

Abstract
Bacillus subtilis mutants lacking ymdB are unable to form biofilms, exhibit a strong overexpression of the flagellin gene hag, and are deficient in SlrR, a SinR antagonist. Here, we report the functional and structural characterization of YmdB, and we find that YmdB is a phosphodiesterase with activity against 2′,3′- and 3′,5′-cyclic nucleotide monophosphates. The structure of YmdB reveals that the enzyme adopts a conserved phosphodiesterase fold with a binuclear metal center. Mutagenesis of a catalytically crucial residue demonstrates that the enzymatic activity of YmdB is essential for biofilm formation. The deletion of ymdB affects the expression of more than 800 genes; the levels of the σD-dependent motility regulon and several sporulation genes are increased, and the levels of the SinR-repressed biofilm genes are decreased, confirming the role of YmdB in regulating late adaptive responses of B. subtilis.
INTRODUCTION
Biofilms are complex communities of bacterial cells that can be established on solid surfaces or air-liquid interfaces. In the case of human pathogens, the formation of biofilms on medical instruments and catheters is a significant problem in hospital-acquired infections. The complex architecture of a biofilm is maintained by an extracellular matrix of exopolysaccharide, protein, and DNA (1, 2). Wild isolates of the Gram-positive model bacterium Bacillus subtilis also have the potential to form architecturally complex colonies and pellicles (3) under laboratory conditions and in their natural environment on the plant rhizosphere (4). Several laboratory strains, such as B. subtilis 168, have lost their ability to form robust biofilms and seem to have adapted to cultivation in liquid culture (5). While some laboratory isolates of B. subtilis strain 168 lack the expression of biofilm-related genes and form flat colonies, others can still form architecturally complex colonies (6, 7).
The control of biofilm formation in B. subtilis is dependent on a molecular switch comprising two transcription repressors and two antirepressors, which participate in a complex network of protein-protein interactions that govern the expression of operons involved in matrix production and autolysis. Central to this regulatory switch is the repressor SinR (8–10), a DNA binding protein that represses the transcription of operons involved in both exopolysaccharide production (3) and the production of the protein component of the matrix (11). SinR activity is controlled by the relative levels of various antagonists, the foremost of which is SinI (8), which causes derepression by binding to SinR. The SinI-SinR complex is no longer able to bind to DNA (12, 13). Although sinI and sinR are adjacent on the chromosome, the transcription profile of the sin operon is under complex regulation to result in an unequal expression profile of the two genes (14, 15). Two additional proteins, SlrR and SlrA, that also influence this switch were discovered recently, and these proteins are homologues of SinR and SinI, respectively (16–18). The complex interplay between these transcription factors and their antagonists, their differential expression profiles, and the feedback loops created by these interactions explain how B. subtilis is able to adapt to challenging environmental conditions by specializing in, for instance, motility, genetic competence, sporulation, or cooperation with other cells to form a biofilm.
YmdB was discovered recently as a factor influencing this decision-making process (19). Mutants lacking ymdB are unable to form biofilms and exhibited a strong expression of the genes in the σD-dependent motility regulon, in particular hag, the gene encoding the flagellin protein. The ymdB mutant had a strong reduction of slrR expression, and the phenotype could partially be relieved by overexpressing slrR (19). It is not known how YmdB exerts its effects on the SlrR-SinR switch. YmdB is homologous to a calcineurin-like phosphodiesterase, DR1281, from Deinococcus radiodurans (20), an enzyme with phosphatase activity against phosphoenolpyruvate and with phosphodiesterase activity against 2′,3′-cyclic AMP (cAMP) (20). To determine the molecular function of YmdB, and thus understand its influence on biofilm formation, we have initiated a structural and functional characterization of YmdB. Here, we show that YmdB has a significant metal ion-dependent phosphodiesterase activity against both 2′,3′- and 3′,5′-cyclic nucleotides, whereas no significant phosphatase activity was detected. Mutagenesis studies revealed that a lack of the phosphodiesterase activity is causal for all phenotypes associated with a ymdB mutant. We have determined the crystal structure of YmdB, the active site of which contains essential metal ions and a phosphate ion that occupies the position of the enzyme's substrate. Finally, a transcriptomic analysis revealed that over 800 mRNAs are affected by ymdB deletion, and the genes affected confirm a central, regulatory role for YmdB in the late adaptive responses of B. subtilis.
MATERIALS AND METHODS
B. subtilis strains and growth conditions.
The B. subtilis strains used in this work are listed in Table 1. They are derived from wild-type laboratory strain 168 or from nondomesticated wild-type strain NCIB3610. B. subtilis was grown in LB medium or in CSE minimal medium containing succinate and glutamate-ammonium as sources of carbon and nitrogen, respectively (21). The medium was supplemented with auxotrophic requirements (at 50 mg/liter) and glucose as indicated. SP, CSE, or MSgg (3) plates were prepared by the addition of 17 g Bacto agar/liter (Difco) to the medium.
TABLE 1.
B. subtilis strains used in this study
| Strain | Genotype or description | Source or referencea |
|---|---|---|
| 168 | trpC2 | Laboratory collection |
| GP583 | trpC2 ΔymdB::spc | 19 |
| GP921 | NCIB3610 ΔymdB::spc | 19 |
| GP922 | trpC2 ΔymdB::cat | 19 |
| GP966 | trpC2 ymdB-cat | LFH-PCR → 168 |
| GP969 | trpC2 ymdBE39Q-cat | LFH-PCR → 168 |
| GP1618 | NCIB3610 ymdB-cat | GP966 → NCIB3610 |
| GP1619 | NCIB3610 ymdBE39Q-cat | GP969 → NCIB3610 |
| NCIB3610 | Wild type | Laboratory collection |
Arrows indicate construction by transformation or transduction. LFH-PCR, long flanking homology PCR.
DNA manipulation and transformation.
B. subtilis chromosomal DNA was isolated as described previously (21). Escherichia coli strain DH5α (22) was used for all cloning experiments. All primer sequences are provided in Table S1 in the supplemental material. Restriction enzymes, T4 DNA ligase, and DNA polymerases were used as recommended by the manufacturers, and the combined chain reaction for site-specific mutagenesis (23) was performed with Accuzyme DNA polymerase and thermostable DNA ligase (Ampligase; Epicentre, WI, USA). DNA fragments were purified from agarose gels by using the QIAquick PCR purification kit (Qiagen, Germany). Plasmid DNA extraction was performed by using standard procedures (22). All plasmid inserts derived from PCR products were verified by DNA sequencing using the dideoxy chain termination method (22).
Standard procedures were used to transform chemically competent E. coli cells (22), and transformants were selected on LB plates containing the appropriate antibiotics. B. subtilis was transformed with plasmid or chromosomal DNA according to a two-step protocol described previously (24). Transformants were selected on SP plates containing chloramphenicol (Cm) (5 μg/ml), kanamycin (Km) (10 μg/ml), spectinomycin (Spc) (100 μg/ml), or erythromycin (Em) plus lincomycin (Lin) (1 μg/ml and 10 μg/ml, respectively).
B. subtilis strain construction.
Strains GP966 and GP969, which express ymdB-cat and ymdBE39Q-cat, respectively (the cat resistance cassette is placed downstream of the wild-type or mutant ymdB alleles to facilitate the introduction of point mutations into the chromosome), were obtained by first amplifying ymdB with primers CD95 and ML196 (ymdB-cat) and with primers CD95 and ML196 and mutagenic primer CD128 (ymdBE39Q-cat). The downstream fragment of ymdB was amplified with primers ML197 and ML198, and the intervening chloramphenicol resistance cassette was derived from plasmid pGEM-cat (25). The PCR products (upstream, cat, and downstream) were mixed and combined by another PCR using outer primers CD95 and ML196. Finally, strains GP966 and GP969 were constructed by transformation of wild-type strain 168 with the combined PCR products (26).
To transfer mutations into the background of nondomesticated wild-type strain NCIB3610, SPP1-mediated phage transduction was used as described previously (19).
Production of recombinant YmdB and YmdBE39Q.
Plasmid pGP172 (27) was used to fuse the ymdB variants to a C-terminal Strep-tag (see Tables S1 and S2 in the supplemental material for details). The PCR products were amplified with primers CD174 and CD93, using chromosomal DNAs of B. subtilis 168 (for the wild-type ymdB gene) and GP969 (for the ymdBE39Q allele) as the templates. The PCR products were digested with SacI and BamHI and cloned into pGP172 linearized with the same enzymes. The resulting plasmids, pGP1916 and pGP1917 (see Tables S1 and S2 in the supplemental material for details), were used for protein production in E. coli. Similarly, C-terminally His6-tagged YmdB was prepared by amplification of B. subtilis 168 genomic DNA with primers NcoFor and BamRev2 (see Table S1 in the supplemental material). The resultant PCR product was ligated into the transfer vector pGEM-T Easy (Promega) and transformed into E. coli DH5α, where the presence of the insert was verified by blue/white screening. For subcloning into pET28a, the ymdB insert was liberated from pGEM-T Easy by restriction using NcoI and BamHI and subsequently ligated into similarly restricted pET28a.
Recombinant YmdB proteins were expressed in E. coli BL21(DE3) strains by isopropyl-β-d-thiogalactopyranoside (IPTG) induction (final concentration, 1 mM) in exponentially growing cultures. Cells were harvested by centrifugation 4 h after induction and were lysed either by sonication or with a French press. After lysis, the crude extracts were centrifuged at 15,000 × g for a minimum of 30 min. Strep-tagged YmdB was purified by Streptactin affinity chromatography (IBA, Göttingen, Germany); the bound proteins were eluted with 2.5 mM desthiobiotin (Sigma-Aldrich). The relevant protein-containing fractions were combined and dialyzed overnight. YmdB-His6 was purified by nickel-immobilized metal affinity chromatography using a His-Trap column (GE Healthcare). Bound proteins were eluted with imidazole before pooling, concentrating, and loading onto a Superdex 200 gel filtration column. The purity of YmdB-His6 was estimated to be >90% by SDS-PAGE.
Enzyme assays.
The assays for phosphatase and phosphodiesterase activities of Strep-tagged YmdB were performed as described previously (20). Phosphatase activity against para-nitrophenol phosphate (pNPP) was assayed in a buffer containing 300 mM Tris-HCl (pH 7.5), 1 mM MnCl2, 1 mM dithiothreitol (DTT), 25 mM pNPP, and different amounts of Strep-tagged YmdB in a total volume of 1 ml. The YmdB phosphodiesterase activity against bis-pNPP was assayed in a buffer containing 300 mM Tris-HCl (pH 8.5), 0.5 mM MnCl2, 1 mM DTT, 0 to 15 mM bis-pNPP, and different amounts of Strep-tagged YmdB in a total volume of 1 ml.
Phosphodiesterase activity with cyclic nucleotides was measured by using a quantitative assay as previously described (20, 28). The activity was assayed in a 400-μl reaction mixture containing 50 mM morpholineethanesulfonic acid (MES)-KOH buffer (pH 6.25), 0.2 mM MnCl2, 0 to 5 mM substrate, and different amounts of Strep-tagged protein (0 to 2 μg).
To detect potential diguanylate cyclase activity of YmdB, assays were performed as follows: 100 nM YmdB in a solution containing 50 mM Tris-HCl buffer (pH 7.8), 10 mM MgCl2, and 0.1% bovine serum albumin (BSA) was preincubated for 10 min at 30°C with shaking at 300 rpm. After addition of 100 μM (each) GTP and MgCl2 (final volume, 100 μl), incubation times of 1 and 18 h, respectively, were chosen. The reactions were stopped by heating the samples for 5 min at 95°C. The samples were centrifuged for 10 min at 14,000 rpm at 4°C. The supernatants were analyzed for cyclic di-GMP (c-di-GMP) by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS), as described previously (29). In parallel, the assays were also performed with the E. coli diguanylate cyclase YdeH (29).
In all kinetic experiments, Km and Vmax were determined by nonlinear curve fitting from Lineweaver-Burk plots.
Analysis of the cyclic dinucleotide pools.
The concentration of cyclic dinucleotides in B. subtilis cells was determined by a LC-MS/MS method essentially as described previously (30, 31). Briefly, cells were cultivated in SP medium, pelleted, and lysed in a Micro Dismembrator S instrument (Sartorius). The resulting cell powder was used to extract the metabolites, and the dried metabolite extracts were dissolved with 200 μl of H2O. After repeated centrifugation and addition of internal standards ([13C,15N]c-di-GMP and [13C,15N]c-di-AMP), part of the extracts was analyzed by LC-MS/MS.
Quantification of cyclic dinucleotides by MS/MS.
Chromatographic separation was performed on a series 200 high-performance liquid chromatography (HPLC) system (PerkinElmer Life Sciences), as described previously (31). Analyte detection was performed on an API 3000 triple-quadrupole mass spectrometer equipped with an electrospray ionization source (AB Sciex), using selected reaction monitoring (SRM) analysis in the positive ionization mode. The SRM transitions labeled “quantifier” were used to quantify the compound of interest, whereas “identifier” SRM transitions were monitored as confirmatory signals. The quantifier SRM transitions were the most intense and were therefore used for quantification.
X-ray crystallography.
YmdB-His6 was buffer exchanged into 10 mM Na-HEPES–100 mM NaCl and concentrated to 10 mg/ml. Crystals of YmdB-His6 were obtained by sparse-matrix screening against a range of commercially available screens. The crystals used for structure determination were obtained from well solutions containing 0.2 M sodium acetate–20% polyethylene glycol 3350 (PEG 3350) and were cryoprotected by soaking for 30 s in well solution supplemented with 20% (vol/vol) ethylene glycol before being loop mounted and plunged into a pool of liquid nitrogen. The crystals belonged to the primitive monoclinic space group P21 and diffracted to 1.64-Å resolution on beamline I02 at the Diamond Light Source. Diffraction data were processed using XDS (32), and the structure was solved by molecular replacement using the program PHASER (33), with the structure of DR1281 from D. radiodurans (20) (PDB accession number 1T70) as the search model. Manual alterations to the model were made in COOT (34) and interspersed with rounds of refinement in PHENIX.REFINE (35) to convergence, with a final Rfactor of 0.159 and a final Rfree of 0.185. The model was restrained to standard bond lengths and angles, with over 96% of residues occupying the most favored regions of the Ramachandran plot. The final YmdB structure superimposes on DR1281 (20) (PDB accession number 1T70), using 247 matched Cα atoms with a root mean square deviation (RMSD) of 1 Å. A summary of the data collection and refinement statistics can be found in Table 2.
TABLE 2.
Data collection and refinement statistics for YmdB

Rmerge = ΣhklΣi|Ii − Im|/ΣhklΣiIi, where Ii and Im are the observed intensity and mean intensity of related reflections, respectively.
Values in parentheses are for the highest-resolution shell, 1.73 to 1.64 Å.
Assays of complex colony formation.
For the analysis of colony architecture, B. subtilis strains were precultured in LB to an optical density at 600 nm (OD600) of 0.6 to 0.8. A total of 1.5 ml of the culture was pelleted and resuspended in 100 μl of sterile supernatant. Ten microliters of this cell suspension was then spotted onto minimal MSgg–1.5% agar plates (3) and incubated at 22°C for 3 to 5 days. The colonies were photographed by using an Olympus SZX12 stereomicroscope.
Analysis of flagellin protein expression.
To monitor the expression patterns of the flagellin protein Hag, the strains were grown in CSE minimal medium with glucose (CSE-Glu medium) and harvested in the logarithmic phase of growth. The cells were disrupted by sonication, and 15 μg crude extract of each culture was loaded onto a 12% sodium dodecyl sulfate-polyacrylamide gel. Following electrophoresis, the gel was stained with Coomassie brilliant blue G-250. The band corresponding to Hag was confirmed by mass spectrometry (19).
Northern blot analysis.
B. subtilis strains 168 (wild type) and GP583 (ymdB mutant) were grown in CSE minimal medium containing 0.5% glucose and were harvested when the culture reached stationary phase. The preparation of total RNA and Northern blot analysis were carried out as described previously (36). Digoxigenin (DIG) RNA probes were obtained by in vitro transcription with T7 RNA polymerase (Roche Diagnostics), using PCR-generated DNA fragments as templates. The primer pairs used to amplify DNA fragments specific for sspB and sigG are listed in Table S1 in the supplemental material. The reverse primer contained a T7 RNA polymerase recognition sequence. In vitro RNA labeling, hybridization, and signal detection were carried out according to the instructions of the manufacturer (DIG RNA labeling kit and detection chemicals; Roche Diagnostics). RNA stability was analyzed as described previously (15). Briefly, rifampin was added to stationary growing cultures (final concentration, 100 μg/ml), and samples were taken at the time points indicated. Quantification was performed by using Image J software v1.42 (37).
Real-time quantitative reverse transcription-PCR.
For real-time quantitative reverse transcription-PCR (qRT-PCR), RNA isolation proceeded as described above. cDNAs were synthesized by using the One-Step RT-PCR kit (Bio-Rad) as described previously (19). qRT-PCR was carried out on an iCycler instrument (Bio-Rad), according to the manufacturer's recommended protocol, by using the primers indicated in Table S1 in the supplemental material. Data analysis and the calculation of expression ratios as fold changes were performed as described previously (19). qRT-PCR experiments were performed in duplicate.
Transcriptome analysis.
Cells were grown in CSE-Glu medium. Samples of wild-type and ymdB mutant strains were harvested by centrifugation (10,397 × g for 1 min at 4°C) at mid-exponential phase and 2 h after the transition point. A total of four independent biological replicates were included. The pellets were frozen immediately in liquid nitrogen and stored at −80°C. RNA extraction was performed with the Macaloid/Roche protocol (38). RNA concentration and purity were assessed by using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). RNA samples were reverse transcribed into cDNA by using the Superscript III reverse transcriptase kit (Invitrogen, Carlsbad, CA, USA) and labeled with Cy3 or Cy5 monoreactive dye (GE Healthcare, Amersham, The Netherlands). Labeled and purified cDNA samples (Nucleospin Extract II; Biokè, Leiden, The Netherlands) were hybridized in Ambion Slidehyb 1 buffer (Ambion Europe Ltd.) at 48°C for 16 h. The arrays were constructed as described previously (39). Slide spotting, slide treatment after spotting, and slide quality control were done as described previously (40). After hybridization, slides were washed for 5 min in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) with 0.5% SDS, twice for 5 min in 1× SSC with 0.25% SDS, and 5 min in 1× SSC 0.1% SDS and then dried by centrifugation (2 min at 2,000 rpm) and scanned with a GenePix 4200AL scanner (Axon Instruments, CA, USA). Fluorescent signals were quantified by using ArrayPro 4.5 (Media Cybernetics Inc., Silver Spring, MD, USA) and further processed and normalized with MicroPrep (39). CyberT (41) was used to perform statistical analysis. Genes with a Bayes P value of 10−5 or lower were considered significantly affected.
Accession numbers.
Microarray data are MIAME compliant, and the raw data have been deposited in the MIAME-compliant Gene Expression Omnibus database (accession no. GSE39142) in accordance with MGED Society instructions (http://www.mged.org/Workgroups/MIAME/miame.html) The atomic coordinates and structure factor amplitudes have been deposited in the PDB under accession no. 4B2O.
RESULTS
YmdB is a phosphodiesterase.
YmdB is annotated in databases as a protein of unknown function or as a putative phosphatase/phosphodiesterase (42), activities that have already been described for DR1281, a 2′,3′-cyclic AMP phosphodiesterase from D. radiodurans with 44% sequence identity to YmdB (20). We therefore tested the potential phosphatase and phosphodiesterase activities of YmdB using purified, recombinant protein. YmdB had negligible enzymatic activity against pNPP, classically used as a synthetic phosphatase substrate, and no activity could be detected with phosphoenolpyruvate as the substrate (Table 3). In contrast, YmdB displayed measureable phosphodiesterase activity against bis-pNPP as the substrate, with a Km of 9.4 × 10−4 M and a Vmax of 105.21 μmol/min/mg (Table 3).
TABLE 3.
Kinetic parameters for enzymatic reactions catalyzed by YmdB variantsa
| Reaction and protein | Km (M) | Mean Vmax (μmol/min/mg) ± SD | Mean kcat (min−1) ± SD | kcat/Km (min−1 M−1) |
|---|---|---|---|---|
| Phosphatase reaction, substrate pNPP | ||||
| WT | ND | ≤0.05 | ||
| E39Q | ND | ≤0.05 | ||
| Phosphatase reaction, substrate PEP | ||||
| WT | ND | |||
| Phosphodiesterase reaction, substrate bis-pNPP | ||||
| WT | 9.40 (±1.8) × 10−4 | 105 ± 27.0 | 23.1 × 103 ± 12.7 × 103 | 2.46 × 107 |
| E39Q | ND | ≤0.05 |
WT, wild type; PEP, phosphoenolpyruvate; ND, not determined.
The phosphodiesterase activity of YmdB is necessary for biofilm formation.
We have shown previously that ymdB mutants do not form biofilms because the corresponding set of genes is not expressed and the flagellin-encoding hag gene is overexpressed (19). In order to assess the phosphodiesterase activity of YmdB in biofilm formation, we constructed two isogenic strains that expressed either wild-type YmdB (GP966) or the E39Q variant (GP969). E39 is predicted by sequence homology to DR1281 (and indeed confirmed by our structural analysis of YmdB [see below]) to coordinate divalent cations, essential for catalysis, in the enzyme's active site. To make sure that the mutation does not affect the stability of the protein variant, we compared the intracellular accumulations of the wild-type and the mutant YmdB proteins by Western blotting. The E39Q substitution did not result in any change of stability (data not shown). Moreover, a corresponding set of strains in the background of nondomesticated wild-type strain NCIB3610 was constructed. As shown in Fig. 1A, wild-type strain B. subtilis NCIB3610 formed rough colonies that are indicative of the expression of biofilm genes. As observed previously, ymdB mutant strain GP921 formed absolutely smooth colonies because of a lack of biofilm gene expression (19). As expected, strain GP1618 formed rough colonies morphologically identical to those of wild-type B. subtilis strain NCIB3610. Strain GP1619, expressing the ymdBE39Q allele, formed smooth colonies that were indistinguishable from those formed by the ymdB deletion mutant (Fig. 1A). As a marker for the expression of motility genes, we made use of the strong overexpression of hag that was detected previously in ymdB mutant strain GP583 and confirmed by mass spectrometry and the use of a hag mutant (19). Strain GP969, carrying the ymdBE39Q allele, resulted in strong overexpression of Hag to levels observed in the ymdB deletion mutant (Fig. 1B).
FIG 1.
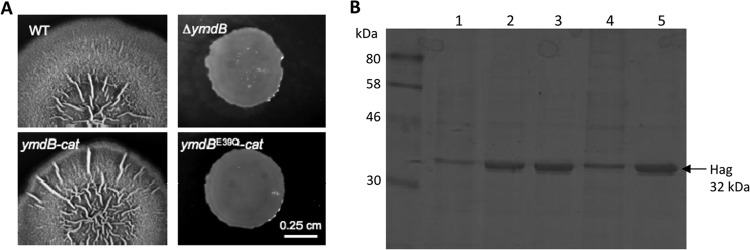
Biofilm formation and flagellin synthesis in ymdB variants. (A) Colony surface architectures of individual colonies grown on MSgg medium are shown. The strains used were as follows: the wild-type (WT) strains (NCIB3610) and the ymdB (GP921), ymdB-cat (GP1618), and ymdBE39Q-cat (GP1619) mutants. The scale bar is 0.25 cm. (B) The expression levels of flagellin (Hag) of different strains are compared. Crude extracts were isolated from B. subtilis strains. Lane 1, 168 (wild type); lane 2, GP583 (ΔymdB::spc); lane 3, GP922 (ΔymdB::cat); lane 4, GP966 (ymdB-cat); lane 5, GP969 (ymdBE39Q-cat). The flagellin protein Hag is indicated with an arrow.
Deletion of ymdB affects 800 mRNAs.
Inactivation of ymdB drastically affects the transcription of motility and biofilm genes (19). Moreover, the presence of the ymdB gene in a transcription unit with rny, encoding the major endoribonuclease RNase Y (19), might imply that YmdB exerts some control over global mRNA accumulation in B. subtilis. Consequently, microarray analyses were performed to compare the transcript profiles of wild-type strain 168 and isogenic ymdB mutant strain GP583 in the exponential and stationary growth phases. The microarray analyses revealed that the levels of over 800 mRNAs were changed by the deletion of ymdB. Microarray comparison of the ymdBE39Q strain (GP969) to its isogenic wild-type strain (GP966) showed that a near-identical set of genes was significantly up- or downregulated in both the exponential and the stationary phases. Among the genes with most strongly increased and decreased expression levels, i.e., >10-fold, were the σD-dependent motility regulon and the SinR-repressed biofilm genes, respectively, a finding that is in agreement with previous results (19) (see Tables S3 and S4 in the supplemental material). A complete list of the mRNAs affected by the loss of ymdB or the ymdBE39Q mutation is provided in the GEO database (accession no. GSE39142).
In addition to the σD regulon, we also observed increased expression levels of a group of operons involved in sugar utilization. Collectively, these operons are under the control of phosphotransferase system-controlled transcriptional activators (the lev, man, and mtl operons, required for fructose, mannose, and mannitol utilization, respectively [43]). The levels of the mRNAs corresponding to these operons were increased in the ymdB mutant in both the exponential and stationary phases. Moreover, a strong accumulation of the mRNA levels of several sporulation-specific genes was observed during the stationary phase in the ymdB mutant. Specifically, many of these genes encode the small acid-soluble proteins that protect the DNA of the spore (44). Most of the mRNAs accumulating during stationary phase depend on the sporulation-specific sigma factors σF and σG, which recognize quite similar promoters (45). σG-dependent gene transcription has been reported previously for a lon protease mutant under nutritional and genetic conditions in which sporulation is prevented (46).
To verify the results concerning the sporulation-specific mRNAs, we performed Northern blot analyses for sspB and the σG gene, sigG. SspB is a small acid-soluble protein with DNA-protective properties (44), and the sigma factor σG is required after engulfment of the prespore in the progression beyond morphological stage III (47). Under the growth conditions used, neither the sspB nor the sigG transcript accumulated to any great extent in the wild-type strain (Fig. 2A), whereas in the ymdB mutant, the levels of both transcripts were significantly enhanced (Fig. 2A), confirming the results of the transcriptome analysis. By Northern blotting, the half-life of the sspB transcript in wild-type B. subtilis is 13 min, whereas in the ymdB null mutant, the sspB transcript half-life is a little longer, at 17 min (Fig. 2B). By qRT-PCR, very comparable half-lives of 12.1 and 16.6 min in the presence and absence of YmdB, respectively, were obtained. When sigG transcript stability was measured by Northern blotting, the half-life was 2.6 min in the presence and 2.8 min in the absence of YmdB (Fig. 2B); by qRT-PCR, the half-lives were 4.1 and 5.5 min, respectively. Therefore, YmdB affects the transcript accumulation, but not the half-life, of sporulation-specific genes in its role as a late adaptive response regulator.
FIG 2.
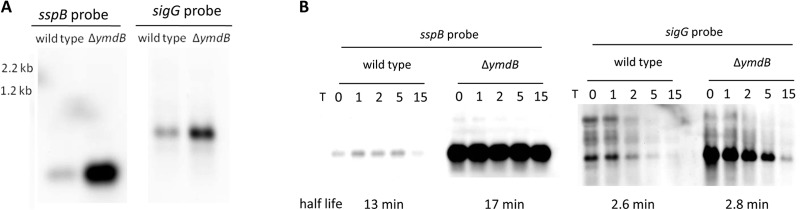
Effect of YmdB on the sporulation genes sspB and sigG. (A) Northern blot analyses to test the effect of YmdB on the expression of sspB and sigG. The transcripts were detected with riboprobes specific for sspB and sigG. (B) The transcript stability of sspB and sigG mRNAs was determined for the wild type (168) and the ymdB mutant (GP583) by Northern blotting. Above each lane is the time (in minutes) after rifampin addition. The half-life of each mRNA transcript is also indicated in minutes. In the lane with the RNA from the ymdB strain, larger amounts of sspB and sigG mRNAs are detectable, but these mRNAs are not stabilized in comparison to the wild type.
Crystal structure of YmdB.
To gain molecular insight into the phosphodiesterase activity of YmdB, its crystal structure was determined and refined to a resolution of 1.64 Å (Fig. 3A). Relevant crystallographic statistics are provided in Table 2. Overall, the electron density map is of excellent quality, such that almost the entire length of all 4 chains in the asymmetric unit could be traced. As expected, YmdB adopts the calcineurin-like fold, a large and functionally diverse family that includes phosphodiesterases, protein (Ser/Thr) phosphatases, and 5′-nucleotidases (48). A VAST search of the Protein Data Bank (PDB) identifies 22 homologous structures above a P value threshold of 10−7, three-quarters of which are unpublished outputs from structural genomics consortia. Common to all members of this family is a conserved αββα architecture (Fig. 3A) that serves as a scaffold for the coordination of a pair of divalent metal ions in the enzyme active site (Fig. 3B and C). In the case of YmdB, both β-sheets are six stranded and are of a mixed parallel/antiparallel nature; the β-sheets are flanked by α-helices on both sides. An extended loop between β11 and β12 forms a flap that protrudes away from the rest of the molecule (Fig. 3A).
FIG 3.

Crystal structure of YmdB. (A) Stereographic ribbon representation of the YmdB monomer, with α-helices in blue and β-sheets in green. Secondary structural elements are labeled according to topology. The active-site environment is also depicted, in stick mode colored by atom type, with bound ions and the proposed catalytic water as spheres. (B) The active site of YmdB, with key amino acids highlighted as sticks and colored by atom type. The final 2mFo-DFc electron density map is contoured at a level of 1.9 σ and is shown as a gray mesh. An anomalous-difference Fourier series, using phases calculated from the refined model, is contoured at a level of 5 σ (pink). The only peaks of this height in the Fourier series correspond to the two Fe ions at the active site. (C) The active site of YmdB in the same orientation as that in panel B, with key amino acids highlighted as sticks and colored by atom type. Hydrogen bonds are shown as dashed black lines. (D) The YmdB dimer, with each chain colored independently. Helix α6 from chain A (blue) packs above the region surrounding strands β9 and β11 in chain B (green).
YmdB quaternary structure.
The four chains in the asymmetric unit of YmdB crystals are virtually identical (∼0.3-Å RMSD on Cα atoms) and associate with each other to form pairs of dimers in a tetrameric D2 assembly with dimensions of 45 Å by 65 Å by 75 Å. The most significant interface links chains A and B (and is duplicated in chains C and D) and is dominated by contacts from β9 to the extended loop linking β11 to β12, which also contains α6. The interface accounts for 1,630 Å2 of interface area (∼14% of the total monomer-accessible surface area) and is a mixture of both polar (10 hydrogen bonds and 2 salt bridges) and nonpolar interactions. The secondary interface that links chains A to C (and chains B to D) is much less extensive, burying only 500 Å2 of accessible surface area, and consists solely of nonpolar interactions. By size exclusion chromatography, YmdB elutes in a major fraction at an expected molecular mass of ∼55 kDa (data not shown), consistent with a dimer of YmdB assembled presumably by the interaction between chains A and B (Fig. 3D).
The catalytic machinery of YmdB.
A pair of divalent metal ions, each coordinated in an octahedral fashion, is located in a shallow cleft between the two β-sheets (Fig. 3B and C). Since there were no metal ions or chelating agents added during purification, the two metal ions must have been retained by YmdB during purification. They have been modeled as iron ions following analysis of purified protein samples by inductively coupled plasma mass spectrometry, which revealed 2 molar equivalents of iron, and the presence of iron in YmdB crystals was confirmed by fluorescence scans prior to data collection (see Fig. S1 in the supplemental material). We suggest that the site contains Fe2+ and Fe3+ ions, in an arrangement described previously for purple acid phosphatases (49), members of the same enzymatic superfamily as YmdB. The Fe2+ ion is coordinated predominantly by soft ligands: the Nδ1 and Nε2 imidazole nitrogens of His175 and His150, respectively; the carboxamide oxygen of Asn67; and a carboxylic oxygen of Glu39. The octahedral coordination is completed by an oxygen from a bound phosphate ion and a water, both of which also provide a ligand to the Fe3+ ion (Fig. 3B and C). His175 is the only outlier in the Ramachandran plot but is very well defined in the electron density and makes several ionic interactions with surrounding residues as well as its Fe2+ ion interaction. The Fe3+ ion is coordinated by a higher proportion of harder-charged oxygen atoms: a carboxylic oxygen of Asp8, the same carboxylate oxygen of Glu39 as that used in the Fe2+ site, the carboxamide oxygen of Asn40, and the Nε2 imidazole nitrogen of His177. The shared water and another oxygen from the same phosphate ion complete the coordination (Fig. 3B and C). All the ligands are between 2.05 Å and 2.21 Å away from the metal ions. Consistent with the Fe ionization states ascribed, the 2mFo-DFc electron density map peak heights in all four copies in the asymmetric unit are significantly lower for the Fe3+ site, between 14.8 and 16.5 σ, than for the Fe2+ ion, between 17.8 and 19.2 σ, as would be expected of an ion with one less electron.
The metal ions and their coordinating residues superimpose closely on other structures from the calcineurin-like metallophosphodiesterase family. The coordinating residues are mostly very well conserved in both chemical identity and position in the protein sequence (Fig. 4A). However, the presence of a water molecule bound to both metal ions is almost universal, reinforcing the idea that once activated to form a hydroxide, this solvent molecule acts as the nucleophile in catalysis. The phosphate is situated directly above the metal ions, in a position that presumably mimics that of the substrate. The geometry of the two Fe ions in relation to the phosphate is highly indicative of an associative in-line mechanism (Fig. 3C). Numerous hydrogen bonds are made by the phosphate to nearby side chains and water molecules, including the invariant His68, a key determinant of 2′,3′-cyclic nucleotide phosphodiesterase activity (50). His68 is part of a conserved His-Asp dyad, which presumably orients the histidine with respect to the leaving group for the donation of a proton from the former to the latter (Fig. 3C).
FIG 4.

The YmdB active site. (A) Stereo view of a comparison of the active site of YmdB (green) to 3 other representative calcineurin-like metallophosphoesterases that contain a dimetal cluster: the putative 5′-nucleotidase from E. coli (PDB accession no. 3IVD) (in pink), the Rv0805 nucleotide phosphodiesterase from Mycobacterium tuberculosis (PDB accession no. 2HY1) (51) (in blue), and the secreted phosphoesterase YfkN from B. subtilis (PDB accession no. 3GVE) (64) (in gray). DR1281 is not used for this comparison because the deposited structure does not contain metal ions. (B) Binding of 2′,3′-cAMP by YmdB. The phosphate anions of YmdB and the model coordinates of 2′,3′-cAMP were superimposed by least-squares minimization. The protein surface is drawn as a semitransparent surface, with the protein backbone drawn as a Cα trace, and the side chains of several likely substrate specificity determinants are drawn. (C) Proposed reaction scheme for YmdB with 2′,3′-cAMP as a representative of the phosphodiesterase activity. A pair of metal ions in the YmdB active site coordinate both the phosphate moiety of the substrate and an active-site water molecule, conserved in other structures of the calcineurin-like phosphodiesterase superfamily, which is perfectly positioned to perform nucleophilic attack on the substrate once activated to form a hydroxyl ion. A key role for Glu39 is confirmed experimentally here; the spatial arrangement of His68 and Asp71 in relation to each other and the bound phosphate is consistent with His68 and Asp71 acting as a classic catalytic dyad, with His68 serving as an acid and protonating the O2′-leaving group to facilitate its displacement.
YmdB and 2′,3′- and 3′,5′-cyclic nucleotides.
Given the homology of YmdB to D. radiodurans DR1281 (20), we determined the kinetic parameters of YmdB against several cyclic mononucleotides. To avoid any redox complications, and because manganese also supports the phosphodiesterase activity of YmdB (see Fig. S2 in the supplemental material), we conducted all biochemical experiments in the presence of manganese. YmdB is active against 2′,3′- and 3′,5′-cyclic mononucleotides, with kinetic parameters described in Table 4. In general terms, the Km values for the 2′,3′-cyclic mononucleotides tested were 4 to 5 times lower than those for their 3′,5′ mononucleotide counterparts; the Vmax values showed the opposite correlation, being 2 to 3 times higher for the 2′,3′-cyclic mononucleotides than for the 3′,5′ variants. The consequent ∼10-fold differences in kcat/Km values indicate that YmdB has a preference for 2′,3′-cyclic mononucleotides, which is perhaps not surprising, as B. subtilis is unable to form either of the signaling nucleotides 3′,5′-cAMP and 3′,5′-cGMP. In all cases, mutation of Glu39 to the isosteric glutamine abolished the phosphodiesterase activity almost completely (Table 4), confirming that Glu39 plays a key role in catalysis because of its role in coordinating correctly the dinuclear metal center (Fig. 3B and C and 4A and B), which will be lost in the glutamine mutant. Using the phosphate ion bound to YmdB as a guide, 2′,3′-cAMP can be positioned into the active site without significant steric clashes to the protein, with the adenine in the energetically favored anti conformation and with the O2′-leaving group in line with the incoming hydroxyl (Fig. 4B and C). Many structures that are homologous to YmdB also contain a phosphate, or a phosphate-containing compound, in a position similar to that observed in YmdB (51–55). With the exception of direct hydrogen bonding to the phosphate oxygens, there are, in general, very few direct contacts between enzyme and substrate (51), and thus, the means by which substrate specificity occurs in the superfamily is obscure.
TABLE 4.
Kinetic parameters for phosphodiesterase activity catalyzed by YmdB
| Protein and substrate | Mean Km (M) ± SD | Mean Vmax (μmol/min/mg) ± SD | Mean kcat (min−1) ± SD | kcat/Km (min−1 M−1) |
|---|---|---|---|---|
| WT YmdB | ||||
| 2′,3′-cAMP | (2.0 ±0.2) × 10−4 | 0.83 ± 0.23 | 15.0 ± 11.0 | 7.5 × 104 |
| 2′,3′-cGMP | (2.9 ±0.6) × 10−4 | 1.02 ± 0.17 | 22.1 ± 11.4 | 7.6 × 104 |
| 3′,5′-cAMP | (1.3 ± 0.3) × 10−3 | 0.45 ± 0.04 | 9.2 ± 2.4 | 7.0 × 103 |
| 3′,5′-cGMP | (8.5 ± 2.1) × 10−4 | 0.37 ± 0.04 | 7.7 ± 2.9 | 9.0 × 103 |
| YmdBE39Q | ||||
| 2′,3′-cAMP | ≤0.05 | |||
| 2′,3′-cGMP | ≤0.05 | |||
| 3′,5′-cAMP | ≤0.05 | |||
| 3′,5′-cGMP | ≤0.05 |
The catalytic properties of several metal-dependent phosphodiesterases against 2′,3′-cAMP have been measured (20, 50, 56). Among the enzymes tested are representatives of the HD phosphohydrolases, the EAL diguanylate phosphodiesterases, the metallo-β-lactamases, and the DHH phosphatases. The Km observed for YmdB against 2′,3′-cAMP (0.2 mM) is toward the lower end of the range of the enzymes tested (Km values of between 0.05 mM and 35 mM), although the YmdB kcat (15 min−1) indicates that it is one of the slowest enzymes tested (kcat values of between 0.13 min−1 and 15,000 min−1). An unusual facet of YmdB is its ability to hydrolyze both 2′,3′- and 3′,5′-cyclic nucleotides; the latter activity is generally not found in phosphodiesterases, including D. radiodurans DR1281 (20). Nonetheless, the finding that a number of characterized enzymes have catalytic properties similar to or better than those of YmdB when tested against physiological substrates other than 2′,3′-cyclic nucleotides suggests that 2′,3′-cAMP is not the primary target of YmdB.
Effect of YmdB on cyclic dinucleotide pools.
If cAMP were not the physiological target of YmdB, cyclic dinucleotides would be an attractive alternative. We considered the secondary messenger cyclic di-GMP, which has been shown to influence biofilm formation in various other bacteria (57). Recently, both c-di-GMP and c-di-AMP have been shown to be present in B. subtilis (31, 58–62), and there are several c-di-GMP and c-di-AMP synthases and hydrolases in the B. subtilis genome, strongly suggesting an important physiological role for these molecules (42). To test the involvement of YmdB in the homeostasis of these molecules, we performed two sets of experiments. First, we determined the intracellular pools of c-di-AMP and c-di-GMP. While the concentration of c-di-AMP (about 15.3 ± 0.21 and 13.4 ± 1.71 ng of c-di-AMP per mg of protein for the wild-type strain and isogenic ymdB mutant strain GP583, respectively) was not affected by the ymdB mutation, a strong effect was observed for c-di-GMP. In the wild-type strain, a concentration of 34.81 (±2.30) ng of c-di-GMP per mg of protein was detected. In contrast, a concentration of only 1.24 (±0.01) ng of c-di-GMP per mg of protein was determined for isogenic ymdB mutant strain GP583.
The reduced concentration of c-di-GMP reported above seems to be counterintuitive for a phosphodiesterase mutant. Therefore, we also tested the cyclic dinucleotide hydrolase activity of YmdB in vitro. To test whether YmdB degrades cyclic dinucleotides, we incubated recombinant YmdB with both cyclic di-AMP and cyclic di-GMP in vitro, but we were unable to find any traces of product formation by thin-layer chromatography (TLC), reverse-phase HPLC, or anion-exchange chromatography (data not shown). This obvious lack of in vitro c-di-GMP phosphodiesterase activity is in good agreement with the in vivo observations. To test whether YmdB might have an unexpected diguanylate cyclase activity, we incubated YmdB with GTP and assayed for c-di-GMP production. In parallel, the experiment was performed with the E. coli diguanylate cyclase YdeH (29). While YdeH exhibited the expected diguanylate cyclase activity, no activity was detectable for YmdB. Thus, the involvement of YmdB in the control of the intracellular c-di-GMP pool seems to be indirect (see Discussion).
DISCUSSION
YmdB belongs to the large (∼15,000 members) calcineurin-like phosphatase/phosphodiesterase family present in all domains of life, representatives of which participate in diverse functions, including phosphodiesterase, phosphomonoesterase, nucleotidase, or nuclease activities. We show here by transcriptome analyses that YmdB is also a key regulator in the decision-making processes of biofilm formation, motility, and sporulation, mutually exclusive late adaptive responses of Bacillus subtilis. YmdB does not participate directly in the network of protein-protein interactions that constitute the Sin/Slr epigenetic switch (19), so the question arises, how does YmdB, a phosphodiesterase, exert global control in this bacterium?
Two possibilities could be taken into consideration. On the one hand, YmdB might be directly implicated in RNA synthesis and/or degradation of the target genes. On the other hand, YmdB might perform an enzymatic reaction that gives rise to the observed transcriptome and phenotypic changes in a rather indirect way.
If YmdB were directly involved in controlling mRNA accumulation, it would most likely act at the level of mRNA stability. The fact that YmdB is encoded in an operon with the essential endonuclease RNase Y lends support for this hypothesis (19). However, the structure of YmdB does not support a role in direct RNA binding. Although other members of the calcineurin-like phosphodiesterases, such as the Mre11 DNA endonuclease, are able to hydrolyze nucleic acids (62), the binding and recognition of nucleic acids are performed primarily by other domains that are not present in YmdB. The recognition and degradation of the transcripts of key regulators could be achieved by the interaction of YmdB with other RNA-responsive macromolecules, and through these interactions, specific substrates are presented to the YmdB active site. However, there is little overlap between the transcripts affected by rny and ymdB disruption. Moreover, there was no difference between wild-type and ymdB mutant strains in the RNase Y-dependent processing of the gapA operon mRNA (data not shown). The transcript stabilities of the sporulation genes sspB and sigG were found to have a very slight (∼1.3-fold) increase in the mRNA half-life in the absence of YmdB, consistent with the modest increase in the half-life (∼1.7-fold) found previously for the monocistronic hag mRNA (19). The transcriptome data suggest that ymdB inactivation results in the coordinated deregulation of complete regulons, indicating that YmdB affects specifically the expression and/or activity of the cognate regulators rather than all of the individual genes. In fact, this has already been demonstrated for the σD- and SinR-controlled motility and biofilm regulons, respectively; reduced expression of the regulator SlrR is responsible for the effects of the ymdB deletion (19).
In conclusion, the effect of the ymdB deletion on mRNA accumulation and, thus, on biofilm formation is likely to be an indirect result of the phosphodiesterase activity of YmdB. While we have determined phosphodiesterase activity for YmdB against both 2′,3′- and 3′,5′-cyclic nucleotides, it is not clear at present if either of these substrates exists to any significant extent in B. subtilis. Neither free 3′,5′-cAMP (63) nor 3′,5′-cAMP-responsive catabolite activator protein (CAP)-like proteins have been found in B. subtilis. Moreover, although some trans-phosphorylation RNA degradation reactions, such as those catalyzed by RNase A and RNase I, produce 2′,3′-cyclic nucleotides (64), there are very few reports of 2′,3′-cyclic nucleotides being isolated from biological systems, perhaps because of the relative susceptibility of these nucleotides to hydrolysis by nonspecific phosphodiesterases (56). The exclusion of cyclic mononucleotides makes cyclic dinucleotides the next attractive potential direct YmdB target. Indeed, c-di-GMP is implicated in the control of biofilm formation in many species (57). Our determination of the pools of cyclic dinucleotides revealed a strong reduction of the c-di-GMP levels in the ymdB mutant. However, this effect in vivo is not supported by the biochemical activity of YmdB in vitro: the protein neither degrades c-di-GMP (as one might assume for a phosphodiesterase) nor produces the cyclic dinucleotide (as would be in perfect agreement with the results of the nucleotide measurements). If not by its direct enzymatic activity, how could YmdB otherwise affect c-di-GMP levels? The transcriptome analysis suggests an answer to this question. In the ymdB mutant, expression levels of the c-di-GMP-specific phosphodiesterases YkoW and YuxH are increased 4.6- and 2.4-fold, respectively. This may result in the decreased intracellular c-di-GMP concentration.
Two recent studies have addressed the role of c-di-GMP in biofilm formation in B. subtilis (58, 59). However, whereas one report suggested an implication of c-di-GMP in biofilm formation (58), the other excluded this possibility. Therefore, the question of whether the alteration of the c-di-GMP pools in the ymdB mutant only parallels the effects on biofilm formation and motility regulon expression or whether there is a causal relation remains open to further investigation.
The observation of increased phosphodiesterase expression with its consequences for the c-di-GMP pool and biofilm formation brings us back to the question of the initial and primary activity of YmdB. The identification of the primary target that is subject to YmdB-mediated hydrolysis will be the major focus of future work.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Christina Herzberg and Annette Garbe for excellent technical assistance and to Julia Busse, Jan Gerwig, and Cedric Blötz for the help with some strain constructions. We thank the Diamond Light Source for access to its synchrotron beamlines and its beamline staff for support during data collection. We are grateful to Kevin Waldron for help with inductively coupled plasma mass spectrometry.
This work was supported by grants from the BBSRC (United Kingdom) to R.J.L., from the DFG through SFB860 (Germany) to J.S., and from the ALW-NWO (The Netherlands) to O.P.K., under the ERANET SysMO1 and SysMO2 initiative.
Footnotes
Published ahead of print 25 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00826-13.
REFERENCES
- 1.Sutherland IW. 2001. The biofilm matrix—an immobilized but dynamic microbial environment. Trends Microbiol. 9:222–227. 10.1016/S0966-842X(01)02012-1 [DOI] [PubMed] [Google Scholar]
- 2.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. 10.1126/science.295.5559.1487 [DOI] [PubMed] [Google Scholar]
- 3.Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. 2001. Fruiting body formation by Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 98:11621–11626. 10.1073/pnas.191384198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Cao S, Chai Y, Clardy J, Kolter R, Guo JH, Losick R. 2012. A Bacillus subtilis sensor kinase involved in triggering biofilm formation on the roots of tomato plants. Mol. Microbiol. 85:418–430. 10.1111/j.1365-2958.2012.08109.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. 2011. Tracing the domestication of a biofilm-forming bacterium. J. Bacteriol. 193:2027–2034. 10.1128/JB.01542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veening JW, Kuipers OP, Brul S, Hellingwerf KJ, Kort R. 2006. Effects of phosphorelay perturbations on architecture, sporulation, and spore resistance in biofilms of Bacillus subtilis. J. Bacteriol. 188:3099–3109. 10.1128/JB.188.8.3099-3109.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovacs AT, Kuipers OP. 2011. Rok regulates yuaB expression during architecturally complex colony development of Bacillus subtilis 168. J. Bacteriol. 193:998–1002. 10.1128/JB.01170-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kearns DB, Chu F, Branda SS, Kolter R, Losick R. 2005. A master regulator for biofilm formation by Bacillus subtilis. Mol. Microbiol. 55:739–749. 10.1111/j.1365-2958.2004.04440.x [DOI] [PubMed] [Google Scholar]
- 9.Chu F, Kearns DB, Branda SS, Kolter R, Losick R. 2006. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol. Microbiol. 59:1216–1228. 10.1111/j.1365-2958.2005.05019.x [DOI] [PubMed] [Google Scholar]
- 10.Newman JA, Rodrigues C, Lewis RJ. 2013. Molecular basis of the activity of SinR protein, the master regulator of biofilm formation in Bacillus subtilis. J. Biol. Chem. 288:10766–10778. 10.1074/jbc.M113.455592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branda SS, Chu F, Kearns DB, Losick R, Kolter R. 2006. A major protein component of the Bacillus subtilis biofilm matrix. Mol. Microbiol. 59:1229–1238. 10.1111/j.1365-2958.2005.05020.x [DOI] [PubMed] [Google Scholar]
- 12.Lewis RJ, Brannigan JA, Smith I, Wilkinson AJ. 1996. Crystallisation of the Bacillus subtilis sporulation inhibitor SinR, complexed with its antagonist, SinI. FEBS Lett. 378:98–100. 10.1016/0014-5793(95)01432-2 [DOI] [PubMed] [Google Scholar]
- 13.Lewis RJ, Brannigan JA, Offen WA, Smith I, Wilkinson AJ. 1998. An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J. Mol. Biol. 283:907–912. 10.1006/jmbi.1998.2163 [DOI] [PubMed] [Google Scholar]
- 14.Gaur NK, Cabane K, Smith I. 1988. Structure and expression of the Bacillus subtilis sin operon. J. Bacteriol. 170:1046–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehnik-Habrink M, Schaffer M, Mäder U, Diethmaier C, Herzberg C, Stülke J. 2011. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol. 81:1459–1473. 10.1111/j.1365-2958.2011.07777.x [DOI] [PubMed] [Google Scholar]
- 16.Chai Y, Norman T, Kolter R, Losick R. 2010. An epigenetic switch governing daughter cell separation in Bacillus subtilis. Genes Dev. 24:754–765. 10.1101/gad.1915010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi K. 2008. SlrR/SlrA controls the initiation of biofilm formation in Bacillus subtilis. Mol. Microbiol. 69:1399–1410. 10.1111/j.1365-2958.2008.06369.x [DOI] [PubMed] [Google Scholar]
- 18.Chai Y, Kolter R, Losick R. 2009. Paralogous antirepressors acting on the master regulator for biofilm formation in Bacillus subtilis. Mol. Microbiol. 74:876–887. 10.1111/j.1365-2958.2009.06900.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diethmaier C, Pietack N, Gunka K, Wrede C, Lehnik-Habrink M, Herzberg C, Hübner S, Stülke J. 2011. A novel factor controlling bistability in Bacillus subtilis: the YmdB protein affects flagellin expression and biofilm formation. J. Bacteriol. 193:5997–6007. 10.1128/JB.05360-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin DH, Proudfoot M, Lim HJ, Choi IK, Yokota H, Yakunin AF, Kim R, Kim SH. 2008. Structural and enzymatic characterization of DR1281: a calcineurin-like phosphoesterase from Deinococcus radiodurans. Proteins 70:1000–1009. 10.1002/prot.21584 [DOI] [PubMed] [Google Scholar]
- 21.Commichau FM, Herzberg C, Tripal P, Valerius O, Stülke J. 2007. A regulatory protein-protein interaction governs glutamate biosynthesis in Bacillus subtilis: the glutamate dehydrogenase RocG moonlights in controlling the transcription factor GltC. Mol. Microbiol. 65:642–654. 10.1111/j.1365-2958.2007.05816.x [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 23.Bi W, Stambrook PJ. 1998. Site-directed mutagenesis by combined chain reaction. Anal. Biochem. 256:137–140. 10.1006/abio.1997.2516 [DOI] [PubMed] [Google Scholar]
- 24.Kunst F, Rapoport G. 1995. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J. Bacteriol. 177:2403–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guérout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336. 10.1016/0378-1119(95)00652-4 [DOI] [PubMed] [Google Scholar]
- 26.Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265. [DOI] [PubMed] [Google Scholar]
- 27.Merzbacher M, Detsch C, Hillen W, Stülke J. 2004. Mycoplasma pneumoniae HPr kinase/phosphorylase. Eur. J. Biochem. 271:367–374. 10.1046/j.1432-1033.2003.03935.x [DOI] [PubMed] [Google Scholar]
- 28.Yakunin AF, Proudfoot M, Kuznetsova E, Savchenko A, Brown G, Arrowsmith CH, Edwards AM. 2004. The HD domain of the Escherichia coli tRNA nucleotidyltransferase has 2′,3′-cyclic phosphodiesterase, 2′-nucleotidase, and phosphatase activities. J. Biol. Chem. 279:36819–36827. 10.1074/jbc.M405120200 [DOI] [PubMed] [Google Scholar]
- 29.Spangler C, Kaever V, Seifert R. 2011. Interaction of the diguanylate cyclase YdeH of Escherichia coli with 2′,(3′)-substituted purine and pyrimidine nucleotides. J. Pharmacol. Exp. Ther. 336:234–241. 10.1124/jpet.110.170993 [DOI] [PubMed] [Google Scholar]
- 30.Spangler C, Böhm A, Jenal U, Seifert R, Kaever V. 2010. A liquid chromatography-coupled tandem mass spectrometry method for quantitation of cyclic di-guanosine monophosphate. J. Microbiol. Methods 81:226–231. 10.1016/j.mimet.2010.03.020 [DOI] [PubMed] [Google Scholar]
- 31.Mehne FMP, Gunka K, Eilers H, Herzberg C, Kaever V, Stülke J. 2013. Cyclic di-AMP homeostasis in Bacillus subtilis: both lack and high level accumulation of the nucleotide are detrimental for cell growth. J. Biol. Chem. 288:2004–2017. 10.1074/jbc.M112.395491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ludwig H, Meinken C, Matin A, Stülke J. 2002. Insufficient expression of the ilv-leu operon encoding enzymes of branched-chain amino acid biosynthesis limits growth of a Bacillus subtilis ccpA mutant. J. Bacteriol. 184:5174–5178. 10.1128/JB.184.18.5174-5178.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abramoff MD, Magalhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int. 11:36–42 http://www.imagescience.org/meijering/publications/download/bio2004.pdf [Google Scholar]
- 38.van Hijum SA, de Jong A, Baerends RJ, Karsens HA, Kramer NE, Larsen R, den Hengst CD, Albers CJ, Kok J, Kuipers OP. 2005. A generally applicable validation scheme for the assessment of factors involved in reproducibility and quality of DNA-microarray data. BMC Genomics 6:77. 10.1186/1471-2164-6-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Hijum SA, de Jong A, Buist G, Kok J, Kuipers OP. 2003. UniFrag and GenomePrimer: selection of primers for genome-wide production of unique amplicons. Bioinformatics 19:1580–1582. 10.1093/bioinformatics/btg203 [DOI] [PubMed] [Google Scholar]
- 40.Kuipers OP, de Jong A, Baerends RJ, van Hijum SA, Zomer AL, Karsens HA, den Hengst CD, Kramer NE, Buist G, Kok J. 2002. Transcriptome analysis and related databases of Lactococcus lactis. Antonie Van Leeuwenhoek 82:113–122. 10.1023/A:1020691801251 [DOI] [PubMed] [Google Scholar]
- 41.Baldi P, Long AD. 2001. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17:509–519. 10.1093/bioinformatics/17.6.509 [DOI] [PubMed] [Google Scholar]
- 42.Mäder U, Schmeisky AG, Flórez LA, Stülke J. 2012. SubtiWiki—a comprehensive community resource for the model organism Bacillus subtilis. Nucleic Acids Res. 40:D1278–D1287. 10.1093/nar/gkr923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stülke J, Hillen W. 2000. Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 54:849–880. 10.1146/annurev.micro.54.1.849 [DOI] [PubMed] [Google Scholar]
- 44.Setlow P. 2007. I will survive: DNA protection in bacterial spores. Trends Microbiol. 15:172–180. 10.1016/j.tim.2007.02.004 [DOI] [PubMed] [Google Scholar]
- 45.Hilbert DW, Piggot PJ. 2004. Compartmentalization of gene expression during Bacillus subtilis spore formation. Microbiol. Mol. Biol. Rev. 68:234–262. 10.1128/MMBR.68.2.234-262.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt R, Decatur AL, Rather PN, Moran CP, Losick R. 1994. Bacillus subtilis Lon protease prevents inappropriate transcription of genes under the control of sporulation transcription factor sigma G. J. Bacteriol. 176:6528–6537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Partridge SR, Errington J. 1993. The importance of morphological events and intercellular interactions in the regulation of prespore-specific gene expression during sporulation in Bacillus subtilis. Mol. Microbiol. 8:945–955. 10.1111/j.1365-2958.1993.tb01639.x [DOI] [PubMed] [Google Scholar]
- 48.Aravind L, Koonin EV. 1998. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 26:3746–3752. 10.1093/nar/26.16.3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richter W. 2002. 3′,5′ cyclic nucleotide phosphodiesterases class III: members, structure, and catalytic mechanism. Proteins 46:278–286. 10.1002/prot.10049 [DOI] [PubMed] [Google Scholar]
- 50.Keppetipola N, Shuman S. 2008. A phosphate-binding histidine of binuclear metallophosphodiesterase enzymes is a determinant of 2′,3′-cyclic nucleotide phosphodiesterase activity. J. Biol. Chem. 283:30942–30949. 10.1074/jbc.M805064200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shenoy AR, Capuder M, Draskovic P, Lamba D, Visweswariah SS, Podobnik M. 2007. Structural and biochemical analysis of the Rv0805 cyclic nucleotide phosphodiesterase from Mycobacterium tuberculosis. J. Mol. Biol. 365:211–225. 10.1016/j.jmb.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 52.Knöfel T, Strater N. 1999. X-ray structure of the Escherichia coli periplasmic 5′-nucleotidase containing a dimetal catalytic site. Nat. Struct. Biol. 6:448–453. 10.1038/8253 [DOI] [PubMed] [Google Scholar]
- 53.Knöfel T, Strater N. 2001. Mechanism of hydrolysis of phosphate esters by the dimetal center of 5′-nucleotidase based on crystal structures. J. Mol. Biol. 309:239–254. 10.1006/jmbi.2001.4656 [DOI] [PubMed] [Google Scholar]
- 54.Chen S, Yakunin AF, Kuznetsova E, Busso D, Pufan R, Proudfoot M, Kim R, Kim SH. 2004. Structural and functional characterization of a novel phosphodiesterase from Methanococcus jannaschii. J. Biol. Chem. 279:31854–31862. 10.1074/jbc.M401059200 [DOI] [PubMed] [Google Scholar]
- 55.Podobnik M, Tyagi R, Matange N, Dermol U, Gupta AK, Mattoo R, Seshadri K, Visweswariah SS. 2009. A mycobacterial cyclic AMP phosphodiesterase that moonlights as a modifier of cell wall permeability. J. Biol. Chem. 284:32846–32857. 10.1074/jbc.M109.049635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rao F, Qi Y, Murugan E, Pasunooti S, Ji Q. 2010. 2′,3′-cAMP hydrolysis by metal-dependent phosphodiesterases containing DHH, EAL, and HD domains is non-specific: implications for PDE screening. Biochem. Biophys. Res. Commun. 398:500–505. 10.1016/j.bbrc.2010.06.107 [DOI] [PubMed] [Google Scholar]
- 57.Jonas K, Melefor O, Römling U. 2009. Regulation of c-di-GMP metabolism in biofilms. Future Microbiol. 4:341–358. 10.2217/fmb.09.7 [DOI] [PubMed] [Google Scholar]
- 58.Chen Y, Chai Y, Guo JH, Losick R. 2012. Evidence for cyclic di-GMP-mediated signaling in Bacillus subtilis. J. Bacteriol. 194:5080–5090. 10.1128/JB.01092-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao X, Mukherjee S, Matthews PM, Hammad LA, Kearns DB, Dann CE. 2013. Functional characterization of core components of the Bacillus subtilis cyclic-di-GMP signaling pathway. J. Bacteriol. 195:4782–4792. 10.1128/JB.00373-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Witte G, Hartung S, Büttner K, Hopfner KP. 2008. Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30:167–178. 10.1016/j.molcel.2008.02.020 [DOI] [PubMed] [Google Scholar]
- 61.Luo Y, Helmann JD. 2012. Analysis of the role of Bacillus subtilis σM in β-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83:623–639. 10.1111/j.1365-2958.2011.07953.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. 2008. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135:97–109. 10.1016/j.cell.2008.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mach H, Hecker M, Mach F. 1984. Evidence for the presence of cyclic adenosine monophosphate in Bacillus subtilis. FEMS Microbiol. Lett. 22:27–30. 10.1111/j.1574-6968.1984.tb00348.x [DOI] [Google Scholar]
- 64.Thompson JE, Venegas FD, Raines RT. 1994. Energetics of catalysis by ribonucleases: fate of the 2′,3′-cyclic phosphodiester intermediate. Biochemistry 33:7408–7414. 10.1021/bi00189a047 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.