

Abstract
Vulvovaginal candidiasis (VVC), caused by Candida albicans, affects women worldwide. Animal and clinical studies suggest that the immunopathogenic inflammatory condition of VVC is initiated by S100 alarmins in response to C. albicans, which stimulate polymorphonuclear neutrophil (PMN) migration to the vagina. The purpose of this study was to extend previous in vitro data and determine the requirement for the alarmin S100A8 in the PMN response and to evaluate pattern recognition receptors (PRRs) that initiate the response. For the former, PMN migration was evaluated in vitro or in vivo in the presence or absence of S100 alarmins initiated by several approaches. For the latter, vaginal epithelial cells were evaluated for PRR expression and C. albicans-induced S100A8 and S100A9 mRNAs, followed by evaluation of the PMN response in inoculated PRR-deficient mice. Results revealed that, consistent with previously reported in vitro data, eukaryote-derived S100A8, but not prokaryote-derived recombinant S100A8, induced significant PMN chemotaxis in vivo. Conversely, a lack of biologically active S100A8 alarmin, achieved by antibody neutralization or by using S100A9−/− mice, had no effect on the PMN response in vivo. In PRR analyses, whereas Toll-like receptor 4 (TLR4)- and SIGNR1-deficient vaginal epithelial cells showed a dramatic reduction in C. albicans-induced S100A8/S100A9 mRNAs in vitro, inoculated mice deficient in these PRRs showed PMN migration similar to that in wild-type controls. These results suggest that S100A8 alarmin is sufficient, but not necessary, to induce PMN migration during VVC and that the vaginal PMN response to C. albicans involves PRRs in addition to SIGNR1 and TLR4, or other induction pathways.
INTRODUCTION
Vulvovaginal candidiasis (VVC) is an opportunistic fungal infection predominantly caused by Candida albicans. The disease affects approximately 75% of healthy women during their reproductive years (1). An additional 5 to 10% of women suffer from recurrent VVC (RVVC), defined as 3 or more episodes of VVC per year. RVVC cases can be either primary, where repetitive idiopathic infections occur with no known predisposing factors, or secondary, where recurrence is the result of the inability to avoid certain known predisposing factors (e.g., use of high-estrogen oral contraception, hormone replacement therapies, frequent antibiotic usage, and diabetes mellitus).
Historically, RVVC had been attributed to a putative local immune deficiency (reviewed in reference 2). However, numerous studies employing a mouse model of Candida vaginitis and clinical studies evaluating women with primary RVVC showed that protection is not mediated by local or systemic adaptive immunity and their associated cytokines/chemokines (2–7). The lack of protection provided by C. albicans-specific adaptive immunity was further supported by evidence of immunomodulation that may regulate such responses (8–10). Instead, epithelial cells appear to mediate innate resistance to C. albicans by inhibiting its growth with little or no associated inflammation via annexin-A1 (11). Symptomatic infection, on the other hand, was shown to be associated with a vaginal cellular infiltrate predominantly consisting of polymorphonuclear neutrophils (PMNs) (12). With no clinical or experimental evidence that the PMNs were protective, these findings suggested that the acute inflammatory response by PMNs in response to C. albicans was instead responsible for the symptoms associated with infection.
More recently, an established experimental mouse model of Candida vaginitis was exploited to further investigate factors/immune mediators involved in the acute PMN response during infection/symptomatic conditions. Proteomic analyses of vaginal secretions in mice under a symptomatic condition identified the alarmins S100A8 and S100A9 as strong candidates for PMN chemotactic factors produced in response to C. albicans (13). Furthermore, the study revealed that S100 alarmins were produced by vaginal epithelial cells following interaction with C. albicans. S100 alarmins are cytosolic and secretory proteins produced by a variety of cell types, including PMNs, monocytes, and epithelial cells (14–18). In VVC, epithelial cell-derived S100 alarmins are suggested to be secreted in response to C. albicans as an initial signal for PMN migration, which becomes amplified further by subsequent production via recruited PMNs in the vaginal cavity. In murine models of infection, the alarmins were shown to exert both antimicrobial and PMN chemotactic properties and have been linked to a variety of inflammatory diseases (16, 19–26). Among the two proteins, antibody neutralization of S100A8 significantly reduced the PMN chemotactic ability of vaginal lavage fluid in vitro (13), suggesting that S100A8 could be a key inflammatory factor associated with the symptomatic condition. However, the functional role of S100A8 in PMN chemotaxis in the vaginal cavity is yet to be confirmed.
Based on the requirement for C. albicans adherence to the vaginal epithelium during the early stage of colonization, the induction of the S100 alarmin response by vaginal epithelial cells likely involves interaction via pattern recognition receptors (PRRs) with C. albicans. Indeed, Toll-like receptor 4 (TLR4) expressed on phagocytes as well as vaginal epithelial cells plays a significant role in proinflammatory responses to C. albicans (27, 28). In addition, TLR2, SIGNR1, dectin-1, and mannose receptor (MR) are also known to be involved in recognition of C. albicans (29–37). The purposes of the present study were to further evaluate the biological role of S100A8 alarmin for the vaginal PMN response in the established mouse model and to identify PRRs involved in the induction of the alarmin response.
MATERIALS AND METHODS
Mice.
Female CBA/J mice, 6 to 10 weeks of age, purchased from Charles River at the National Cancer Institute (NCI, Frederick, MD), were used throughout the studies unless indicated otherwise. Breeders of S100A9−/− mice, which express normal S100A8 mRNA but lack both S100A8 and S100A9 at the protein level, were obtained from Thomas Vogl (University of Münster, Muenster, Germany). Age-matched C57BL/6 mice (NCI) were tested in parallel as the wild-type strain. For evaluation of PRRs, TLR4-defective female C3H/HeJ mice were purchased from NCI. Mannose receptor-negative (MR−/−) female mice (C57BL/6 background) were obtained from Judy Teale (University of Texas, San Antonio, TX). Breeders of SIGNR1−/− (BALB/c background) were obtained from Andrew McKenzie (MRC Laboratory of Molecular Biology, Cambridge, United Kingdom). Age-matched C3H/HeOuJ (Jackson Laboratories, Bar Harbor, ME), C57BL/6 (NCI), and BALB/c (NCI) mice were tested in parallel as wild-type strains for C3H/HeJ, MR−/−, and SIGNR1−/− mice, respectively. Of note, previous studies by several groups reported no difference in experimental vaginal C. albicans burden among several genetically distinct mouse strains, including those used in the present study (13, 38, 39). All animals were housed and handled according to institutionally recommended guidelines. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the LSU Health Sciences Center, New Orleans, LA.
Vaginal C. albicans inoculation.
Vaginal inoculation with C. albicans in mice was conducted as previously described (7, 40). Briefly, mice were administered 0.1 mg of β-estradiol 17-valerate (Sigma Chemical Co., St. Louis, MO) in 100 μl of sesame oil (Sigma) by subcutaneous injection 72 h prior to inoculation, and then again weekly until the completion of the study. Estrogen-treated (estrogenized) mice were intravaginally inoculated by introducing 20 μl of phosphate-buffered saline (PBS) containing 5 × 104 C. albicans (3153A) blastoconidia into the vaginal lumen. Inoculation was conducted exclusively using blastoconidia from a stationary-phase culture (cultured for 12 to 18 h at 30°C in Phytone-peptone broth with 0.1% glucose). Uninoculated control mice were estrogenized and given PBS intravaginally. Groups of 5 to 10 mice were evaluated either longitudinally or terminally at specific time points postinoculation. Upon euthanasia or under anesthesia, vaginal lavage samples were collected using 100 μl of sterile PBS with gentle aspiration and agitation for ∼30 s. To assess vaginal fungal burden, serial dilutions of the vaginal lavage fluid were cultured on Sabouraud-dextrose agar plates (BD Diagnostics, Sparks, MD) supplemented with gentamicin (Invitrogen, Carlsbad, CA). CFU were enumerated after incubation for 48 h at 34°C. Supernatants of the remaining lavage fluids were sterilely filtered and stored at −70°C until use.
Quantification of vaginal PMNs.
Smear preparations of 10 μl vaginal lavage fluid collected from each inoculated and uninoculated mouse were stained using the Papanicolaou technique (Pap smear). PMNs, if present, were identified by their trilobed nuclear morphology and were the predominant cell type among other infiltrating leukocytes as previously confirmed (13, 41). PMNs were enumerated in 5 nonadjacent high-powered fields (×400) per mouse by light microscopy and averaged. Previous studies confirmed that the Pap smear technique was as efficient and consistent as PMN quantification by flow cytometry or direct microscopic counts using a hemocytometer (13).
Construction of mouse S100A8- and S100A9-expressing strains of C. albicans.
S100A8- and S100A9-expressing C. albicans strains were constructed and provided by Glen Palmer (42). Briefly, a gene fusion containing the S100A8 or S100A9 open reading frame (ORF) tagged with a leader peptide of C. albicans secreted aspartyl protease 2 (SAP2, amino acids 1 to 56) was constructed to facilitate secretion of the S100 proteins by the C. albicans strains. This gene fusion was placed in an expression cassette under the control of the C. albicans ACT1 promoter, and the construct was transformed into C. albicans strain CAI4 (ura3−/−) to reconstitute the URA3 gene at its native locus, negating the positional effect of URA3 expression. While entering the endoplasmic reticulum, followed by transport to the Golgi apparatus, the SAP2 leader peptide of the fusion protein is cleaved by Kex2 protease at a Lys-Arg dipeptide, liberating the intact S100A8 or S100A9, which is then transported to the cell surface for exocytosis (see Fig. S1 in the supplemental material). A control strain was constructed by transforming the vector alone into the wild-type C. albicans strain CAI4 and tested in parallel with S100A8- and S100A9-expressing strains. Growth curve evaluation indicated no change in growth rates of all modified strains in vitro. Secretion of the target proteins were validated by Western blotting and enzyme-linked immunosorbent assay (ELISA), confirming the presence of S100A8 or S100A9 in culture supernatants (∼400 ng/ml and ∼200 ng/ml, respectively) (42). Prior to use, C. albicans strains were cultured overnight to reach the stationary growth phase in yeast extract-peptone-dextrose (YPD) medium at 30°C with shaking. Culture supernatants were obtained by centrifugation at 800 × g for 5 min and sterilely filtered prior to storage at −80°C.
Immunocytochemistry and immunohistochemistry.
For immunocytochemical analysis of PRR expression on vaginal epithelial cells, cellular fractions of vaginal lavage fluid from inoculated and uninoculated estrogen-treated mice were cytospun onto glass slides using a cytospin 4 cytocentrifuge (Thermo Fisher Scientific, Rockford, IL) at 1,000 rpm for 5 min. Slides were fixed in ice-cold acetone for 5 min and stored at −20°C until use. For immunohistochemical analysis of PRR expression in vaginal epithelia, vaginae from estrogenized inoculated or uninoculated mice were excised and placed in Tissue-Tek cryomolds (Miles Corp., Elkhart, ID) containing optimal cutting temperature medium (Sakura Finetek USA, Torrance, CA) in an orientation that allowed cross-sectional cutting. Tissues were stored at −70°C and cut in 6-μm sections. Tissue sections were collected on glass slides and fixed in ice-cold acetone for 5 min. Upon hydration, all immunostaining steps were performed using the cell and tissue staining kit containing horseradish peroxidase (HRP)-conjugated 3-amino-9-ethylcarbazole (AEC; R&D Systems) with appropriate primary and secondary antibodies. First, cell or tissue specimens were blocked with peroxidase, goat serum, avidin, and biotin blocking buffers, followed by an overnight incubation with monoclonal rat anti-mouse antibody specific for TLR4, TLR2, MR, SIGNR1, or dectin-1 (10 μg/ml; R&D Systems) or an isotype control (rat IgG2a or IgG2b) at 4°C. The slides were washed and incubated with biotinylated anti-rat IgG for 1 h. The slides were washed and incubated with streptavidin-HRP for 30 min. Finally, the slides were washed and reacted with AEC chromogen substrate. Specimens were counterstained with CAT hematoxylin (Biocare Medical, Concord, CA), preserved in aqueous mounting medium (R&D Systems), and observed by light microscopy. For quantitative analyses of immunocytochemical data, the numbers of positively stained epithelial cells were enumerated in five nonadjacent fields per sample at 100×, and the percent positively stained epithelial cells was calculated.
Primary cell culture and in vitro C. albicans infection.
Mouse primary vaginal epithelial cell cultures were established from vaginal tissue explants obtained from estrogenized uninoculated mice deficient in MR, TLR4 (C3H/HeJ), and SIGNR1 as well as C57BL/6, C3H/HeOuJ, and BALB/c mice as wild-type controls, respectively. Vaginal explants were placed in the centers of wells in a 12-well culture plate (Corning) containing 500 μl keratinocyte serum-free medium (KSFM; Invitrogen) supplemented with 50 μg/ml bovine pituitary extract, 0.1 ng/ml epidermal growth factor, 100 U/ml penicillin, and 100 μg/ml streptomycin and cultured at 37°C with 5% CO2. Upon reaching near confluence, epithelial cells were inoculated with 1 × 105 C. albicans in a volume of 1 ml KSFM for 24 h. This time point was identified as optimal for S100 alarmin mRNA induction following preliminary evaluation of S100A8 and S100A9 expression from 6 to 48 h postinoculation (data not shown).
Gene expression analysis.
Vaginal epithelial cells were harvested from the cultures described above by incubation in 250 μl trypsin-EDTA (0.05%; Invitrogen) for 1 min at 37°C. In select experiments, vaginal epithelial cells were harvested by dispase treatment of vaginae excised from inoculated mice as previously described (13). The cells were washed in PBS and subjected to total RNA extraction using the RNeasy minikit (Qiagen, Valencia, CA). Synthesis of cDNA from 10 ng of total RNA was completed using 20 U SuperScript III reverse transcriptase with 5 mM dithiothreitol (Invitrogen), GeneAmp 10× PCR buffer II with 5 mM MgCl2 (Applied Biosystems, Foster City, CA), 1 mM deoxynucleoside triphosphates (GE Healthcare), and 20 U of RNasin RNase inhibitor (Promega, Madison, WI) in a total volume of 10 μl and primed with random hexamers in a 96-well thermal cycler (25°C for 10 min, 48°C for 30 min, and 95°C for 5 min). Real-time PCR was performed by using TaqMan gene expression assays predesigned for mouse S100A8 and S100A9 genes (assay IDs Mn01220132_g1 and Mm00656925_m1, respectively; Applied Biosystems) and Brilliant II quantitative PCR master mix (Stratagene, La Jolla, CA) in a total reaction volume of 25 μl. The PCR products were detected in 45 consecutive cycles (95°C for 15 s and 60°C for 1 min) in an iCycler IQ detection system (Bio-Rad, Hercules, CA). Signals were normalized to β-actin RNA content. Normalized data were used to quantify relative expression levels of S100A8 and S100A9 mRNA using the ΔΔCT method. The results are expressed as the fold increase over expression in epithelial cells cultured alone (for in vitro inoculation) or epithelial cells from estrogenized uninoculated mice (for in vivo inoculation).
Intravaginal administration of S100A8 and S100A9.
Recombinant mouse S100A8 and S100A9 (Escherichia coli-derived; available by custom order from R&D Systems) or culture supernatants containing C. albicans-derived S100A8 or S100A9 were intravaginally administered to estrogenized mice. Recombinant S100A8 and S100A9 (10 μg/ml, the maximal concentration possible) in PBS or vaginal gel formulation (semisolidified with 3% carboxymethylcellulose in PBS; Sigma) were delivered into the vaginal lumen in a volume of 20 μl using a pipette or a microdispenser. S100A8- and S100A9-expressing C. albicans culture supernatants (approximately 400 ng/ml) in YPD medium or vaginal gel formulation in YPD medium were administered following the same procedures. Culture supernatants were used neat or at various dilutions to establish a dose-response range or concentrated to reach the target concentration. Mice were treated daily for 2 days starting at 72 h after subcutaneous estrogen administration. Vaginal PMNs were evaluated daily beginning at 48 h after the initial intravaginal treatment.
Intravaginal antibody neutralization. (i) Anti-S100A8 and S100A9 antibody administration. (a) Intravaginal neutralization of S100A8 and S100A9.
To neutralize S100 alarmins in the vaginal cavity, estrogenized mice were treated with anti-S100A8 and -S100A9 antibodies intravaginally. Estrogenized inoculated mice were administered polyclonal anti-S100A8, anti-S100A9, or both (100 μg/ml; R&D Systems) in 20 μl PBS or vaginal gel formulation on day 1, 2, and 3 postinoculation as described above. Control animals were inoculated and received isotype antibodies in parallel. Vaginal fungal burden and PMNs were evaluated on day 0 (preinoculation) and 4 and 7 days postinoculation.
(b) Confirmation of the neutralizing capacity of anti-S100A8 and anti-S100A9 antibodies.
To determine the efficacy of anti-S100A8 and anti-S100A9 antibodies to neutralize the target proteins, C. albicans culture supernatants containing S100A8 and S100A9 were pretreated with the respective antibodies and tested by ELISA with modifications to the standard protocol described previously (13). Briefly, S100A8 or S100A9 culture supernatants were incubated with anti-S100A8 or anti-S100A9 antibodies used above at various concentrations for 1 h at 37°C. Following incubation, antibody-treated culture supernatants were transferred to 96-well enzyme immunoassay/radioimmunoassay plates (Costar) coated with polyclonal goat anti-mouse S100A8 or S100A9 antibodies (1 μg/ml) as a capture antibody. Serially diluted recombinant mouse S100A8 or S100A9 was included as a protein standard. The plates were further incubated with primary antibodies (monoclonal rat anti-mouse S100A8 or S100A9; 2 μg/ml) followed by a secondary antibody (biotinylated anti-rat IgG; 0.05 μg/ml) and streptavidin-horseradish peroxidase. Colorimetric reaction by tetramethylbenzidine (TMB) and sulfuric acid (2 N) was measured at 450 nm on a microplate reader (Labsystems, Helsinki, Finland). Detected amounts of unbound S100 proteins in the antibody-treated culture supernatants were calculated and expressed as nanograms per microliter.
(ii) Vaginal SIGNR1-TLR4 double deficiency. (a) Preliminary in vitro evaluation of blocking TLR4 biological activity by antibody neutralization.
To test the efficacy of anti-TLR4 antibody to block TLR4 and its activity in proinflammatory responses, primary mouse macrophages were treated with anti-TLR4 antibody and evaluated for proinflammatory interleukin 1β (IL-1β) response to a lipopolysaccharide (LPS) challenge. Briefly, mouse peritoneal exudate cells were subjected to Ficoll-Paque density gradient centrifugation as described above for PMN isolation. Macrophage-enriched cells were enumerated, and 1 × 106 cells were seeded in a 12-well plate. Then the cells were incubated in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% fetal bovine serum (FBS) for 2 h at 37°C with 5% CO2, and nonadherent cells were removed by washing the wells with sterile PBS. Resulting adherent cells were incubated with monoclonal anti-mouse TLR4 antibody (100 μg/ml; R&D Systems) or isotype control for 1 h. After being washed with PBS, the cells were incubated in the DMEM-FBS medium containing LPS (100 ng/ml) for 18 h. Following incubation, the cells were washed with PBS and harvested by treatment with 250 μl of 0.05% trypsin-EDTA for 5 min at 37°C with 5% CO2. The cells were washed twice in PBS, cytospun onto glass slides, and subjected to immunocytochemistry as described for vaginal epithelial cells using polyclonal anti-mouse IL-1β–HRP conjugate (0.4 μg/ml; R&D Systems) as the detection antibody. Inhibition of LPS-induced IL-1β production by anti-TLR4 antibody-treated macrophages confirmed the efficacy of the antibody in TLR4 neutralization in vitro (see Fig. S2 in the supplemental material).
(b) In vivo versus ex vivo competitive binding by anti-TLR4 antibodies.
The efficacy of TLR4 blocking by intravaginal antibody neutralization was confirmed by in vivo and ex vivo competitive binding using unlabeled and biotinylated anti-TLR4 antibodies, respectively. First, the presence of TLR4 on SIGNR1−/− vaginal epithelium was confirmed by immunohistochemical staining of vaginal tissue sections from estrogenized uninoculated SIGNR1−/− mice (see Fig. S3A in the supplemental material). Second, estrogenized SIGNR1−/− mice were intravaginally treated with unlabeled anti-TLR4 or isotype control antibody (100 μg/ml; eBioscience, San Diego, CA) in 20 μl of PBS at 12-h intervals over a 24-h period (3 treatments total). Two days after the last treatment, vaginae were excised and tissue section preparations were stained with biotinylated anti-TLR4 antibody by immunohistochemistry as described above. Successful TLR4 blocking was confirmed by negative staining with biotinylated anti-TLR4 antibody (i.e., intravaginal blocking of TLR4 by the original anti-TLR4 antibody) compared to positive staining on tissues collected from mice given isotype control antibody (i.e., no in vivo blocking; staining by ex vivo anti-TLR4 antibody) (see Fig. S3B in the supplemental material).
(c) Treatment of SIGNR1/mice with anti-TLR4 antibody during infection.
To induce a SIGNR1-TLR4 double deficiency in the vaginal cavity, estrogenized inoculated SIGNR1−/− mice were treated intravaginally with anti-TLR4 antibody as described above, starting at 4 h prior to inoculation, and treatments were repeated at 16 and 24 h postinoculation. Vaginal lavage fluids were evaluated for the presence of PMNs and fungal burden on days 4 postinoculation. In addition, vaginal lavage fluids from inoculated anti-TLR4 antibody-treated SIGNR1−/− mice were examined for S100A8 and PMN migration at 24, 36, and 96 h postinoculation to confirm reduced or delayed PMN migration (see Fig. S4A in the supplemental material) (P = 0.041), S100A8 production (see Fig. S4B in the supplemental material) (P = 0.018), and S100A8 mRNA expression by vaginal epithelial cells (see Fig. S4B, inset).
RESULTS
PMN chemotactic ability of eukaryote-derived S100 alarmins.
Based on the lack of in vitro PMN chemotactic activity by E. coli-derived recombinant S100 proteins (data not shown), together with potent in vitro chemotactic activity of C. albicans-derived S100A8 and S100A9 on PMNs and macrophages, respectively (42), we employed these C. albicans strains as a source of eukaryote-derived recombinant S100 proteins (see Fig. S1 in the supplemental material). Accordingly, we administered the culture supernatants containing the S100 alarmins into the vaginal cavities of mice and evaluated the mice for vaginal PMN migration. For this, estrogenized uninoculated mice were administered vaginal gel preparations containing S100A8, S100A9, or wild-type culture supernatants, while separate groups of mice received the commercial recombinant S100A8 (rS100A8) or rS100A9 in similar gel preparations. The vaginal lavage fluids were observed for the presence of PMNs at various time points posttreatment. Similar to in vitro data, results showed substantial vaginal PMN migration in mice treated with S100A8-containing culture supernatant (P = 0.048 at 48 h) but not in those treated with S100A9-containing supernatants and wild-type culture supernatants or with commercial rS100A8 or rS100A9 (Fig. 1A). YPD medium alone also showed no PMN migration (data not shown). When serially diluted, the vaginal PMN response in mice given S100A8 culture supernatant exhibited a dose-dependent effect in the PMN chemotactic activity (Fig. 1B).
FIG 1.

In vivo PMN chemotactic ability of culture supernatants containing C. albicans-derived S100 alarmins. (A) PMN migration induced by S100 alarmins of C. albicans or E. coli origin. Overnight culture supernatants of S100A8, S100A9, and wild-type C. albicans were semisolidified with carboxymethylcellulose (3%) to reach a consistency of vaginal gel formulation. The vaginal gel preparations were administered intravaginally to estrogenized uninoculated mice once daily for 2 days in a volume of 20 μl per mouse using a microdispenser. Gel preparations of E. coli-derived recombinant S100A8 (rS100A8), S100A9 (rS100A9), or PBS (gel control) were tested in parallel. Vaginal lavage samples were collected prior to treatment and then daily after the last treatment. Vaginal PMNs were quantified by identifying PMNs by Pap smear staining and enumerating PMNs in 5 high-powered fields (×400 magnification) per mouse, and values were averaged. (B) Dose-dependent effects of S100A8-containing culture supernatant in vaginal PMN migration. Estrogenized uninoculated mice were treated with culture supernatant from S100A8-producing C. albicans at various dilutions. Vaginal lavage fluids were evaluated for PMNs at 48 h postadministration. The figure presents cumulative data from two repeats. *, P < 0.05 compared to the estrogenized untreated group (0 h). SEM, standard error of the mean.
Requirement of S100 alarmins in the vaginal PMN response. (i) Intravaginal neutralization of S100 alarmins.
Based on the abrogation of PMN migration following in vitro neutralization of S100A8 (13), attempts were made to similarly inhibit PMN migration in vivo in inoculated mice. The first approach was antibody neutralization, where mice inoculated with C. albicans were given several intravaginal treatments with anti-S100A8, anti-S100A9, or a combination of the two antibodies and evaluated for vaginal PMN migration. Unlike in in vitro neutralization, PMN migration in anti-S100A8- and/or anti-S100A9-treated and isotype antibody-treated mice was similar (Fig. 2). Results did not differ whether the antibodies were given in PBS or the gel formulation (data not shown). Of note, the neutralizing capacity of the anti-S100A8 and anti-S100A9 antibodies was confirmed by abrogation of detection of the respective recombinant proteins by ELISA in a dose-dependent manner (see Fig. S5 in the supplemental material).
FIG 2.
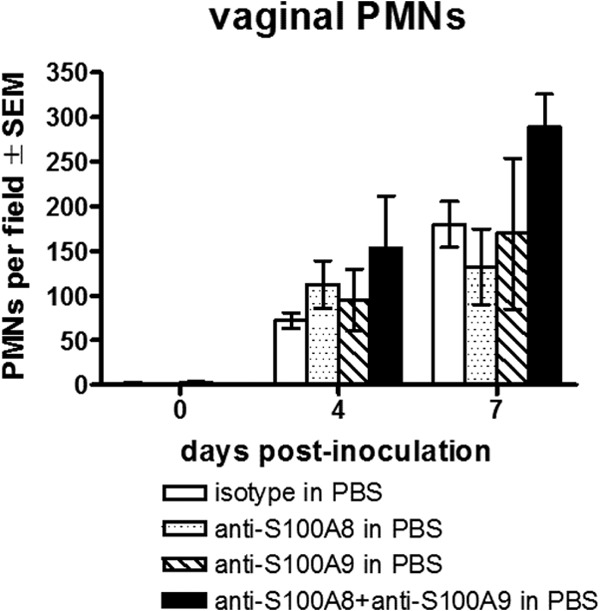
Antibody neutralization of S100A8 and S100A9 during infection. Estrogenized inoculated mice were intravaginally treated with anti-S100A8 (100 μg/ml), anti-S100A9 (100 μg/ml), or a combination of the two antibodies in a volume of 20 μl on days 1, 2, and 3 postinoculation. Vaginal lavage samples were collected prior to inoculation and on days 4 and 7 postinoculation. Vaginal PMNs were quantified by identifying PMNs by Pap smear staining and enumerating PMNs in 5 high-powered fields (magnification, ×400) per mouse and averaged. The figure presents cumulative data from three repeats with 5 mice per group. SEM, standard error of the mean.
(ii) Inoculation of S100 alarmin-deficient mice.
To further investigate the requirement for S100 alarmins in vaginal PMN migration, S100 alarmin-deficient mice (S100A9−/−, i.e., lacking both S100A8 and S100A9 proteins) were inoculated with C. albicans and evaluated for the PMN response. As shown in Fig. 3A, although S100 alarmin-deficient mice exhibited a reduction in vaginal fungal burden compared to wild-type mice (P < 0.0001), PMN migration was equivalent between the two strains (Fig. 3B).
FIG 3.
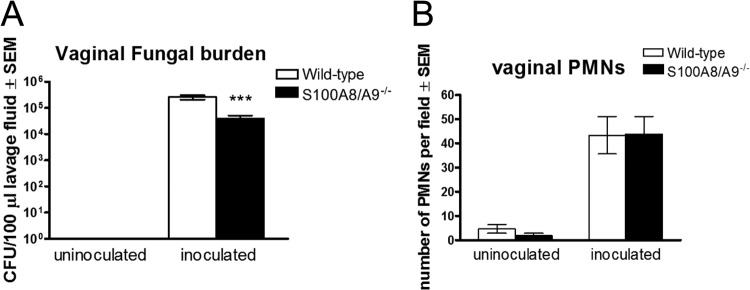
Role of S100 alarmins in the PMN response during infection. Quantification of vaginal Candida burden (A) and vaginal PMNs (B). The number of CFU per 100 μl of lavage fluids from estrogenized S100A8- and S100A9-deficient or C57BL/6 wild-type mice inoculated with C. albicans was assessed on day 4 postinoculation. PMNs in vaginal lavage fluid were identified by Pap smear staining and enumerated in 5 high-powered fields (magnification, ×400) per mouse. The figure presents cumulative data from three repeats with 10 mice per group. ***, P < 0.0001 compared to the wild-type control group. SEM, standard error of the mean.
Identification of PRRs involved in the induction of S100 alarmins. (i) PRR expression by vaginal epithelial cells during infection.
We previously reported that C. albicans adherence to vaginal epithelium at early stages of colonization is crucial for the induction of S100 alarmins and ensuing PMN response (13). Based on this result, we were interested in which host PRR(s) was required for the S100 response. First, it was important to identify/confirm which PRRs were expressed by vaginal epithelial cells and to examine any modulation as a result of C. albicans interaction. For this, epithelial cells from vaginal lavage fluid of uninoculated and day 4-inoculated mice were evaluated for selected PRRs with known interactions with C. albicans, namely, TLR4, TLR2, MR, SIGNR1, and dectin-1 (43–47). As summarized in Table 1, expression of SIGNR1 (P = 0.022) and MR (P = 0.003) was increased on vaginal epithelial cells from inoculated mice experiencing symptomatic conditions (high PMNs) compared to those with asymptomatic conditions (low PMNs) (13) and those from uninoculated mice. Expression of TLR4, though expressed on vaginal epithelial cells, remained unchanged following inoculation (P = 0.874). TLR2 and dectin-1 were not detected on vaginal epithelial cells under any condition. Representative positive and negative immunohistochemical images for SIGNR1 (positive), dectin-1 (negative), and isotype control (negative) are shown in Fig. S6 in the supplemental material.
TABLE 1.
PRR expression on vaginal epithelial cells 4 days postinoculationa
| PRR | % positive cells per field (mean ± SEM) |
Pb | ||
|---|---|---|---|---|
| Inoculated, high PMNs | Inoculated, low PMNs | Uninoculated | ||
| SIGNR1 | 30.7 ± 5.9 | 9.97 ± 3.4 | 8.99 ± 1.7 | 0.022 |
| Mannose receptor | 2.6 ± 0.6 | 0.21 ± 0.02 | 0.408 ± 0.20 | 0.003 |
| TLR4 | 5.98 ± 1.9 | 6.49 ± 2.5 | 3.02 ± 1.2 | 0.87 |
Results are representative of 3 experiments with 4 to 6 mice per group.
Statistical analyses (Student's t test) were performed on data from the inoculated groups comparing animals with high PMNs to those with low PMNs. No positively stained epithelial cells were detected for TLR2 or dectin-1.
(ii) In vitro screening by PRR-deficient cell cultures.
Based on data showing positive expression of SIGNR1, MR, and TLR4 in vaginal epithelial cells following inoculation, we next focused on examining their role in the S100 alarmin induction and subsequent PMN response. As an initial screen prior to in vivo studies, primary vaginal epithelial cell cultures from PRR-deficient mice were established and observed for S100 alarmin induction following inoculation in vitro. Results showed that epithelial cells from MR−/− mice responded to C. albicans with S100A8 and S100A9 mRNAs similarly to wild-type mice. In contrast, epithelial cells from TLR4-defective and SIGNR1−/− mice showed negligible upregulation in S100A8 and S100A9 mRNAs in response to C. albicans (Fig. 4).
FIG 4.
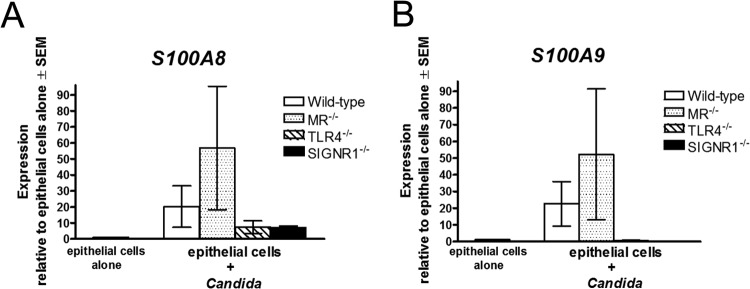
In vitro screening of S100 alarmin response by PRR-deficient vaginal epithelial cells. Primary vaginal epithelial cell cultures derived from wild-type, MR−/−, TLR4-deficient, and SIGNR1−/− vaginal tissue explants were inoculated with C. albicans, and S100A8 (A) and S100A9 (B) mRNA expression was quantified by real-time PCR 24 h following inoculation. The expression of the target genes was normalized to that of the β-actin gene and expressed as the fold increase over expression in epithelial cells cultured alone in parallel. The figure presents cumulative data from two repeats. SEM, standard error of the mean.
(iii) In vivo evaluation of PRR-deficient mice.
Based on the in vitro screening that identified TLR4 and SIGNR1 as candidate PRRs important for the S100 alarmin induction, the possibility of a role for TLR4 and SIGNR1 in the PMN response was further investigated in vivo. For this, TLR4-defective C3H/HeJ mice and SIGNR1−/− mice were inoculated with C. albicans together with the respective wild-type mice (C3H/HeOuJ and BALB/c, respectively) and evaluated for vaginal PMN migration. Results showed that both vaginal fungal burden (Fig. 5A and C) and PMN migration (Fig. 5B and D) were similar in the different mouse strains.
FIG 5.

Role of TLR4 and SIGNR1 in the PMN response following inoculation with C. albicans. (A and C) Quantification of vaginal Candida burden. The numbers of CFU per 100 μl of lavage fluids from estrogenized inoculated C3H/HeJ (TLR4-defective) (A), SIGNR1−/− (C) or appropriate wild-type strains of mice were assessed 4 days postinoculation. (B and D) Quantification of PMNs in vaginal lavage fluids. PMNs from estrogenized inoculated C3H/HeJ (TLR4-defective) (B), SIGNR1−/− (D), or the wild-type strains were identified by Pap smear staining and enumerated in 5 high-powered fields (magnification, ×400) per mouse and averaged. (E and F) Inoculation of TLR4-neutralized SIGNR1−/− mice with C. albicans. Estrogenized SIGNR1−/− mice were treated with anti-TLR4 or isotype control antibodies (100 μg/ml in PBS) in a volume of 20 μl at −4, 16, and 24 h postinoculation. Vaginal lavage fluids were collected on day 4 postinoculation, and vaginal fungal burden (E) and vaginal PMN migration (F) were assessed. The figure presents cumulative data from two or three repeat experiments with 6 to 10 mice per group. SEM, standard error of the mean.
(iv) Evaluation of the PMN response in TLR4-SIGNR1 doubly deficient conditions.
In consideration of the in vivo results showing no change in PMNs in the absence of TLR4 or SIGNR1, we then proceeded to investigate potential effects of TLR4-SIGNR1 double deficiency on PMN migration during infection. To induce a TLR4-SIGNR1 deficiency, our approach was to neutralize vaginal TLR4 activity in SIGNR1−/− mice using anti-TLR4 antibodies. Following confirmation of a TLR4-SIGNR1 deficiency (see Fig. S2 and S3 in the supplemental material) and further supported by reduced/lagging S100A8 production and PMN migration under the doubly deficient condition compared to those in the wild type (see Fig. S4 in the supplemental material), estrogen-treated inoculated doubly deficient and wild-type mice were evaluated for vaginal fungal burden and PMN migration during the standard observation time (day 4 postinoculation). Results showed that vaginal fungal burden and PMN migration were not modulated by the TLR4/SIGNR1 double deficiency (Fig. 5E and F). Similar results were observed at day 7 postinoculation (data not shown).
DISCUSSION
Based on previously reported in vitro data indicating S100A8 as a strong candidate for the chemotactic factor associated with the PMN-mediated acute vaginal inflammatory response (13), the current studies further investigated the biological role of S100A8 in vivo. Experimental questions included (i) whether S100A8 was sufficient to induce PMN chemotaxis into the vaginal cavity, (ii) whether S100A8 was necessary for the PMN response, and (iii) identification of candidate PRRs involved in recognition of C. albicans to signal the induction of S100 alarmins by vaginal epithelial cells.
Taking into account the lack of chemotactic function by commercial recombinant S100 proteins made in bacterial expression systems, we employed a eukaryotic expression system whereby C. albicans was engineered to secrete S100 alarmins. Precedent for this comes from Cryptococcus neoformans engineered to secrete mouse IFN-γ (48). These S100A8 and S100A9 alarmin-expressing C. albicans constructs were recently validated for production, secretion, and chemotactic function (42).
The use of S100 alarmin-containing C. albicans culture supernatants permitted great flexibility in both in vitro and in vivo experimental designs, where the amount of S100 alarmins could be adjusted accordingly to achieve desired concentrations. With this design, consistent with our previous in vitro antibody neutralization results (13) and more recent in vitro PMN chemotaxis data (42), C. albicans-derived S100A8, but not S100A9, induced substantial PMN chemotaxis in vivo in a dose-dependent manner following intravaginal administration. These results suggest that S100A8 is sufficient to mediate the PMN response in the vaginal cavity. Of note, we recognize that factors in the culture supernatants may play a role along with S100A8 as a codependent pathogen-associated molecular pattern (PAMP). However, since little to no PMN migration was induced by culture supernatants from the wild-type or S100A9-producing strain, we contend that the effects of other Candida-derived molecules on vaginal inflammation are minimal and certainly are not independent of S100A8. It is interesting that the rS100 proteins derived from bacterial expression vectors were not equally chemotactic. Possible explanations include the need for posttranslational modifications, which is often the case in eukaryotic hosts, or a suboptimal oxidation state (49, 50). Conversely, the eukaryote-derived S100 proteins did not appear to be compromised by low oxidation states, as evidenced by the positive in vitro chemotactic activity of both proteins (42).
A strong biological role of C. albicans-derived S100A8 in PMN migration prompted a series of in vivo studies to examine whether S100 alarmins were also necessary for the vaginal PMN response. Unlike the previous in vitro antibody neutralization study (13), in vivo administration of anti-S100A8 in buffer or gel formulation had no effect on PMN migration during infection. This could have been due to low binding efficiency of the antibodies in vivo despite effective in vitro blocking (see Fig. S5 in the supplemental material). Moreover, we could not rule out the lack of effective neutralization of biological activity by the antibodies despite sufficient binding. However, in lieu of using additional designs to evaluate biological neutralization (i.e., immunoprecipitation), we proceeded to employ S100A9−/− mice, which are deficient in both S100A8 and S100A9 proteins. Similar to the antibody neutralization results, the S100 alarmin-deficient mice exhibited a PMN response similar to that of wild-type mice following inoculation. Taken together, these data suggest that S100 alarmins are not a strict requirement for the induction of the PMN response and that other factors are likely involved during in vivo infection. Although previous work by our laboratory showed no apparent difference in the levels of classical inflammatory cytokines/chemokines compared between symptomatic and asymptomatic conditions (5, 51, 52), our current data suggest involvement of as-yet-unidentified classical or nonclassical mediators in the inflammatory response. Future crossover in vitro chemotactic studies utilizing PMNs from S100A9−/− or wild-type mice stimulated by vaginal lavage fluid from the reciprocal infected mice may provide evidence for an alternative PMN chemotactic mediator(s). It will also be interesting to further dissect the differential mechanisms in PMN chemotactic activity by receptor analyses, including RAGE (receptor for advanced glycation end products), a known receptor for S100 alarmins, and CXCR2, a receptor for a wide array of chemokines that mediate PMN migration. Interestingly, despite the lack of differences in PMN migration between S100A9−/− and wild-type mice, the S100A9−/− mice had reduced fungal burden. This could be due to potential alarmin-dependent effects on immunoregulation, where growth of C. albicans is affected by antifungal host responses that may become active in the absence of alarmins. Future studies are under way to examine this further. Finally, studies to evaluate the role of the inflammasome (53, 54) during infection may shed light, especially based on data from experimental oral C. albicans infection (55).
Taking into account previous reports showing that early C. albicans adherence to the vaginal epithelium is required for the S100 alarmin-mediated PMN response, we next investigated PRR expression on vaginal epithelial cells and their potential role for the induction of PMN migration. Among several PRRs known for C. albicans recognition, preliminary immunocytochemical analysis of PRR expression profile on vaginal epithelial cells from inoculated mice allowed us to prioritize PRRs for the subsequent in vitro and in vivo evaluations. For in vitro screening, we utilized primary vaginal epithelial cell cultures derived from PRR-deficient mice and monitored them for S100 alarmin mRNA expression following C. albicans challenge. With this approach, two strong PRR candidates, TLR4 and SIGNR1, were identified. However, despite the clear abrogation of the S100 alarmin mRNAs by TLR4- and SIGNR1-deficient epithelial cells in response to C. albicans in vitro, the lack of these PRRs individually failed to modulate the PMN response in vivo. Thus, compensatory processes are suggested. Indeed, documented evidence for cooperative functions by multiple PRRs in recognition of microbial PAMPs has been reported (56, 57). However, attempts to address this by creating and testing a TLR4-SIGNR1-deficient condition still failed to identify the limits for the PRRs involved in the PMN response. Thus, the in vivo PMN response appears to be compensated by PRRs in addition to TLR4 and SIGNR1. We cannot exclude the possibility, however, that despite the blocking of TLR4 in vivo, the biological activity remained intact. We feel that this is unlikely based on in vitro data showing abrogation of LPS-TLR4-mediated IL-1β production by macrophages using the same anti-TLR4 antibody (see Fig. S2 in the supplemental material). Moreover, evaluation of early (24 to 36 h) S100A8 production (mRNA and protein) and PMN migration showed a significant lag of both parameters following inoculation under the TLR4-SIGNR1-deficient condition compared to wild-type (see Fig. S4 in the supplemental material). Hence, the epithelial cell response was compromised in the absence of TLR4-SIGNR1 signaling. The subsequent PMN migration and presence of S100A8 later after inoculation (96 h) provide additional evidence for compensatory PRRs and the secretion of another PMN chemotactic factor(s) in the overall response, together with PMN-derived S100A8 production. Future studies will include evaluation of other PRRs (i.e., complement receptor 3, Fcγ receptor, formyl peptide receptor) or groups of PRRs via adaptor proteins (i.e., MyD88 and CARD9) and identification of other nontraditional chemotactic factors.
In the present study, we further investigated a biological role for S100 alarmins in the immunopathogenesis of C. albicans vaginal infection. The results indicated that S100 alarmins are sufficient but not necessary for the PMN response during infection. Furthermore, while TLR4 and SIGNR1 appear to be important PRRs responsible for the activation of vaginal epithelial cells leading to the S100 alarmin response, PMN migration appears to involve additional compensatory PRRs and chemotactic factors. Despite less-than-optimal results for the mechanisms surrounding S100 alarmin response in VVC, these danger-signaling molecules still appear to be important mediators contributing to symptomatic infection. Better understanding of the functional role of S100 alarmins in C. albicans-host interactions within the vaginal cavity could lead to development of novel strategies for immunotherapies or diagnostic targets. Clinical studies are under way to explore these possibilities.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alison Quayle and Lyndsey Baiamonte for technical assistance with establishing primary vaginal epithelial cell cultures, and Judy Teale for kindly providing us with MR−/− mice.
This work was supported by research grant R01 AI32556 (PLF) from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH), and in part by Louisiana Vaccine Center and South Louisiana Institute for Infectious Disease Research sponsored by the Louisiana Board of Regents.
Footnotes
Published ahead of print 9 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00861-13.
REFERENCES
- 1.Sobel JD, Faro S, Force R, Foxman B, Ledger WJ, Nyirjesy PR, Reed BD, Summers PR. 1998. Vulvovaginal candidiasis: epidemiologic, diagnostic, and therapeutic considerations. Am. J. Obstet. Gynecol. 178:203–211. 10.1016/S0002-9378(98)80001-X [DOI] [PubMed] [Google Scholar]
- 2.Fidel PL., Jr 2002. Distinct protective host defenses against oral and vaginal candidiasis. Med. Mycol. 40:359–375. 10.1080/mmy.40.4.359.375 [DOI] [PubMed] [Google Scholar]
- 3.Black CA, Eyers FM, Russell A, Dunkley ML, Clancy RL, Beagley KW. 1999. Increased severity of Candida vaginitis in BALB/c nu/nu mice versus the parent strain is not abrogated by adoptive transfer of T cell enriched lymphocytes. J. Reprod. Immunol. 45:1–18. 10.1016/S0165-0378(99)00017-0 [DOI] [PubMed] [Google Scholar]
- 4.Fidel PL, Jr, Lynch ME, Redondo-Lopez V, Sobel JD, Robinson R. 1993. Systemic cell-mediated immune reactivity in women with recurrent vulvovaginal candidiasis (RVVC). J. Infect. Dis. 168:1458–1465. 10.1093/infdis/168.6.1458 [DOI] [PubMed] [Google Scholar]
- 5.Taylor BN, Saavedra M, Fidel PL., Jr 2000. Local Th1/Th2 cytokine production during experimental vaginal candidiasis. Med. Mycol. 38:419–431 [DOI] [PubMed] [Google Scholar]
- 6.Fidel PL, Jr, Sobel JD. 1996. Immunopathogenesis of recurrent vulvovaginal candidiasis. Clin. Microbiol. Rev. 9:335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fidel PL, Jr, Sobel JD. 1999. Murine models of Candida vaginal infections, p 741–748 In Zak O, Sande M. (ed), Experimental models in antimicrobial chemotherapy, 2nd ed. Academic Press Ltd., London, United Kingdom [Google Scholar]
- 8.Fidel PL, Jr, Barousse M, Espinosa T, Chesson RR, Dunlap K. 2003. Local immune responsiveness following intravaginal challenge with Candida antigen in adult women at different stages of the menstrual cycle. Med. Mycol. 41:97–109. 10.1080/mmy.41.2.97.109 [DOI] [PubMed] [Google Scholar]
- 9.Fidel PL, Jr, Wormley FL, Jr, Chaiban J, Chesson RR, Lounev V. 2001. Analysis of the CD4 protein on human vaginal CD4+ T cells. Am. J. Reprod. Immunol. 45:200–204. 10.1111/j.8755-8920.2001.450402.x [DOI] [PubMed] [Google Scholar]
- 10.LeBlanc DM, Barousse MM, Fidel PL., Jr 2006. A role for dendritic cells in immunoregulation during experimental vaginal candidiasis. Infect. Immun. 74:3213–3221. 10.1128/IAI.01824-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lilly EA, Yano J, Fidel PL., Jr 2010. Annexin-A1 identified as the oral epithelial cell anti-Candida effector moiety. Mol. Oral Microbiol. 25:293–304. 10.1111/j.2041-1014.2010.00579.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fidel PL, Jr, Barousse M, Espinosa T, Ficarrai M, Sturtevant J, Martin DH, Quayle AJ, Dunlap K. 2004. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect. Immun. 72:2939–2946. 10.1128/IAI.72.5.2939-2946.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yano J, Lilly E, Barousse M, Fidel PL., Jr 2010. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect. Immun. 78:5126–5137. 10.1128/IAI.00388-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gebhardt C, Németh J, Angel P, Hess J. 2006. S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 72:1622–1631. 10.1016/j.bcp.2006.05.017 [DOI] [PubMed] [Google Scholar]
- 15.Kumar RK, Yang Bilson ZS, Thliveris S, Cooke BE, Geczy CL. 2001. Dimeric S100A8 in human neutrophils is diminished after phagocytosis. J. Leukoc. Biol. 70:59–64 http://www.jleukbio.org/content/70/1/59.long [PubMed] [Google Scholar]
- 16.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. 2003. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J. Immunol. 170:3233–3242 http://www.jimmunol.org/content/170/6/3233.long [DOI] [PubMed] [Google Scholar]
- 17.Ross KF, Herzberg MC. 2001. Calprotectin expression by gingival epithelial cells. Infect. Immun. 69:3248–3254. 10.1128/IAI.69.5.3248-3254.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foell D, Wittkowski H, Ren Z, Turton J, Pang G, Daebritz J, Ehrchen J, Heidemann J, Borody T, Roth J, Clancy R. 2008. Phagocyte-specific S100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. J. Pathol. 216:183–192. 10.1002/path.2394 [DOI] [PubMed] [Google Scholar]
- 19.Zimmer DB, Cornwall EH, Landar A, Song W. 1995. The S100 protein family: history, function, and expression. Brain Res. Bull. 37:417–429. 10.1016/0361-9230(95)00040-2 [DOI] [PubMed] [Google Scholar]
- 20.Sohnle PG, Hahn BL, Santhanagopalan V. 1996. Inhibition of Candida albicans growth by calprotectin in the absence of direct contact with the organisms. J. Infect. Dis. 174:1369–1372. 10.1093/infdis/174.6.1369 [DOI] [PubMed] [Google Scholar]
- 21.Urban CF, Ermert DSM, Abu-Abed U, Goosman C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5:e1000639. 10.1371/journal.ppat.1000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandal K, Rouleau P, Boivin A, Ryckman C, Talbot M, Tessier PA. 2003. Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J. Immunol. 171:2602–2609 http://www.jimmunol.org/content/171/5/2602.long [DOI] [PubMed] [Google Scholar]
- 23.Cornish CJ, Devery JM, Poronnik P, Lackmann M, Cook DI, Geczy CL. 1996. S100 protein CP-10 stimulates myeloid cell chemotaxis without activation. J. Cell. Physiol. 166:427–437. 10.1002/(SICI)1097-4652(199602)166:2<427::AID-JCP21>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- 24.Devery JM, King NJ, Geczy CL. 1994. Acute inflammatory activity of the S100 protein CP-10: activation of neutrophils in vivo and in vitro. J. Immunol. 152:1888–1897 [PubMed] [Google Scholar]
- 25.Kocher M, Kenny PA, Farram E, Abdul Majid KB, Finlay-Jones JJ, Geczy CL. 1996. Functional chemotactic factor CP-10 and MRP-14 are abundant in murine abscesses. Infect. Immun. 64:1342–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foell D, Frosch M, Sorg C, Roth J. 2004. Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin. Chim. Acta 344:37–51. 10.1016/j.cccn.2004.02.023 [DOI] [PubMed] [Google Scholar]
- 27.Pivarcsi A, Nagy I, Koreck A, Kis K, Kenderessy-Szabo A, Szell M, Dobozy A, Kemeny L. 2005. Microbial compounds induce the expression of pro-inflammatory cytokines, chemokines and human beta-defensin-2 in vaginal epithelial cells. Microbes Infect. 7:1117–1127. 10.1016/j.micinf.2005.03.016 [DOI] [PubMed] [Google Scholar]
- 28.Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, Schaller M. 2007. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J. Clin. Invest. 117:3664–3672. 10.1172/JCI28115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor PR, Brown GD, Herre J, Williams DL, Willment JA, Gordon S. 2004. The role of SIGNR1 and the beta-glucan receptor (dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J. Immunol. 172:1157–1162 http://www.jimmunol.org/content/172/2/1157.long [DOI] [PubMed] [Google Scholar]
- 30.Geraldino TH, de Vito E, Custódio LA, Conchon-Costa I, Gaziri LC, Felipe I, Loyola W, Bonifácio KL. 2010. Increased tumour necrosis factor-alpha production, higher mannose receptor activity and ability to kill Candida by concanavalin-A-activated macrophages. FEMS Immunol. Med. Microbiol. 59:11–17. 10.1111/j.1574-695X.2010.00655.x [DOI] [PubMed] [Google Scholar]
- 31.Heinsbroek SE, Taylor PR, Martinez FO, Martinez-Pomares L, Brown GD, Gordon S. 2008. Stage-specific sampling by pattern recognition receptors during Candida albicans phagocytosis. PLoS Pathog. 4:e1000218. 10.1371/journal.ppat.1000218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, Kullberg BJ, Torensma R, Williams DL, Figdor CG. 2008. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 283:20590–20599. 10.1074/jbc.M709334200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dupasquier M, Stoitzner P, van Oudenaren A, Romani N, Leenen PJ. 2004. Macrophages and dendritic cells constitute a major subpopulation of cells in the mouse dermis. J. Investig. Dermatol. 123:876–879. 10.1111/j.0022-202X.2004.23427.x [DOI] [PubMed] [Google Scholar]
- 34.Kang YS, Yamazaki S, Iyoda T, Pack M, Bruening SA, Kim JY, Takahara K, Inaba K, Steinman RM, Park CG. 2003. SIGN-R1, a novel C-type lectin expressed by marginal zone macrophages in spleen, mediates uptake of the polysaccharide dextran. Int. Immunol. 15:177–186. 10.1093/intimm/dxg019 [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y, Kawasaki H, Hsu SC, Lee RT, Yao X, Plunkett B, Fu J, Yang K, Lee YC, Huang SK. 2010. Oral tolerance to food-induced systemic anaphylaxis mediated by the C-type lectin SIGNR1. Nat. Med. 16:1128–1133. 10.1038/nm.2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yáñez A, Flores A, Murciano C, O'Conner JE, Gonzalbo D, Gil ML. 2010. Signalling through TLR2/MyD88 induces differentiation of murine bone marrow stem and progenitor cells to functional phagocytes in response to Candida albicans. Cell Microbiol. 12:114–128. 10.1111/j.1462-5822.2009.01382.x [DOI] [PubMed] [Google Scholar]
- 37.Marakalala MJ, Vautier S, Potrykus J, Walker LA, Shepardson KM, Hopke A, Mora-Montes HM, Kerrigan A, Netea MG, Murray GI, MacCallum DM, Wheeler R, Munro CA, Gow NA, Cramer RA, Brown AJ, Brown GD. 2013. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS Pathog. 9:e1003315. 10.1371/journal.ppat.1003315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fidel PL, Jr, Cutright JL, Sobel JD. 1995. Effects of systemic cell-mediated immunity on vaginal candidiasis in mice resistant and susceptible to Candida albicans infections. Infect. Immun. 63:4191–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Black CA, Eyers FM, Dunkley ML, Clancy RL, Beagley KW. 1999. Major histocompatibility haplotype does not impact the course of experimentally induced murine vaginal candidiasis. Lab. Anim. Sci. 49:668–672 [PubMed] [Google Scholar]
- 40.Fidel PL, Jr, Lynch ME, Sobel JD. 1993. Candida-specific cell-mediated immunity is demonstrable in mice with experimental vaginal candidiasis. Infect. Immun. 61:1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saavedra M, Taylor B, Lukacs N, Fidel PL., Jr 1999. Local production of chemokines during experimental vaginal candidiasis. Infect. Immun. 67:5820–5829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnston DA, Yano J, Fidel PL, Jr, Eberle KE, Palmer GE. 2013. Engineering Candida albicans to secrete a host immunomodulatory factor. FEMS Microbiol. Lett. 346:131–139. 10.1111/1574-6968.12211 [DOI] [PubMed] [Google Scholar]
- 43.Netea MG, Brown GD, Kullberg BJ, Gow NA. 2008. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6:67–78. 10.1038/nrmicro1815 [DOI] [PubMed] [Google Scholar]
- 44.Reid DM, Gow NA, Brown GD. 2009. Pattern recognition: recent insights from Dectin-1. Curr. Opin. Immunol. 21:30–37. 10.1016/j.coi.2009.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8:31–38. 10.1038/ni1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Veerdonk FL, Kullberg BJ, van der Meer JW, Gow NA, Netea MG. 2008. Host-microbe interactions: innate pattern recognition of fungal pathogens. Curr. Opin. Microbiol. 11:305–312. 10.1016/j.mib.2008.06.002 [DOI] [PubMed] [Google Scholar]
- 47.Willment JA, Brown GD. 2008. C-type lectin receptors in antifungal immunity. Trends Microbiol. 16:27–32. 10.1016/j.tim.2007.10.012 [DOI] [PubMed] [Google Scholar]
- 48.Wormley FL, Jr, Perfect JR, Steele C, Cox GM. 2007. Protection against cryptococcosis by using a murine gamma interferon-producing Cryptococcus neoformans strain. Infect. Immun. 75:1453–1462. 10.1128/IAI.00274-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison CA, Raftery MJ, Walsh J, Alewood P, Lismaa SE, Thliveris S, Geczy CL. 1999. Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J. Bio. Chem. 274:8561–8569. 10.1074/jbc.274.13.8561 [DOI] [PubMed] [Google Scholar]
- 50.Sroussi HY, Berline J, Palefski JM. 2007. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J. Leukoc. Biol. 81:818–824. 10.1189/jlb.0706433 [DOI] [PubMed] [Google Scholar]
- 51.Barousse M, Van Der Pol BJ, Fortenberry D, Orr D, Fidel PL., Jr 2004. Vaginal yeast colonization, prevalence of vaginitis, and associated local immunity in adolescents. Sex. Transm. Infect. 80:48–53. 10.1136/sti.2002.003855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yano J, Kolls JK, Happel KI, Wormley FL, Jr, Wozniak KL, Fidel PL., Jr 2012. The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS One 7:e46311. 10.1371/journal.pone.0046311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lev-Sagie A, Prus D, Linhares I, Lavy Y, Ledger W, Witkin S. 2009. Polymorphism in a gene coding for the inflammasome component NALP3 and recurrent vulvovaginal candidiasis in women with vulvar vestibulitis syndrome. Am. J. Obstet. Gynecol. 200:e1–e6. 10.1016/j.ajog.2008.10.039 [DOI] [PubMed] [Google Scholar]
- 54.Joly S, Sutterwala F. 2010. Fungal pathogen recognition by the NLRP3 inflammasome. Virulence 1:276–280. 10.4161/viru.1.4.11482 [DOI] [PubMed] [Google Scholar]
- 55.Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, Fotzgerald KA, Hise AG. 2011. A novel role for the NLRC4 inflammasome in mucosal defense against the fungal pathogen Candida albicans. PLoS Pathog. 7:e1002379. 10.1371/journal.ppat.1002379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagaoka K, Takahara K, Tanaka K, Yoshida H, Steinman RM, Saitoh S, Akashi-Takamura S, Miyake K, Kang YS, Park CG, Inaba K. 2005. Association of SIGNR1 with TLR4–MD-2 enhances signal transduction by recognition of LPS in gram-negative bacteria. Int. Immunol. 17:827–836. 10.1093/intimm/dxh264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Netea MG, Gow NAR, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T, Jacobs L, Buurman ET, Gijzen K, Williams DL, Torensma R, McKinnon A, MacCallum DM, Odds FC, van der Meer JWM, Brown AJP, Kullberg BJ. 2006. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Invest. 116:1642–1650. 10.1172/JCI27114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.