

Abstract
AMP-activated protein kinase (AMPK) has been shown to activate p53 in response to metabolic stress. However, the underlying mechanisms remain unclear. Here we show that metabolic stresses induce AMPK-mediated phosphorylation of human MDMX on Ser342 in vitro and in cells, leading to enhanced association between MDMX and 14-3-3. This markedly inhibits p53 ubiquitylation and significantly stabilizes and activates p53. By striking contrast, no phosphorylation of MDM2 by AMPK was noted. AMPK-mediated MDMX phosphorylation, MDMX–14-3-3 binding, and p53 activation were drastically reduced in mouse embryo fibroblasts harboring endogenous MDMX with S341A (mouse homologue of human serine 342), S367A, and S402A (mouse homologue of human serine 403) mutations. Moreover, deficiency of AMPK prevented MDMX–14-3-3 interaction and p53 activation. The activation of p53 through AMPK-mediated MDMX phosphorylation and inactivation was further confirmed by using cell and animal model systems with two AMPK activators, metformin and salicylate (the active form of aspirin). Together, the results unveil a mechanism by which metabolic stresses activate AMPK, which, in turn, phosphorylates and inactivates MDMX, resulting in p53 stabilization and activation.
INTRODUCTION
The p53 tumor suppressor executes its antitumor functions primarily via its transcriptional activity to induce the expression of protein-encoding genes responsible for p53-dependent apoptosis, cell growth arrest, differentiation, and senescence (1) as well as its ability to induce apoptosis and autophagy by transcription-independent mechanisms (2). Since these cellular functions are detrimental to cells, p53 is often tightly monitored by a pair of partner proteins, MDM2 (called HDM2 in humans) and MDMX (also called MDM4), in normally growing cells (3–5). MDM2 and MDMX act as a complex during early embryogenesis (6–10) to ubiquitylate p53 and mediate its proteosomal turnover as well as inactivate its activity in a negative-feedback fashion (10–12), and cooperatively or individually restrain the p53 level to maintain the normal development and function of different tissues (13–16), by binding to p53, inhibiting its transcriptional activity and/or enhancing its ubiquitination. Hence, to activate p53, cells need to trigger different cellular mechanisms or pathways that block the MDM2-MDMX-p53 feedback loop through modifications of one of these proteins in response to a variety of stresses (17, 18). For instance, DNA damage signals can induce p53 by activating the ATM-Chk2 or ATR-Chk1 pathway that leads to phosphorylation of p53, MDMX, and MDM2 (19–21). Of relevance to MDMX, Ser367 phosphorylation by Chk2 or Chk1 triggers interaction between MDMX and 14-3-3, leading to MDMX inactivation and p53 activation (19, 21, 22). The importance of 14-3-3 binding to Ser367-phosphorylated MDMX for p53 activation by DNA damage was further emphasized in an animal knock-in study in which three serines, including Ser341, Ser367, and Ser402 (23), were mutated into alanines. This mutant MDMX exhibits substantially reduced 14-3-3, decreases p53 activation, and renders mice very radioresistant (20, 23, 24) and less sensitive to hypoxia (25) signals. Also, oncogenic stress can activate p53 by inducing the expression of ARF, enhancing the interaction of ARF with MDM2 and thus inactivating MDM2 activity (26, 27). In addition, ribosomal stress signals have been shown to activate p53 by elevating the binding of several ribosomal proteins with MDM2 and reducing MDM2 activity (28–32) and MDMX activity (33, 34). Therefore, these studies firmly demonstrate that genotoxic, oncogenic, ribosomal stress, and hypoxia signals turn on p53 by directly blocking the inhibitory effects of MDM2 and MDMX.
Previously, metabolic stresses, such as glucose deprivation, which elevates the intracellular level of AMP, or treatment with 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), a cell-permeative AMPK inducer, were shown to activate AMPK via phosphorylation by LKB1 and other kinases (35, 36). AMPK activation also activates p53 to provoke a cell cycle checkpoint (37, 38). Although AMPK reportedly phosphorylates p53 on serine 15 (39), this phosphorylation is not likely to be sufficient for p53 activation (40, 41). Hence, the mechanism by which p53 is activated by metabolic stress remains in question. We therefore set out to investigate the underlying mechanisms using a combined biochemical, cellular, and genetic approach.
MATERIALS AND METHODS
Cell lines and drugs.
Human osteosarcoma cells (U2OS), human embryonic kidney (HEK) epithelial cells (HEK293), human lung adenocarcinoma cells (H1299), and human colon cancer cells (HCT116) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 U/ml of penicillin, and 0.1 mg/ml of streptomycin at 37°C in 5% CO2. Wild-type (WT) mouse embryonic fibroblasts (MEFs), MDMX triple-mutant (MDMX-3SA) MEFs (23), and AMPKα1−/− α2−/− double-knockout (AMPKα−/−) MEFs (42) were cultured in DMEM supplemented with 15% FBS, 100 U/ml of penicillin, 0.1 mg/ml of streptomycin, 1× MEM nonessential amino acids (Cellgro), and 1× β-mercaptoethanol (Millipore). AICAR, metformin (MET), and sodium salicylate (salicylate [SAL]) were purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO) (AICAR, 1 M) or in water (metformin, 1 M, and salicylate, 2 M). AMP-activated protein kinase (AMPK) was purchased from Upstate (catalog number 14-305).
Antibodies, reagents, and plasmids.
Polyclonal anti-p-ACC, anti-p-AMPK, anti-cleaved procyclic acidic repetitive protein (PARP), and anti-p53 (1C12) were purchased from Cell Signaling. Monoclonal anti-p53 (DO-1) was from Santa Cruz. Of note, the anti-p53 antibodies were specific to p53, as they did not detect any band in p53-null cells, and also the anti-PARP antibody was specific to the short cleaved PARP fragment (data not shown). Antiactin, polyclonal anti-p21 (M-19), goat polyclonal anti-MDMX (D-19), and rabbit polyclonal anti-14-3-3γ (C-16) antibodies were purchased from Santa Cruz. Mouse monoclonal anti-14-3-3γ (CG31) was from Neomarker. Mouse monoclonal anti-MDM2 antibodies 4B11 and 2A10 were described previously (43). Mouse monoclonal anti-MDMX (8C6), mouse monoclonal anti-MDMX (7A8), and rabbit polyclonal anti-p-S342 antibodies and pcDNA3–c-myc–MDMX, c-myc-MDMX–S367A, HA–MDMX-S342A, and HA–MDMX-S342A/S367A plasmids were gifts from Jiandong Chen (H. Lee Moffitt Comprehensive Cancer Center, Tampa, FL). AMPKα1 small interfering RNAs (siRNAs) were purchased from Life Technologies (108454 and s100), as were AMPKα2 siRNAs (50583 and s11058), which were used for Fig. 4 and for Fig. S4 in the supplemental material. Mutation vectors c-myc–MDMX-S403A, c-myc–MDMX-S342A/S403A, and c-myc–MDMX-S342A/S367A/S403A were generated from c-myc–MDMX, c-myc-S342A, c-myc-S342A/S367A vectors by changing S403 to A, respectively. His-MDMX, His–MDMX-S342A, His–MDMX-S367A, His–MDMX-S403A, His–MDMX-S342A/S367A, His–MDMX-S342A/S403A, and His–MDMX-S342A/S367A/S403A were generated from c-myc–MDMX, HA–MDMX-S342A, c-myc–MDMX-S367A, c-myc–MDMX-S403A, HA–MDMX-S342A/S367A, c-myc–MDMX-S342A/S403A, and c-myc–MDMX-S342A/S367A/S403A by subcloning the insertion into His (3.0) vectors. A human ACC alpha fragment was cloned into pGEXKG at the XbaI and HindIII sites with the 5′ primer TCTAGATGAACCATCTCCCTTGGCC and the 3′ primer AAGCTTAAGATCTTTTATTTCCCCCAAAGCG by PCR from a mammalian expression plasmid, and this expression vector was named GST-ACC.
FIG 4.

AMPK enhances MDMX and 14-3-3γ interaction. (A and B) HCT116 cells were transfected with a 50 nM final concentration of AMPK siRNA (108454 and 50583) and scramble siRNA using BIONTEX siRNA transfection reagent according to the manufacturer's instructions. At 48 h posttransfection, cells were treated with AICAR (1 mM) for 6 h before harvest. Eighty-microgram quantities of whole-cell lysates were used for WB analysis with the indicated antibodies (A) and co-IP (B) with anti-14-3-3γ polyclonal antibodies (c-16) followed by WB analysis with polyclonal anti-14-3-3γ and anti-MDMX antibodies (Bethyl Laboratories). (C and D) Wild-type MEFs and AMPKα1 and AMPKα2 double-knockout cells were treated with AICAR (1 mM) for 6 h before harvest. Eighty-microgram quantities of whole-cell lysates were used for WB analysis (C) with the indicated antibodies and for co-IP (D) with anti-14-3-3γ polyclonal antibody (c-16) followed by WB analysis with polyclonal anti-14-3-3γ and anti-MDMX antibodies.
Transient transfection.
Cells were transfected with combined plasmids using TransFectin lipid reagent (Bio-Rad) and posttransfection treated (see figure legends for the amount of plasmids used, treatment method, and time points). Cells were harvested, lysed in lysis buffer (50 mM Tris-HCl [pH 8.0], 5 mM EDTA, 150 mM NaCl, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride [PMSF]), and centrifuged for 5 min at 13,200 rpm. Supernatants were taken out for straight Western blotting (WB) or immunoprecipitation assay.
Immunoprecipitation assay.
Cell lysates were incubated with antibodies (lysate amounts, antibodies, and protein agarose are indicated in figure legends) for 3 h at 4°C and then with protein agarose beads for 1 more hour. The beads were washed twice intensively with lysis buffer and once with 1× SNNTE buffer (50 mM Tris-HCl [pH 8.0], 5 mM EDTA, 500 mM NaCl, 1% NP-40, 5% sucrose, and 1 mM PMSF), then boiled in 1× SDS loading buffer, and analyzed by SDS-PAGE and WB with anti-p-S342 antibodies as well as other antibodies as indicated in figure legends.
In vitro phosphopeptide analysis and phosphorylation site mapping.
For in vitro MDMX phosphorylation analysis, the kinase reaction mixture containing 200 ng of protein, 6 mM AMP, 25 mM dithiothreitol (DTT), 7.5 ng of AMPK, 60 mM NaHEPES, 120 mM NaCl, 0.5 μg/ml of leupeptin, 12 μM benzamidine, 0.5 μM pepstatin A, 6 mM ATP, and 2 μl of [32P]ATP was incubated for 1 h at room temperature and boiled in 1× SDS loading buffer. The samples were analyzed by SDS-PAGE. Phosphorylated proteins were detected by autoradiography.
For phosphorylation site mapping, His-MDMX was expressed in and purified from Escherichia coli through nickel beads. Purified His-MDMX proteins were phosphorylated by AMPK in vitro as described above. The mixture without AMPK was used as a nonphosphorylated control. Two hundred nanograms of nonphosphorylated control and 200 ng of phosphorylated MDMX were analyzed using collision-induced dissociation (CID) fragmentation on an LTQ Velos.
Animal studies.
The C57BL/6 MDMX3SA/3SA knock-in (MDMX-3SA) mouse line was described previously (23) and maintained with MDMX3SA/+ heterozygotes. Five-month-old male MDMX3SA/3SA and wild-type (WT) littermates were used for the in vivo experiments based on the previous study (44). Briefly, mice were fasted overnight, followed by a 2-h refeeding period, and then intraperitoneally injected with 250 mg/kg (of body weight) of salicylate (dissolved in phosphate-buffered saline [PBS]) or with PBS as a negative control. After injection, mice were no longer allowed access to any food, and they were sacrificed by euthanasia 1.5 h later. Blood glucose levels were analyzed after fasting, after refeeding, and before sacrifice. Liver tissues were harvested, freshly frozen in liquid nitrogen, and stored at −80°C for further immunoprecipitation and Western blot analysis.
All animal experiments were conducted in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals (45) and were approved by the Institutional Animal Care and Use Committee at Tulane University School of Medicine.
Statistics.
Data were reported as means ± standard errors of the means (SEM), with n being the sample size. Comparisons among groups were analyzed by using one-way analysis of variance (ANOVA). Probability values of P <0.05 are considered to be statistically significant.
RESULTS
Metabolic stress activates AMPK and induces p53 protein levels through inhibition of p53 ubiquitylation.
We confirmed that AMPK activation by glucose deprivation (GD) or AICAR is correlated with p53 induction and apoptosis in multiple human cancer cell lines (HCT116, H460, and U2OS) as detected by Western blotting (WB) analysis (Fig. 1A and B and data not shown). Next, we tested whether AMPK activation affects p53 stability since previous studies only assessed p53 S15 phosphorylation and mRNA induction in response to AMPK activation (39, 46). Indeed, both AICAR treatment and glucose deprivation of HCT116 (Fig. 1C) or U2OS (see Fig. S1 in the supplemental material) cells significantly extended the half-life of p53. Because glucose deprivation may induce ribosomal stress in addition to metabolic stress (47, 48) (data not shown), we decided to use AICAR as a potentially more restrictive stressor in the following experiments. Consistent with the results in Fig. 1C, AICAR treatment of HCT116 markedly reduced ubiquitylation of either exogenous p53 by ectopically expressed MDM2/MDMX or endogenous p53 by endogenous MDM2/MDMX (Fig. 1D and E). These results, also obtained with other human cancer cells (data not shown), demonstrate that AICAR activation of AMPK can increase p53 stability and protein levels by inhibiting MDM2/MDMX-mediated p53 ubiquitylation. As described below, it is highly unlikely that AICAR-mediated p53 activation involves DNA damage induction.
FIG 1.
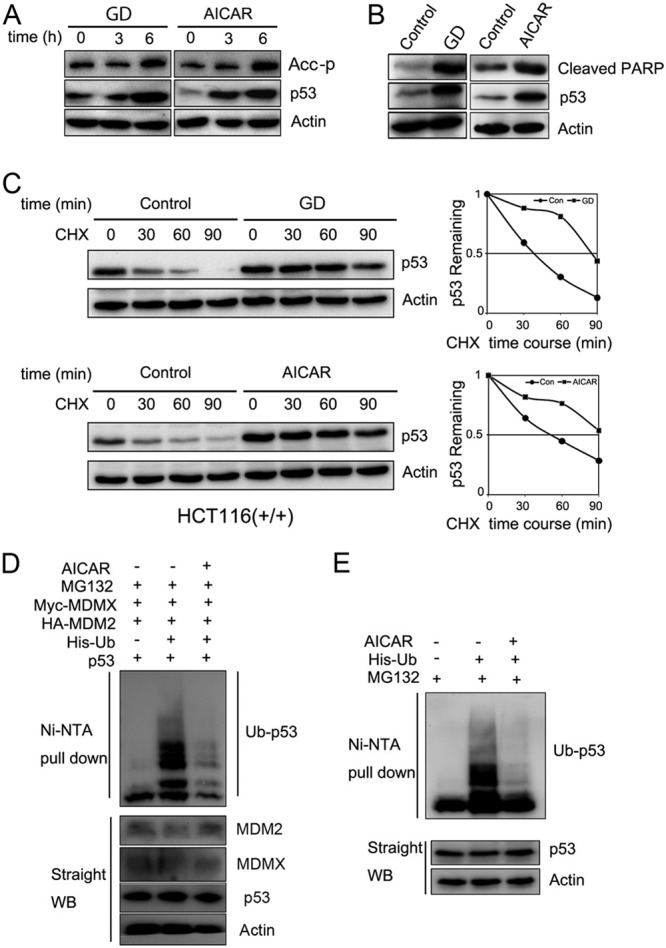
Metabolic stress activates AMPK and induces p53 protein levels through inhibition of p53 ubiquitylation. (A and B) HCT116 cells were cultured in glucose-depleted DMEM or treated with AICAR (1 mM) and harvested at different time points as indicated (6 h in panel B). Thirty-microgram quantities of cell lysates were loaded for WB analysis with the indicated antibodies. (C) HCT116 cells [HCT116(+/+)] were cultured in glucose-depleted DMEM or treated with AICAR (1 mM) for 6 h, followed by cycloheximide (CHX; 50 μg/μl) treatment. Cell lysates (30 μg) were loaded for WB analysis with anti-p53 (DO-1) and anti-β-actin antibodies. Quantitative results were analyzed with ImageJ software and plotted with Excel. (D and E) H1299 cells were transfected with the indicated plasmids. The transfected cells were treated with MG132 (20 μM) and AICAR (1mM) for 6 h before they were harvested. The expression of total p53, MDM2, MDMX, and actin proteins is shown. Ubiquitinated p53 (Ub-p53) proteins were detected with anti-p53 (DO-1) antibody. Ni-NTA, nickel-nitrilotriacetic acid.
AMPK phosphorylates serine 342 of MDMX in vitro and in cells.
Since phosphorylation of MDM2 and MDMX is an important antecedent of p53 activation, we tested whether AMPK also mediates phosphorylation and inactivation of MDM2 and/or MDMX. We conducted an in vitro kinase assay using AMPK and purified His-MDM2 or His-MDMX as substrates. Surprisingly, AMPK appeared to selectively phosphorylate MDMX, as no MDM2 phosphorylation was observed under identical conditions (Fig. 2A). We then used this in vitro kinase assay to identify MDMX residues between amino acids 324 and 432 as containing the AMPK target sites (Fig. 2B). Interestingly, three serines, S342, S367, and S403, within this domain were previously shown to be phosphorylated by ATM-Chk2 and ATR-Chk1 in response to DNA damage signals (19, 21, 22). To determine whether AMPK also targets the same residues, we again conducted the same AMPK kinase assay using purified MDMX proteins with single and multiple mutations at these three residues. Interestingly, S342, rather than S367 or S403, appeared to be the predominant AMPK target, as the S342A mutation almost completely abolished AMPK-mediated MDMX phosphorylation in vitro (Fig. 2C). We verified this result by mass spectrometric (MS) analysis of AMPK-phosphorylated MDMX in vitro, which revealed that S342 is the major phosphorylated site in the peptide 337-LTHSLSpTSDITAIPEK-352 (Fig. 2D). We further confirmed this MS result by WB analysis (Fig. 2E) using antibodies specific to phosphorylated S342 MDMX (24) and with another AMPK substrate, acetyl coenzyme A (acetyl-CoA) carboxylase (ACC), phosphorylated at Ser79 as a positive control (Fig. 2E). Moreover, while the S367A mutation did not affect AMPK-mediated MDMX phosphorylation, the S342A mutation effectively eliminated phosphorylation (Fig. 2F). Ectopically expressed MDMX S367A, but not S342A, was still phosphorylated by endogenous AMPK in response to AICAR treatment in H1299 cells, as detected using anti-S342 antibodies but not anti-S367 antibodies (Fig. 2G) (19, 22). AICAR treatment of mouse embryo fibroblasts (MEFs) also induced phosphorylation of endogenous MDMX S342 but not S367 (Fig. 3D and 4C). Together, these results demonstrate that AMPK can directly phosphorylate S342 of MDMX in vitro and in the cellular response to AICAR treatment.
FIG 2.

AMPK phosphorylates serine 342 of MDMX in vitro and in cells. (A) Five hundred nanograms of His-MDMX, His-MDM2, and GST-ACC was incubated with purified AMPK as described in Materials and Methods. Phosphorylated proteins were detected by autoradiography. (B) A series of MDMX deletions (d) were incubated with purified AMPK and detected by autoradiography. (C) Wild-type MDMX and the indicated mutants (m) were incubated with purified AMPK and detected by autoradiography. (D) AMPK and 200 ng of purified His-MDMX proteins were subjected to an in vitro kinase assay and analyzed by mass spectrometry. The spectrum shows that serine 342 is phosphorylated in the peptide 337-LTHSLSpTSDITAIPEK-352. b ions, fragmentation ions containing the amino terminus of the peptide; y ions, fragmentation ions containing the carboxy terminus of the peptide. (E and F) Five hundred nanograms each of the indicated proteins was incubated with AMPK. Proteins were then loaded for WB analysis and detected by the indicated antibodies. (G) H1299 cells were transfected with indicated plasmids. At 42 h posttransfection, cells were treated with AICAR (1 mM) for 6 h. Thirty-microgram quantities of whole-cell lysates were loaded for WB analysis with the indicated antibodies.
FIG 3.

Mutations of MDMX impair AMPK-mediated MDMX phosphorylation and p53 activation. (A) H1299 cells were transfected with the indicated plasmids. Five-hundred-microgram quantities of cell lysates were used for immunoprecipitation (IP) with anti-Flag antibodies, and precipitates were analyzed by WB with anti-Myc antibodies. Fifty-microgram quantities of cell lysates were loaded for straight WB analysis with the indicated antibodies. (B) Wild-type MEFs and MDMX-3SA mutant MEFs were treated with AICAR (1 mM) for 6 h. One-milligram quantities of cell lysates were used for IP with anti-MDMX polyclonal antibodies (Bethyl Laboratories), and precipitates were analyzed by WB with polyclonal anti-14-3-3γ antibodies (c-16). Eighty-microgram quantities of whole-cell lysates were analyzed by straight WB. (C and D) MEFs were treated with AICAR (1 mM) for 6 h before harvest. Eighty-microgram quantities of cell lysates were analyzed by WB with the indicated antibodies. (E) MEFs were treated with AICAR (1 mM) for 6 h, followed by CHX (50 μg/μl) treatment. Eighty-microgram quantities of cell lysates were analyzed by WB with the indicated antibodies. Quantitative results were analyzed with ImageJ software and plotted with Excel.
As AICAR also affects purine biosynthesis (49), we investigated whether the above effects resulted from induction of a DNA damage response. The following data indicate that this is highly unlikely. First, AICAR treatment did not result in induction of DNA damage-associated γ-H2AX foci (see Fig. S2A and B in the supplemental material). Second, MDMX S367 is phosphorylated in response to DNA damage (21, 22, 24), but AICAR treatment did not result in detectable MDMX S367 phosphorylation (Fig. 2G and 3D). Finally, AICAR induced p53 in human cells defective in ATM (AT−/− cells) (see Fig. S2C in the supplemental material), and this result is consistent with a previous report showing that p53 is activated by AICAR even when ATM is inhibited by KU-55933 (50).
Mutations of MDMX impair AMPK-mediated MDMX phosphorylation, MDMX–14-3-3 interactions, and p53 activation.
Since MDMX phosphorylation at both S367 and S342 is critical for MDMX binding to 14-3-3 (21, 22, 24), we determined if either or both of these two residues affect MDMX binding to 14-3-3 in response to AICAR treatment. First, we observed that AICAR indeed induced the binding of ectopic MDMX to each of the exogenous 14-3-3 isoforms, except 14-3-3σ, as tested in H1299 cells (see Fig. S3 in the supplemental material). This result is consistent with previous reports (19, 21, 22, 24). Then, we tested if either S342A or S367A mutant MDMX affects this AICAR-responsive interaction between MDMX and 14-3-3. S342A drastically reduced its ability to bind to 14-3-3γ (of note, we mainly tested 14-3-3γ in this study because its other isoforms bind to MDMX similarly). Even though S367 was not phosphorylated by AMPK in response to AICAR (Fig. 2 to 4), the S367A mutation also abolished the interaction of MDMX with 14-3-3γ (Fig. 3A). These results were consistent with the previous studies showing that both S367 and S342 are critical for MDMX-14-3-3 binding (19, 21, 22, 24) and also suggested that phosphorylation at either of the two serine residues is sufficient to initiate the effective interaction of MDMX with 14-3-3 in response to different stress signals.
We extended these analyses to endogenous MDMX–14-3-3 interactions. As shown in Fig. 3B, MDMX–14-3-3 binding was markedly enhanced when wild-type (WT) mouse embryo fibroblasts (MEFs) were treated with AICAR, whereas this binding was completely inhibited when MEFs expressing endogenous alleles mutated to encode MDMX-3SA (S341A, S367A, and S402A) (23) were used regardless of AICAR treatment. Consistent with this result, p53 and p21 levels as well as S342 phosphorylation, but not S367 phosphorylation, were induced by AICAR treatment of WT but not 3SA MEFs (Fig. 3C and D). The induction of p53 levels in WT but not 3SA MEFs was due to increased p53 half-life (Fig. 3E). Taken together, these results demonstrate that AICAR can activate AMPK, which, in turn, phosphorylates S342 of MDMX to increase the interaction of S342-phosphorylated MDMX with 14-3-3, which decreases MDM2/MDMX ubiquitin ligase activity (22, 24). As a result, p53 is stabilized and activated as a transcriptional factor.
AICAR-mediated MDMX–14-3-3 interaction enhancement is dependent on AMPK.
Next, we determined if the AICAR–MDMX–14-3-3–p53 pathway is truly dependent on AMPK activity. To do so, we knocked down endogenous AMPK levels in HCT116 cells and confirmed that p53 induction by AICAR was significantly impaired (Fig. 4A) (39, 46). Then, we tested if AICAR can induce MDMX–14-3-3 interaction when AMPK is depleted. Consistent with the above result (Fig. 4A), knockdown of endogenous AMPK using a specific siRNA prevented the interaction between endogenous MDMX and 14-3-3 proteins, while coimmunoprecipitation revealed that this interaction was induced by AICAR in cells transfected with a scrambled siRNA control (Fig. 4B). Another set of specific AMPKα1/α2 siRNAs was used to further confirm the involvement of AMPK in AICAR-induced binding of MDMX to 14-3-3 (see Fig. S4 in the supplemental material). Again, in accordance with these results (Fig. 4A and B; see also Fig. S4 in the supplemental material), the increase in p53 level, MDMX S342 phosphorylation, ACC (a classical substrate of AMPK [51]) phosphorylation, and interaction of MDMX with 14-3-3 were all severely impaired in ampk-alpha1 and alpha2 double-knockout MEFs, compared to those in WT MEFs (Fig. 4C and D). Furthermore, AICAR-induced G2 arrest in WT MEFs was no longer evident in either 3SA or ampk-null MEFs (Fig. 5), which was p53 dependent, as this G2 arrest was not evident in p53 knockout (p53−/−) MEFs (see Fig. S5 in the supplemental material). Hence, these results demonstrate that AMPK is essential for AICAR-mediated induction of MDMX phosphorylation at S342, MDMX–14-3-3 binding, p53 activation, and cell cycle arrest.
FIG 5.
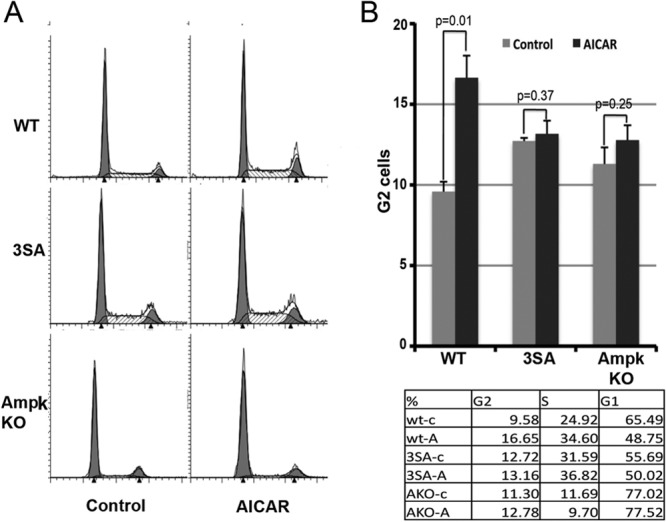
AMPK induces cell cycle arrest in wild-type MEFs but not in 3SA cells or AMPK KO cells. Three types of MEFs (wild type, 3SA, and AMPK KO) were treated with AICAR (1 mM) or DMSO as a negative control for 24 h and stained with propidium iodide for cell cycle analysis by fluorescence-activated cell sorting (A). For statistical analysis, P values were evaluated by one-way ANOVA (B).
AMPK-mediated MDMX phosphorylation and binding to 14-3-3 account for p53 activation by metformin and/or salicylate in cells and in animals.
As an essential regulator of the metabolic system, AMPK has been a worthwhile therapeutic target for some disorders, such as type 2 diabetes (52–54). To determine if AMPK-mediated MDMX phosphorylation and subsequent p53 activation can also apply to the actions of some related clinical drugs, we performed further cell culture and animal experiments using two classic drugs, metformin and salicylate (the active form of aspirin), which have been demonstrated to activate AMPK (44, 55, 56). Cells were treated with metformin or salicylate based on previous studies (44, 57, 58). As expected, both metformin and salicylate activated AMPK and p53 in a time-dependent manner in HCT116 colon cancer cells and reached the peak at 6 to 12 h (Fig. 6A). Also, these drugs induced a remarkably enhanced binding of MDMX to 14-3-3 (Fig. 6B). The AMPK–MDMX–14-3-3–p53 pathway was activated as well by treatment with metformin for 12 h in WT MEFs. However, the induction of p53 level was moderate, and no obvious binding between MDMX and 14-3-3 was detected in the metformin-treated MDMX-3SA MEFs (Fig. 6C). These results indicate that metformin and salicylate can activate p53 through AMPK-mediated MDMX phosphorylation and binding to 14-3-3.
FIG 6.

Metformin or salicylate activates p53 mainly through AMPK-mediated MDMX phosphorylation and binding to 14-3-3 in vitro and in vivo. (A) Both metformin and salicylate activate AMPK and p53 in a dose-dependent manner. HCT116 colon cancer cells were treated with 10 mM metformin or 10 mM salicylate and harvested at the indicated time points. Forty micrograms of protein was loaded into each lane to measure the expression level and activation of AMPK, p53, and their targets. (B) Metformin or salicylate remarkably enhanced the binding of endogenous MDMX to 14-3-3γ in HCT116 cells treated with metformin or salicylate for 12 h. Seven hundred micrograms of total protein per sample was incubated with 2 μg of anti-14-3-3γ (Ab-2) for 4 h at 4°C. (C) Metformin increases p53 level and activity by activating AMPK and enhancing the binding of MDMX to 14-3-3γ in WT but not MDMX-3SA MEFs. Both cell lines were treated with 10 mM metformin for 12 h. Forty micrograms of total protein per sample was used for Western blot analysis and 1.2 mg of total protein for co-IP experiments. For IP analysis, lysates were incubated with 6 μg of anti-MDMX (D-19) for 4 h at 4°C. (D) Salicylate activates p53 mainly through AMPK-mediated MDMX phosphorylation and binding to 14-3-3γ in mouse livers. WT or MDMX-3SA mice were treated with salicylate for 1.5 h, and liver tissues were harvested for WB and co-IP analysis, as described in Materials and Methods. The conditions for WB and co-IP analysis were similar to those described for panel C. (E) A model demonstrates that some metabolic stresses activate p53 by AMPK phosphorylating MDMX at S342, further inducing the binding of 14-3-3 to phosphorylated MDMX, which, in turn, inhibits the activity of MDMX.
We wanted to determine whether the above results obtained with in vitro models could be extended to more physiological settings. We therefore treated wild-type or MDMX-3SA mice with salicylate. Before drug administration, mice (3 per group) underwent a fasting-refeeding period (see Materials and Methods) to ensure initiation of the study with consistent and low levels of endogenous AMPK. As indicated in Fig. 6D, salicylate induced AMPK activation in both WT and MDMX-3SA mouse liver tissues. However, p53 levels were dramatically enhanced only in WT mouse livers after treatment with this drug, whereas the induction of p53 and p21 levels was hardly evident in MDMX-3SA mouse livers (Fig. 6D). Consistent with this result, salicylate only induced the MDMX–14-3-3 interaction in WT, and not mutant, mouse livers (Fig. 6D, bottom). These results not only verify the aforementioned AMPK-mediated p53 activation in vivo but also demonstrate that the clinically used drugs that act as AMPK activators can also activate p53 by inducing AMPK-mediated MDMX phosphorylation and subsequent binding to 14-3-3.
DISCUSSION
Our study demonstrates that a key element of p53 activation by metabolic stress requires phosphorylation of MDMX at S342 by AMPK (Fig. 5C). To our surprise, even though MDMX S367 was not phosphorylated by AMPK in vitro (Fig. 2) or in vivo (Fig. 3 and 4), its mutation to alanine (S367A) still completely blocked the interaction between MDMX and 14-3-3, also abrogating p53 activation in response to metabolic stress. This result was consistent with previous studies (21, 22, 24), again verifying that both S342 and S367 are crucial for 14-3-3 to bind to MDMX in response to stress signals. Because only S342 was phosphorylated by AMPK (Fig. 2 to 4), phosphorylation at this specific residue could be sufficient to initiate the effective binding of cellular 14-3-3 molecules to the two binding motifs (S342 and S367) within MDMX. This might mean that once binding of 14-3-3 to one of these S342 and S367 sites occurs, this action would promote its binding to the other site within MDMX in a so-called “inducible” fashion, though this inducible mechanism needs to be further studied using purified proteins in vitro. DNA damage signals have previously been shown to activate p53 by activating the ATM-Chk2 or ATR-Chk1 kinase cascade, which, in turn, leads to S342 and S367 phosphorylation of MDMX and subsequent MDMX–14-3-3 binding (21, 22, 24). Although glucose deprivation also activates AMPK (46) and induces p53 stability (Fig. 1), this nutrient stress might utilize multiple signaling pathways to activate p53, as it has been shown and believed that glucose deprivation causes ribosomal stress (47, 48) and we have preliminary evidence (data not shown) suggesting that reducing RPL11 levels impairs p53 induction by glucose deprivation (data not shown). Of note, the p53 protein level was relatively lower in the AMPKα1/α2-KO MEF cells than in wild-type MEFs used in this study (Fig. 4C). This might be partially due to the fact that loss of both of the α1 and α2 subunits of AMPK could reduce the sensitivity of p53 to cell culture stress and partially due to the uneven protein loading as suggested by the internal β-actin control (Fig. 4C). A lower level of p53 does not necessarily suggest that this p53 form could not be inducible, as a previous study showed that the basal level of p53 was barely visible in both control and AMPKα1/α2 double-knockdown U2OS cells; however, p53 was still activated by glucose deprivation in control cells but not in AMPK knockdown cells (46). This result demonstrates that p53 is still activated even if its basal level is nearly undetectable by Western blotting, and AMPK is indeed involved in metabolic stress-mediated p53 activation, which is consistent with our current results. Another study showed a higher basal level of p53 in AMPKα1-KO MEFs (59). However, this result is not contradictory with our observation, as AMPKα1-KO MEFs still contain AMPKα2, which might compensate for the loss of AMPKα1 function and could be one reason for the higher p53 basal level, whereas AMPKα1/α2-KO MEFs are clearly deficient in both of the α subunits.
Metformin, as the first-line medication for metabolic syndrome and type 2 diabetes, has been identified to promote insulin sensitivity primarily by activating AMPK (55, 56, 60–62). Some reports suggest that metformin can activate p53 by both AMPK-dependent (63, 64) and independent (65, 66) mechanisms to induce cell apoptosis and growth arrest and therefore may be beneficial for cancer therapy. Salicylate is an ancient cyclooxygenase inhibitor, derivatives of which have been used for treating diverse disorders (67, 68). Recent findings imply a promising application of salicylate in type 2 diabetes (69, 70) and cancer prevention (71), which may be associated with its newly discovered role in AMPK activation (44). In this study, we used in vitro and in vivo approaches to verify that both metformin and salicylate upregulate p53 through the AMPK/phospho-MDMX–14-3-3 pathway revealed here. As these drugs failed to induce p53 in metformin- or sodium salicylate-treated MDMX-3SA MEFs and mouse livers (Fig. 6), we infer that AMPK-mediated MDMX phosphorylation is a central node in p53 activation by such agents. As an energy sensor, AMPK is activated by increases in AMP/ATP and ADP/ATP ratios and further restrains energy-consuming processes to regulate energy homeostasis (72, 73). When cells are under low-energy status, AMPK activation maintains energy homeostasis for normal cellular events but also induces p53 to restrict cell growth rates to save more energy and to limit induction of potential damage in energy-rich environments. Considering the potential fine-tuning of MDMX on p53 activity, which is supported by the fact that deletion of MDMX is more subtle and moderate than MDM2 deficiency in some tissues (13, 14, 16), the involvement of MDMX in AMPK-mediated p53 activation in response to energy shortage suggests that the MDMX–14-3-3–p53 pathway may play a major role for cells to respond to some mild stresses, which allows the cells to have more chances to recover, rather than to undergo rapid death.
In summary, our findings as described here reveal MDMX as an important sensor of metabolic stress that mediates p53 activation via a pathway involving AMPK and 14-3-3 proteins. This can at least partially interpret the activation of p53 by the AMPK activators metformin and salicylate and provide additional evidence for the potential application of these classic drugs to cancer therapy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Larry David for mass spectrometric analysis of phosphorylated MDMX, Jiandong Chen for offering MDMX mutant plasmids, and the members in the Lu laboratory for active discussion.
H.L. was supported by NIH-NCI grants CA127724, CA095441, CA129828, and CA 172468. G.M.W. was supported by NIH-NCI grant CA 061449.
Footnotes
Published ahead of print 4 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00670-13.
REFERENCES
- 1.Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, Levine AJ. 2010. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2:a001198. 10.1101/cshperspect.a001198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson TM, Meade K, Pathak N, Marques MR, Attardi LD. 2008. Knockin mice expressing a chimeric p53 protein reveal mechanistic differences in how p53 triggers apoptosis and senescence. Proc. Natl. Acad. Sci. U. S. A. 105:1215–1220. 10.1073/pnas.0706764105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barak Y, Juven T, Haffner R, Oren M. 1993. mdm2 expression is induced by wild type p53 activity. EMBO J. 12:461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. 1992. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245. 10.1016/0092-8674(92)90644-R [DOI] [PubMed] [Google Scholar]
- 5.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG. 1996. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 15:5349–5357 [PMC free article] [PubMed] [Google Scholar]
- 6.Huang L, Yan Z, Liao X, Li Y, Yang J, Wang ZG, Zuo Y, Kawai H, Shadfan M, Ganapathy S, Yuan ZM. 2011. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. U. S. A. 108:12001–12006. 10.1073/pnas.1102309108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. 2007. RING domain-mediated interaction is a requirement for MDM2's E3 ligase activity. Cancer Res. 67:6026–6030. 10.1158/0008-5472.CAN-07-1313 [DOI] [PubMed] [Google Scholar]
- 8.Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. 2011. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl. Acad. Sci. U. S. A. 108:11995–12000. 10.1073/pnas.1102241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, Pavletich N, Prives C. 2007. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 26:90–101. 10.1038/sj.emboj.7601465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uldrijan S, Pannekoek WJ, Vousden KH. 2007. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 26:102–112. 10.1038/sj.emboj.7601469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwakuma T, Lozano G. 2003. MDM2, an introduction. Mol. Cancer Res. 1:993–1000 http://mcr.aacrjournals.org/content/1/14/993 [PubMed] [Google Scholar]
- 12.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. 1993. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362:857–860. 10.1038/362857a0 [DOI] [PubMed] [Google Scholar]
- 13.Francoz S, Froment P, Bogaerts S, De Clercq S, Maetens M, Doumont G, Bellefroid E, Marine JC. 2006. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl. Acad. Sci. U. S. A. 103:3232–3237. 10.1073/pnas.0508476103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, Lozano G. 2006. Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol. Cell. Biol. 26:192–198. 10.1128/MCB.26.1.192-198.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilliard S, Aboudehen K, Yao X, El-Dahr SS. 2011. Tight regulation of p53 activity by Mdm2 is required for ureteric bud growth and branching. Dev. Biol. 353:354–366. 10.1016/j.ydbio.2011.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Fernandez-Garcia B, Lozano G. 2007. Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation 115:2925–2930. 10.1161/CIRCULATIONAHA.107.689901 [DOI] [PubMed] [Google Scholar]
- 17.Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622. 10.1016/j.cell.2009.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wade M, Wang YV, Wahl GM. 2010. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 20:299–309. 10.1016/j.tcb.2010.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LeBron C, Chen L, Gilkes DM, Chen J. 2006. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. EMBO J. 25:1196–1206. 10.1038/sj.emboj.7601032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, Teunisse A, Migliorini D, Kitabayashi I, Marine JC, Prives C, Shiloh Y, Jochemsen AG, Taya Y. 2005. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol. Cell. Biol. 25:9608–9620. 10.1128/MCB.25.21.9608-9620.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pereg Y, Lam S, Teunisse A, Biton S, Meulmeester E, Mittelman L, Buscemi G, Okamoto K, Taya Y, Shiloh Y, Jochemsen AG. 2006. Differential roles of ATM- and Chk2-mediated phosphorylations of Hdmx in response to DNA damage. Mol. Cell. Biol. 26:6819–6831. 10.1128/MCB.00562-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin Y, Dai MS, Lu SZ, Xu Y, Luo Z, Zhao Y, Lu H. 2006. 14-3-3gamma binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. EMBO J. 25:1207–1218. 10.1038/sj.emboj.7601010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YV, Leblanc M, Wade M, Jochemsen AG, Wahl GM. 2009. Increased radioresistance and accelerated B cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell 16:33–43. 10.1016/j.ccr.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. 2005. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 24:3411–3422. 10.1038/sj.emboj.7600812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JH, Jin Y, He G, Zeng SX, Wang V, Wahl GM, Lu H. 2012. Hypoxia activates the tumor suppressor p53 by inducing ATR-Chk1 kinase cascade-mediated phosphorylation and consequent 14-3-3gamma inactivation of MDMX. J. Biol. Chem. 287:20898–20903. 10.1074/jbc.M111.336875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda R, Yasuda H. 1999. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 18:22–27. 10.1093/emboj/18.1.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Xiong Y, Yarbrough WG. 1998. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92:725–734. 10.1016/S0092-8674(00)81401-4 [DOI] [PubMed] [Google Scholar]
- 28.Dai MS, Shi D, Jin Y, Sun XX, Zhang Y, Grossman SR, Lu H. 2006. Regulation of the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J. Biol. Chem. 281:24304–24313. 10.1074/jbc.M602596200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. 2003. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3:577–587. 10.1016/S1535-6108(03)00134-X [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, Xiao H, Chai SC, Hoang QQ, Lu H. 2011. Hydrophilic residues are crucial for ribosomal protein L11 (RPL11) interaction with zinc finger domain of MDM2 and p53 protein activation. J. Biol. Chem. 286:38264–38274. 10.1074/jbc.M111.277012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Lu H. 2009. Signaling to p53: ribosomal proteins find their way. Cancer Cell 16:369–377. 10.1016/j.ccr.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. 2003. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 23:8902–8912. 10.1128/MCB.23.23.8902-8912.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilkes DM, Chen L, Chen J. 2006. MDMX regulation of p53 response to ribosomal stress. EMBO J. 25:5614–5625. 10.1038/sj.emboj.7601424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Gu W. 2011. A critical role for noncoding 5S rRNA in regulating Mdmx stability. Mol. Cell 43:1023–1032. 10.1016/j.molcel.2011.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. 2003. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2:28. 10.1186/1475-4924-2-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. 2003. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13:2004–2008. 10.1016/j.cub.2003.10.031 [DOI] [PubMed] [Google Scholar]
- 37.Lee DH, Lee TH, Jung CH, Kim YH. 2012. Wogonin induces apoptosis by activating the AMPK and p53 signaling pathways in human glioblastoma cells. Cell. Signal. 24:2216–2225. 10.1016/j.cellsig.2012.07.019 [DOI] [PubMed] [Google Scholar]
- 38.Thoreen CC, Sabatini DM. 2005. AMPK and p53 help cells through lean times. Cell Metab. 1:287–288. 10.1016/j.cmet.2005.04.009 [DOI] [PubMed] [Google Scholar]
- 39.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18:283–293. 10.1016/j.molcel.2005.03.027 [DOI] [PubMed] [Google Scholar]
- 40.Ashcroft M, Kubbutat MH, Vousden KH. 1999. Regulation of p53 function and stability by phosphorylation. Mol. Cell. Biol. 19:1751–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chao C, Saito S, Anderson CW, Appella E, Xu Y. 2000. Phosphorylation of murine p53 at Ser-18 regulates the p53 responses to DNA damage. Proc. Natl. Acad. Sci. U. S. A. 97:11936–11941. 10.1073/pnas.220252297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 2006. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell. Biol. 26:5336–5347. 10.1128/MCB.00166-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng X, Chen L, Jost CA, Maya R, Keller D, Wang X, Kaelin WG, Jr, Oren M, Chen J, Lu H. 1999. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 19:3257–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG. 2012. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336:918–922. 10.1126/science.1215327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 46.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. 2008. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 283:3979–3987. 10.1074/jbc.M705232200 [DOI] [PubMed] [Google Scholar]
- 47.Deisenroth C, Zhang Y. 2011. The ribosomal protein-Mdm2-p53 pathway and energy metabolism: bridging the gap between feast and famine. Genes Cancer 2:392–403. 10.1177/1947601911409737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang F, Hamanaka RB, Bobrovnikova-Marjon E, Gordan JD, Dai MS, Lu H, Simon MC, Diehl JA. 2006. Ribosomal stress couples the unfolded protein response to p53-dependent cell cycle arrest. J. Biol. Chem. 281:30036–30045. 10.1074/jbc.M604674200 [DOI] [PubMed] [Google Scholar]
- 49.Mentzer RM, Jr, Ely SW, Lasley RD, Berne RM. 1988. The acute effects of AICAR on purine nucleotide metabolism and postischemic cardiac function. J. Thorac. Cardiovasc. Surg. 95:286–293 [PubMed] [Google Scholar]
- 50.Maclaine NJ, Hupp TR. 2009. The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging (Albany NY) 1:490–502 http://www.impactaging.com/papers/v1/n5/full/100047.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witters LA, Nordlund AC, Marshall L. 1991. Regulation of intracellular acetyl-CoA carboxylase by ATP depletors mimics the action of the 5′-AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 181:1486–1492. 10.1016/0006-291X(91)92107-U [DOI] [PubMed] [Google Scholar]
- 52.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. 2006. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 3:403–416. 10.1016/j.cmet.2006.05.005 [DOI] [PubMed] [Google Scholar]
- 53.Yu LF, Qiu BY, Nan FJ, Li J. 2010. AMPK activators as novel therapeutics for type 2 diabetes. Curr. Top. Med. Chem. 10:397–410. 10.2174/156802610790980611 [DOI] [PubMed] [Google Scholar]
- 54.Zhang BB, Zhou G, Li C. 2009. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9:407–416. 10.1016/j.cmet.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Wang Y, Bao C, Xu Y, Shen H, Chen J, Yan J, Chen Y. 2012. Metformin interacts with AMPK through binding to gamma subunit. Mol. Cell. Biochem. 368:69–76. 10.1007/s11010-012-1344-5 [DOI] [PubMed] [Google Scholar]
- 56.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108:1167–1174. 10.1172/JCI13505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karnevi E, Said K, Andersson R, Rosendahl AH. 2013. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 13:235. 10.1186/1471-2407-13-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarfstein R, Friedman Y, Attias-Geva Z, Fishman A, Bruchim I, Werner H. 2013. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS One 8:e61537. 10.1371/journal.pone.0061537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang S, Song P, Zou MH. 2012. Inhibition of AMP-activated protein kinase alpha (AMPKalpha) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1. J. Biol. Chem. 287:8001–8012. 10.1074/jbc.M111.315812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SK, Lee JO, Kim JH, Kim SJ, You GY, Moon JW, Jung JH, Park SH, Uhm KO, Park JM, Suh PG, Kim HS. 2011. Metformin sensitizes insulin signaling through AMPK-mediated PTEN down-regulation in preadipocyte 3T3-L1 cells. J. Cell. Biochem. 112:1259–1267. 10.1002/jcb.23000 [DOI] [PubMed] [Google Scholar]
- 61.Ou HY, Cheng JT, Yu EH, Wu TJ. 2006. Metformin increases insulin sensitivity and plasma beta-endorphin in human subjects. Horm. Metab. Res. 38:106–111. 10.1055/s-2006-925128 [DOI] [PubMed] [Google Scholar]
- 62.Palomba S, Falbo A, Russo T, Manguso F, Tolino A, Zullo F, De Feo P, Orio F., Jr 2007. Insulin sensitivity after metformin suspension in normal-weight women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 92:3128–3135. 10.1210/jc.2007-0441 [DOI] [PubMed] [Google Scholar]
- 63.Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF, Bost F. 2010. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 70:2465–2475. 10.1158/0008-5472.CAN-09-2782 [DOI] [PubMed] [Google Scholar]
- 64.Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G, Tsakiridis T. 2013. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 108:2021–2032. 10.1038/bjc.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. 2011. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 71:4366–4372. 10.1158/0008-5472.CAN-10-1769 [DOI] [PubMed] [Google Scholar]
- 66.Janjetovic K, Harhaji-Trajkovic L, Misirkic-Marjanovic M, Vucicevic L, Stevanovic D, Zogovic N, Sumarac-Dumanovic M, Micic D, Trajkovic V. 2011. In vitro and in vivo anti-melanoma action of metformin. Eur. J. Pharmacol. 668:373–382. 10.1016/j.ejphar.2011.07.004 [DOI] [PubMed] [Google Scholar]
- 67.Ekinci D, Senturk M, Kufrevioglu OI. 2011. Salicylic acid derivatives: synthesis, features and usage as therapeutic tools. Expert Opin. Ther. Pat. 21:1831–1841. 10.1517/13543776.2011.636354 [DOI] [PubMed] [Google Scholar]
- 68.Higgs GA, Salmon JA, Henderson B, Vane JR. 1987. Pharmacokinetics of aspirin and salicylate in relation to inhibition of arachidonate cyclooxygenase and antiinflammatory activity. Proc. Natl. Acad. Sci. U. S. A. 84:1417–1420. 10.1073/pnas.84.5.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fleischman A, Shoelson SE, Bernier R, Goldfine AB. 2008. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31:289–294. 10.2337/dc07-1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. 2010. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med. 152:346–357. 10.7326/0003-4819-152-6-201003160-00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elwood PC, Gallagher AM, Duthie GG, Mur LA, Morgan G. 2009. Aspirin, salicylates, and cancer. Lancet 373:1301–1309. 10.1016/S0140-6736(09)60243-9 [DOI] [PubMed] [Google Scholar]
- 72.Hardie DG, Scott JW, Pan DA, Hudson ER. 2003. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 546:113–120. 10.1016/S0014-5793(03)00560-X [DOI] [PubMed] [Google Scholar]
- 73.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. 2006. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 281:32207–32216. 10.1074/jbc.M606357200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.