

Abstract
Archaea produce membrane lipids that typically possess fully saturated isoprenoid hydrocarbon chains attached to the glycerol moiety via ether bonds. They are functionally similar to, but structurally and biosynthetically distinct from, the fatty acid-based membrane lipids of bacteria and eukaryotes. It is believed that the characteristic lipid structure helps archaea survive under severe conditions such as extremely low or high pH, high salt concentrations, and/or high temperatures. We detail here the first successful production of an intact archaeal membrane lipid, which has fully saturated isoprenoid chains, in bacterial cells. The introduction of six phospholipid biosynthetic genes from a methanogenic archaeon, Methanosarcina acetivorans, in Escherichia coli enabled the host bacterium to synthesize the archaeal lipid, i.e., diphytanylglyceryl phosphoglycerol, while a glycerol modification of the phosphate group was probably catalyzed by endogenous E. coli enzymes. Reduction of the isoprenoid chains occurred only when archaeal ferredoxin was expressed with geranylgeranyl reductase, suggesting the role of ferredoxin as a specific electron donor for the reductase. This report is the first identification of a physiological reducer for archaeal geranylgeranyl reductase. On the other hand, geranylgeranyl reductase from the thermoacidophilic archaeon Sulfolobus acidocaldarius could, by itself, replace both its orthologue and ferredoxin from M. acetivorans, which indicated that an endogenous redox system of E. coli reduced the enzyme.
INTRODUCTION
One of the most striking biochemical features of the organisms of the domain Archaea is a specific membrane lipid (1, 2). The archaeal membrane lipids are fundamentally analogous to bacterial (and eukaryotic) glycerolipids, but possess very unique structures. First, the hydrocarbon chains of the archaeal lipids are fully saturated isoprenoid alcohols that usually possess 20 carbon atoms. Furthermore, the isoprenoid chains are connected via ether bonds to the sn-2 and -3 positions of the glycerol moiety of the lipid. Such a structure is typical of archaeal lipid, but it is distinct from that of bacterial glycerolipids where fatty acids are connected via ester bonds to the sn-1 and -2 positions of the glycerol moiety. Modification of the isoprenoid chains—intra- or intermolecular head-to-head condensation, cyclization to form 5- or 6-membered rings, or site-specific hydroxylation—largely expands the structural variation of the archaeal lipids, while they share, with a few exceptions, polar-head group structures with bacterial membrane lipids. Of course, the archaeal biosynthetic machinery utilized to produce such characteristic compounds is totally different from what is required for bacterial lipid biosynthesis. Archaea require sn-glycerol-1-phosphate (G1P) dehydrogenase for membrane lipid biosynthesis because the precursor of the glycerol moiety of archaeal lipids is G1P, which is the enantiomer of its bacterial (and eukaryotic) counterpart sn-glycerol-3-phosphate. To G1P, two geranylgeranyl groups from geranylgeranyl diphosphate (GGPP) are transferred to form digeranylgeranylglyceryl phosphate (DGGGP), which is considered a common precursor of most archaeal lipids with various isoprenoid-chain structures. Generally, all the carbon-carbon double bonds in the precursor are reduced by geranylgeranyl reductase (GGR).
Recent reports have introduced the creation of recombinant Escherichia coli cells that synthesize biosynthetic precursors of archaeal membrane lipids. Lai et al. (3) and Yokoi et al. (4) have constructed E. coli strains that harbor four archaeal lipid biosynthetic genes, i.e., G1P dehydrogenase, GGPP synthase, geranylgeranylglyceryl phosphate synthase, and DGGGP synthase, from a hyperthermophile, Archaeoglobus fulgidus, and a mesophilic methanogen, Methanosarcina acetivorans, respectively. The former strain reportedly produced DGGGP, while the latter yielded its alcoholic form, digeranylgeranylglycerol (DGGGOH), and a phosphatidylglycerol-type derivative, digeranylgeranylglyceryl phosphoglycerol (DGGGP-Gro). However, the bacterial production of “intact” archaeal membrane lipids with fully saturated isoprenoid chains has never been reported, and neither has it been found in nature. Phospholipids possessing the unsaturated digeranylgeranylglycerol core structure are reportedly very rare in archaea (5).
In the present study, we constructed E. coli strains that can produce an intact archaeal lipid, diphytanylglyceryl phosphoglycerol (DPhGP-Gro) (Fig. 1). To achieve this, the gene of a putative GGR was cloned from M. acetivorans, although the putative reductase showed no individual activity in E. coli cells. Coexpression of a ferredoxin-like protein, which is encoded just downstream from the GGR gene in the genome of M. acetivorans, was found necessary to activate GGR, suggesting that the protein connects the reductase with endogenous redox systems in E. coli. This is the first report of the identification of a physiological electron donor for archaeal GGR. On the other hand, previously isolated GGR from a thermoacidophilic archaeon Sulfolobus acidocaldarius (6) catalyzed the partial reduction of archaeal lipid precursors in E. coli without help from archael ferredoxins, suggesting its relatively broad preference toward electron donors. The effect of the archaeal lipid production on the growth of the host bacterial cells was evaluated because they were supposed to possess an unnatural archaeal-bacterial chimeric lipid membrane.
FIG 1.

Schematic views of the biosynthetic pathway of an intact archaeal phospholipid in E. coli cells and the artificial operons contained in the plasmids used in the present study. Asterisks on the GGPP synthase gene in pBAD-ALB6mut indicate mutagenesis. Abbreviations: G1P, sn-glycerol-1-phosphate; GGPP, geranylgeranyl diphosphate; DGGGP, digeranylgeranylglyceryl phosphate; DGGGP-Gro, digeranylgeranylglyceryl phosphoglycerol; GGR, geranylgeranyl reductase; DPhGP-Gro, diphytanylglyceryl phosphoglycerol.
MATERIALS AND METHODS
General procedures.
Restriction enzyme digestions, transformations, and other standard molecular biological techniques were carried out as described by Sambrook et al. (7).
Construction of plasmids containing archaeal phospholipid biosynthetic genes.
The chromosomal DNA of M. acetivorans was prepared as described in a previous study (4). The MA1485-MA1484 tandem genes encoding a ferredoxin-like protein and a GGR homologue, respectively, were amplified either simultaneously or separately with the primers shown in Table 1 and KOD DNA polymerase (Toyobo, Japan), using the genome of M. acetivorans as a template. The gene of S. acidocaldarius GGR, saci_0986, was also amplified with the primers in Table 1, using the plasmid pET16b-Saci_0986 (8) as a template. In each forward primer for amplification, a designed Shine-Dalgarno sequence, 5′-GAAAGGAG-3′, was placed upstream of the initiation codon. Using an In-Fusion Advantage PCR cloning kit (TaKaRa, Japan) according to the manufacturer's instructions, an amplified DNA fragment containing both MA1485-MA1484 genes, each of the genes, or saci_0986 was ligated into the plasmid pBAD-ALB4 (4), which contained four archaeal genes that were sufficient for the biosynthesis of DGGGP, to construct pBAD-ALB4-MA1485/4, pBAD-ALB4-MA1484, pBAD-ALB4-MA1485, and pBAD-ALB4-SaGGR (Fig. 1). A DNA fragment containing MA1485, which had been amplified with a different primer pair, was ligated with pBAD-ALB4-MA1484 to construct pBAD-ALB6, which contained MA1484 and MA1485 genes—in that order. Before the In-Fusion cloning, all of the plasmids were cleaved at the SalI site just downstream from the artificial operon for archaeal phospholipid biosynthesis.
TABLE 1.
Primers used for plasmid construction
| Function | Primer |
|
|---|---|---|
| Orientation | Sequence (5′–3′)a | |
| Amplification of saci_0986 to construct pBAD-ALB4-SaGGR | Forward | CATAAGTTGAGTCGAAAGGAGATATATTATGAAGGAACTTAAATATGACGTTC |
| Reverse | ATGCCTGCAGGTCGACCTAACTTAAACTTTTGTTAAACTCTG | |
| Amplification of MA1485-MA1484 to construct pBAD-ALB4-MA1485/4 | Forward | CATAAGTTGAGTCGAAAGGAGAATTATATGGTGTGGCATTATAC |
| Reverse | ATGCCTGCAGGTCGACTTATGTCCTGTTCTTCATC | |
| Amplification of MA1484 to construct pBAD-ALB4-MA1484 | Forward | CATAAGTTGAGTCGAAAGGAGATATATTATGAAGGATATCTATGATGTTC |
| Reverse | ATGCCTGCAGGTCGACTTATGTCCTGTTCTTCATC | |
| Amplification of MA1485 to construct pBAD-ALB4-MA1485 | Forward | CATAAGTTGAGTCGAAAGGAGAATTATATGGTGTGGCATTATACTAATG |
| Reverse | ATGCCTGCAGGTCGACGAGAACATCATAGATATCCTTC | |
| Amplification of MA1485 to construct pBAD-ALB6 | Forward | CAGGACATAAGTCGAAAGGAGATATTATATGGTGTGGCATTATACTAATG |
| Reverse | ATGCCTGCAGGTCGACGAGAACATCATAGATATCCTTC | |
| Mutagenesis of pBAD-MA0606 | Sense | CACTCTGATCCATGCCGCCATTATGGATAAGG |
| Antisense | CCTTATCCATAATGGCGGCATGGATCAGAGTG | |
| Amplification of five genes from M. acetivorans to construct pBAD-ALB6mut | Forward | ACAATCTAGAGTCGAAGGAAGATTATAATG |
| Reverse | ATGCCTGCAGGTCGACG | |
Recognition sites for the restriction enzyme SalI are underlined.
Mutagenesis for the construction of a nonproducer of archaeal phospholipids.
M. acetivorans GGPP synthase gene MA0606 in the plasmid pBAD-MA0606, which is an ancestor plasmid of pBAD-ALB6 and contains MA0606, was mutagenized to replace aspartate 85 and aspartate 86 residues of the enzyme with alanine, using a QuikChange mutagenesis kit (Stratagene, USA) according to the manufacturer's protocol and the primer pair shown in Table 1. The mutagenized plasmid was digested with SalI. The downstream portion of the artificial operon in pBAD-ALB6, containing MA0961, MA3969, MA3686, MA1484, and MA1485, was amplified with the primers shown in Table 1. The amplified fragment was ligated with the SalI-digested plasmid by the In-Fusion method to construct pBAD-ALB6mut, which contained an operon composed of the same six archaeal genes and had the same sequence as pBAD-ALB6, with the exception of the mutation of the encoded GGPP synthase (Fig. 1). The levels of enzyme expression were confirmed by SDS–10% PAGE.
Lipid extraction from E. coli harboring the plasmids.
E. coli Top10 (Life Technologies, USA) was transformed with each of the plasmids constructed above, or pBAD-ALB4, and was cultured statically at 37°C for 45 h in 1.5 liters of Luria-Bertani (LB) medium supplemented with 100 mg of ampicillin/liter and 0.02% l-arabinose. Lipid samples for the liquid chromatography-electrospray ionization mass spectrometry (LC/ESI-MS) analysis were extracted from the harvested cells using a method developed by Lai et al. (3) with slight modification, as described elsewhere (4).
Lipid extraction from M. acetivorans.
M. acetivorans was cultured in 1.2 liters of JCM 385 medium and harvested during the stationary phase. Total lipid extracts from 1.5 g of M. acetivorans cell pellets were prepared using the Bligh-Dyer method (9). The chloroform layer was recovered and evaporated to dryness at 55°C under a stream of nitrogen gas, and the residual was provided for two-phase partitioning with the same volume of n-pentane and H2O. The pentane layer was recovered and evaporated, and the residual was dissolved with 1.5 ml of methanol–2-propanol (1:1) for analysis by LC/ESI-MS.
LC/ESI-MS analysis.
ESI-MS was performed with an Esquire 3000 ion trap system (Bruker Daltonics, USA) under the same conditions as described previously (4). The system was connected to an Agilent 1100 Series HPLC (Agilent Technologies, USA). The compounds eluted from a COSMOSIL packed column 5C18-AR-II (2.0 by 150 mm; Nacalai, Japan) were detected by UV absorption at 210 nm and ESI-MS with the positive mode. The mobile phase consisted of methanol, 2-propanol, and 100 mg of sodium acetate liter−1 (7:2:1). The flow rate was 0.2 ml min−1. After each analysis, the column was washed with 2-propanol for 20 min at a flow rate of 0.2 ml min−1 and was then equilibrated with the mobile phase for 30 min before the next analysis. It should be noted that the elution time of the compounds analyzed with the protocol used was sensitive to the conditions of the pretreatment of the column, i.e., wash and equilibration, and sometimes fluctuated. Column aging also largely affected the retention time.
Growth analysis and microscopic observation.
E. coli Top10 harboring pBAD18, pBAD-ALB4, pBAD-ALB6, or pBAD-ALB6mut was cultivated at 37°C in 9 ml of LB medium supplemented with 100 mg of ampicillin/liter and 0.02% l-arabinose in a glass tube, and the turbidity of the culture was monitored by determining the optical density at 600 nm (OD600). When cultivation started, the OD600 of the culture was set to 0.02 by diluting the preculture of each strain in LB medium supplemented only with 100 mg of ampicillin/liter. The shape of the cells in the culture was observed using a phase-contrast BH2 microscope (Olympus, Japan) equipped with a SPlan100PL objective lens and a digital camera. To simultaneously show many cells during microscopic analysis, the culture was concentrated 10-fold by centrifugal precipitation and resuspension of the cells. Direct cell counting was performed with the same microscope and a bacterial counter (Sunlead Glass Corporation, Japan).
Extraction of inclusion bodies from E. coli harboring pBAD-ALB4.
E. coli cells harvested from 4 ml of cultures were suspended in 200 μl of lysis buffer (50 mM Tris-HCl [pH 8.0], 200 mM NaCl, 5% Triton X-100) containing 1 mM EDTA. The cell suspension was mixed with 20 μl of 10 mg of lysozyme/ml in 50 mM Tris-HCl (pH 8.0), followed by incubation at 37°C for 30 min, followed in turn by DNase I digestion to remove viscosity. After the mixture was centrifuged at 6,000 × g for 15 min at 4°C, the precipitates were recovered, washed twice with 200 μl of lysis buffer, and then suspended in 60 μl of lysis buffer for microscopic observation.
RESULTS
Analysis of archaeal lipids produced in GGR-expressing E. coli.
In our previous study (4), the introduction of four genes from M. acetivorans, i.e., the genes of G1P dehydrogenase, GGPP synthase, geranylgeranylglyceryl phosphate synthase, and DGGGP synthase, on the plasmid pBAD-ALB4, into E. coli resulted in the accumulation of DGGGP-Gro and DGGGOH in the bacterial cells. In the LC/ESI-MS analysis of the lipid extracted from E. coli cells harboring pBAD-ALB4, ions with m/z values of typically 836 and 660, which correspond to [DGGGP-Gro+2Na]+ and [DGGGOH+Na]+, respectively, were detected (see Fig. S1 and S2 in the supplemental material). We had tried to synthesize intact archaeal lipids with fully saturated isoprenoid chains in E. coli by introducing GGR genes into pBAD-ALB4 (Fig. 1). We first constructed a plasmid pBAD-ALB4-SaGGR by introducing the previously isolated gene of GGR from S. acidocaldarius. In addition, we searched for a GGR gene from the genome of M. acetivorans because we were concerned that the enzyme from the thermoacidophile would not work properly in the cells of E. coli. The orthologue of GGR is encoded in MA1484, and a ferredoxin-like protein is encoded in MA1485 just upstream from MA1484. The supposed operon MA1485-MA1484 was amplified and cloned into pBAD-ALB4 to give pBAD-ALB4-MA1485/4, which contains six genes from M. acetivorans. However, reduction of the isoprenoid chains of the archaeal lipids produced in E. coli cells was not observed with the sample-preparation protocol we had used previously, in which E. coli was grown aerobically and induced for enzyme expression at the mid-log-growth phase (data not shown). Thus, we changed the protocol to optimize the intracellular environment for the reducing reaction. Reduction was observed in the cells grown semianaerobically and induced from at an early growth phase. By our LC/ESI-MS analysis, conversion of DGGGP-Gro to a fully reduced product was implied in the cells harboring pBAD-ALB4-MA1485/4. An extracted ion peak of m/z 852.8 (with ±0.5 width), which corresponds to [DGGGP-Gro +2Na+16H]+, namely, [DPhGP-Gro+2Na]+, emerged at a longer retention time than that of DGGGP-Gro (Fig. 2A). Tandem MS (MS/MS) analysis of this ion yielded fragment ions corresponding well with the expected fragmentation pattern of the parent ion: m/z 834.6 with [DPhGP-Gro+2Na-H2O]+, m/z 778.5 with [diphytanylglyceryl phosphate+2Na]+, m/z 698.4 with [diphytanylglycerol+2Na–H]+, and m/z 676.4 with [diphytanylglycerol+Na]+ (Fig. 3). We also performed LC/ESI-MS analysis of lipid extracted from M. acetivorans to confirm the retention time of DPhGP-Gro, which was reportedly found in the cells of a relative strain, Methanosarcina barkeri (10). An ion with the m/z of 852.9 corresponding to [DPhGP-Gro+2Na]+ was eluted at a retention time that was similar to that detected in the analysis of lipid extract from E. coli (Fig. 2B). MS/MS analysis of the ion gave almost the same fragmentation pattern as that shown in Fig. 3 (data not shown). To our surprise, an ion likely derived from DGGGP-Gro was also detected in the M. acetivorans lipid extract, even though the existence of such unsaturated lipids in Methanosarcina species had never been reported. These data clearly proved the formation of the fully reduced product DPhGP-Gro in the E. coli cells. In the LC/ESI-MS analysis, partially saturated lipids derived from DGGGP-Gro were also detected (see Fig. S1 in the supplemental material). Ion peaks of the intermediates eluted at gradually longer retention times as more double bonds were saturated.
FIG 2.
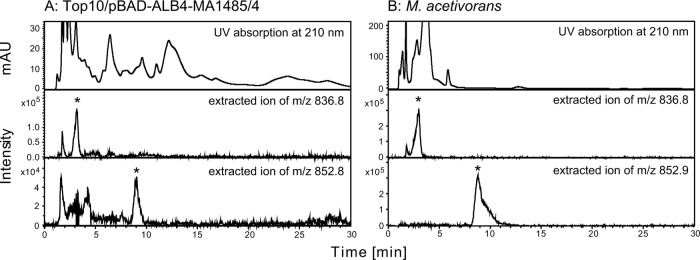
LC/ESI-MS analysis of DGGGP-Gro-derived archaeal phospholipid extracted from E. coli cells harboring pBAD-ALB4-MA1485/4. (A) Elution profiles of UV detection at 210 nm (top panel) and extracted ions (lower panels) in the LC/ESI-MS analysis of the lipid extracted from the E. coli strain Top10 harboring pBAD-ALB4-MA1485/4. The middle and bottom panels show the extracted ion profiles corresponding to [DGGGP-Gro+2Na]+ and [DPhGP-Gro+2Na]+, respectively. The ion peaks supposedly derived from archaeal lipids are marked by asterisks. (B) LC/ESI-MS analysis of lipid extracted from M. acetivorans was performed under the same conditions. The top, middle, and bottom panels show the elution profiles of UV detection at 210 nm, extracted ion corresponding to [DGGGP-Gro+2Na]+, and that corresponding to [DPhGP-Gro+2Na]+, respectively.
FIG 3.
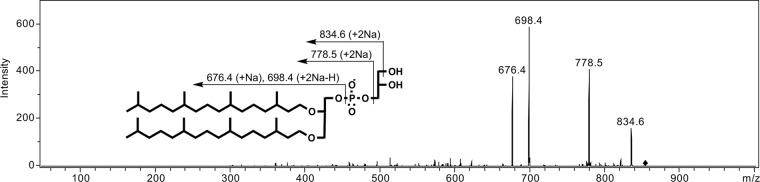
MS/MS profile of the ion corresponding to [DPhGP-Gro+2Na]+. The m/z of the parent ion was 852.8 in this analysis. The expected fragmentation patterns are also indicated.
On the other hand, in the cells harboring pBAD-ALB4-SaGGR, only a very slight reduction occurred: DGGGP-Gro-derived lipids, in which only a few double bonds were saturated, were scarcely detected (data not shown). However, an obvious reduction was observed with DGGGOH-derived lipids. By LC/ESI-MS analysis, we detected DGGGOH-derived ions, in which up to four double bonds were saturated (see Fig. S2 in the supplemental material). Also in the case of DGGGOH, the level of reduction was significantly lower in the cells harboring pBAD-ALB4-SaGGR than in those harboring pBAD-ALB4-MA1485/4, which produces almost fully saturated lipid. (Formation of the fully reduced product, i.e., diphytanylglycerol, was unidentifiable because its ion peak overlaps another, which derived from an unknown compound that was also extracted from E. coli harboring pBAD-ALB4.) The reason for the lower activity of S. acidocaldarius GGR was unclear but might have been due to the absence of the ferredoxin-like protein or because its optimal reaction temperature was probably higher than the growth temperature of E. coli.
Identification of proteins required for GGR activity.
We then tried to identify the molecule that has the GGR activity in the cells harboring pBAD-ALB4-MA1485/4. New plasmids derived from pBAD-ALB4 were constructed (Fig. 1). The insertion of the GGR orthologue gene MA1484 or the ferredoxin homologue gene MA1485 into pBAD-ALB4 gave pBAD-ALB4-MA1484 and pBAD-ALB4-MA1485, respectively. The E. coli cells harboring these plasmids did not produce saturated archaeal lipids. The UV and extracted ion profiles of lipid from the cells harboring pBAD-ALB4-MA1484 or pBAD-ALB4-MA1485 were almost identical to those of the cells harboring pBAD-ALB4 (see Fig. S1 in the supplemental material). Thus, we cloned the MA1485 gene into pBAD-ALB4-MA1484 to construct pBAD-ALB6 to confirm whether the observed lipid reduction arises from the coexistence of GGR and its reducer, or from a change in the translation efficiency of MA1484 caused by its longer 5′ untranslated region in the pBAD-ALB4-MA1485/4 construct than that in pBAD-ALB4-MA1484. The plasmid pBAD-ALB6, which contained MA1484 and MA1485, in that order, was sufficient to synthesize DPhGP-Gro in the transformed E. coli cells as well as pBAD-ALB4-MA1485/4 (see Fig. S1 in the supplemental material). These results support the idea that MA1484 and MA1485 encode GGR and ferredoxin, respectively, and that the latter can supply electrons to the former from endogenous redox systems in E. coli. This is the first evidence that ferredoxin can reduce archaeal GGR, for which most of the physiological reducing agents have remained undiscovered.
Phenotypes of E. coli producing archaeal lipids.
Given the fact that the height of the peak corresponding to DPhGP-Gro in the extracted ion profile reflects the lipid amount, the efficiency of the reducing reaction seems higher in the cells harboring pBAD-ALB6 than in those harboring pBAD-ALB4-MA1485/4 (see Fig. S1 in the supplemental material), probably because the Shine-Dalgarno sequence upstream from MA1484 was optimized in the former construct (Fig. 1). Thus, we used pBAD-ALB6 to evaluate the effects of the formation of the intact archaeal lipid DPhGP-Gro on the phenotypes of E. coli. E. coli cells harboring each of the plasmids, i.e., pBAD-ALB6, pBAD-ALB4, or the empty vector pBAD18, were cultivated in a medium supplemented with the inducer arabinose from the beginning, and their growth curves were compared. All of the strains grew similarly in the exponential phase, but the turbidity of the cultures differed in the stationary phase (Fig. 4A). Direct microscopic cell counting showed that the difference in optical density was not due to cell density because the culture of the cells harboring pBAD18 had higher cell density comparing with the other cultures (data not shown). Through the stationary phase, the strains producing the archaeal-type lipids showed a higher optical density than the control strain harboring the empty vector. Microscopic analysis of the cells revealed that particles were formed only in the cells of the lipid producers (Fig. 4B to D). We assumed that the particles must arise from an inclusion body that is composed of highly expressed archaeal enzymes, or from lipid droplets formed by the archaeal-type lipids. Therefore, we constructed a control E. coli strain in which the archaeal enzymes were expressed but archaeal lipids were not produced. To achieve this, we mutagenized the plasmid pBAD-ALB6 to inactivate GGPP synthase, which supplies the substrate for the other archaeal prenyltransferases, i.e., geranylgeranylglyceryl phosphate synthase and DGGGP synthase (Fig. 1). By this mutagenesis, the aspartate residues at positions 85 and 86, which are highly conserved among all-trans prenyl diphosphate synthases and are known to be critical for activity (11, 12), were replaced with alanine. Neither DGGGP-Gro nor the reduced archaeal lipids were detected in lipid extract from the cells transformed with pBAD-ALB6mut (data not shown). In contrast, within the detection limits of SDS-PAGE, the protein expression profiles of E. coli harboring the new plasmids pBAD-ALB6mut or pBAD-ALB6 seemed identical with the exception of the protein band of the mutated GGPP synthase, which showed a slightly faster migration (Fig. 4E). The expression profiles showed that G1P dehydrogenase was expressed at the same levels in the cells harboring pBAD-ALB6 or pBAD-ALB6mut, suggesting that all of the archaeal genes downstream from the GGPP synthase gene were expressed without being affected by mutagenesis. The strain harboring pBAD-ALB6mut grew as fast as the other strains in the exponential phase, and its optical density slightly exceeded those of the strains harboring pBAD-ALB6 and pBAD-ALB4 in the stationary phase (Fig. 4A). Microscopic observation revealed the formation of particles in the cells of the lipid nonproducer strain as in those of the producers (Fig. 4F). These data show that the particles, which likely cause a difference in optical density in the stationary phase, are inclusion bodies formed by some of the archaeal enzymes. The fact that the particles could be collected using the standard protocol for the recovery of inclusion bodies also supports this conclusion (see Fig. S3 in the supplemental material).
FIG 4.

Effect of the production of archaeal lipids on the growth and morphology of the host E. coli cells. (A) Growth curves of E. coli harboring the plasmids. (B to D) Photographs of the cells of E. coli harboring pBAD18, pBAD-ALB4 and pBAD-ALB6, respectively. (E) SDS-PAGE analysis of the crude extracts from E. coli expressing the archaeal enzymes. Lane 1, molecular marker; lanes 2 to 5, E. coli harboring pBAD18, pBAD-ALB4, pBAD-ALB6mut, and pBAD-ALB6, respectively. Arrowheads and asterisks indicate protein bands corresponding to GGPP synthase and G1P dehydrogenase, respectively. (F) Photograph of the cells of E. coli harboring pBAD-ALB6mut.
DISCUSSION
A prominent result from the present study was the discovery of a proteinaceous reducer for archaeal GGR, which is specific for the biosynthesis of archaeal membrane lipid. This finding enabled the first production of intact archaeal phospholipid in E. coli cells. Except for Thermoplasma acidophilum GGR, which reportedly acts with ∼1.7 mM NADPH (13), the biological reducing agents for the other archaeal GGRs (6, 14), as well as homologous menaquinone-specific prenyl reductase from Archaeoglobus fulgidus (15), remain undiscovered. To reduce only a small portion of the substrate, GGR from Sulfolobus acidocaldarius requires a very high concentration (≥10 mM) of NADH (8). Thus far, neither biotic nor abiotic general reducing agents, other than sodium dithionite, have been found to activate the enzymes from S. acidocaldarius and A. fulgidus in vitro (unpublished data). These facts imply the existence of a specific physiological reducer for archaeal GGR, which enables an efficient electron supply for lipid biosynthesis. In the present study, we clearly showed that the reduction of M. acetivorans GGR (MA1484) is mediated by ferredoxin (MA1485) encoded next to the gene of GGR in the genome. Besides, recombinantly expressed M. acetivorans GGR failed to exert activity on DGGGP in vitro when dithionite was used as a reducing agent (unpublished data). The C-terminal half of the archaeal ferredoxin, which contains eight cysteine residues, is highly homologous to putative 4Fe-4S ferredoxins from E. coli and thus is likely to be the substrate of NADPH:ferredoxin reductase in E. coli cells. However, the fact that the expression of GGR without ferredoxin results in nonreduction of DGGGP-Gro indicates that E. coli ferredoxins cannot replace the archaeal homologue. The situation is different with S. acidocaldarius GGR, which alone can reduce the archaeal lipid precursors, more effectively for DGGGOH than for DGGGP-Gro, in E. coli cells. The reducing activity of S. acidocaldarius GGR suggests that the enzyme can be reduced by endogenous ferredoxin or some reducing agent in E. coli. This property is similar to that of A. fulgidus prenyl reductase, which can effectively reduce menaquinone in E. coli cells without help from other archaeal proteins (15). Our next focus will be to determine whether the ferredoxin from M. acetivorans is specific for GGR. The strength of the interaction between them should be evaluated in the future. Moreover, their cocrystal structure will convey much information.
Another noteworthy result from the present study is the creation of bacterial cells in which intact archaeal membrane lipids are synthesized. Thanks to the discovery of GGR-specific ferredoxin, the first in vivo production of an intact archaeal lipid with fully saturated isoprenoid chains, i.e., DPhGP-Gro, was enabled in bacterial cells. The glycerol modification of the phosphate group was supposedly catalyzed by endogenous enzymes of E. coli, suggesting that archaeal lipids with other polar head group structures, including diphytanylglycerol, were also synthesized at least in small amounts. It should be noted that the name archaetidylglycerol is incorrect as a designation for DPhGP-Gro, because the term “archaetidyl” (16) corresponds to “phosphatidyl,” which is used for bacterial glycerophospholipids, and is a generic name used for diether-type archaeal lipids whether they are saturated or unsaturated, while the term “archaeol” has recently come to mean diphytanylglycerol. Defective properties with an organism that possesses a bacterial-archaeal chimeric lipid membrane were expected from the evolutionary hypotheses stating that the instability of a chimeric membrane of the last universal common ancestor has caused a current “lipid divide” situation, which clearly divides archaea and other organisms according to their use of different types of lipids (17, 18). In contrast, our data from the growth analysis of E. coli cells showed no significant effects of the production of the archaeal lipid on the host. However, we cannot exclude the possibility that the content of the archaeal lipid might be too low to exert phenotypic effects. Only ∼60 μg of DGGGP-Gro was reportedly extracted from 1 g of wet E. coli cells harboring pBAD-ALB4 (4), suggesting that it accounts for less than 1% of the entire lipid in E. coli.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by MEXT KAKENHI grants 23108531 and 25108712.
Footnotes
Published ahead of print 8 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00927-13.
REFERENCES
- 1.Koga Y, Morii H. 2007. Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol. Mol. Biol. Rev. 71:97–120. 10.1128/MMBR.00033-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koga Y, Morii H. 2005. Recent advances in structural research on ether lipids from archaea including comparative and physiological aspects. Biosci. Biotechnol. Biochem. 69:2019–2034. 10.1271/bbb.69.2019 [DOI] [PubMed] [Google Scholar]
- 3.Lai D, Lluncor B, Schroder I, Gunsalus RP, Liao JC, Monbouquette HG. 2009. Reconstruction of the archaeal isoprenoid ether lipid biosynthesis pathway in Escherichia coli through digeranylgeranylglyceryl phosphate. Metab. Eng. 11:184–191. 10.1016/j.ymben.2009.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yokoi T, Isobe K, Yoshimura T, Hemmi H. 2012. Archaeal phospholipid biosynthetic pathway reconstructed in Escherichia coli. Archaea 2012:ID438931. 10.1155/2012/438931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishihara M, Morii H, Matsuno K, Ohga M, Stetter KO, Koga Y. 2002. Structural analysis by reductive cleavage with LiAlH4 of an allyl ether choline-phospholipid, archaetidylcholine, from the hyperthermophilic methanoarchaeon Methanopyrus kandleri. Archaea 1:123–131. 10.1155/2002/682753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato S, Murakami M, Yoshimura T, Hemmi H. 2008. Specific partial reduction of geranylgeranyl diphosphate by an enzyme from the thermoacidophilic archaeon Sulfolobus acidocaldarius yields a reactive prenyl donor, not a dead-end product. J. Bacteriol. 190:3923–3929. 10.1128/JB.00082-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 8.Sasaki D, Fujihashi M, Iwata Y, Murakami M, Yoshimura T, Hemmi H, Miki K. 2011. Structure and mutation analysis of archaeal geranylgeranyl reductase. J. Mol. Biol. 409:543–557. 10.1016/j.jmb.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 9.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 10.Nishihara M, Koga Y. 1995. Two new phospholipids, hydroxyarchaetidylglycerol and hydroxyarchaetidylethanolamine, from the archaea Methanosarcina barkeri. Biochim. Biophys. Acta 1254:155–160. 10.1016/0005-2760(94)00178-2 [DOI] [PubMed] [Google Scholar]
- 11.Joly A, Edwards PA. 1993. Effect of site-directed mutagenesis of conserved aspartate and arginine residues upon farnesyl diphosphate synthase activity. J. Biol. Chem. 268:26983–26989 [PubMed] [Google Scholar]
- 12.Song L, Poulter CD. 1994. Yeast farnesyl-diphosphate synthase: site-directed mutagenesis of residues in highly conserved prenyltransferase domains I and II. Proc. Natl. Acad. Sci. U. S. A. 91:3044–3048. 10.1073/pnas.91.8.3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishimura Y, Eguchi T. 2006. Biosynthesis of archaeal membrane lipids: digeranylgeranylglycerophospholipid reductase of the thermoacidophilic archaeon Thermoplasma acidophilum. J. Biochem. 139:1073–1081. 10.1093/jb/mvj118 [DOI] [PubMed] [Google Scholar]
- 14.Murakami M, Shibuya K, Nakayama T, Nishino T, Yoshimura T, Hemmi H. 2007. Geranylgeranyl reductase involved in the biosynthesis of archaeal membrane lipids in the hyperthermophilic archaeon Archaeoglobus fulgidus. FEBS J. 274:805–814. 10.1111/j.1742-4658.2006.05625.x [DOI] [PubMed] [Google Scholar]
- 15.Hemmi H, Takahashi Y, Shibuya K, Nakayama T, Nishino T. 2005. Menaquinone-specific prenyl reductase from the hyperthermophilic archaeon Archaeoglobus fulgidus. J. Bacteriol. 187:1937–1944. 10.1128/JB.187.6.1937-1944.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishihara M, Morii H, Koga Y. 1987. Structure determination of a quartet of novel tetraether lipids from Methanobacterium thermoautotrophicum. J. Biochem. 101:1007–1015 [DOI] [PubMed] [Google Scholar]
- 17.Koga Y. 2011. Early evolution of membrane lipids: how did the lipid divide occur? J. Mol. Evol. 72:274–282. 10.1007/s00239-011-9428-5 [DOI] [PubMed] [Google Scholar]
- 18.Wachtershauser G. 2003. From pre-cells to Eukarya: a tale of two lipids. Mol. Microbiol. 47:13–22. 10.1046/j.1365-2958.2003.03267.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.