | + | − |
| XF141 | − | + | + | − |
| Xanthomonas | ||||
| EC-12 | + | + | + | + |
| Jal-4 | − | − | + | + |
| Presidio-4 | − | − | + | + |
| North 40 | − | − | + | − |
| Ft. Basinger | − | − | + | − |
| Block 22 | − | − | + | − |
Ability to form individual plaques.
FIG 1.
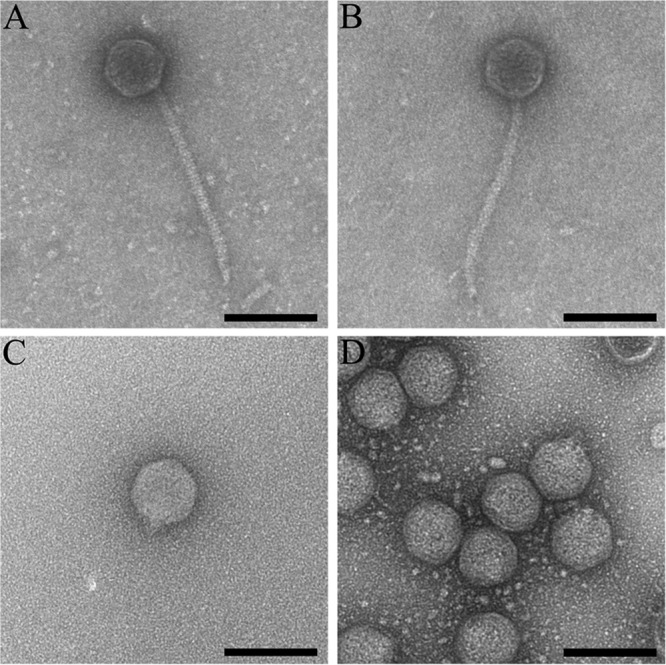
Morphology of phages Sano, Salvo, Prado, and Paz. Transmission electron micrographs show Sano (A), Salvo (B), Prado (C), and Paz (D) CsCl-purified phage particles. Samples were negatively stained with 2% (wt/vol) aqueous uranyl acetate, as defined in Materials and Methods. Bars, 100 nm.
Phages Salvo and Paz plated efficiently to X. fastidiosa strain Temecula 1 and Xanthomonas strain EC-12 (Table 4). However, there was an ∼2- to 3-log reduction in efficiency for phages Sano and Prado on the same hosts (Table 4). Adsorption rate constants for the four phages on X. fastidiosa strain Temecula 1 were on the order of ∼4 × 10−12 ml cell−1 min−1, which is much lower than those for most phages previously reported but similar to that for phage Xfas53 (5). In contrast, the rate constants for Xanthomonas strain EC-12 were ∼100-fold higher, at ∼5 × 10−10 ml cell−1 min−1. The burst size of all four phages was ∼102 PFU/cell at ∼52 min with Xanthomonas strain EC-12 as the host (Table 2). The extremely slow adsorption to X. fastidiosa strain Temecula 1 made it unfeasible to determine burst size with this host.
TABLE 4.
Influence of bacterial host on efficiency of plating
| Production host | Indicator host | Mean efficiency of plating ± SDa |
|||
|---|---|---|---|---|---|
| Sano | Salvo | Prado | Paz | ||
| Temecula 1 | Temecula 1 | 1.0 | 1.0 | 1.0 | 1.0 |
| Temecula 1 | EC-12 | (1.2 ± 0.2) × 10−2 | (1.4 ± 0.5) × 10−1 | (2.3 ± 0.2) × 10−3 | (3.1 ± 0.3) × 10−1 |
| EC-12 | EC-12 | 1.0 | 1.0 | 1.0 | 1.0 |
| EC-12 | Temecula 1 | (1.4 ± 0.7) × 10−3 | (2.0 ± 0.2) × 10−1 | (9.5 ± 0.5) × 10−2 | (3.2 ± 0.3) × 10−1 |
Data shown are the means of triplicate independent experiments ± standard deviations.
Genomics of siphophages Sano and Salvo.
The general characteristics of the phage genomes are summarized in Table 5, and complete annotations with supporting evidence are provided in Table S2 in the supplemental material. The genomes of Sano and Salvo were found to be 56.1 and 55.6 kb, encoding 77 and 72 genes, respectively. Both have 12-nt 5′-protruding single-stranded cos ends. Having cos ends makes it unlikely that Sano and Salvo are capable of generalized transduction.
TABLE 5.
General features of Sano, Salvo, Prado, and Paz phage genomes
| Feature | Value for phage |
|||
|---|---|---|---|---|
| Sano | Salvo | Prado | Paz | |
| Genome size (bp) | 56,147 | 55,601 | 43,940 | 43,869 |
| GC content (%) | 62.3 | 63.0 | 63.0 | 60.2 |
| Predicted no. of genes | 77 | 72 | 52 | 51 |
| Coding density (%) | 95.4 | 96.9 | 96.0 | 93.9 |
| Genomic termini | 12-bp 5′ overhang | 12-bp 5′ overhang | 619-bp DTRa | 675-bp DTR |
DTR, direct terminal repeats.
When analyzed by pairwise DNA MegaBLAST, the genomes of phages Sano and Salvo can be aligned over ∼69% of their length, with an average ∼80% sequence identity in aligned regions. Sano and Salvo contain 58 homologous genes, with amino acid sequence similarities ranging from ∼13 to 94%, and have retained genome synteny (Fig. 2; see also Table S2 in the supplemental material). Despite no direct evidence of transcriptional boundaries or rho-independent terminators, breaks between genes are not large enough to contain promoters. Therefore, the arrangement of genes on the two strands suggests that the genomes are organized into four transcriptional units. Genes found in transcriptional units I (starting at the left end; reverse strand) and II (forward strand) primarily encode proteins of unknown function and show very limited similarity to other phage genes in the NCBI database. The lysis cassette, structural module, and DNA metabolism module are all encoded in transcriptional unit III (reverse strand), whereas transcriptional unit IV (forward strand) encodes only a few genes, including a primase.
FIG 2.

Synteny of siphophages Sano and Salvo compared to Burkholderia phage Nazgul, Burkholderia phage AH2, Enterobacter phage Enc34, enteric bacterium phage Chi, and Providencia phage Redjac. Predicted genes are represented as boxes. Functional modules are represented by different colors (color key at the bottom right). Functionally equivalent genes are connected by solid lines (lysis genes are omitted for clarity). Homologous genes between Sano and Salvo are connected by dotted lines, and similarity at the amino acid level is indicated by color (color key at the bottom left). Apparent transcriptional units are indicated by Roman numerals. A distinct divergent transcriptional module in AH2 is indicated by a red box.
When analyzed by BLASTp, Sano and Salvo are most closely related to Burkholderia phage BcepNazgul (GenBank accession number NC_005091) (here referred to as Nazgul); however, the relationship is distant. Of the 77 predicted Sano genes, only 23 show homology to Nazgul, ranging from ∼11 to 48% sequence identity at the amino acid level. Homologs are primarily located in the structural and DNA metabolism regions (excluding tail fiber and assembly proteins). However, despite the low observed total homology, phages Sano and Salvo are syntenic with Nazgul (Fig. 2).
As defined previously by Casjens (29), Nazgul does not belong to any existing phage type because of its unique architecture and low homology to previously described phages. We therefore propose that Nazgul is the founder of a new phage type, to which both Sano and Salvo belong. Reannotation of the genomes of Burkholderia phage AH2 (30) and the closely related Enterobacter phage Enc34 (31), enteric bacteria phage Chi (32), and Providencia phage Redjac (33) indicates that they are all members of the Nazgul-like phage type (Fig. 2).
In the following sections, except where significant differences exist between Sano and Salvo, only the relationships between the proteins of phage Sano and their homologs are discussed in detail (Fig. 2; see also Table S2 in the supplemental material).
Transcriptional units I and II.
Most proteins encoded in transcriptional units I and II do not have homologs in the database. Despite this, 24 of the 38 proteins encoded in Sano transcriptional units I and II have homologs in Salvo, with ∼13 to 94% sequence identity at the amino acid level (Fig. 2; see also Table S2 in the supplemental material). Only one protein (Sano gp28) has a homolog in phage Nazgul (Nazgul gp34).
Lysis genes.
Phages of Gram-negative hosts have genes coding for three functional classes of lytic proteins: holins, endolysins, and spanins (34, 35). In addition, many phages encode an antiholin, a specific inhibitor of holin. Analysis of the genes of the Nazgul-like phage type suggested that all four functional classes are represented although with unexpected and novel variations. As with many phage genomes, the lysis proteins are clustered in lysis cassettes for the Nazgul-like phage type. Figure 3 shows the lysis cassettes of Sano, Salvo, Nazgul, AH2, and Chi; Redjac and Enc34 are not shown because the lysis gene arrangements are essentially identical to that of Chi. In each case, the lysis genes are the last cistrons of transcriptional unit III, which includes a long series of morphogenesis genes, except for AH2, which has a single novel gene inserted before the convergence point with unit III (Fig. 2 and 3). Except for Sano, the lysis cassettes have the order most commonly found in phages of Gram-negative hosts, with the genes encoding the holins and antiholins first, followed by the endolysin and then the i-spanin/o-spanin gene pair. In Sano, the endolysin gene is the first gene of the cassette. Beyond this synteny, however, the Nazgul-like family exhibits extreme diversity and mosaicism in its lysis genes, in that among the 24 lysis genes in the five cassettes shown in Fig. 3, 16 are unrelated to any of the others. Although the spanin genes fall into only two groups (see below), the five endolysin genes lack similarity with each other. Moreover, the nine genes encoding integral membrane proteins encode seven proteins with no detectable sequence similarity, presumably including at least three holins and four antiholins, as explained below.
FIG 3.

Lysis cassettes in the Nazgul-like phage type. Black boxes indicate unrelated genes of unknown function; gray and brown indicate o-spanin and i-spanin genes. Shades of gray and brown indicate sequence homology; diagonal hatching indicates endolysin genes. No sequence similarity is present among the five endolysin genes shown. The blue box in the Salvo endolysin gene indicates the N-terminal lipoprotein domain; vertical hatching indicates genes encoding proteins with at least one TMD, potentially holins or antiholins. Pink and yellow indicate sequence homology. Topological models of Sano, Salvo, Nazgul, AH2, and Chi membrane proteins are shown at the right. Genes are drawn to scale.
Endolysins.
The endolysin genes (Fig. 3, diagonal hatching) in these five phages can be identified independently of their proximity to the other lysis genes although with various degrees of confidence. gp38 of Nazgul is very similar to the P endolysin of the paradigm coliphage 186; P is a transglycosylase of the well-characterized lambda R family. gp43 of Sano is closely related to the endolysin of a Pseudomonas phage, PaP1, which has been shown to have muralytic activity, although its enzymatic type is not known (36). Gene 42 of AH2 has many related genes in prophage elements although not to any identified endolysins. However, HHpred analysis reveals weak similarity to eukaryotic and prokaryotic lysozyme (glycosidase) proteins. The Chi/Redjac/Enc34 endolysins were similar to proteins designated lysis protein A in a Salmonella phage, iEPS5 (37). Although all of these endolysins are distinct and belong to at least two enzymatic classes, they lack signal sequences or membrane domains and are thus soluble endolysins that require canonical holins for release across the cytoplasmic membrane (see below). gp36 of phage Salvo stands in stark contrast and represents an endolysin of unprecedented structure. The last ∼170 residues of gp36, although lacking any similarity to individual proteins by BLAST analysis, nevertheless are strongly predicted by HHpred analysis to be related to a number of experimentally and structurally characterized phage endolysins, including the classical phage T4 lysozyme. However, the first ∼100 residues are unrelated to any sequence in the database. Moreover, the N terminus has a strongly predicted outer membrane (OM) lipoprotein signal sequence, indicating that this endolysin is exported by the sec translocon and, after processing by signal peptidase II and localization by the Lol system, becomes tethered to the inner leaflet of the OM as a 275-amino-acid (aa) mature lipoprotein. In support of this interpretation, gp42 has two pairs of Cys residues in the periplasmic domain; Cys residues in pairs are always found in the catalytic domains of SAR endolysins, presumably to provide stabilizing intramolecular disulfide linkages necessary for the protease-rich environment of the periplasm (38). In contrast, the cytoplasmic endolysins of the other phages of the Nazgul-like family have no Cys residues, except for the Redjac endolysin, which has a single Cys; this supports the notion that these are all cytoplasmic endolysins.
Spanins.
Most phages of Gram-negative hosts have two-component spanins, comprised of an integral inner membrane (IM) protein, or i-spanin, and an OM lipoprotein, or o-spanin, that together form a complex that spans the periplasm by C-terminal interactions (39). Genes for two spanin proteins can be arranged in three distinct architectures (39). All 7 members of the Nazgul-like phage type encode two-component spanins (Fig. 3). The sequences fall into two families, with Sano and Salvo comprising one family and the other five members forming the second, as shown most clearly in Fig. 4 for the mature o-spanin lipoproteins. Surprisingly, in both families, there are representatives of two spanin gene architecture classes. In the Sano/Salvo family, the former has its spanin genes in the separated class, whereas in the latter, the spanin genes are overlapped. Similarly, the AH2 genes are entirely separated, whereas the four spanin gene pairs with similarity to the AH2 genes are in the overlapped architecture. These are the first two examples of spanin gene families that include members from different architectural classes (see Discussion).
FIG 4.

Alignments of o-spanin proteins from the Sano/Salvo and Nazgul/AH2/Chi/Enc34/Redjac families. Alignments using the Feng-Doolittle (77) progressive algorithm are shown for the Sano/Salvo (A) and Nazgul/AH2/Chi/Enc34/Redjac (B) families of o-spanin proteins. Residues are highlighted with RASMOL (78) colors. Consensus residues were determined based on 100% identity.
Holins and antiholins.
Absent similarity to a known holin, the general practice is to assign holin function to any integral membrane protein encoded by a gene clustered with the more easily identified endolysin and spanin genes, especially if the putative holin shares topology with one of the three established holin topologies (40, 41). Among the Nazgul-like phages, the Chi/Enc34/Redjac subset is the simplest for holin annotation, since there is only a single gene encoding an integral membrane protein immediately upstream of the endolysin and spanin genes, the most common arrangement for lysis cassettes. This protein, gp02 in Chi (Fig. 3), has a single transmembrane domain (TMD) and is arranged in an N-in, C-out topology, making it the first member of topology class III that is not related to the phage T4 T holin family. The other four members of the Nazgul-like phage type each have genes for two integral membrane proteins clustered with the lysis genes. Among these eight proteins, seven different membrane topologies are represented. Both of the two membrane proteins encoded by Nazgul have homologs in AH2 (Fig. 3, yellow and pink), but otherwise, all of these proteins are unrelated and novel. Although Nazgul gp39 has a predicted topology consistent with the most common holin topology, class I (three TMDs with N-out and C-in topology), its AH2 homolog lacks TMD1. Since it has been shown for two paradigm class I holins, lambda S105 and P2 Y, that the first TMD is required for function, it seems unlikely that these proteins encode holins. In contrast, Nazgul gp40 and its homolog AH2 gp44 both have class III topology, like the Chi holin noted above. The lysis cassettes of Sano and Salvo each encode two novel integral membrane proteins, and because of ambiguity as to the identity of the holin or antiholin, we have annotated all four genes (Sano genes 41 and 42 and Salvo genes 37 and 38) “holin-antiholin.”
Structural genes.
There is limited amino acid sequence homology between the tail fiber and assembly genes of Sano and Nazgul (see Table S2 in the supplemental material). However, Sano genes 44 to 52 can be presumptively assigned the functions of tail fiber and assembly proteins based on synteny to similar genes in Nazgul (genes 41 to 49) (Fig. 2). Sano gp53 is a tail tape measure protein based on the conserved tape measure domain (InterPro IPR013491). In most siphophages and myophages, the reading frames upstream of the tape measure protein encode the functional homologs of the lambda tail assembly chaperones G and G-T. These represent alternate translational products related by programmed −1 frameshifts, and both products have been shown to be required for tail assembly (42). In Sano, the G- and G-T-equivalent frames are genes 55 and 54; manual inspection reveals a translational slippery sequence, 5′-GGGAAAC-3′, near the end of Sano gene 55. This sequence is conserved in Salvo.
Genomics of podophages Prado and Paz.
The general characteristics of the phage genomes are summarized in Table 5, and complete annotations with supporting evidence are provided in Table S3 in the supplemental material. The genomes of Prado and Paz were found to be 43.9 and 43.8 kb, encoding 52 and 51 genes, respectively. The genomes of Prado and Paz contain nonpermutated direct terminal repeats of 619 and 675 bp, respectively.
Prado and Paz are clearly phiKMV-like based on genome organization and protein homology with other phiKMV-like phages. Prado is most similar to Caulobacter phage Cd1 (GenBank accession number GU393987), with 26 homologous genes ranging from ∼8 to 55% amino acid similarity. Similar to T7-like phages, phiKMV-like phage genomes consist of three functional gene clusters encoded on the forward strand. Class I consists of early genes with functions to overcome host restriction and to convert the metabolism of the host cell to the production of phage proteins. Class II encodes proteins involved in DNA metabolism. Class III encodes structural and assembly proteins, DNA packaging proteins, and lysis proteins. The phiKMV-like phages are distinguished from T7-like phages in that they encode a single-subunit RNA polymerase (RNAP) at the end of the class II gene cluster rather than in the early genomic region (43). Phages Prado and Paz share no significant DNA homology (∼4%) in comparisons by pairwise DNA MegaBLAST. Despite the divergence in DNA sequence, Prado and Paz contain 43 homologous genes with amino acid similarities ranging from ∼17 to 79% and have the canonical phiKMV-like gene order (Fig. 5; see also Table S3 in the supplemental material).
FIG 5.

Synteny of podophages Prado and Paz compared to Pseudomonas phage phiKMV and Caulobacter phage Cd1. Predicted genes are represented as boxes. Functional modules are represented by different colors (color key at the bottom right). Functionally equivalent genes are connected by solid lines. Homologous genes between Prado and Paz are connected by dotted lines, and similarity at the amino acid level is indicated by color (color key at the bottom left).
In the following sections, except where significant differences exist between Prado and Paz, only the relationship between Prado proteins and database homologs are discussed in detail (Fig. 5; see also Table S3 in the supplemental material).
Early genes.
Early genes are involved in host conversion and have been reported to protect phage from bacterial defense mechanisms or alter host cell mechanisms (44). When comparing phages of the same phage type, phage proteins that interact most intimately with the bacterial host have been reported to be the most diverse (45). Five out of twelve Prado early genes have homologs in Paz. Only two early gene proteins, Prado gp02 and gp11, have homologs in other phiKMV-like phages, namely, Cd1 gp01 and gp09, respectively.
DNA metabolism.
Phage Prado DNA metabolism genes follow the canonical phiKMV-like gene order of DNA primase (gene 13), DNA helicase (gene 14), DNA polymerase A (gene 17), 5′-3′ exonuclease (gene 19), DNA exonuclease VII (gene 20), ATP-dependent DNA ligase (gene 26), and a single-subunit RNAP (gene 28) (Fig. 5). Prado gp21 and gp22 do not have homologs in Cd1 but do have homologs in other phiKMV-like phages, such as Ralstonia phage RSB1 gp22 and gp24, respectively. The Prado gene encoding single-subunit RNAP is located at the right end of the DNA metabolism gene cluster, a distinguishing characteristic of all reported phiKMV-like phages. However, it has been noted that the position of the DNA ligase gene can vary within phiKMV-like phages (46). In phiKMV, the DNA ligase is encoded at the beginning of the DNA metabolism genes, whereas in Cd1 and Salvo, the DNA ligase is encoded directly before the RNA polymerase (Fig. 5).
The DNA metabolism region of Prado also shares high homology to several Xanthomonas phages, including CP1 (GenBank accession number NC_019933), Xop411 (accession number NC_009543), Xp10 (accession number NC_004902), OP1 (accession number NC_007709), phiL7 (accession number NC_012742), and CP2 (accession number NC_020205). These phages are non-phiKMV-like yet have homology and preserved gene order characteristic of phiKMV-like DNA metabolism regions, including a single-subunit RNAP located at the end of the region.
Structural and lysis genes.
As for the DNA metabolism genes noted above, Prado's structural and lysis gene region follows the phiKMV-like gene order. It begins immediately downstream of the single-subunit RNAP, with three small genes for conserved proteins of hypothetical function (genes 29 to 31) that are homologs of Cd1 genes 27 to 29. Next in sequence are genes encoding a head-tail connector (gene 32), scaffold protein (gene 33), major capsid protein (gene 34), tail tube A (gene 36), tail tube B (gene 37), and three internal virion proteins (genes 38 to 40). Prado gp40 possesses a C-terminal lysozyme-like domain (IPR023346) identical to that found in Cd1 gp38. However, Prado gp40 is ∼300 aa larger than any previously reported homolog in phiKMV-like phages.
There is diversity within the tail fiber proteins of the phiKMV-like phages in terms of both sequence homology and components (46). In phiKMV-like phages, tail structural proteins are found immediately downstream of the internal virion proteins. Similarly, genes for the Prado tail fiber proteins (genes 41 to 44) are found in the same location. Prado gp41 contains a T7 tail fiber protein domain (IPR005604), and gp43 and gp44 have homologs of tail fiber proteins found in phiKMV-like phages. It appears, however, that Prado genes 41 to 44 may have been acquired through horizontal gene transfer with a non-phiKMV-like phage. The four genes are found as a cluster and are homologs of genes 26 to 29 in Xanthomonas phage CP2, with ∼31 to 41% identity.
The lysis cassette of phage phiKMV has been studied in detail and is organized with genes encoding holin, endolysin, i-spanin (formerly Rz), and o-spanin (formerly Rz1) immediately downstream of TerS and TerL (47). The lysis cassette of Prado is similar to that of phiKMV, except that the predicted holin is found upstream of the endolysin, separated by three genes, encoding TerS (gene 46), TerL (gene 47), and a hypothetical protein (gene 48) (Fig. 5). This configuration, with lysis and terminase genes intermingled, was previously observed for phage SP6 and the phiKMV-like phage JG068 (48, 49).
Prado gene 45 encodes a putative type II holin based on the presence of two TMDs (N-in, C-in topology). TMD2 (residues 31 to 48) is the most hydrophobic of the two. TMD1 (residues 13 to 29) is not predicted by TMHMM and has characteristics of a SAR domain, with a high percentage of weakly hydrophobic or polar residues. The class II topology and the presence of a SAR domain followed by a typical TMD suggest that gp45 is a pinholin, similar to phiKMV gp45 (47). Prado gene 49 encodes an endolysin, based on homology to other phiKMV-like phage endolysins in the database. Prado endolysin gp49 exhibits an N-terminal hydrophobic domain rich in weakly hydrophobic residues that is characteristic of a SAR endolysin (50). A glycoside hydrolase domain (IPR002196) is predicted by InterProScan and contains an E-8 aa-D-8 aa-T catalytic triad, similar to the E-8 aa-D-5 aa-T and E-9 aa-D-6 aa-T catalytic triads found in phage T4 protein E and phiKMV gp45, respectively (38, 47). Prado spanins were identified by characteristic gene orientation and proximity to the endolysin. Prado gp52 is homologous with HslV family proteins of Xanthomonas phages OP1 (gp51), Xp10 (gp51), and Xop411 (gp51), with ∼38 to 42% identity, but does not contain the N-terminal hydrolase domain present in the other phage proteins.
Phage receptor site identification.
In Gram-negative bacteria, OM proteins, pili, flagella, oligosaccharides, or lipopolysaccharides can act as phage receptors (51). Our preliminary results (not shown) demonstrated that two transposon insertion mutants of X. fastidiosa strain Temecula 1, TM1 (unpiliated) (26) and “tonB1 mutant” (hyperpiliated) (52), were resistant to the four phages, indicating the phages may be type IV pilus dependent. To confirm these observations, we generated type IV pilus mutants (in-frame deletions of pilA) in X. fastidiosa strain Temecula 1 and Xanthomonas strain EC-12 and tested for phage sensitivity. PilA is the major subunit of type IV pili and is required for type IV pilus formation (53). Wild-type X. fastidiosa strain Temecula 1 and Xanthomonas strain EC-12 exhibited twitching motility (indicative of pilus retraction) (Fig. 6A and D, respectively) and were sensitive to all four phages. The ΔpilA derivatives XF15-1 and EC-12-1 were devoid of twitching motility (Fig. 6B and E, respectively) and were resistant to all four phages. Twitching motility and phage sensitivity were restored in XF15-1 and EC-12-1 by complementation of pilA in trans (Fig. 6C and F, respectively), providing evidence that the phages are type IV pilus dependent.
FIG 6.

Bacterial colony morphology as an indicator of twitching motility. Colonies of the indicated strains were grown on the appropriate solid medium modified with 1.2% agar and incubated at 28°C (X. fastidiosa [5 days] and Xanthomonas [18 h]), as described in Materials and Methods. (A) Wild-type X. fastidiosa strain Temecula 1; (B) Temecula 1 ΔpilA strain; (C) Temecula 1 ΔpilA strain complemented chromosomally in trans with pilA; (D) wild-type Xanthomonas strain EC-12; (E) EC-12 ΔpilA strain; (F) EC-12 ΔpilA strain complemented with pilA-containing plasmid pSJA107. Bars, 0.1 mm.
Lysogeny.
To examine the potential for lysogeny, 40 phage-insensitive isolates of X. fastidiosa strain Temecula 1 and Xanthomonas strain EC-12 each were recovered following a challenge by phage Sano, Salvo, Prado, or Paz. PCR using phage-specific primers did not detect the presence of phage lysogens in resistant isolates, indicating that resistance was not due to lysogeny. Additionally, we examined the potential for abortive lysogeny using infection at a high MOI and measuring survival (27). As shown in Table 6, following infection with Sano or Prado, there was no significant difference between predicted and actual survivors, indicating that phage infection at a high MOI did not lead to the establishment of repression. Together, these results indicate that there is no evidence for lysogeny or repression, supporting the conclusion that the four phages are virulent.
TABLE 6.
Predicted bacterial survivors based on MOIactual compared to measured bacterial survivors of Xanthomonas strain EC-12 following exposure to phage Sano or Pradoa
| Phage | Replicate | MOIactual | Predicted % surviving cells | Measured % surviving cells | Fold difference of survivors vs prediction |
|---|---|---|---|---|---|
| Sano | 1 | 6.51 | 0.15 | 0.12 | 0.80 |
| 2 | 5.57 | 0.38 | 0.25 | 0.65 | |
| 3 | 5.99 | 0.30 | 0.24 | 0.80 | |
| Prado | 1 | 5.40 | 0.45 | 0.38 | 0.80 |
| 2 | 5.39 | 0.45 | 0.49 | 1.08 | |
| 3 | 5.52 | 0.40 | 0.37 | 0.92 |
Predicted survivors were calculated from the Poisson distribution for the measured MOIactual. Data shown are from three independent replicate experiments.
DISCUSSION
We have previously reported the isolation and propagation of the first temperate phage of X. fastidiosa, Xfas53. Here we report the isolation and propagation of the first virulent phages able to infect X. fastidiosa and Xanthomonas. Phages were isolated from plant extracts that contained Xanthomonas spp. but not X. fastidiosa. We have more recently isolated phage directly from the petioles of grapevines that plate to both X. fastidiosa isolates and Xanthomonas. Both genera are members of the Xanthomonadaceae and are associated with a broad range of plant hosts (54, 55). Xylella and Xanthomonas share a common origin and have diverged, with X. fastidiosa having a reduced genome that may reflect a xylem-limited lifestyle (56, 57). Despite their divergence, both have retained type IV pili (58–62), which have been previously identified as phage receptors (63–70). Phages Sano, Salvo, Prado, and Paz are type IV pilus dependent.
Sano and Salvo are siphophages with isometric heads and noncontractile tails, whereas Prado and Paz are podophages with isometric heads and short stubby tails (Fig. 1). The adsorption rate of the four phages to X. fastidiosa strain Temecula 1 was similar to that observed previously for Xfas53 (5) but was ∼100-fold higher for Xanthomonas strain EC-12. The observed difference in rate constants between hosts may be due to possible differences in the affinity for the type IV pilus of X. fastidiosa and Xanthomonas spp. or potential differences in type IV pilus retraction speed/frequency that may affect irreversible binding to a secondary receptor site(s). The genomes of Sano, Salvo, Prado, and Paz have an average GC content of ∼62.1%, which is similar to that of sequenced Xanthomonas spp. (∼63.6 to 65.3%) and Xanthomonas phages (∼60%) but significantly higher than those of X. fastidiosa strains (∼49 to 55.6%) (54, 57), suggesting that the primary hosts in the plant environment are Xanthomonas spp. We speculate that overlapping plant host ranges and shared environmental niches, along with the conservation of type IV pili, may have led to the selection of phages able to infect both genera. Restriction-modification and clustered regularly interspaced short palindromic repeat (CRISPR)-cas systems have been identified in some Xanthomonas spp. (71, 72), with X. fastidiosa exhibiting only the former (73). The observed weak inhibition of efficiency of plating (Table 4) suggests that either the above-mentioned systems are relatively inefficient in both hosts or the phages are subverting them, as has been demonstrated for phage T7 (74).
Phages Prado and Paz are members of the phiKMV-like phage type, a large group of virulent phage, and presumably have diverged from a common phiKMV-like ancestor due to genetic drift. However, the DNA metabolism regions of Prado and Paz share homology to several non-phiKMV-like (based on genome architecture) Xanthomonas phages. Additionally, the tail fibers of Prado and Paz share the highest homology with the non-phiKMV-like phage CP2. The observed homology in individual genes and gene clusters between Prado and Paz with non-phiKMV-like Xanthomonas phages is indicative of horizontal gene transfer between these two phage groups and further demonstrates the mosaic nature of phages.
We propose that phage Nazgul is the founder of a new phage type, the Nazgul-like phages. Phages Sano and Salvo are members of this new phage type. Analysis indicates that Burkholderia phage AH2, Enterobacter phage Enc34, enteric bacterium phage Chi, and Providencia phage Redjac are Nazgul-like (Fig. 2). It would be expected that closely related phages would share a similar infection lifestyle (i.e., temperate or virulent). However, closely related virulent and temperate mycobacteriophage and siphophage of P. aeruginosa have been reported (75, 76). Phage AH2 was not previously defined as temperate (30), but there is clear evidence that it may be a temperate phage. AH2 is predicted to encode three excisionase proteins (genes 30, 31, and 69), an integrase (gene 37), and a CI-like repressor (gene 70). Homologs of these genes are not present in the other Nazgul-like phages. In fact, AH2 genes 30, 31, and 37 are harbored in two adjacent transcriptional units (based on coding strand position) (boxed in red in Fig. 2) not present in other Nazgul-like phages. Additionally, there is no clear evidence that the small proteins with helix-turn-helix DNA-binding domains of phages Nazgul, Sano, and Salvo (gp73, gp75, and gp70, respectively) are Cro-like proteins, as annotated in the other Nazgul-like phages (30–33). Rather, these proteins may be involved in transcriptional regulation. The lysogeny experiments conducted with phages Sano and Salvo support the conclusion that the phages are virulent. Together, this suggests that the Nazgul-like phages, excepting AH2, have diverged to become virulent, due to possible deletions and modifications of lysogeny-related genes.
Despite the rather complete synteny of the Nazgul-like phage-type genomes, the lysis cassettes are examples of pronounced mosaicism. Among the seven genomes, there are at least four novel, unrelated holins represented. Two of these (gp02 of Chi, with homologs in Redjac and AH2, and gp40 in Nazgul, with a gp44 homolog in AH2) can be identified with confidence and represent holins with class III topology. These are the first holins of class III that are not related to the T holin of the paradigm phage T4. Moreover, all of the holins except for Sano must be canonical “large-hole” holins, rather than pinholins, since the respective endolysins lack secretory or membrane localization signals and thus require large holin-mediated lesions for escape across the bilayer (34).
In the Nazgul-like phage type, the most surprising finding is that the Salvo endolysin gene 36 has a strong lipobox signal (Fig. 3), which would mandate that the enzyme is secreted through the translocon, modified as a lipoprotein, and sorted to the inner leaflet of the OM. Heretofore, only two modes of endolysin regulation have been characterized in phages of Gram-negative hosts: canonical (cytoplasmic) endolysins, which are simply sequestered fully active in the cytoplasm until being liberated by holin-mediated hole formation, and SAR endolysins, which are secreted in a membrane-tethered, inactive state by the sec system and activated by release from the bilayer after pinholin-dependent membrane depolarization. Neither of these strategies would apply to gp36, since there is no obvious way that a holin could alter the IM in a way that would directly affect the OM attachment of the gp36 endolysin molecules. It seems unlikely that gp36 endolysin accumulates as a fully active, OM-attached endolysin throughout the morphogenesis period without causing premature lysis, so a novel mode of holin-mediated endolysin control may be in place.
The second most surprising finding concerns the spanin genes of the Nazgul-like phage type. There are two distinct families of i-spanin/o-spanin gene pairs, the Sano/Salvo family and the Nazgul family, shared by the other four phages (Fig. 4). Unexpectedly, both of these families have members in the overlapped and separated architectural classes (Fig. 2 and 3). The Salvo o-spanin gene is overlapped by its i-spanin gene, whereas in Sano, the genes are separated. Similarly, the AH2 genes are separated, but the homologs of the AH2 i-spanin and o-spanin genes in the other phages are all overlapped. Besides this being the first instances where spanin gene families have members that cross an architectural class boundary (39), the alignments of the homologs of differing classes may provide a clue to the evolutionary process that underlies the unexpected gene arrangements. When the two gene pairs are aligned, it can be seen that the homologous regions of the i-spanins and o-spanins of Sano and Salvo are separated by ∼100 bp in the Sano-separated genes (Fig. 7). The most parsimonious interpretation is that a deletion that crossed the gene boundaries flanking this intervening DNA resulted in the overlapped architecture seen in Salvo. Paradoxically, the deletion, which presumably included the DNA encoding the lipobox of the mature lipoprotein, resulted in a fusion to the +1 reading frame of the upstream i-spanin gene and a consequent extension of the o-spanin by 8 N-terminal residues. The structures examined here suggest that the overlapped gene architecture evolved by a process of successive deletion and reading frame fusion and support our previous hypothesis that the spanin gene architectures originated with the “normal” separated gene format, in which both proteins could evolve independently (39).
FIG 7.
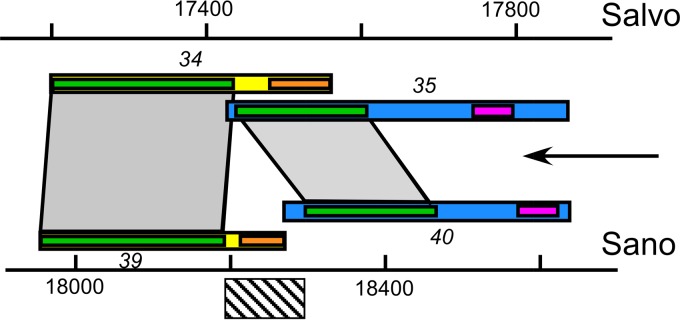
Spanin gene architecture. Shown is a putative deletion (diagonal-hatched bar) that could convert the Sano-separated spanin gene pair into a Salvo-like overlapped gene pair. Gray areas connecting green domains show homologous domains of Salvo and Sano i-spanins (blue bars) and o-spanins (yellow bars). Orange sub-bars indicate lipoprotein signal sequences in o-spanins. Pink sub-bars indicate the TMD in i-spanin. The arrow indicates the direction of transcription.
In medicine and agriculture, the search for innovative solutions to control disease is an ongoing process. Phage therapy offers a novel and environmentally acceptable treatment for the control of disease caused by X. fastidiosa. This is the first report of the propagation and characterization of virulent phages for X. fastidiosa. The fact that virulent phages for X. fastidiosa can now be easily propagated in a surrogate host (Xanthomonas) that grows in a standard medium allows the efficient isolation, characterization, and propagation of virulent phages that can be implemented as biocontrol agents for prevention or treatment of disease caused by X. fastidiosa. The reported phages are therefore attractive candidates for the development of phage cocktails to control disease caused by X. fastidiosa.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by a grant to the Center for Phage Technology and Texas A&M AgriLife Research from Otsuka Pharmaceutical Co., Ltd.
We thank Thomas J. Burr for kindly providing strains. We thank Michele Igo for kindly providing plasmids used in this study. We thank Jason Gill for his critical reading of the manuscript and Guichun Yao for her technical assistance. We thank Tsutomu Nishida for his support throughout this project.
Footnotes
Published ahead of print 8 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01080-13.
REFERENCES
- 1.Chatterjee S, Almeida RPP, Lindow S. 2008. Living in two worlds: the plant and insect lifestyles of Xylella fastidiosa. Annu. Rev. Phytopathol. 46:243–271. 10.1146/annurev.phyto.45.062806.094342 [DOI] [PubMed] [Google Scholar]
- 2.Stokstad E. 2012. Agriculture field research on bees raises concern about low-dose pesticides. Science 335:1555. 10.1126/science.335.6076.1555 [DOI] [PubMed] [Google Scholar]
- 3.Whitehorn PR, O'Connor S, Wackers FL, Goulson D. 2012. Neonicotinoid pesticide reduces bumble bee colony growth and queen production. Science 336:351–352. 10.1126/science.1215025 [DOI] [PubMed] [Google Scholar]
- 4.Jones JB, Jackson LE, Balogh B, Obradovic A, Iriarte FB, Momol MT. 2007. Bacteriophages for plant disease control. Annu. Rev. Phytopathol. 45:245–262. 10.1146/annurev.phyto.45.062806.094411 [DOI] [PubMed] [Google Scholar]
- 5.Summer EJ, Enderle CJ, Ahern SJ, Gill JJ, Torres CP, Appel DN, Black MC, Young R, Gonzalez CF. 2010. Genomic and biological analysis of phage Xfas53 and related prophages of Xylella fastidiosa. J. Bacteriol. 192:179–190. 10.1128/JB.01174-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill JJ, Hyman P. 2010. Phage choice, isolation, and preparation for phage therapy. Curr. Pharm. Biotechnol. 11:2–14. 10.2174/138920110790725311 [DOI] [PubMed] [Google Scholar]
- 7.Hill BL, Purcell AH. 1995. Multiplication and movement of Xylella fastidiosa within grapevine and 4 other plants. Phytopathology 85:1368–1372. 10.1094/Phyto-85-1368 [DOI] [Google Scholar]
- 8.Davis MJ, French WJ, Schaad NW. 1981. Axenic culture of the bacteria associated with phony disease of peach and plum leaf scald. Curr. Microbiol. 6:309–314. 10.1007/BF01566883 [DOI] [Google Scholar]
- 9.Hansen JB, Olsen RH. 1978. Isolation of large bacterial plasmids and characterization of P2 I compatibility group plasmids Pmg1 and Pmg5. J. Bacteriol. 135:227–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamad MA, Zajdowicz SL, Holmes RK, Voskuil MI. 2009. An allelic exchange system for compliant genetic manipulation of the select agents Burkholderia pseudomallei and Burkholderia mallei. Gene 430:123–131. 10.1016/j.gene.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertani G. 1951. Studies on lysogenesis. 1. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62:293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bextine B, Child B. 2007. Xylella fastidiosa genotype differentiation by SYBR Green-based QRT-PCR. FEMS Microbiol. Lett. 276:48–54. 10.1111/j.1574-6968.2007.00910.x [DOI] [PubMed] [Google Scholar]
- 13.Summer EJ. 2009. Preparation of a phage DNA fragment library for whole genome shotgun sequencing. Methods Mol. Biol. 502:27–46. 10.1007/978-1-60327-565-1_4 [DOI] [PubMed] [Google Scholar]
- 14.Lukashin AV, Borodovsky M. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26:1107–1115. 10.1093/nar/26.4.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. 10.1093/bioinformatics/16.10.944 [DOI] [PubMed] [Google Scholar]
- 16.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST plus: architecture and applications. BMC Bioinformatics 10:421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi HY, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJA, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40:D306–D312. 10.1093/nar/gkr948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248. 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229. 10.1093/nar/gkq1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12:1652–1662. 10.1110/ps.0303703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 22.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 23.Clokie MRJ, Kropinski AM. 2009. Bacteriophages: methods and protocols. Humana Press, New York, NY [Google Scholar]
- 24.Schwartz M. 1975. Reversible interaction between coliphage lambda and its receptor protein. J. Mol. Biol. 99:185–201. 10.1016/S0022-2836(75)80167-7 [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto A, Young GM, Igo MM. 2009. Chromosome-based genetic complementation system for Xylella fastidiosa. Appl. Environ. Microbiol. 75:1679–1687. 10.1128/AEM.00024-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Hao G, Galvani CD, Meng Y, De La Fuente L, Hoch HC, Burr TJ. 2007. Type I and type IV pili of Xylella fastidiosa affect twitching motility, biofilm formation and cell-cell aggregation. Microbiology 153:719–726. 10.1099/mic.0.2006/002311-0 [DOI] [PubMed] [Google Scholar]
- 27.Gill JJ, Summer EJ, Russell WK, Cologna SM, Carlile TM, Fuller AC, Kitsopoulos K, Mebane LM, Parkinson BN, Sullivan D, Carmody LA, Gonzalez CF, LiPuma JJ, Young R. 2011. Genomes and characterization of phages Bcep22 and BcepIL02, founders of a novel phage type in Burkholderia cenocepacia. J. Bacteriol. 193:5300–5313. 10.1128/JB.05287-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasman LM, Kasman A, Westwater C, Dolan J, Schmidt MG, Norris JS. 2002. Overcoming the phage replication threshold: a mathematical model with implications for phage therapy. J. Virol. 76:5557–5564. 10.1128/JVI.76.11.5557-5564.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casjens SR. 2008. Diversity among the tailed-bacteriophages that infect the Enterobacteriaceae. Res. Microbiol. 159:340–348. 10.1016/j.resmic.2008.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch KH, Stothard P, Dennis JJ. 2012. Comparative analysis of two phenotypically-similar but genomically-distinct Burkholderia cenocepacia-specific bacteriophages. BMC Genomics 13:223. 10.1186/1471-2164-13-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kazaks A, Dislers A, Lipowsky G, Nikolajeva V, Tars K. 2012. Complete genome sequence of the Enterobacter cancerogenus bacteriophage Enc34. J. Virol. 86:11403–11404. 10.1128/JVI.01954-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH, Shin H, Choi Y, Ryu S. 2013. Complete genome sequence analysis of bacterial-flagellum-targeting bacteriophage Chi. Arch. Virol. 158:2179–2183. 10.1007/s00705-013-1700-0 [DOI] [PubMed] [Google Scholar]
- 33.Onmus-Leone F, Hang J, Clifford RJ, Yang Y, Riley MC, Kuschner RA, Waterman PE, Lesho EP. 2013. Enhanced de novo assembly of high throughput pyrosequencing data using whole genome mapping. PLoS One 8:e61762. 10.1371/journal.pone.0061762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young R. 7 October 2013. Phage lysis: do we have the hole story? Curr. Opin. Microbiol. pii:S1369-5274(13)00150-1. 10.1016/j.mib.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang IN, Young R. 2006. Phage lysis, p 104–125 In Calendar R. (ed), The bacteriophages, 2nd ed. Oxford University Press, New York, NY [Google Scholar]
- 36.Sun WZ, Tan YL, Jia M, Hu XM, Rao XC, Hu FQ. 2010. Functional characterization of the endolysin gene encoded by Pseudomonas aeruginosa bacteriophage PaP1. Afr. J. Microbiol. Res. 4:933–939 http://www.academicjournals.org/journal/AJMR/article-abstract/78B3FB61328 [Google Scholar]
- 37.Choi Y, Shin H, Lee JH, Ryu S. 2013. Identification and characterization of a novel flagellum-dependent Salmonella-infecting bacteriophage, iEPS5. Appl. Environ. Microbiol. 79:4829–4837. 10.1128/AEM.00706-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuty GF, Xu M, Struck DK, Summer EJ, Young R. 2010. Regulation of a phage endolysin by disulfide caging. J. Bacteriol. 192:5682–5687. 10.1128/JB.00674-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Summer EJ, Berry J, Tran TAT, Niu L, Struck DK, Young R. 2007. Rz/Rz1 lysis gene equivalents in phages of gram-negative hosts. J. Mol. Biol. 373:1098–1112. 10.1016/j.jmb.2007.08.045 [DOI] [PubMed] [Google Scholar]
- 40.Young R. 2002. Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 4:21–36 http://www.horizonpress.com/backlist/jmmb/v/v4/03.pdf [PubMed] [Google Scholar]
- 41.Wang IN, Smith DL, Young R. 2000. Holins: the protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 54:799–825. 10.1146/annurev.micro.54.1.799 [DOI] [PubMed] [Google Scholar]
- 42.Xu J, Hendrix RW, Duda RL. 30 July 2013. Chaperone-protein interactions that mediate assembly of the bacteriophage lambda tail to the correct length. pii:S0022-2836(13)00478-6. 10.1016/j.jmb.2013.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ceyssens PJ, Lavigne R, Mattheus W, Chibeu A, Hertveldt K, Mast J, Robben J, Volckaert G. 2006. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: establishment of the phi KMV subgroup within the T7 supergroup. J. Bacteriol. 188:6924–6931. 10.1128/JB.00831-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roucourt B, Lavigne R. 2009. The role of interactions between phage and bacterial proteins within the infected cell: a diverse and puzzling interactome. Environ. Microbiol. 11:2789–2805. 10.1111/j.1462-2920.2009.02029.x [DOI] [PubMed] [Google Scholar]
- 45.Casjens SR, Thuman-Commike PA. 2011. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411:393–415. 10.1016/j.virol.2010.12.046 [DOI] [PubMed] [Google Scholar]
- 46.Adriaenssens EM, Ceyssens PJ, Dunon V, Ackermann HW, Van Vaerenbergh J, Maes M, De Proft M, Lavigne R. 2011. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses.” Appl. Environ. Microbiol. 77:3443–3450. 10.1128/AEM.00128-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briers Y, Peeters LM, Volckaert G, Lavigne R. 2011. The lysis cassette of bacteriophage phiKMV encodes a signal-arrest-release endolysin and a pinholin. Bacteriophage 1:25–30. 10.4161/bact.1.1.14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobbins AT, George M, Basham DA, Ford ME, Houtz JM, Pedulla ML, Lawrence JG, Hatfull GF, Hendrix RW. 2004. Complete genomic sequence of the virulent Salmonella bacteriophage SP6. J. Bacteriol. 186:1933–1944. 10.1128/JB.186.7.1933-1944.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lynch KH, Abdu AH, Schobert M, Dennis JJ. 2013. Genomic characterization of JG068, a novel virulent podovirus active against Burkholderia cenocepacia. BMC Genomics 14:574. 10.1186/1471-2164-14-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu M, Struck DK, Deaton J, Wang IN, Young R. 2004. A signal-arrest-release sequence mediates export and control of the phage P1 endolysin. Proc. Natl. Acad. Sci. U. S. A. 101:6415–6420. 10.1073/pnas.0400957101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindberg AA. 1973. Bacteriophage receptors. Annu. Rev. Microbiol. 27:205–241. 10.1146/annurev.mi.27.100173.001225 [DOI] [PubMed] [Google Scholar]
- 52.Cursino L, Li Y, Zaini PA, De La Fuente L, Hoch HC, Burr TJ. 2009. Twitching motility and biofilm formation are associated with tonB1 in Xylella fastidiosa. FEMS Microbiol. Lett. 299:193–199. 10.1111/j.1574-6968.2009.01747.x [DOI] [PubMed] [Google Scholar]
- 53.Craig L, Pique ME, Tainer JA. 2004. Type IV pilus structure and bacterial pathogenicity. Nat. Rev. Microbiol. 2:363–378. 10.1038/nrmicro885 [DOI] [PubMed] [Google Scholar]
- 54.Moreira LM, De Souza RF, Digiampietri LA, Da Silva ACR, Setubal JC. 2005. Comparative analyses of Xanthomonas and Xylella complete genomes. Omics 9:43–76. 10.1089/omi.2005.9.43 [DOI] [PubMed] [Google Scholar]
- 55.Vauterin L, Yang P, Alvarez A, Takikawa Y, Roth DA, Vidaver AK, Stall RE, Kersters K, Swings J. 1996. Identification of non-pathogenic Xanthomonas strains associated with plants. Syst. Appl. Microbiol. 19:96–105 [Google Scholar]
- 56.Pieretti I, Royer M, Barbe V, Carrere S, Koebnik R, Cociancich S, Couloux A, Darrasse A, Gouzy J, Jacques MA, Lauber E, Manceau C, Mangenot S, Poussier S, Segurens B, Szurek B, Verdier V, Arlat M, Rott P. 2009. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10:616. 10.1186/1471-2164-10-616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monteiro-Vitorello CB, De Oliveira MC, Zerillo MM, Varani AM, Civerolo E, Van Sluys MA. 2005. Xylella and Xanthomonas mobil'omics. Omics 9:146–159. 10.1089/omi.2005.9.146 [DOI] [PubMed] [Google Scholar]
- 58.Mhedbi-Hajri N, Jacques MA, Koebnik R. 2011. Adhesion mechanisms of plant-pathogenic Xanthomonadaceae. Adv. Exp. Med. Biol. 715:71–89. 10.1007/978-94-007-0940-9_5 [DOI] [PubMed] [Google Scholar]
- 59.Wang L, Makino S, Subedee A, Bogdanove AJ. 2007. Novel candidate virulence factors in rice pathogen Xanthomonas oryzae pv. oryzicola as revealed by mutational analysis. Appl. Environ. Microbiol. 73:8023–8027. 10.1128/AEM.01414-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim SH, So BH, Wang JC, Song ES, Park YJ, Lee BM, Kang HW. 2008. Functional analysis of pilQ gene in Xanthomanas oryzae pv. oryzae, bacterial blight pathogen of rice. J. Microbiol. 46:214–220. 10.1007/s12275-007-0173-9 [DOI] [PubMed] [Google Scholar]
- 61.Darsonval A, Darrasse A, Durand K, Bureau C, Cesbron S, Jacques MA. 2009. Adhesion and fitness in the bean phyllosphere and transmission to seed of Xanthomonas fuscans subsp fuscans. Mol. Plant Microbe Interact. 22:747–757. 10.1094/MPMI-22-6-0747 [DOI] [PubMed] [Google Scholar]
- 62.Qian W, Jia YT, Ren SX, He YQ, Feng JX, Lu LF, Sun QH, Ying G, Tang DJ, Tang H, Wu W, Hao P, Wang LF, Jiang BL, Zeng SY, Gu WY, Lu G, Rong L, Tian YC, Yao ZJ, Fu G, Chen BS, Fang RX, Qiang BQ, Chen Z, Zhao GP, Tang JL, He CZ. 2005. Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15:757–767. 10.1101/gr.3378705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chibeu A, Ceyssens PJ, Hertveldt K, Volckaert G, Cornelis P, Matthijs S, Lavigne R. 2009. The adsorption of Pseudomonas aeruginosa bacteriophage phi KMV is dependent on expression regulation of type IV pili genes. FEMS Microbiol. Lett. 296:210–218. 10.1111/j.1574-6968.2009.01640.x [DOI] [PubMed] [Google Scholar]
- 64.Bradley DE. 1973. Basic characterization of a Pseudomonas aeruginosa pilus-dependent bacteriophage with a long noncontractile tail. J. Virol. 12:1139–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bradley DE, Robertson D. 1968. The structure and infective process of a contractile Pseudomonas aeruginosa bacteriophage. J. Gen. Virol. 3:247–254. 10.1099/0022-1317-3-2-247 [DOI] [PubMed] [Google Scholar]
- 66.Bradley DE. 1973. The adsorption of the Pseudomonas aeruginosa filamentous bacteriophage Pf to its host. Can. J. Microbiol. 19:623–631. 10.1139/m73-103 [DOI] [PubMed] [Google Scholar]
- 67.Byrne M, Kropinski AM. 2005. The genome of the Pseudomonas aeruginosa generalized transducing bacteriophage F116. Gene 346:187–194. 10.1016/j.gene.2004.11.001 [DOI] [PubMed] [Google Scholar]
- 68.Wang PW, Chu L, Guttman DS. 2004. Complete sequence and evolutionary genomic analysis of the Pseudomonas aeruginosa transposable bacteriophage D3112. J. Bacteriol. 186:400–410. 10.1128/JB.186.2.400-410.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Budzik JM, Rosche WA, Rietsch A, O'Toole GA. 2004. Isolation and characterization of a generalized transducing phage for Pseudomonas aeruginosa strains PAO1 and PA14. J. Bacteriol. 186:3270–3273. 10.1128/JB.186.10.3270-3273.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roncero C, Darzins A, Casadaban MJ. 1990. Pseudomonas aeruginosa transposable bacteriophages D3112 and B3 require pili and surface growth for adsorption. J. Bacteriol. 172:1899–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu YJ, Yang MT. 2007. A novel restriction-modification system from Xanthomonas campestris pv. vesicatoria encodes a m4C-methyltransferase and a nonfunctional restriction endonuclease. FEMS Microbiol. Lett. 272:83–90. 10.1111/j.1574-6968.2007.00738.x [DOI] [PubMed] [Google Scholar]
- 72.Semenova E, Nagornykh M, Pyatnitskiy M, Artamonova II, Severinov K. 2009. Analysis of CRISPR system function in plant pathogen Xanthomonas oryzae. FEMS Microbiol. Lett. 296:110–116. 10.1111/j.1574-6968.2009.01626.x [DOI] [PubMed] [Google Scholar]
- 73.Matsumoto A, Igo MM. 2010. Species-specific type II restriction-modification system of Xylella fastidiosa temecula1. Appl. Environ. Microbiol. 76:4092–4095. 10.1128/AEM.03034-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eskin B, Lautenberger JA, Linn S. 1973. Host-controlled modification and restriction of bacteriophage T7 by Escherichia coli B. J. Virol. 11:1020–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. 1998. Genome structure of mycobacteriophage D29: implications for phage evolution. J. Mol. Biol. 279:143–164. 10.1006/jmbi.1997.1610 [DOI] [PubMed] [Google Scholar]
- 76.Kim S, Rahman M, Seol SY, Yoon SS, Kim J. 2012. Pseudomonas aeruginosa bacteriophage PA1Ø requires type IV pili for infection and shows broad bactericidal and biofilm removal activities. Appl. Environ. Microbiol. 78:6380–6385. 10.1128/AEM.00648-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feng DF, Doolittle RF. 1996. Progressive alignment of amino acid sequences and construction of phylogenetic trees from them. Methods Enzymol. 266:368–382. 10.1016/S0076-6879(96)66023-6 [DOI] [PubMed] [Google Scholar]
- 78.Sayle RA, Milnerwhite EJ. 1995. Rasmol—biomolecular graphics for all. Trends Biochem. Sci. 20:374–376. 10.1016/S0968-0004(00)89080-5 [DOI] [PubMed] [Google Scholar]
- 79.Van Sluys MA, de Oliveira MC, Monteiro-Vitorello CB, Miyaki CY, Furlan LR, Camargo LE, da Silva AC, Moon DH, Takita MA, Lemos EG, Machado MA, Ferro MI, da Silva FR, Goldman MH, Goldman GH, Lemos MV, El-Dorry H, Tsai SM, Carrer H, Carraro DM, de Oliveira RC, Nunes LR, Siqueira WJ, Coutinho LL, Kimura ET, Ferro ES, Harakava R, Kuramae EE, Marino CL, Giglioti E, Abreu IL, Alves LM, do Amaral AM, Baia GS, Blanco SR, Brito MS, Cannavan FS, Celestino AV, da Cunha AF, Fenille RC, Ferro JA, Formighieri EF, Kishi LT, Leoni SG, Oliveira AR, Rosa VE, Jr, Sassaki FT, Sena JA, de Souza AA, Truffi D, Tsukumo F, Yanai GM, Zaros LG, Civerolo EL, Simpson AJ, Almeida NF, Jr, Setubal JC, Kitajima JP. 2003. Comparative analyses of the complete genome sequences of Pierce's disease and citrus variegated chlorosis strains of Xylella fastidiosa. J. Bacteriol. 185:1018–1026. 10.1128/JB.185.3.1018-1026.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhattacharyya A, Stilwagen S, Reznik G, Feil H, Feil WS, Anderson I, Bernal A, D'Souza M, Ivanova N, Kapatral V, Larsen N, Los T, Lykidis A, Selkov E, Walunas TL, Purcell A, Edwards RA, Hawkins T, Haselkorn R, Overbeek R, Kyrpides NC, Predki PF. 2002. Draft sequencing and comparative genomics of Xylella fastidiosa strains reveal novel biological insights. Genome Res. 12:1556–1563. 10.1101/gr.370702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. 10.1016/0378-1119(85)90120-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.