

Abstract
Although fosfomycin is an old antibiotic, it has resurfaced with particular interest. The antibiotic is still effective against many pathogens that are resistant to other commonly used antibiotics. We have found that fosfomycin resistance of enterohemorrhagic Escherichia coli (EHEC) O157:H7 is controlled by the bacterial two-component signal transduction system CpxAR. A cpxA mutant lacking its phosphatase activity results in constitutive activation of its cognate response regulator, CpxR, and fosfomycin resistance. We have shown that fosfomycin resistance requires CpxR because deletion of the cpxR gene in the cpxA mutant restores fosfomycin sensitivity. We have also shown that CpxR directly represses the expression of two genes, glpT and uhpT, which encode transporters that cotransport fosfomycin with their native substrates glycerol-3-phosphate and glucose-6-phosphate, and repression of these genes leads to a decrease in fosfomycin transport into the cpxA mutant. However, the cpxA mutant had an impaired growth phenotype when cultured with glycerol-3-phosphate or glucose-6-phosphate as a sole carbon substrate and was outcompeted by the parent strain, even in nutrient-rich medium. This suggests a trade-off between fosfomycin resistance and the biological fitness associated with carbon substrate uptake. We propose a role for the CpxAR system in the reversible control of fosfomycin resistance. This may be a beneficial strategy for bacteria to relieve the fitness burden that results from fosfomycin resistance in the absence of fosfomycin.
INTRODUCTION
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 is a food-borne pathogen that can cause hemolytic-uremic syndrome (HUS), which can be fatal (1, 2). However, the efficacy of antibiotic treatment for this infectious disease is controversial because in vitro experiments suggest that some antibiotics promote the release of Shiga-like toxins (verotoxins) produced by the bacterium and may increase the risk of HUS development (3–5). On the other hand, animal studies and clinical trials have shown that treatment with fosfomycin at an early time point during the course of infection decreased HUS development and mortality (6–8). Furthermore, the antibiotic is effective against some multidrug-resistant (MDR) pathogens because it has no structural relationship with other known antibiotics; hence, there is no cross-resistance (9, 10). A shortage of new antimicrobial agents is a critical issue at present, and together with the spread of MDR pathogens, the use of fosfomycin to treat infections is being revisited (11).
Fosfomycin is an antagonist of the UDP-N-acetylglucosamine-3-enolpyruvyltransferase MurA, which transfers phosphoenolpyruvate (PEP) to the 3′-hydroxyl group of UDP-N-acetylglucosamine. Fosfomycin is a PEP analog that inhibits MurA activity and results in interference of the first step in peptidoglycan biosynthesis (12). Mechanisms for fosfomycin resistance are encoded on plasmids and the chromosome. Resistance to fosfomycin is conferred by the production of the plasmid-encoded enzymes FosA, FosB, FosC, and FosX, which leads to inactivation of fosfomycin (13–16). Chromosomal mutations that lead to the expression of MurA variants that have impaired fosfomycin binding or result in overexpression of MurA also confer resistance to fosfomycin. Substitution of the Cys115 residue in MurA, which forms a covalent bond with fosfomycin, leads to a significant increase in the MIC (17–19). In addition, a MurA-overexpressing clinical isolate has been reported, although the mutation site was uncharacterized (20). In E. coli, UhpT, a glucose-6-phosphate (G6P) transporter, and GlpT, a glycerol-3-phosphate (alternatively called alpha-glycerophosphate [alpha-GP]) transporter, are involved in the uptake of fosfomycin, and mutations in the genes encoding these transporters can confer fosfomycin resistance (21, 22). Additionally, mutations in genes encoding the regulators of uhpT expression, UhpA and CyaA, confer resistance because bacteria with these mutations do not activate uhpT expression and, as a consequence, have reduced uptake of fosfomycin (17, 23). Chromosomal mutations that increase fosfomycin resistance rely on defects in GlpT, UhpT, and/or MurA production or native biological function. Therefore, fosfomycin resistance is believed to be associated with a high biological cost to the cell. In support of this hypothesis, mutants that are resistant to fosfomycin can be frequently isolated in vitro (24, 25). However, epidemiologic data indicate that susceptibility rates have remained relatively stable since the introduction of this agent in clinical practice (26–28).
Bacteria have the ability to sense and adapt to environmental stress. CpxAR is a pair of proteins that makes up a two-component system (TCS) that responds to a number of environmental cues (29). CpxA is a sensor kinase that senses bacterial envelope stress and transfers a phosphoryl group to its cognate response regulator, CpxR. Phosphorylated CpxR activates the expression of genes encoding a subset of proteins involved in envelope maintenance, including a periplasmic protease, chaperones, and peptidoglycan enzymes (30). Although CpxAR has been characterized largely as a sensor of bacterial envelope stress, CpxAR has also been implicated in drug resistance. We previously found that CpxR overexpression confers moderate resistance to novobiocin, β-lactams, and deoxycholate (31, 32). In other studies, constitutive phosphorylation of CpxR decreases aminoglycoside and hydroxyurea susceptibility (33). The role of CpxAR in resistance to these compounds remains unclear.
We are interested in determining if there is a reversible mechanism of resistance control for fosfomycin. This may be beneficial for bacteria because they can perhaps relieve the fitness burden conferred by fosfomycin resistance in fosfomycin-free circumstances. Resistance control is achieved by the alteration of drug uptake attributed to the production of the transporters GlpT and UhpT. Activation of the Cpx pathway reduced production of GlpT and UhpT and elevated fosfomycin resistance but led to a defect in the uptake of carbon substrates during growth and reduced biological fitness. We therefore propose a regulatory model of reversible fosfomycin resistance and carbon substrate uptake operated by CpxAR.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Unless otherwise indicated, all bacteria were grown in LB (Luria-Bertani) medium. For marker selection and maintaining plasmids, antibiotics were added to growth medium at following concentrations: 150 μg/ml for ampicillin and 15 μg/ml for chloramphenicol. For growth experiments, EHEC strains were grown at 37°C with shaking for 12 to 16 h. The pellets were washed twice with carbon-free M9 minimal medium (M9 salts, 2 mM MgSO4, 0.1 mM CaCl2, and 2 μM FeSO4) and resuspended in half the volume with the same medium. The cell suspensions were diluted into fresh medium (M9 minimal medium containing 0.5% glycerol-3-phosphate, glucose-6-phosphate, or galactose as a sole carbon source) at a 1:100 ratio. Bacteria were grown at 37°C with shaking, and cell growth was monitored by determining the absorbance at 600 nm.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotypea | Reference or source |
|---|---|---|
| Strains | ||
| HH-H7-008 | Parent strain (tnaA lacZI deletion from EHEC O157:H7 [RIMD 0509952]) | 34 |
| HH-H7-040 | cpxA mutant from HH-H7-008 | This work |
| HH-H7-062 | cpxR mutant from HH-H7-008 | This work |
| HH-H7-093 | cpxAR double mutant from HH-H7-008 | This work |
| MG1655 | E. coli K-12; wild type; reporter strain | 35 |
| Rosetta(DE3) | T7 expression strain; Cmr | Novagen/EMD Bioscience |
| Plasmids | ||
| pKO3 | Temperature-sensitive vector for gene targeting of sacB; Cmr | 36 |
| pTrc99A | Vector for IPTG-inducible expression; Apr | 31 |
| pTrc99cpxA | CpxA complement expression plasmid; Apr | This work |
| pTrc99nlpE | NlpE overexpression plasmid; Apr | This work |
| pTrc99glpT | GlpT overexpression plasmid; Apr | This work |
| pTrc99uhpT | UhpT overexpression plasmid; Apr | This work |
| pQE80 | Vector for expression of His-tagged protein; Apr | Qiagen |
| pQE80cpxR | N-terminal His6-CpxR overexpression plasmid; Apr | This work |
Apr, ampicillin resistance; Cmr, chloramphenicol resistance.
Cloning and mutant constructions.
In-frame deletions of cpxA and cpxR were constructed by sequence overlap extension PCR according to a strategy described previously (36), with primer pairs cpxA-delta1/cpxA-delta2 and cpxA-delta3/cpxA-delta4 for cpxA and primer pairs cpxR-delta1/cpxR-delta2 and cpxR-delta3/cpxR-delta4 for cpxR (Table 2). The upstream flanking DNA included 450 bp, the first 3 amino acid codons for cpxA, and the first 6 amino acid codons for cpxR. The downstream flanking DNA included the last 2 amino acid codons for cpxA and the last 9 amino acid codons for cpxR, the stop codon, and 450 bp of DNA. These deletion constructs were ligated into BamHI- and SalI-digested temperature-sensitive vector pKO3 and introduced into HH-H7-008, the parent strain (34). We selected sucrose-resistant/chloramphenicol-sensitive colonies at 30°C and confirmed the resulting mutant strains by using PCR analysis and DNA sequencing. We also constructed a cpxAR double deletion mutant by same method, using primer pairs cpxR-delta1/cpxAR-delta2 and cpxAR-delta3/cpxA-delta4.
TABLE 2.
Primers used in this study
| Primer | DNA sequence (5′–3′) | Use |
|---|---|---|
| cpxA-delta1 | GCGGGATCCTGAACTTGATCGCGTTCTCG | cpxA mutant construction |
| cpxA-delta2 | CTACAAATGCGGAGTTTAACTCCGGCCTATCATGAAGCAGAAACC | cpxA mutant construction |
| cpxA-delta3 | GATGGTTTCTGCTTCATGATAGGCCGGAGTTAAACTCCGCATTTG | cpxA mutant construction |
| cpxA-delta4 | GCGGTCGACAATTCAGGTCAGCCAGCCGC | cpxA mutant construction |
| cpxR-delta1 | GCGGGATCCCTGCCTGTGCGCGCACAGC | cpxR mutant construction |
| cpxR-delta2 | GCAGAAACCATCAGGTAGCCGCGTAACAGGATTTTATTCATTG | cpxR mutant construction |
| cpxR-delta3 | TAAACAATGAATAAAATCCTGTTACGCGGCTACCTGATGGTTTC | cpxR mutant construction |
| cpxR-delta4 | GCGGTCGACTGCTGGCCGGACGAATCAG | cpxR mutant construction |
| cpxAR-delta2 | CTACAAATGCGGAGTTTAACTCCGTAACAGGATTTTATTCATTG | cpxAR double mutant construction |
| cpxAR-delta3 | TAAACAATGAATAAAATCCTGTTACGGAGTTAAACTCCGCATTTG | cpxAR double mutant construction |
| pTrc-cpxA-F | GCGCCATGGTAGGCAGCTTAACCGC | pTrc99cpxA construction |
| pTrc-cpxA-R | GCGGGATCCTTAACTCCGCTTATACAGCG | pTrc99cpxA construction |
| pTrc-nlpE-F | GCGCCATGGTGAAAAAAGCGATAGTG | pTrc99nlpE construction |
| pTrc-nlpE-R | GCGGGATCCTTACTGCCCCAAACTACTG | pTrc99nlpE construction |
| pTrc-glpT-F | GCGGAATTCTTGAGTATTTTTAAACCAGCGC | pTrc99glpT construction |
| pTrc-glpT-R | GCGAAGCTTTTAGCCTCCGTTGCGTTTTTGC | pTrc99glpT construction |
| pTrc-uhpT-F | GCGGAATTCCTGGCTTTCTTAAACCAGGTC | pTrc99uhpT construction |
| pTrc-uhpT-R | GCGAAGCTTTTATGCCACTGTCAACTGCTG | pTrc99uhpT construction |
| pQE-cpxR-F | GCGGGATCCAATAAAATCCTGTTAGTTGATG | pQE80cpxR construction |
| pQE-cpxR-R | GCGAAGCTTTCATGAAGCAGAAACCATCAG | pQE80cpxR construction |
| glpT-PF | GCGGCGGCCGCTCACTTGATTGCGAGTCGCG | Probe preparation for gel shift assay |
| glpT-PR | GCGAAGCTTTGAAAGCCTCCGTGGCCCGTG | Probe preparation for gel shift assay |
| uhpT-PF | GCGGCGGCCGCGCTTGTTTGCTTATCTGGGG | Probe preparation for gel shift assay |
| uhpT-PR | GCGAAGCTTGGGTTACTCCTGAAATGAATAC | Probe preparation for gel shift assay |
| cpxP-PF | GCGGCGGCCGCTAATAGGGAAGTCAGCTCTC | Probe preparation for gel shift assay |
| cpxP-PR | GCGAAGCTTCATTTGCTCCCAAAATCTTTC | Probe preparation for gel shift assay |
| rhlR-PF | GCGGGATCCGACCAAGTCCCCGTGTCGTG | Probe preparation for gel shift assay |
| rhlR-PR | GCGGGATCCTCGCCATCATCCTGAGCATC | Probe preparation for gel shift assay |
| rrsA-qPCR-F | CGGTGGAGCATGTGGTTTAA | Quantitative real-time PCR |
| rrsA-qPCR-R | GAAAACTTCCGTGGATGTCAAGA | Quantitative real-time PCR |
| rpoD-qPCR-F | CAAGCCGTGGTCGGAAAA | Quantitative real-time PCR |
| rpoD-qPCR-R | GGGCGCGATGCACTTCT | Quantitative real-time PCR |
| glpT-qPCR-F | TGCCCGCAGGTTTGATTC | Quantitative real-time PCR |
| glpT-qPCR-R | CCATGGCACAAAGCCCATA | Quantitative real-time PCR |
| uhpT-qPCR-F | AAGCCGACCCTGGACCTT | Quantitative real-time PCR |
| uhpT-qPCR-R | ACGGTTTGAACCACATTTTGC | Quantitative real-time PCR |
| murA-qPCR-F | CACAATTTCCGGCGCTAAA | Quantitative real-time PCR |
| murA-qPCR-R | GCCAGTAGAGCGGCAAAAAG | Quantitative real-time PCR |
| cpxP-qPCR-F | TGGAGACAATGCATCGTCTTG | Quantitative real-time PCR |
| cpxP-qPCR-R | GCGCGCACAGCGTTTT | Quantitative real-time PCR |
To construct cpxA, nlpE, glpT, and uhpT expression plasmids pTrc99cpxA, pTrc99nlpE, pTrc99glpT, and pTrc99uhpT, respectively, each gene was amplified with the primer pairs shown in Table 2. The product was digested with NcoI and BamHI for cpxA and nlpE or EcoRI and HindIII for glpT and uhpT and ligated into similarly digested plasmid pTrc99A. His6-CpxR expression plasmid pQE80-cpxR was constructed by ligating the cpxR gene, PCR amplified with primers pQE-cpxR-F and pQE-cpxR-R, into BamHI- and HindIII-digested plasmid pQE80 (Table 2). The resulting E. coli construct produces CpxR as an N-terminally hexahistidine-tagged protein in the presence of IPTG (isopropyl-β-d-thiogalactopyranoside). All constructs were confirmed by DNA sequencing.
Drug susceptibility assays.
MIC assays were performed by serial agar dilution methods as described previously (31), with minor modifications. Bacteria were grown for 20 h at 37°C in LB medium without shaking. Five microliters of 100-fold-diluted cultures (∼5,000 cells) was inoculated onto an LB agar plate containing antibiotics and incubated for 16 h at 37°C. The MICs were determined as the lowest concentration at which growth was inhibited. To examine bacterial growth rates in fosfomycin-containing broth, a 100-fold dilution of a culture left standing overnight was inoculated into fresh LB broth in the presence or absence of fosfomycin. Bacteria were grown at 37°C with shaking, and CFU were counted.
RNA extraction and quantitative real-time PCR analyses.
Bacteria were grown to the mid-logarithmic growth phase (optical density at 600 nm [OD600] of ∼0.7) in LB medium. NlpE was overexpressed in the wild-type parent harboring pTrc99nlpE grown with 0.01 mM IPTG. Indole was added to LB medium at a concentration of 1 mM. For time course experiments, the wild-type parent and ΔcpxA strains were grown with 0.2 μg/ml of fosfomycin for 1, 2, 3, and 4 h. Total RNA extraction and cDNA synthesis were performed by using the SV Total-RNA isolation system and the GoScript reverse transcription system according to the manufacturer's instructions (Promega Corp., Madison, WI). Real-time PCR mixtures included 2.5 ng cDNA and 200 nM primers in SYBR Select master mix (Applied Biosystems, Foster City, CA) and were run on an ABI Prism 7900HT Fast real-time PCR system. Constitutively expressed rrsA and rpoD genes were used as an internal control. Primers are listed in Table 2. Amplification plot and melting-curve data are available upon request.
Overexpression and purification of His6-CpxR.
His6-CpxR was expressed in and purified from Escherichia coli Rosetta(DE3) (Novagen/EMD Bioscience, Philadelphia, PA). Bacteria containing recombinant plasmids were grown at 37°C to an OD600 of 0.4 in LB medium, 0.5 mM IPTG was then added, and culture growth was continued for 3 h. Cells were harvested and stored at −80°C overnight. The cell pellet was suspended in lysis buffer (20 mM Tris [pH 7.9], 500 mM NaCl, and 10% glycerol) and lysed by sonication. The lysate was centrifuged, and the resulting supernatant was mixed with Ni-nitrilotriacetic acid (NTA) agarose (Qiagen, Valencia, CA) for 1 h. The agarose was washed with 50 mM imidazole, and His6-CpxR was then eluted with 200 mM imidazole. Purified protein was desalted with Zeba Desalt spin columns (Thermo Scientific, Rockford, IL) and then eluted with buffer for gel shift assays (20 mM Tris [pH 7.5], 50 mM KCl, 1 mM dithiothreitol, and 10% glycerol). The protein was >95% pure, as estimated by SDS-PAGE and Coomassie brilliant blue staining. Protein concentration was determined by using a Bio-Rad protein assay (Bio-Rad, Hercules, CA). Proteins were stored at −80°C.
Gel shift assays.
To assess CpxR binding to glpT and uhpT promoter sequences in gel shift assays, we used 321-bp DNA probes containing the 300-bp region upstream of the glpT and uhpT start codons, respectively. We also used a 220-bp DNA fragment from the 200-bp region upstream of the cpxP start codon as a positive control and a 323-bp DNA fragment from the Pseudomonas aeruginosa rhlR gene as a nonspecific control probe. Purified CpxR was phosphorylated with 50 mM carbamoyl phosphate (Sigma-Aldrich, St. Louis, MO) and 10 mM magnesium chloride for 1 h at 30°C. We then expected that at least some of the CpxR protein would be phosphorylated in the in vitro reaction. The probe DNA fragments (0.30 pmol) were mixed with phosphorylated His6-CpxR in a 10-μl reaction mixture containing 20 mM Tris (pH 7.5), 50 mM KCl, 1 mM dithiothreitol, and 10% glycerol. After incubation for 20 min at room temperature, samples were separated by electrophoresis on a 5% nondenaturing acrylamide–Tris-glycine-EDTA (10 mM Tris [pH 8.0], 380 mM glycine, and 1 mM EDTA) gel in Tris-glycine-EDTA buffer at 4°C. The gel was incubated in 10,000-fold-diluted SYBR green I nucleic acid stain (Lonza, Walkersville, MD), and DNA was visualized under UV light at 300 nm.
Fosfomycin active transport assays.
Assays to test fosfomycin accumulation in bacterial cells were conducted as previously described, with some modifications (37). Bacteria were grown in 20 ml of LB medium to late-logarithmic phase and resuspended in 1 ml of LB medium. This suspension was incubated for 60 min at 37°C in the presence of 2 mg of fosfomycin per ml and then washed three times with hypertonic buffer (10 mM Tris [pH 7.3], 0.5 mM MgCl2, and 150 mM NaCl) to remove the antibiotic. Cells were resuspended in 0.5 ml of distilled water and plated onto LB agar to determine the number of CFU/ml. The bacterial resuspension was boiled at 100°C for 3 min to release the fosfomycin. After centrifugation, the antibiotic concentration in the supernatant was determined by a disc diffusion assay. In this assay, sterilized assay discs (13 mm; Whatman, Florham Park, NJ) were saturated with 0.1 ml of the supernatant and deposited onto LB agar plates overlaid with a 1:10 dilution of a culture of E. coli MG1655 as a reporter strain grown overnight (35). Commercial fosfomycin was used as a standard (Wako Pure Chemical Industries, Ltd., Osaka, Japan). The fosfomycin concentration in supernatants was quantified by the diameter (mm) of inhibitory rings on the LB agar culture and is represented as μg per 107 cells.
Competition assays.
The parent and mutant strains were grown in 3 ml of LB medium with shaking at 37°C overnight. Cultures grown overnight and diluted 1:100 were mixed at a ratio of 1 to 1 in 3 ml of LB medium and grown with shaking at 37°C. After 2, 3, 6, and 24 h of growth, subpopulations of the mixed culture were estimated from 50 independent colonies on serially diluted plates. The wild-type and mutant strains were distinguished by PCR.
RESULTS
Activation of the CpxAR pathway confers fosfomycin resistance in EHEC.
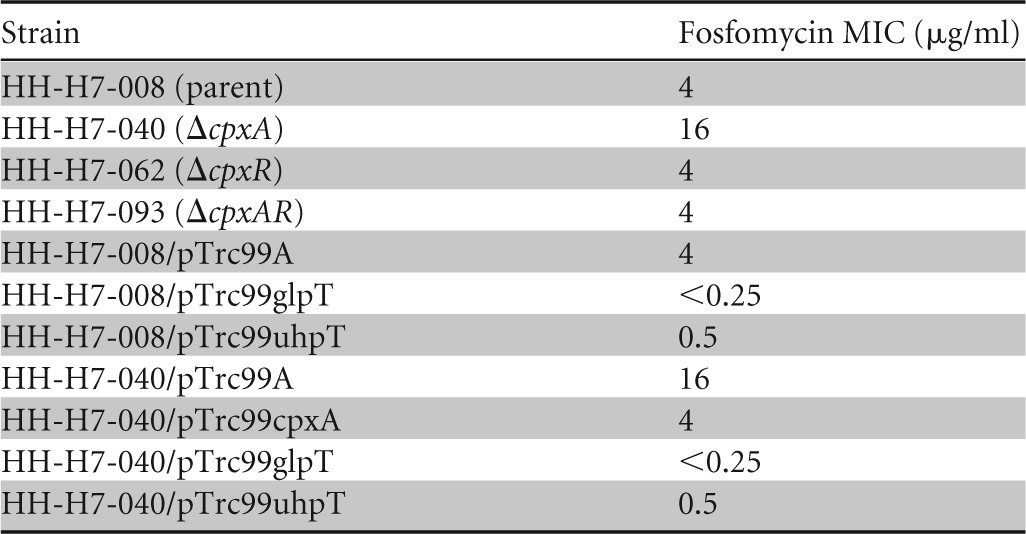
Previous work with nonpathogenic E. coli K-12 suggested that the CpxAR TCS is involved in resistance to antibiotics, including β-lactams (31, 32). To further investigate the CpxAR function in bacterial drug resistance, we determined the role of CpxAR in antibiotic resistance of toxin-producing pathogenic E. coli strains. We constructed cpxA and cpxR gene deletion mutants of EHEC O157:H7, which produces a Shiga-like toxin, and determined the MICs on LB agar containing various drugs for the wild-type parent and the cpxA and cpxR deletion mutant strains. We found that the cpxA mutant had a lower susceptibility to fosfomycin than the parent (MICs of 4 μg/ml for the parent strain versus 16 μg/ml for the cpxA mutant), and unlike the cpxA mutant, deletion of cpxR did not affect susceptibility to fosfomycin (Table 3). Deletion of cpxA is proposed to increase the amount of phosphorylated CpxR because the phosphatase activity of CpxA prevents CpxR from nonspecific phosphorylation and consequently causes constitutive activation of the CpxAR regulon (38). To examine whether or not the increase in fosfomycin resistance in the cpxA mutant can be attributed to CpxR activation, we constructed a cpxAR double deletion mutant and compared MICs of the antibiotic between the parent and the cpxAR mutant strains. As expected, deletion of both the cpxA and cpxR genes no longer exhibited increased resistance to fosfomycin, as determined by MIC (Table 3). We demonstrated that wild-type susceptibility to fosfomycin could be restored in the cpxA mutant when complemented with a cpxA-expressing plasmid, pTrc99cpxA (Table 3).
TABLE 3.
Fosfomycin MICs for EHEC O157:H7 and cpxAR mutant derivatives
We also compared the growth rates of the wild-type parent and cpxA mutant strains grown in LB medium with and without 0.78 μg/ml of fosfomycin. The OD600 and CFU/ml were similarly increased between the wild-type parent and cpxA mutant strains in the absence of fosfomycin (Fig. 1A and B). When fosfomycin was present, the CFU/ml of the wild-type parent immediately decreased to 10% in first 1 h and subsequently increased until 3 h. After this period, the number of CFU/ml again largely decreased. On the other hand, the cpxA mutant grew without any inhibition of growth until 3 h, even when fosfomycin was present; the growth was slightly inhibited beyond 3 h. The growth experiments indicate that the wild-type parent was more sensitive to fosfomycin than the cpxA mutant (Fig. 1A and B). This is consistent with the results of MIC experiments. Therefore, we can conclude that the constitutively activated CpxAR pathway increases the resistance of EHEC O157:H7 to fosfomycin.
FIG 1.
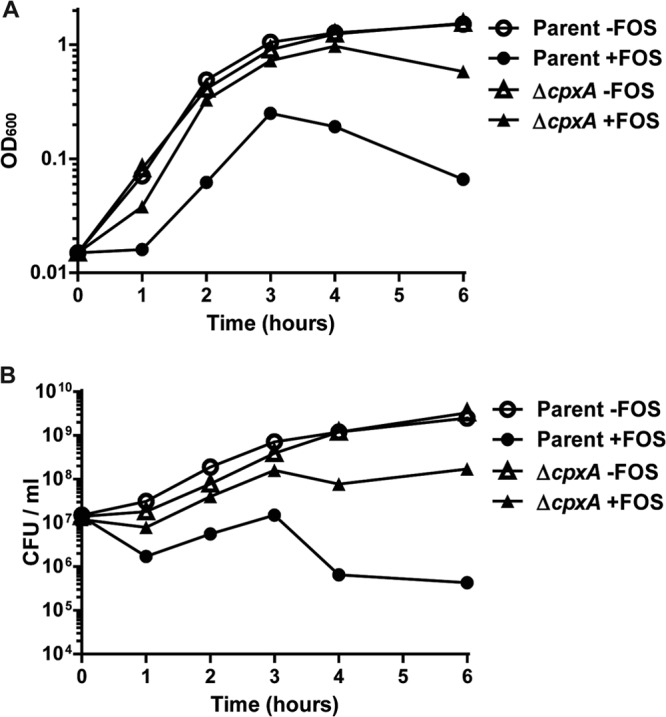
Cell growth of the wild-type parent and ΔcpxA strains in the presence or absence of 0.78 μg/ml fosfomycin (FOS). The growth of these strains was monitored by determining the absorbance at 600 nm (A), and the numbers of live cells are reported as CFU/ml (B). The experiment was repeated at least three times, and similar results were obtained.
Activation of the CpxAR pathway represses glpT and uhpT expression and results in reduced fosfomycin uptake.
GlpT and UhpT transporters and the MurA target enzyme are determinants for susceptibility to fosfomycin. CpxAR may affect the expression of the glpT, uhpT, and/or murA gene and result in fosfomycin resistance. To test this hypothesis, we compared the levels of glpT, uhpT, and murA transcription between the parent and the cpxA mutant strains by quantitative PCR (qPCR) analysis. We found that levels of both glpT and uhpT expression in the cpxA mutant were 8- and 2.5-fold lower than those in the parent, respectively, whereas no significant difference between these strains in levels of murA expression were seen (The murA transcript level in the cpxA mutant was 1.2-fold lower than that in the parent) (Fig. 2A). In addition to a deletion in cpxA, overproduction of NlpE, an outer membrane lipoprotein, or indole can activate the CpxAR pathway (39–41). We observed that the introduction of an NlpE overexpression plasmid and exogenous indole addition to the parent strain showed significantly reduced glpT and uhpT expression levels (Fig. 2B and C). This indicates that the CpxAR pathway either directly or indirectly controls the expression of glpT and uhpT.
FIG 2.
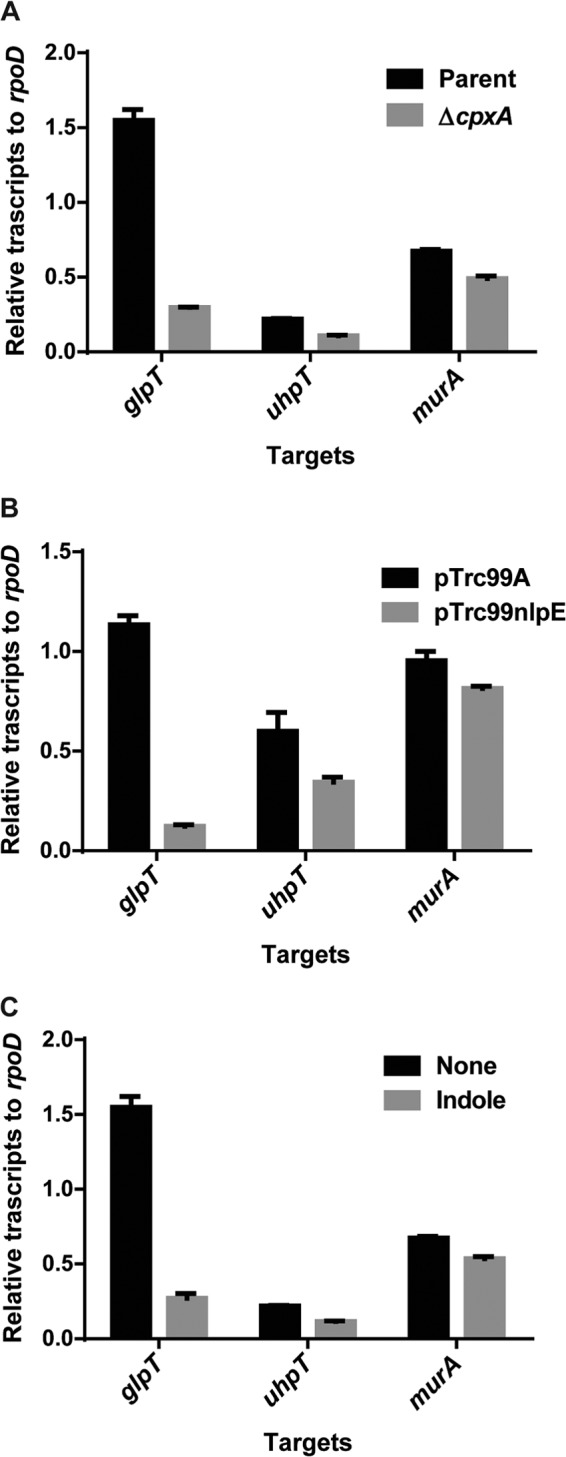
Transcript levels of the glpT, uhpT, and murA genes in the wild-type parent and cpxA mutant strains (A), the wild-type parent strain harboring pTrc99A (vector control) or pTrc99nlpE (NlpE expression plasmid) with 0.01 mM IPTG (B), and the wild-type parent strain grown with or without 1 mM indole (C). Transcript levels of glpT, uhpT, and murA are shown as relative values compared those of rpoD (housekeeping gene). Data plotted are the means of two biological replicates, and error bars indicate ranges.
To determine if CpxR directly regulates the expression of glpT and uhpT, we purified CpxR as an N-terminally hexahistidine-tagged protein and examined its ability to bind the promoters of glpT and uhpT by gel shift assays. We used a 321-bp region upstream of the glpT and uhpT genes as a probe. CpxR bound to both glpT and uhpT promoter DNA fragments. We also observed binding of the CpxR protein to cpxP promoter DNA used as a positive control but not to a similarly sized nonspecific probe from the region upstream of the rhlR gene from P. aeruginosa (Fig. 3). The mobility of glpT and cpxP promoter DNA fragments by electrophoresis was completely shifted in the presence of 20 pmol of CpxR. On the other hand, an unshifted uhpT promoter DNA fragment was still observed at the same concentration of CpxR. This suggests that CpxR has a higher affinity for the glpT promoter than the uhpT promoter, which is in agreement with results of qPCR analyses showing that CpxR represses glpT to a higher degree than uhpT. These observations indicate that CpxR functions as a repressor of glpT and uhpT gene expression. CpxR is proposed to bind to a tandem repeated GTAAA sequence that is separated by a 5-base spacer (GTAAA-[N]5-GTAAA) (42, 43). We found a similar sequence 146 bp upstream of the glpT translational start site (GTAAA-tctta-TTTAA) and 109 bp upstream of the uhpT translational start site (GCAAA-actaa-GAAAT). (Underlining indicates conserved nucleotides in the CpxR binding motif, and lowercase letters indicate nucleotides corresponding to the [N]5 spacer region.)
FIG 3.
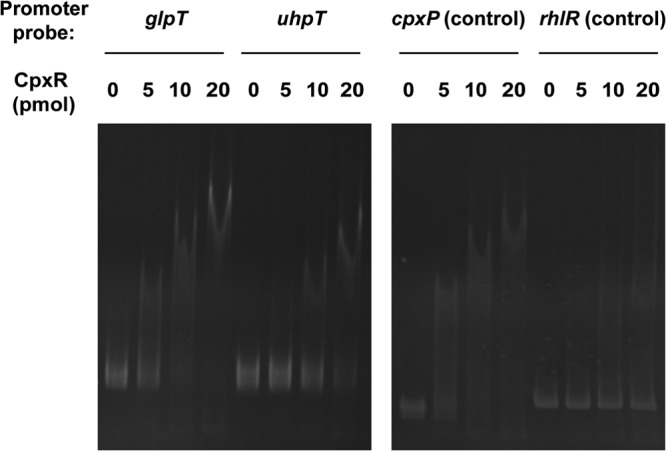
Gel shift assay showing binding of CpxR to the glpT and uhpT promoters. CpxR protein (0, 5, 10, or 20 pmol) was added to reaction mixtures containing 0.3 pmol of DNA probe. DNAs upstream of cpxP and rhlR were used as the positive control and nonbinding (negative) control, respectively. Reaction mixtures were separated on polyacrylamide gels. Free and CpxR-bound DNAs were visualized by SYBR green I staining under UV light at 300 nm.
Suppression of GlpT and UhpT production in the cpxA mutant may impair fosfomycin uptake by cells, which leads to an increase in fosfomycin resistance. To test this hypothesis, we measured intracellular fosfomycin levels of the wild-type parent and cpxA mutant strains by fosfomycin active transport assays, as described in Materials and Methods. The mutant showed lower fosfomycin accumulation than the parent (Fig. 4). The level in the cpxA mutant was 2% of the parental level (1.1 ± 0.3 μg per 107 cells for the ΔcpxA mutant versus 66.0 ± 17.3 μg per 107 cells for the parent).
FIG 4.
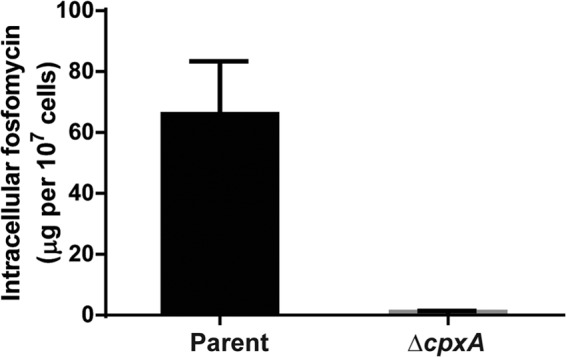
Intracellular accumulation of fosfomycin in the wild-type parent and ΔcpxA strains. Accumulation among these strains is shown as amounts of fosfomycin (μg) in 107 cells. Data plotted are the means from three independent experiments; error bars indicate standard deviations.
We also demonstrated that overexpression of the glpT and uhpT genes from an IPTG-inducible promoter in the cpxA mutant resulted in hypersensitivity to fosfomycin (Table 3). This result supports our proposed model that the elevated fosfomycin resistance through the CpxAR pathway is attributed to downregulation of the glpT and uhpT genes.
CpxAR-activated cells have limited glycerol-3-phosphate and glucose-6-phosphate uptake.
Since GlpT and UhpT transport glycerol-3-phosphate and glucose-6-phosphate, respectively, into cells, we predicted that the constitutive activation of the CpxAR pathway should lead to a growth defect for bacterial cells grown with glycerol-3-phosphate or glucose-6-phosphate as a sole carbon source. As shown in Fig. 5A and B, the cpxA mutant had a growth defect compared to the wild-type parent strain when grown in M9 minimal medium with glycerol-3-phosphate or glucose-6-phosphate as a sole carbon source (Fig. 5A and B). These data suggest a limited availability of glycerol-3-phosphate and glucose-6-phosphate as carbon sources in CpxAR-activated cells. However, the effect of the cpxA mutation on the growth defect in minimal medium supplemented only with glucose-6-phosphate was not drastic. This is in agreement with our above-described data from qPCR and gel shift assays. There was no significant difference in growth between the cpxA mutant and the wild-type parent strain when grown in M9 minimal medium with galactose, which does not depend on GlpT and UhpT transport (Fig. 5C).
FIG 5.

Cell growth of the wild-type parent and ΔcpxA strains in M9 minimal medium with 0.5% (wt/vol) glycerol-3-phosphate (glycerol-3P) (A), glucose-6-phosphate (glucose-6P) (B), or galactose (C) as a sole carbon source. The growth of these strains was monitored by determining the absorbance at 600 nm. The experiment was repeated twice, and similar results were obtained.
Constitutive activation of the CpxAR pathway confers a fitness burden on EHEC cells.
The defect that we observed in the cpxA mutant's ability to utilize certain carbon sources may confer a fitness burden on the cells. To investigate this hypothesis, we cocultured the wild-type parent and cpxA mutant strains and examined their subpopulations in competitive growth. For primary cultures, strains were separately grown in LB medium overnight, and their final CFU per ml were determined to be equivalent in the resulting cultures (approximately 4.5 × 109 CFU/ml). The same number of CFU/ml of stationary-phase cultures was coinoculated into fresh LB medium and grown for 3 or 6 h. After this period of growth, we determined that the percentage of the cpxA mutant in the total population compared to the wild-type parent was approximately 15% and became <4% after 24 h (Fig. 6). The cpxA mutant was outcompeted even in nutrient-rich medium, indicating that this mutant is less fit under these conditions.
FIG 6.
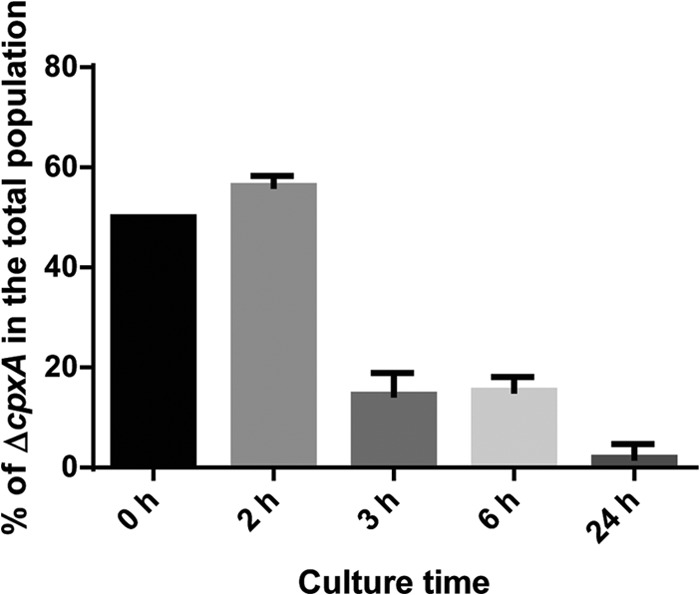
Coculture of the wild-type parent and ΔcpxA strains in LB medium. The same numbers of CFU of strains grown overnight were coinoculated into fresh LB medium and grown. After 2, 3, 6, and 24 h, percentages of ΔcpxA CFU in the total colonies tested were determined. Data plotted are the means from two independent experiments; error bars indicate ranges.
Fosfomycin activates the CpxAR pathway and represses glpT expression.
To gain insights into the physiological relevance of CpxAR-induced fosfomycin resistance, we tested whether fosfomycin can activate the CpxAR pathway and induce fosfomycin resistance. We measured transcript levels of cpxP (a gene that is activated by CpxAR), glpT, and uhpT in the wild-type parent and cpxA mutant strains grown in LB medium with 0.2 μg/ml of fosfomycin by qPCR. Cell growth was not affected with this concentration of fosfomycin (data not shown). The level of cpxP expression in the wild-type parent strain was highest 2 and 3 h after the addition of fosfomycin, while the cpxP expression level was high in the cpxA mutant even in the absence of fosfomycin (Fig. 7A). Expression of glpT in the wild-type parent strain was repressed by the addition of fosfomycin and remained the same in the cpxA mutant with and without the addition of fosfomycin (Fig. 7B). Growth assays depicted in Fig. 1A and B show that the wild-type parent strain grew even in the presence of fosfomycin, 1 to 3 h after the addition of fosfomycin. This appears to be consistent with the timing of glpT repression. Unlike glpT, the effect of fosfomycin on the repression of uhpT was not detected in this assay (data not shown). GlpT is more likely responsible for CpxAR-induced fosfomycin resistance. Thus, fosfomycin activates the CpxAR pathway and induces resistance to fosfomycin by repressing the expression of glpT.
FIG 7.
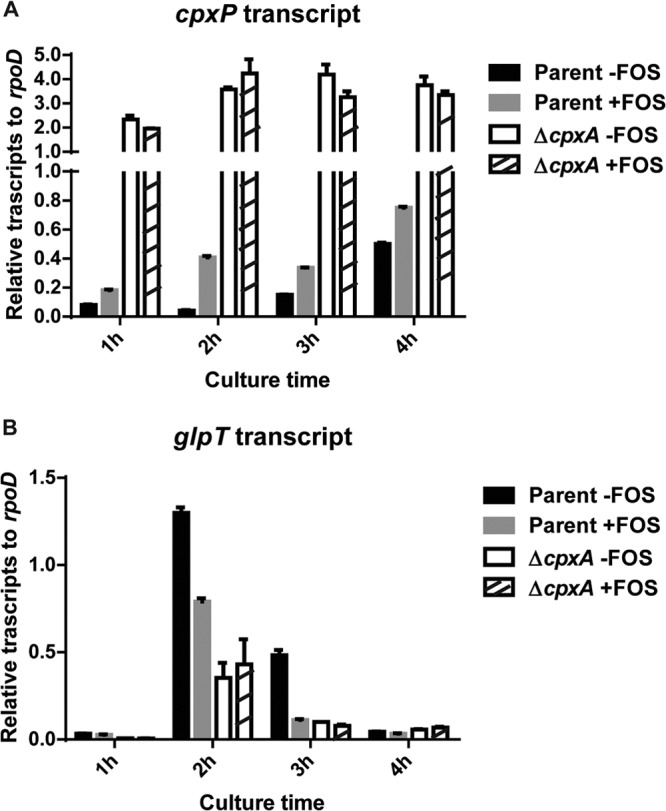
Transcript levels of the cpxP (A) and glpT (B) genes in the wild-type parent and cpxA mutant strains grown in LB medium with or without 0.2 μg/ml of fosfomycin (FOS) for 1, 2, 3, or 4 h. These transcript levels are shown as values relative to those of rpoD (housekeeping gene). Data plotted are the means of two biological replicates, and error bars indicate ranges.
DISCUSSION
Fosfomycin is classified as an old antibiotic, but the problem of overcoming antimicrobial resistance has reinvigorated interest in its use. Fosfomycin is not affected by development of cross-resistance in MDR pathogens such as extended-spectrum β-lactamase (ESBL) producers because of its structure, which is unrelated to the structure of any other traditional antibiotics (9, 10). The Clinical and Laboratory Standards Institute (CLSI) reported that fosfomycin has a lower antibacterial activity for E. coli species than other commonly used antibiotics, such as β-lactams and fluoroquinolones (53). However, fosfomycin offers a great advantage, because so far, cross-resistance to other antibiotics has not been reported.
Fosfomycin resistance is known to be conferred by mutations in the genes encoding GlpT, UhpT, and MurA. Since GlpT and UhpT mediate glycerol-3-phosphate and glucose-6-phosphate uptake, and MurA functions as a peptidoglycan biosynthetic enzyme, fosfomycin resistance may come at a high biological cost. This may account for the low frequency of clinical isolates resistant to fosfomycin despite its prevalent use (44, 45). Our work indicates that there is indeed a trade-off between fosfomycin resistance and biological fitness in EHEC. When fosfomycin is present, resistant strains will have an advantage over susceptible members. However, we predict that under conditions of subinhibitory levels of fosfomycin, resistant strains would be outcompeted by their susceptible counterparts because of the fitness burden conferred by resistance.
Our primary interest was to determine if there is a reversible mechanism of resistance to fosfomycin. It would be physiologically beneficial for bacteria to have a reversible mechanism regulating fosfomycin resistance. In this study, we found the mechanism by which the CpxAR TCS reversibly controls fosfomycin resistance. Our results suggest that the activation of the CpxAR pathway increases fosfomycin resistance because there is reduced expression of glpT and uhpT, which leads to a decrease in the amount of intracellular fosfomycin. However, cells that have a constitutively active CpxAR regulon are less competitive than the wild-type parent strain in the absence of fosfomycin. These data suggest that reversible activation of the CpxAR regulon could allow transient fosfomycin resistance without the long-term biological cost attributed to carbon source uptake.
We also found that fosfomycin activates the CpxAR pathway and leads to the repression of glpT expression but not uhpT expression. This result suggests that fosfomycin activation of CpxAR could be clinically relevant and that GlpT transport is primarily responsible for the resistance mechanism. However, induction does not render wild-type cells resistant because the cpxAR double mutant presented a resistance phenotype that was similar to that of the wild type in our MIC assays. Therefore, CpxAR may be not the sole system responsible for the induction of fosfomycin resistance. Our data demonstrate that CpxAR contributes to fosfomycin resistance via the activation of CpxR.
The CpxAR system was originally characterized as a sensor of bacterial envelope stress caused by misfolded envelope proteins and membrane damage (29). In response to this stress, the CpxAR system activates a subset of genes that contribute to envelope rearrangement and protein refolding and degradation (30). A link between CpxAR-activated envelope stress adaptations and antibiotic resistance was recently proposed. CpxAR activation confers resistance to aminoglycosides (33). It has been hypothesized that aminoglycosides lead to protein mistranslations and the generation of reaction oxygen species (ROS), and CpxAR-induced proteases and membrane proteins may relieve the bacteria from membrane perturbations caused by the drug (46, 47). Bactericidal agents stimulate the oxidation of NADH through the electron transport chain, which is dependent on the tricarboxylic acid (TCA) cycle, and hyperactivation of the electron transport chain promotes ROS formation, causing stress-induced cell death. Similar to aminoglycoside antibiotics, hydroxyurea also promotes ROS formation and causes cell death (48). CpxAR activation alleviates ROS-induced envelope damage and confers resistance to hydroxyurea. Our study represents yet another mechanism of CpxAR-mediated drug resistance other than alleviation of ROS-induced envelope stress. Fosfomycin is produced by Streptomyces species (49). As another physiology aspect, the CpxAR response might also contribute to a part of the innate biodefense against fosfomycin produced by Streptomyces species for free-living E. coli species, including EHEC, in nature.
The involvement of the Cpx system in biological fitness and virulence was recently discussed. CpxAR activity is required for optimal growth of uropathogenic E. coli (UPEC) in bladder cells (50). This observation implies the contribution of CpxAR to the biological fitness of UPEC in bladder cells, although the precise mechanism has not been elucidated. However, in other studies, Cpx-induced stress responses attenuate expression of LEE (locus of enterocyte effacement) proteins, components of the type III secretion system that contributes to the virulence in enteropathogenic E. coli (EPEC) (51, 52). It is possible that the activation of the CpxAR pathway imposes energy costs because it simultaneously induces the production of proteins to get rid of envelope stress, such as periplasmic proteases, chaperones, and some membrane-associated proteins. Therefore, CpxAR-active strains appear to have high metabolic costs and are less virulent.
The CpxAR pathway could confer transient antibiotic resistance and counteract the long-term biological cost of losing nutrient importers. We suggest that reversible mechanisms of resistance may be an important clinical issue to keep in mind as new antibiotics are developed or antibiotics already in use are repurposed.
ACKNOWLEDGMENTS
This study was kindly supported by JSPS KAKENHI Grant-in-Aid for Young Scientists (B) grant number 25870115, JST Program; Improvement of Research Environment for Young Researchers, Japanese Ministry of Education, Culture, Sport, Science and Technology (Gunma University operation grants); the Inamori Foundation; the Institute for Fermentation, Osaka (IFO); and the Japanese Ministry of Health, Labor and Welfare (H24-Shinkou-Ippan-010).
Footnotes
Published ahead of print 25 October 2013
REFERENCES
- 1.Karmali MA. 1989. Infection by verocytotoxin-producing Escherichia coli. Clin. Microbiol. Rev. 2:15–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086. 10.1016/S0140-6736(05)71144-2 [DOI] [PubMed] [Google Scholar]
- 3.Proulx F, Seidman E. 1999. Is antibiotic therapy of mice and humans useful in Escherichia coli O157:H7 enteritis? Eur. J. Clin. Microbiol. Infect. Dis. 18:533–534. 10.1007/s100960050343 [DOI] [PubMed] [Google Scholar]
- 4.Safdar N, Said A, Gangnon RE, Maki DG. 2002. Risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 enteritis: a meta-analysis. JAMA 288:996–1001. 10.1001/jama.288.8.996 [DOI] [PubMed] [Google Scholar]
- 5.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. 2000. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 342:1930–1936. 10.1056/NEJM200006293422601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda K, Ida O, Kimoto K, Takatorige T, Nakanishi N, Tatara K. 1999. Effect of early fosfomycin treatment on prevention of hemolytic uremic syndrome accompanying Escherichia coli O157:H7 infection. Clin. Nephrol. 52:357–362 [PubMed] [Google Scholar]
- 7.Kurioka T, Yunou Y, Harada H, Kita E. 1999. Efficacy of antibiotic therapy for infection with Shiga-like toxin-producing Escherichia coli O157:H7 in mice with protein-calorie malnutrition. Eur. J. Clin. Microbiol. Infect. Dis. 18:561–571. 10.1007/s100960050348 [DOI] [PubMed] [Google Scholar]
- 8.Takeda T, Yoshino K, Uchida H, Ikeda N, Tanimura M. 1998. Early use of fosfomycin for Shiga toxin-producing Escherichia coli O157 infection reduces the risk of hemolytic-uremic syndrome, p 385–387 In Kaper JB, O'Brien AD. (ed), Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. ASM Press, Washington, DC [Google Scholar]
- 9.Dinh A, Salomon J, Bru JP, Bernard L. 2012. Fosfomycin: efficacy against infections caused by multidrug-resistant bacteria. Scand. J. Infect. Dis. 44:182–189. 10.3109/00365548.2011.616221 [DOI] [PubMed] [Google Scholar]
- 10.Falagas ME, Kastoris AC, Kapaskelis AM, Karageorgopoulos DE. 2010. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum beta-lactamase producing, Enterobacteriaceae infections: a systematic review. Lancet Infect. Dis. 10:43–50. 10.1016/S1473-3099(09)70325-1 [DOI] [PubMed] [Google Scholar]
- 11.Michalopoulos AS, Livaditis IG, Gougoutas V. 2011. The revival of fosfomycin. Int. J. Infect. Dis. 15:e732–e739. 10.1016/j.ijid.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 12.Brown ED, Vivas EI, Walsh CT, Kolter R. 1995. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J. Bacteriol. 177:4194–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia P, Arca P, Evaristo Suarez J. 1995. Product of fosC, a gene from Pseudomonas syringae, mediates fosfomycin resistance by using ATP as cosubstrate. Antimicrob. Agents Chemother. 39:1569–1573. 10.1128/AAC.39.7.1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rigsby RE, Fillgrove KL, Beihoffer LA, Armstrong RN. 2005. Fosfomycin resistance proteins: a nexus of glutathione transferases and epoxide hydrolases in a metalloenzyme superfamily. Methods Enzymol. 401:367–379. 10.1016/S0076-6879(05)01023-2 [DOI] [PubMed] [Google Scholar]
- 15.Roberts AA, Sharma SV, Strankman AW, Duran SR, Rawat M, Hamilton CJ. 2013. Mechanistic studies of FosB: a divalent-metal-dependent bacillithiol-S-transferase that mediates fosfomycin resistance in Staphylococcus aureus. Biochem. J. 451:69–79. 10.1042/BJ20121541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma SV, Jothivasan VK, Newton GL, Upton H, Wakabayashi JI, Kane MG, Roberts AA, Rawat M, La Clair JJ, Hamilton CJ. 2011. Chemical and chemoenzymatic syntheses of bacillithiol: a unique low-molecular-weight thiol amongst low G + C Gram-positive bacteria. Angew. Chem. Int. Ed. Engl. 50:7101–7104. 10.1002/anie.201100196 [DOI] [PubMed] [Google Scholar]
- 17.Takahata S, Ida T, Hiraishi T, Sakakibara S, Maebashi K, Terada S, Muratani T, Matsumoto T, Nakahama C, Tomono K. 2010. Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int. J. Antimicrob. Agents 35:333–337. 10.1016/j.ijantimicag.2009.11.011 [DOI] [PubMed] [Google Scholar]
- 18.Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. 1996. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry 35:4923–4928. 10.1021/bi952937w [DOI] [PubMed] [Google Scholar]
- 19.Venkateswaran PS, Wu HC. 1972. Isolation and characterization of a phosphonomycin-resistant mutant of Escherichia coli K-12. J. Bacteriol. 110:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horii T, Kimura T, Sato K, Shibayama K, Ohta M. 1999. Emergence of fosfomycin-resistant isolates of Shiga-like toxin-producing Escherichia coli O26. Antimicrob. Agents Chemother. 43:789–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Argast M, Ludtke D, Silhavy TJ, Boos W. 1978. A second transport system for sn-glycerol-3-phosphate in Escherichia coli. J. Bacteriol. 136:1070–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadner RJ, Winkler HH. 1973. Isolation and characterization of mutations affecting the transport of hexose phosphates in Escherichia coli. J. Bacteriol. 113:895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nilsson AI, Berg OG, Aspevall O, Kahlmeter G, Andersson DI. 2003. Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob. Agents Chemother. 47:2850–2858. 10.1128/AAC.47.9.2850-2858.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denamur E, Bonacorsi S, Giraud A, Duriez P, Hilali F, Amorin C, Bingen E, Andremont A, Picard B, Taddei F, Matic I. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J. Bacteriol. 184:605–609. 10.1128/JB.184.2.605-609.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellington MJ, Livermore DM, Pitt TL, Hall LM, Woodford N. 2006. Mutators among CTX-M beta-lactamase-producing Escherichia coli and risk for the emergence of fosfomycin resistance. J. Antimicrob. Chemother. 58:848–852. 10.1093/jac/dkl315 [DOI] [PubMed] [Google Scholar]
- 26.Oteo J, Orden B, Bautista V, Cuevas O, Arroyo M, Martinez-Ruiz R, Perez-Vazquez M, Alcaraz M, Garcia-Cobos S, Campos J. 2009. CTX-M-15-producing urinary Escherichia coli O25b-ST131-phylogroup B2 has acquired resistance to fosfomycin. J. Antimicrob. Chemother. 64:712–717. 10.1093/jac/dkp288 [DOI] [PubMed] [Google Scholar]
- 27.Schito GC. 2003. Why fosfomycin trometamol as first line therapy for uncomplicated UTI? Int. J. Antimicrob. Agents 22(Suppl 2):79–83. 10.1016/S0924-8579(03)00231-0 [DOI] [PubMed] [Google Scholar]
- 28.Shimizu M, Shigeobu F, Miyakozawa I, Nakamura A, Suzuki M, Mizukoshi S, O'Hara K, Sawai T. 2000. Novel fosfomycin resistance of Pseudomonas aeruginosa clinical isolates recovered in Japan in 1996. Antimicrob. Agents Chemother. 44:2007–2008. 10.1128/AAC.44.7.2007-2008.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 9:387–398. 10.1101/gad.9.4.387 [DOI] [PubMed] [Google Scholar]
- 30.Raivio TL. 2005. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol. Microbiol. 56:1119–1128. 10.1111/j.1365-2958.2005.04625.x [DOI] [PubMed] [Google Scholar]
- 31.Hirakawa H, Nishino K, Hirata T, Yamaguchi A. 2003. Comprehensive studies of drug resistance mediated by overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Bacteriol. 185:1851–1856. 10.1128/JB.185.6.1851-1856.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirakawa H, Nishino K, Yamada J, Hirata T, Yamaguchi A. 2003. Beta-lactam resistance modulated by the overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Antimicrob. Chemother. 52:576–582. 10.1093/jac/dkg406 [DOI] [PubMed] [Google Scholar]
- 33.Mahoney TF, Silhavy TJ. 2013. The Cpx stress response confers resistance to some, but not all, bactericidal antibiotics. J. Bacteriol. 195:1869–1874. 10.1128/JB.02197-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirakawa H, Kodama T, Takumi-Kobayashi A, Honda T, Yamaguchi A. 2009. Secreted indole serves as a signal for expression of type III secretion system translocators in enterohaemorrhagic Escherichia coli O157:H7. Microbiology 155:541–550. 10.1099/mic.0.020420-0 [DOI] [PubMed] [Google Scholar]
- 35.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. 10.1126/science.277.5331.1453 [DOI] [PubMed] [Google Scholar]
- 36.Link AJ, Phillips D, Church GM. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 179:6228–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leon J, Garcia-Lobo JM, Ortiz JM. 1982. Fosfomycin resistance plasmids do not affect fosfomycin transport into Escherichia coli. Antimicrob. Agents Chemother. 21:608–612. 10.1128/AAC.21.4.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirakawa H, Inazumi Y, Masaki T, Hirata T, Yamaguchi A. 2005. Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol. Microbiol. 55:1113–1126. 10.1111/j.1365-2958.2004.04449.x [DOI] [PubMed] [Google Scholar]
- 40.Raffa RG, Raivio TL. 2002. A third envelope stress signal transduction pathway in Escherichia coli. Mol. Microbiol. 45:1599–1611. 10.1046/j.1365-2958.2002.03112.x [DOI] [PubMed] [Google Scholar]
- 41.Snyder WB, Davis LJ, Danese PN, Cosma CL, Silhavy TJ. 1995. Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J. Bacteriol. 177:4216–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Wulf P, Mcguire AM, Liu X, Lin EC. 2002. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J. Biol. Chem. 277:26652–26661. 10.1074/jbc.M203487200 [DOI] [PubMed] [Google Scholar]
- 43.Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J. Bacteriol. 191:1798–1815. 10.1128/JB.00798-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karageorgopoulos DE, Wang R, Yu XH, Falagas ME. 2012. Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J. Antimicrob. Chemother. 67:255–268. 10.1093/jac/dkr466 [DOI] [PubMed] [Google Scholar]
- 45.Marchese A, Gualco L, Debbia EA, Schito GC, Schito AM. 2003. In vitro activity of fosfomycin against gram-negative urinary pathogens and the biological cost of fosfomycin resistance. Int. J. Antimicrob. Agents 22(Suppl 2):53–59. 10.1016/S0924-8579(03)00230-9 [DOI] [PubMed] [Google Scholar]
- 46.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8:423–435. 10.1038/nrmicro2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690. 10.1016/j.cell.2008.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC. 2009. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell 36:845–860. 10.1016/j.molcel.2009.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grif K, Dierich MP, Pfaller K, Miglioli PA, Allerberger F. 2001. In vitro activity of fosfomycin in combination with various antistaphylococcal substances. J. Antimicrob. Chemother. 48:209–217. 10.1093/jac/48.2.209 [DOI] [PubMed] [Google Scholar]
- 50.Debnath I, Norton JP, Barber AE, Ott EM, Dhakal BK, Kulesus RR, Mulvey MA. 2013. The Cpx stress response system potentiates the fitness and virulence of uropathogenic Escherichia coli. Infect. Immun. 81:1450–1459. 10.1128/IAI.01213-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacRitchie DM, Acosta N, Raivio TL. 2012. DegP is involved in Cpx-mediated posttranscriptional regulation of the type III secretion apparatus in enteropathogenic Escherichia coli. Infect. Immun. 80:1766–1772. 10.1128/IAI.05679-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacRitchie DM, Ward JD, Nevesinjac AZ, Raivio TL. 2008. Activation of the Cpx envelope stress response down-regulates expression of several locus of enterocyte effacement-encoded genes in enteropathogenic Escherichia coli. Infect. Immun. 76:1465–1475. 10.1128/IAI.01265-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clinical and Laboratory Standards Institute 2011. Performance standards for antimicrobial susceptibility testing; 20th informational supplement. CLSI document M100-S21 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]