

Abstract
Toxoplasma gondii is an obligate intracellular parasite that forms a lifelong infection within the central nervous system of its host. The T. gondii genome encodes six members of the patatin-like phospholipase family; related proteins are associated with host-microbe interactions in bacteria. T. gondii patatin-like protein 1 (TgPL1) was previously determined to be necessary for parasites to suppress nitric oxide and prevent degradation in activated macrophages. Here, we show that in the rapidly replicating tachyzoite stage, TgPL1 is localized within vesicles inside the parasite that are distinct from the dense granules; however, in the encysted bradyzoite stage, TgPL1 localizes to the parasitophorous vacuole (PV) and cyst wall. While we had not previously seen a defect of the TgPL1 deletion mutant (ΔTgPL1) during acute and early chronic infection, the localization change of TgPL1 in bradyzoites caused us to reevaluate the ΔTgPL1 mutant during late chronic infection and in a toxoplasmic encephalitis (TE) mouse model. Mice infected with ΔTgPL1 are more resistant to TE and have fewer inflammatory lesions than mice infected with the wild type and ΔTgPL1 genetically complemented with TgPL1. This increased resistance to TE could result from several contributing factors. First, we found that ΔTgPL1 bradyzoites did not convert back to tachyzoites readily in tissue culture. Second, a subset of cytokine levels were higher in ΔTgPL1-infected mice, including gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and monocyte chemotactic protein 1 (MCP-1). These studies suggest that TgPL1 plays a role in the maintenance of chronic T. gondii infection.
INTRODUCTION
Patatin-like phospholipases (PLPs) belong to a diverse family of proteins that are found in a variety of biological systems. The founding member of this family is patatin, a major component of the soluble glycosylated protein found in potatoes (1). Patatin localizes to the plant storage vacuole (2) but exhibits phospholipase A2 (PLA2) activity when it relocalizes to the cytoplasm under stress conditions (3, 4). Plants induce PLA2 activity in response to stresses such as infection, using the fatty acid and lysophospholipid products as signaling molecules to mount antimicrobial responses (5). Other plant species have patatin homologs that also appear to respond to infection (6, 7). PLPs are also encoded in a number of bacterial genomes and have been associated with severe disease (8). The genomes of host-associated bacteria, including commensals and pathogens, have a higher frequency of PLPs than the genomes of free-living bacteria, indicating a role in host-microbe interactions (9). The genome of the obligate intracellular parasite Toxoplasma gondii encodes six predicted PLPs, the functions of which have not been thoroughly characterized.
Throughout the T. gondii life cycle, the parasite is exposed to many types of stresses within its host. When T. gondii parasites enter the host through the digestive tract, they are confronted with digestive enzymes in the stomach, which they use as a signal to egress from the cysts, escape the acidic environment, and replicate in the peritoneal cells outside the stomach (10). As the parasite replicates in these tissues, the host mounts an immune response, whereupon the parasite is assaulted by a variety of antimicrobial responses, including nitric oxide (NO), reactive oxygen species, and engulfment by phagocytic cells (11, 12). The parasite is able to reduce these antimicrobial responses and to hijack immune cells, such as macrophages and dendritic cells, to disseminate to distal sites in the body (13, 14). Stress from the immune response likely drives the rapidly dividing tachyzoite form to transition to the latent, slow-growing bradyzoite form (15). Immunocompromised individuals often suffer disease symptoms due to the reactivation of a latent infection in the form of encephalitis (16). While pharmaceuticals are effective against the tachyzoite form of T. gondii, novel therapeutics that combat the chronic stage of infection before reactivation are sorely needed.
The T. gondii gene TgPL1 has homology to PLPs and is known to be important for parasite survival in activated macrophages. TgPL1 is localized to punctate vesicles within tachyzoites grown in fibroblasts (17) but changes localization to outside the parasite but within the parasitophorous vacuole (PV) in activated macrophages (18). In this study, we see that TgPL1 localizes to punctate spots in tachyzoites, but in bradyzoites, TgPL1 localizes to the PV and cyst wall. While we had not previously seen a defect of the TgPL1 deletion (ΔTgPL1) mutant during acute and early chronic infection, the localization change caused us to further assess the ΔTgPL1 mutant in a toxoplasmic encephalitis (TE) mouse model. Within tissue cysts, TgPL1 is involved in regulating the host immune response during chronic infection, as ΔTgPL1-infected mice succumb less frequently to encephalitis, with fewer inflammatory lesions and higher cytokine levels.
MATERIALS AND METHODS
T. gondii strains and cell culture.
The parental wild-type T. gondii strain was Pru, in which the HPT gene was replaced with firefly luciferase under the control of the tubulin promoter (18). The ΔTgPL1 mutant and the genetically complemented strains were generated as previously described (18). Parasites were passaged in confluent human foreskin fibroblasts (HFFs) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 1% penicillin-streptomycin.
Plasmid construction.
The plasmid with a hemagglutinin (HA)-tagged version of TgPL1 (pTgPL1-SacHA) was constructed as described previously (19). A fragment containing the three in-frame start codons of TgPL1 was digested from pEndoTgPL1HA (17) using StuI and SacI and subcloned into pBSII SK+ using SacI and NotI. The first two in-frame start codons were mutagenized to isoleucines using site-directed mutagenesis with the primer 5′-TCCCTTAAAAACGAACATCATCAAGGGTAACAGCGTGG-3′ and its reverse complement. The third in-frame start codon was removed using the primer 5′-CTGTTCTCCTTCTGTATCGACACACGCTCCAGT-3′ and its reverse complement. These mutagenized fragments were then subcloned into a version of pTgPL1-SacHA that lacked the dihydrofolate reductase (DHFR) gene. The DHFR gene from pDHFR-TSC3 (20) was reintroduced by blunt ligation into the EcoRI site. The construct in which the first two start codons were mutagenized is called pTgPL1HAM12QC, and the construct in which the third start codon was mutagenized is called pTgPL1HAM3QC.
Mouse infections.
In 4 independent experiments, 10- to 12-week-old C57BL/6 mice were intraperitoneally infected with 103 tachyzoites for each parasite strain tested. Plaque assays were performed in HFFs immediately after inoculation to confirm parasite counts and viability. Experiments with more than 2-fold differences in plaques between strains were discarded. The surviving mice were sacrificed at 4, 6, 8, and 12 weeks postinfection to determine the cyst burden, as previously described (17). T. gondii mouse bioassays were performed by withholding food from gamma interferon−/− (IFN-γ−/−) mice overnight and then feeding each mouse half of a freshly harvested brain from chronically infected mice. The mice were sacrificed if paralysis, tremors, or severe head tilt, balance, and coordination problems were apparent. Animals were treated in compliance with guidelines set by the Institutional Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health.
For TgPL1 localization from in vivo bradyzoites, 10- to 12-week-old C57BL/6 mice were intraperitoneally infected with 2 × 103 tachyzoites expressing TgPL1-HA. After 22 days, the mice were sacrificed, their brains were removed and ground with a mortar and pestle, and the bradyzoite cysts were purified with a Percoll gradient (21). The purified cysts were stained for immunofluorescence assay (IFA), as described below.
Serum cytokine levels.
Serum samples were obtained by tail bleed at 4 and 6 weeks postinfection in 2 independent experiments or 8 weeks postinfection in 3 independent experiments. Levels of interleukin 12p70 (IL-12p70), IFN-γ, tumor necrosis factor alpha (TNF-α), monocyte chemotactic protein 1 (MCP-1), IL-6, and IL-10 in serum were measured using a BD Cytometric Bead Array (CBA) Mouse Inflammation kit (BD Biosciences, San Jose, CA).
Bradyzoite differentiation.
HFFs were seeded in 25-cm2 tissue culture flasks or on glass coverslips in 24-well tissue culture plates and grown in 10% FBS, 2 mM l-glutamine, and 1% penicillin-streptomycin in DMEM. The confluent HFFs were infected with 4 × 106 tachyzoites for the 25-cm2 tissue culture flasks or 105 tachyzoites for the 24-well plates. The parasites were allowed to invade for 3 h. After invasion, the medium was replaced with bradyzoite development medium (0.5% FBS, 0.5% penicillin-streptomycin, and 21 mM HEPES in RPMI 1640 without bicarbonate, pH 8.0). The parasites were grown in bradyzoite development medium for 5 days and harvested as indicated. For reactivation studies, bradyzoites were syringe passed first through a 27-gauge needle and then a 30-gauge needle before passage onto HFFs and growth under tachyzoite conditions for 24, 48, or 72 h.
TgPL1 mRNA analysis.
RNA was extracted from tachyzoites and bradyzoites using TRIzol and treated with Invitrogen amplification grade DNase I prior to cDNA synthesis using random hexamers and SuperScript III (Invitrogen, Carlsbad, CA). The predicted intron splice junction in bradyzoites was confirmed by PCR of cDNA with primers 5′-GCTCTGCCACATTCTCGTTC-3′ and 5′-TTGCCTCTTCGTCCATAGGC-3′. For quantitative PCR (qPCR), primer efficiencies (E) for alpha-tubulin 1A, BAG1, and TgPL1 were calculated using the slopes of a 1:10 dilution series (neat through 104) and the following formula: E = 10−1/slope. The efficiencies were 1.93, 1.98, and 2.04, respectively. The reactions used Bio-Rad iTaq Universal SYBR green Supermix and 300-nm primers and were analyzed on an Applied Biosystems StepOnePlus Real-Time PCR system with an extension temperature of 60°C for 60 s. Relative expression was calculated using the 2−ΔΔCt method. Three biological replicates were used. Each biological replicate was conducted in duplicate on each plate and run twice.
Primer sequences were as follows: Tub1A F, 5′-GACGACGCCTTCAACACCTTCTTT-3′; Tub1A Rev, 5′-AGTTGTTCGCAGCATCCTCTTTCC-3′; BAG1 F, 5′-GATGACGTAACCATAGAAGTCGACAAC-3′; BAG1 Rev, 5′-GCAAAATAACCGGACACTCGCTCAGTC-3′; TgPL1 F, 5′-CCGTGAACTGAAGTGGGACG-3′; and TgPL1 Rev, 5′-AAAACCTCCACAGGCTCTCGC-3′.
Western analysis.
Protein samples were harvested from parasites grown in a single 25-cm2 tissue culture flask for tachyzoites and in three 25-cm2 tissue culture flasks for bradyzoite conditions, as described above. Lysates were separated on 10% polyacrylamide gels, transferred to polyvinylidene difluoride (PVDF) membranes, and incubated with mouse anti-HA or rabbit anti-SAG1, anti-BAG1, or anti-β-tubulin antibodies to detect the indicated proteins. Horseradish peroxidase-conjugated donkey anti-mouse and donkey anti-rabbit secondary antibodies were used (Jackson Laboratories, Bar Harbor, ME). The membranes were incubated with ECL Plus detection reagent (GE Healthcare) and visualized by exposure to film.
Immunofluorescence assay.
T. gondii expressing HA-tagged TgPL1 was used for all experiments. Tachyzoites were grown in HFFs on glass coverslips for 24 h, and bradyzoites were grown under bradyzoite development conditions for 5 days before they were washed with PBS and fixed in 3% formaldehyde. Blocking, permeabilization, and staining were performed as previously described (19). Mouse anti-HA monoclonal 16B12 (MMS-101P-200; Covance, Princeton, NJ) was used to detect TgPL1 and costained with rabbit anti-GRA4, rabbit anti-GRA-7, rabbit anti-BAG-1, or biotinylated Dolichos biflorus agglutinin (DBA). Alexa Fluor 488-conjugated donkey anti-mouse was used to visualize the HA, while Alexa Fluor 633-conjugated goat anti-rabbit or streptavidin was used to visualize the costains (Invitrogen, Carlsbad, CA). No fluorescence signal was seen with any of the secondary antibodies alone (data not shown). Coverslips were mounted onto slides using VectaShield mounting medium containing 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Serial image stacks (0.2-μm z increment) were taken at ×100 magnification (PlanApo oil immersion; 1.4 numerical aperture [NA]) using a motorized Zeiss Axioplan III equipped with a rear-mounted excitation filter wheel, a triple-pass (DAPI/fluorescein isothiocyanate [FITC]/Texas Red) emission cube, differential interference contrast optics, and a Hamamatsu ORCA-AG charge-coupled-device (CCD) camera operated by OpenLabs 4.0 software (Improvision, Lexington, MA). Fluorescence images were deconvolved by a constrained iterative algorithm, pseudocolored, and merged using the Volocity software package (Perkin Elmer, Downers Grove, IL).
Determination of brain inflammatory foci.
In 2 independent experiments, three C57BL/6 mice were infected with 103 parasites of each strain (a total of 6 mice per strain) and allowed to establish a chronic infection for 8 weeks. The brains were perfused with saline followed by 4% paraformaldehyde before removal and fixation overnight in 4% paraformaldehyde. Coronal samples 5 μm thick were taken every 100 μm, providing 36 brain sections per animal. The sections were stained with hematoxylin and eosin and examined by light microscopy on the 4× objective, and aggregates of 10 or more inflammatory cells were enumerated.
RESULTS
TgPL1 is secreted from the parasite to localize with the PV and cyst wall in bradyzoites.
Due to its homology to plant patatin, which changes localization under stress conditions, we were not surprised to see TgPL1 localize to the PV when parasites were grown in macrophages (18). We therefore wanted to examine if TgPL1 changed localization under the stress of bradyzoite-inducing conditions. After 5 days under bradyzoite conditions, TgPL1 partially colocalizes with the cyst wall marker DBA and the dense-granule proteins GRA4 and GRA7 (Fig. 1). Similar to the localization change seen for TgPL1 in activated macrophages (18), the localization of TgPL1 to the PV was not readily apparent until the parasites had been under bradyzoite development conditions for at least 5 days. TgPL1 does not colocalize with BAG1, a bradyzoite-specific heat shock protein that is localized in the parasite cytoplasm (Fig. 1). We had previously seen that TgPL1 did not colocalize with any markers of secretory organelles, including dense granules (17), so we reexamined the localization of TgPL1 with GRA4 in tachyzoites. Intracellular tachyzoites were allowed to invade and divide in confluent fibroblast monolayers for 24 h before fixation. As we had seen previously, TgPL1 does not colocalize with GRA4 (Fig. 1). These results show that in tachyzoites, TgPL1 is intracellular but does not colocalize with the dense granules. In bradyzoites, TgPL1 is secreted from the parasite to the PV space.
FIG 1.

TgPL1 localizes within the parasite in tachyzoites but on the cyst wall and PV space in bradyzoites. Tachyzoites were grown on coverslips containing confluent HFFs for 24 h before fixation. Bradyzoites were grown in confluent HFFs on coverslips under low CO2 and high pH for 5 days. The parasites were stained for HA-tagged TgPL1 (green) and costained for dense-granule proteins GRA4 and GRA7, a cytoplasmic bradyzoite-specific heat shock protein (BAG1), or DBA as a marker of the bradyzoite cyst wall (all costain red). Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (blue in the merged images). DIC, differential interference contrast. Scale bar = 5 μm. The scale is identical for all bradyzoite images.
To examine whether the TgPL1 localization change occurs during chronic infection, we stained bradyzoite cysts purified from mouse brains 22 days after infection for TgPL1. Within in vivo bradyzoite cysts, most TgPL1 is present on the cyst wall, partially colocalizing with DBA (Fig. 2).
FIG 2.

TgPL1 localizes to the cyst wall and PV space in bradyzoite cysts from mouse brains. C57BL/6 mice were infected with HA-tagged TgPL1 containing parasites for 22 days. Bradyzoite cysts from the brains were partially purified by Percoll gradient. The cysts were incubated with mouse anti-HA antibody and Alexa Fluor 488-conjugated donkey anti-mouse to detect TgPL1 (green) and biotinylated DBA and Alexa Fluor 633-conjugated streptavidin to visualize the cyst (red).
TgPL1 begins translation at one of the first two in-frame methionines in tachyzoites and bradyzoites.
The mechanism of the localization change of TgPL1 between the tachyzoite and bradyzoite stages is intriguing. The length of time required for TgPL1 to change locations, 5 days either in activated macrophages or under bradyzoite development conditions, suggests that new protein synthesis may occur. TgPL1 has three potential initiator methionines near the predicted translation start site (Fig. 3A). The first two AUGs are adjacent and directly followed by a predicted endoplasmic reticulum (ER)-type signal sequence. The third AUG is found directly after the predicted cleavage site for the signal sequence. To examine the start codon usage for TgPL1, site-directed mutagenesis was employed to remove the first two initiator methionines (M1 and -2) or the third (M3) of HA-tagged TgPL1 (Fig. 3A). While clones containing WT and M3 versions of TgPL1 showed robust expression, none of the clones containing the M1-2 plasmid expressed TgPL1 (Fig. 3B; see Fig. S1 in the supplemental material). Likewise, under bradyzoite conditions, the M1-2 mutant does not produce TgPL1 protein, in contrast to clones containing the WT and the M3 mutant (Fig. 3B). TgPL1 expressed from the M3 mutant also appears to have a size similar to that of WT TgPL1. The WT and M3 proteins also appear to be equally abundant in tachyzoites and bradyzoites, as confirmed by qPCR analysis (Fig. 3C). PCR analysis of the TgPL1 mRNA from tachyzoites and bradyzoites showed that the message is not alternatively spliced in the two stages (data not shown). These results indicate that TgPL1 is translated from one of the first two methionines in both tachyzoites and bradyzoites and that the open reading frame does not change between stages.
FIG 3.

TgPL1 translates from one of the first two ATG codons. (A) Genomic region encompassing the potential start codons. The start codons are in boldface, and a predicted ER signal peptide is labeled with a black bar. At the top is the wild-type sequence with all three potential methionines. In the middle is the M1-2 construct, which lacks the first two start codons. At the bottom is the M3 construct, which lacks the third start codon. (B) Clones of WT, M1-2, and M3 were grown under tachyzoite conditions or bradyzoite conditions for 3 days. The parasites were assessed for protein expression by Western analysis with anti-HA (TgPL1) or anti-β-tubulin (β-TUB). No TgPL1 protein was present when the first two methionines were mutated (M1-2), even on long exposures (data not shown). (C) Relative expression of BAG1 and TgPL1 normalized against tubulin 1A using the 2−ΔΔCt method between tachyzoites and bradyzoites. BAG1 was used to determine the switch efficiency. A relative expression value greater than 1 indicates higher expression in bradyzoites than tachyzoites. Three biological replicates and two technical replicates were performed.
ΔTgPL1 parasites form bradyzoite cysts in vitro and in vivo.
Because TgPL1 expression localizes to the cyst wall, we determined whether the TgPL1 gene is necessary for cyst formation. In vitro cyst formation was assayed by culturing parasites under low-CO2 and high-pH conditions for 2, 3, or 5 days. No differences were seen in the ability of ΔTgPL1 to form cysts at any of the time points when assayed for DBA expression and cyst wall formation (data not shown). In vivo cyst formation was assayed using C57BL/6 mice infected with 103 tachyzoites (a nonlethal dose that allows brain cyst formation) of either WT, ΔTgPL1, or ΔTgPL1 genetically complemented with the TgPL1 genomic locus (complement). At 4, 6, and 8 weeks postinfection, brains were collected, and the numbers of cysts per brain were determined. Cyst numbers from WT-, ΔTgPL1-, and complement-infected mice were similar (Fig. 4).
FIG 4.
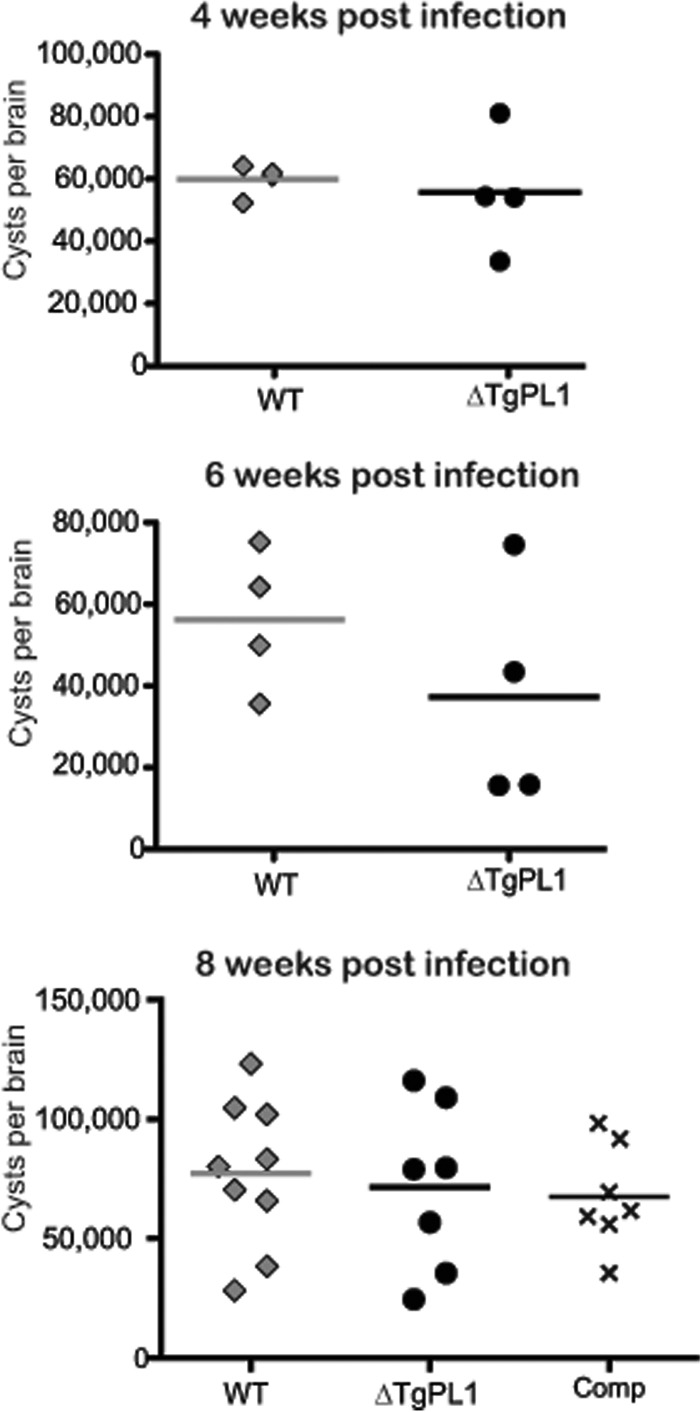
ΔTgPL1 does not have reduced cyst numbers in vivo. C57BL/6 mice were infected with 1,000 tachyzoites of the wild type (diamonds), ΔTgPL1 (circles), or complemented ΔTgPL1 (Comp; ×). Mice were sacrificed at 4, 6, 8, or 12 weeks postinfection, and cyst burdens were assessed by fluorescence microscopy. Each symbol represents cyst counts from an individual mouse, and the horizontal bars indicate the mean of each group. No significant differences in cyst numbers were seen between WT-, ΔTgPL1-, and complement-infected mice.
Mice infected with ΔTgPL1 parasites are less susceptible to TE than WT-infected mice.
To discover whether the TgPL1 gene has a role in the development of TE, C57BL/6 mice were infected with 103 WT, ΔTgPL1, or complement tachyzoites and allowed to progress into chronic infection. Survival rates for mice infected with all three strains were similar during late acute and early chronic infection prior to 7 weeks postinfection. Between 7 and 11 weeks, all of the WT-infected mice were sacrificed due to encephalitic symptoms, including paralysis, tremors, head tilt, and/or severe balance and coordination problems (Fig. 5A). In contrast, the majority of ΔTgPL1-infected mice were asymptomatic and survived past 12 weeks postinfection. Mice infected with the complement strain were symptomatic and needed to be sacrificed at rates similar to those for WT-infected mice. The differences in mortality rates between those infected with WT and ΔTgPL1 (P < 0.0001), ΔTgPL1, and complement (P = 0.0147) were statistically significant but were not significant between WT and complement strains. When we examined the histology of the WT-infected brains, we saw a large number of inflammatory lesions, another indication of encephalitis. These data indicate that TgPL1 may contribute to the induction of TE by 12 weeks postinfection.
FIG 5.

ΔTgPL1-infected mice have delayed TE and reduced brain inflammatory foci. (A) C57BL/6 mice were infected with 1,000 tachyzoites and monitored for survival. The graph is a compilation of 4 independent experiments, with 17 total mice infected with WT parasites, 18 mice infected with ΔTgPL1 parasites, and 12 mice infected with complemented ΔTgPL1 (comp) parasites. For statistical analysis, log rank (Mantel-Cox) values for WT versus ΔTgPL1 had a P value of <0.0001 and for ΔTgPL1 versus comp they had a P value of 0.0147, and differences were not significant for WT versus comp. (B) WT (W), ΔTgPL1 (Δ), or complemented ΔTgPL1 (C) parasites were grown as tachyzoites (Tachy) or bradyzoites (Brady) for 5 days. The bradyzoites were syringe lysed and grown under tachyzoite conditions for 24 (B→T 24 hr), 48 (B→T 48 hr), or 72 (B→T 72 hr) hours. The parasites were assessed for protein expression by Western analysis with anti-SAG1 (SAG1), anti-BAG1 (BAG1), or anti-β-tubulin (β-tub) as a loading control. Shown is a representative immunoblot from three independent experiments. (C) Example of a section of a wild-type-infected brain at 8 weeks postinfection. Sections were stained with hematoxylin and eosin and then scanned using light microscopy to identify aggregates of 10 or more inflammatory cells. The arrowheads indicate examples of foci. Scale bar = 50 μm. (D) Three C57BL/6 mice from two independent experiments (a total of 6 mice per strain) were infected with either WT (diamonds), TgPL1 (circles), or complemented (Comp) (triangles) parasites and allowed to establish a chronic infection for 8 weeks. After fixation, 5-μm sections were taken every 100 μm, providing 36 total sections per animal. Each symbol represents the total number of inflammatory loci counted in the 36 sections for each individual mouse, and the horizontal bars indicate the mean of each group. Significance was analyzed by one-way ANOVA. ***, P < 0.0001.
ΔTgPL1-infected mice maintain viable cysts at 12 weeks postinfection.
One possible reason why the ΔTgPL1-infected mice did not succumb to TE was that the mice had cleared the tissue cysts from the brain. To examine this possibility, the cyst burden was assessed in the surviving mice at 12 weeks postinfection. Cyst burdens in the surviving ΔTgPL1-infected mice ranged from 20,000 to 65,000 cysts per brain, which was comparable to cyst burdens in the ΔTgPL1-infected mice at 4, 6, and 8 weeks postinfection (Fig. 4). Another explanation for the lack of TE in ΔTgPL1-infected mice was that the parasites within the cysts were no longer viable and therefore were unable to cause disease. To assay the viability of the ΔTgPL1 cysts, we performed a mouse bioassay in IFN-γ−/− mice, which are highly susceptible to T. gondii. All mice succumbed to infection at day 7 postinfection, indicating that ΔTgPL1 cysts are infectious at 12 weeks postinfection (data not shown).
ΔTgPL1 parasites do not convert from bradyzoites to tachyzoites efficiently.
Another possible explanation for the reduced TE in ΔTgPL1-infected mice is that ΔTgPL1 parasites do not readily revert from bradyzoite cysts into tachyzoites. To examine this hypothesis, we cultured WT, ΔTgPL1, or complement parasites under bradyzoite induction conditions for 5 days, syringe lysed the cysts, and grew them under tachyzoite conditions. Western blot analysis of the tachyzoite-specific surface antigen SAG1 showed that while ΔTgPL1 parasites express SAG1 normally when grown continuously as tachyzoites, SAG1 is not readily induced in ΔTgPL1 bradyzoites grown under tachyzoite conditions, even after 72 h (Fig. 5B). Conversely, BAG1 was highly induced in ΔTgPL1 bradyzoites and was still abundant when ΔTgPL1 bradyzoites were grown for 72 h under tachyzoite conditions (Fig. 5B). In fact, BAG1 protein was apparent in ΔTgPL1 tachyzoites when the blot was overexposed, despite the fact that the parasites had been continuously grown as tachyzoites for many passages (data not shown). These results indicate that certain stage-specific genes are misregulated in ΔTgPL1 parasites and that ΔTgPL1 parasites may be defective in bradyzoite-to-tachyzoite reactivation.
ΔTgPL1-infected mice have fewer inflammatory lesions at 8 weeks postinfection.
We had seen inflammatory lesions in the brains of WT-infected mice, most likely resulting from reactivation of bradyzoite cysts. As ΔTgPL1 parasites may be defective in bradyzoite reactivation, we determined the number of inflammatory lesions in WT-, ΔTgPL1-, or complement-infected mice. Eight weeks after infection, the brains were perfused, fixed, serially sectioned, and stained with hematoxylin and eosin. Sections throughout the whole brain were analyzed by counting the foci of inflammatory cells per section (Fig. 5C). Mice infected with WT or ΔTgPL1-complemented parasites had statistically significantly higher numbers of inflammatory lesions per brain section than ΔTgPL1-infected brains (Fig. 5D). No inflammatory lesions were seen in uninfected, age-matched control mice.
ΔTgPL1-infected mice have higher cytokine levels at 8 weeks postinfection.
To understand why ΔTgPL1-infected mice had fewer inflammatory lesions in their brains, we measured the levels of cytokines associated with chronic T. gondii infection (IL-12p70, IFN-γ, TNF-α, MCP-1, IL-6, and IL-10). There were no significant differences in the serum levels of any of these cytokines between WT- and ΔTgPL1-infected mice at 4 or 6 weeks postinfection (see Fig. S2 and S3 in the supplemental material). At 8 weeks postinfection, the average levels of IFN-γ, TNF-α, MCP-1, and IL-6 were significantly higher in ΔTgPL1-infected than in WT-infected mice (Fig. 6). These cytokine levels were also higher in ΔTgPL1-infected than in complement-infected mice, but the differences did not reach the significance cutoff. These results indicate that high cytokine levels are maintained in the absence of TgPL1, which prevents the encephalitic state seen in C57BL/6 mice.
FIG 6.

ΔTgPL1-infected mice have higher cytokine levels at 8 weeks postinfection. The graphs are compilations of 3 independent experiments, with 3 uninfected age-matched control mice (uninf) (squares), 10 mice infected with WT parasites (diamonds), 12 mice infected with ΔTgPL1 parasites (circles), and 7 mice infected with complemented ΔTgPL1 parasites (comp) (triangles). Sera were collected at 8 weeks postinfection, and serum cytokine levels were assessed by cytometric bead array for the interleukin 12 heterodimer (IL-12p70) (A), TNF-α (B), IFN-γ (C), MCP-1 (D), IL-10 (E), and IL-6 (F). Each symbol represents cytokine levels from an individual mouse, and the horizontal bars indicate the mean of each group. *, P < 0.05; **, P < 0.01.
DISCUSSION
The development of TE in mice has been well characterized, but the contributions of specific parasite factors involved in the disease have not been determined. In response to stress, the TgPL1 protein changes its localization to the host-parasite interface and alters the parasite's interactions with the host cell. To our knowledge, this is the first report to identify a specific T. gondii gene involved in the development of encephalitis. ΔTgPL1-infected animals are less susceptible to TE and have fewer brain inflammatory lesions and higher serum IFN-γ, TNF-α, MCP-1, and IL-6 levels than WT-infected controls. ΔTgPL1 parasites also appear to be defective in bradyzoite-to-tachyzoite reactivation. These data suggest that in WT parasites, TgPL1 plays a role in reactivation and in reducing cytokine levels during chronic infection, which contribute to the development of TE.
Mice with the H-2b haplotype (which includes C57BL/6 mice) develop inflammatory changes in their brains during chronic infection with T. gondii, whereas mice with the H-2a or H-2d haplotype do not (22). Examining cytokine levels in WT- and ΔTgPL1-infected C57BL/6 mice leading up to the onset of TE gives insights into which immunological processes are likely to be subverted by TgPL1. IFN-γ (23), TNF-α (24), and IL-6 (25) have been shown to be directly involved in resistance to TE. Likewise, MCP-1 is transcribed to high levels in the brains of chronically infected mice (26), and MCP-1−/− mice have more severe neuropathology early in chronic infection than wild-type mice (27). While the serum IL-12 and IL-10 levels did not consistently drop in ΔTgPL1-infected animals as they do for WT-infected mice at 8 weeks postinfection, both of these cytokines have also been previously shown to be critical for limiting inflammation during TE (28, 29). The consistency of our findings with previous results suggests that ΔTgPL1 parasites will be a useful model for studies of TE in H-2b haplotype mice.
We examined multiple possible reasons why ΔTgPL1-infected mice did not succumb to TE. First, we determined whether ΔTgPL1-infected mice clear the tissue cysts over time. Cyst counts performed at 12 weeks postinfection showed cyst burdens comparable to those at 4, 6, and 8 weeks postinfection. Another explanation was that ΔTgPL1 cysts were no longer viable, but a mouse bioassay concluded that ΔTgPL1 cysts were infectious. A third possibility is that ΔTgPL1 cysts are defective in reactivation. While reactivation was grossly examined in the bioassay in IFN-γ−/− mice, these mice are so susceptible to T. gondii that a single viable parasite is lethal, and thus, subtle defects in reactivation would be missed. Therefore, we examined the reactivation of tissue culture bradyzoites by Western blotting for the stage-specific proteins SAG1 and BAG1. When stress-induced bradyzoites were grown under tachyzoite conditions, SAG1 was readily induced in WT and ΔTgPL1-complemented parasites, but not in ΔTgPL1 parasites. Expression of BAG1 was higher in ΔTgPL1 bradyzoites than in WT and ΔTgPL1-complemented bradyzoites and was still apparent after 72 h of growth under tachyzoite conditions. These studies indicate that suppression and induction of certain genes may be defective during ΔTgPL1 bradyzoite-to-tachyzoite stage transition. Future genome-wide expression analyses will elucidate the extent of the development and/or reactivation defect present in ΔTgPL1.
TgPL1 has homology to PLPs (17), which may suggest a mechanism for this modulation. PLPs typically have lipid hydrolase activity, most notably phospholipase A2 activity, which results in lysophospholipid and fatty acid production (30). Myristic and palmitic acids, which are abundant in T. gondii, have been found to suppress production of TNF-α in macrophages (31). However, TgPL1 lacks one of the catalytic residues that is necessary for PLA2 activity in other homologs (17) and has been shown not to have PLA2 activity (18). Because the other active-site residues are conserved, TgPL1 may be involved in binding phospholipids. Lipid binding activity would allow TgPL1 to sequester molecules that could lead to cytokine production or perhaps to act as a coenzyme that delivers phospholipids to other PLA2 enzymes.
Plant patatin changes location from within vesicles to the cytoplasm in response to stress (5), but the timing and mechanism of this localization change have not been elucidated. TgPL1 is mostly within the parasite 1 day after macrophage activation, but after 5 days under macrophage activation conditions, TgPL1 is largely localized to the PV (18). Similarly, after 5 days under bradyzoite development conditions, TgPL1 is mostly found in the PV and colocalizes with DBA (Fig. 1). This delayed response may indicate that new protein synthesis occurs, so we examined if TgPL1 initiates at different methionines in the tachyzoite and bradyzoite stages. These studies indicate that the mechanism of TgPL1 localization change is not due to differences in the translation start site, as TgPL1 begins from one of the first two initiator methionines in both tachyzoites and bradyzoites. The lack of TgPL1 expression when these two codons were mutagenized could be due to a nonoptimal sequence context for the third ATG. The optimal start codon sequence context for T. gondii is GNCAAAATGG (32). The sequence context around the third ATG is TTCTGTATGT, which has only one of the preferred T. gondii base pairs in those positions. The second ATG has the best sequence context (GACATGATGA), which has optimal residues at 3 of the designated positions. Alternatively, the M1-2 mutant may be mislocalized and/or misfolded and then degraded by the parasite. In any case, here we have shown that the open reading frame of TgPL1 does not change between tachyzoites and bradyzoites. It may be that a subset of T. gondii vesicles are delivered to the PV space under stress conditions. We have seen partial colocalization of TgPL1 with some of the plant-like vacuole markers (data not shown), but none of the resident plant-like vacuole proteins we have examined so far change location in response to stress. It is possible that the partial colocalization of plant-like vacuole markers with TgPL1 in tachyzoites is due to shared compartmentalization during early trafficking but that TgPL1 localizes to an uncharacterized vesicle distinct from the plant-like vacuole. We plan to characterize TgPL1 trafficking in the future. Understanding the mechanism of the TgPL1 localization change in bradyzoites will undoubtedly uncover interesting cell biology.
This study identifies a role for the T. gondii gene TgPL1 in the development of TE. We show that a ΔTgPL1 mutant is able to develop viable cysts in the brains of C57BL/6 mice, similar to WT parasites. In contrast to WT-infected mice, which succumb to TE by 11 weeks, the ΔTgPL1-infected mice survive past 12 weeks and maintain viable cysts. This phenotype correlates with the loss of IFN-γ, TNF-α, IL-6, and MCP-1 in WT-infected mice but with maintenance of these cytokines in ΔTgPL1-infected animals. Further studies will elucidate the mechanism by which TgPL1 alters cytokine production during chronic infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Sibley for anti-β-tubulin and anti-GRA antibodies, Louis Weiss for the anti-BAG1 antibody, John Boothroyd for the anti-SAG1 antibody, Jay Bangs for the use of his microscope, and Jenny Gumperz for the use of her flow cytometer.
This research was supported by NIH National Research Service Award GM072125 (C.T.M.), NSF Doctoral Fellowship DGE-0718123 (K.J.P.), a fellowship from The Hartwell Foundation (MSN123175 to L.A.M.), and American Heart Association Award 0840059N (L.J.K.).
Footnotes
Published ahead of print 25 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00444-13.
REFERENCES
- 1.Park WD, Blackwood C, Mignery GA, Hermodson MA, Lister RM. 1983. Analysis of the heterogeneity of the 40,000 molecular weight tuber glycoprotein of potatoes by immunological methods and by NH(2)-terminal sequence analysis. Plant Physiol. 71:156–160. 10.1104/pp.71.1.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonnewald U, Studer D, Rocha-Sosa M. 1989. Immunocytochemical localization of patatin, the major glycoprotein in potato (Solanum tuberosum L.) tubers. Planta 178:176–183. 10.1007/BF00393192 [DOI] [PubMed] [Google Scholar]
- 3.Senda K, Yoshioka H, Doke N, Kawakita K. 1996. A cytosolic phospholipase A2 from potato tissues appears to be patatin. Plant Cell Physiol. 37:347–353. 10.1093/oxfordjournals.pcp.a028952 [DOI] [PubMed] [Google Scholar]
- 4.Kawakita K, Senda K, Doke N. 1993. Factors affecting in vitro activation of potato phospholipase A2. Plant Sci. 92:183–190. 10.1016/0168-9452(93)90205-E [DOI] [Google Scholar]
- 5.Laxalt AM, Munnik T. 2002. Phospholipid signalling in plant defence. Curr. Opin. Plant Biol. 5:332–338. 10.1016/S1369-5266(02)00268-6 [DOI] [PubMed] [Google Scholar]
- 6.La Camera S, Geoffroy P, Samaha H, Ndiaye A, Rahim G, Legrand M, Heitz T. 2005. A pathogen-inducible patatin-like lipid acyl hydrolase facilitates fungal and bacterial host colonization in Arabidopsis. Plant J. 44:810–825. 10.1111/j.1365-313X.2005.02578.x [DOI] [PubMed] [Google Scholar]
- 7.Sonnewald U, von Schaewen A, Willmitzer L. 1990. Expression of mutant patatin protein in transgenic tobacco plants: role of glycans and intracellular location. Plant Cell. 2:345–355. 10.1105/tpc.2.4.345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sitkiewicz I, Stockbauer KE, Musser JM. 2007. Secreted bacterial phospholipase A2 enzymes: better living through phospholipolysis. Trends Microbiol. 15:63–69. 10.1016/j.tim.2006.12.003 [DOI] [PubMed] [Google Scholar]
- 9.Banerji S, Flieger A. 2004. Patatin-like proteins: a new family of lipolytic enzymes present in bacteria? Microbiology 150:522–525. 10.1099/mic.0.26957-0 [DOI] [PubMed] [Google Scholar]
- 10.Jacobs L, Remington JS, Melton ML. 1960. The resistance of the encysted form of Toxoplasma gondii. J. Parasitol. 46:11–21. 10.2307/3275325 [DOI] [PubMed] [Google Scholar]
- 11.Denkers EY, Kim L, Butcher BA. 2003. In the belly of the beast: subversion of macrophage proinflammatory signalling cascades during Toxoplasma gondii infection. Cell Microbiol. 5:75–83. 10.1046/j.1462-5822.2003.00258.x [DOI] [PubMed] [Google Scholar]
- 12.Lang C, Groß U, Lüder CGK. 2007. Subversion of innate and adaptive immune responses by Toxoplasma gondii. Parasitol. Res. 100:191–203. 10.1007/s00436-006-0306-9 [DOI] [PubMed] [Google Scholar]
- 13.Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gâtel D, Tardieux I. 2006. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107:309–316. 10.1182/blood-2005-02-0666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert H, Vutova PP, Adams WC, Lore K, Barragan A. 2009. The Toxoplasma gondii-shuttling function of dendritic cells is linked to the parasite genotype. Infect. Immun. 77:1679–1688. 10.1128/IAI.01289-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Black M, Boothroyd J. 2000. Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 64:607–623. 10.1128/MMBR.64.3.607-623.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss LM, Dubey JP. 2009. Toxoplasmosis: a history of clinical observations. Int. J. Parasitol. 39:895–901. 10.1016/j.ijpara.2009.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mordue DG, Scott-Weathers CF, Tobin CM, Knoll LJ. 2007. A patatin-like protein protects Toxoplasma gondii from degradation in activated macrophages. Mol. Microbiol. 63:482–496. 10.1111/j.1365-2958.2006.05538.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobin CM, Knoll LJ. 2012. A patatin-like protein protects Toxoplasma gondii from degradation in a nitric oxide-dependent manner. Infect. Immun. 80:55–61. 10.1128/IAI.05543-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tobin C, Pollard A, Knoll L. 2010. Toxoplasma gondii cyst wall formation in activated bone marrow-derived macrophages and bradyzoite conditions. J. Vis. Exp. 42:2091. 10.3791/2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donald RG, Roos DS. 1993. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl. Acad. Sci. U. S. A. 90:11703–11707. 10.1073/pnas.90.24.11703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blewett DA, Miller JK, Harding J. 1983. Simple technique for the direct isolation of toxoplasma tissue cysts from fetal ovine brain. Vet. Rec. 112:98–100. 10.1136/vr.112.5.98 [DOI] [PubMed] [Google Scholar]
- 22.Suzuki Y, Joh K, Orellana MA, Conley FK, Remington JS. 1991. A gene(s) within the H-2D region determines the development of toxoplasmic encephalitis in mice. Immunology 74:732–739 [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240:516–518. 10.1126/science.3128869 [DOI] [PubMed] [Google Scholar]
- 24.Gazzinelli RT, Eltoum I, Wynn TA, Sher A. 1993. Acute cerebral toxoplasmosis is induced by in vivo neutralization of TNF-alpha and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J. Immunol. 151:3672–3681 [PubMed] [Google Scholar]
- 25.Suzuki Y, Rani S, Liesenfeld O, Kojima T, Lim S, Nguyen TA, Dalrymple SA, Murray R, Remington JS. 1997. Impaired resistance to the development of toxoplasmic encephalitis in interleukin-6-deficient mice. Infect. Immun. 65:2339–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen X, Kudo T, Payne L, Wang X, Rodgers L, Suzuki Y. 2010. Predominant interferon-gamma-mediated expression of CXCL9, CXCL10, and CCL5 proteins in the brain during chronic infection with Toxoplasma gondii in BALB/c mice resistant to development of toxoplasmic encephalitis. J. Interferon Cytokine Res. 30:653–660. 10.1089/jir.2009.0119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robben PM, LaRegina M, Kuziel WA, Sibley LD. 2005. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 201:1761–1769. 10.1084/jem.20050054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yap G, Pesin M, Sher A. 2000. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 165:628–631 [DOI] [PubMed] [Google Scholar]
- 29.Wilson EH, Hunter CA. 2004. The role of astrocytes in the immunopathogenesis of toxoplasmic encephalitis. Int. J. Parasitol. 34:543–548. 10.1016/j.ijpara.2003.12.010 [DOI] [PubMed] [Google Scholar]
- 30.Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. 1999. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell 97:349–360. 10.1016/S0092-8674(00)80744-8 [DOI] [PubMed] [Google Scholar]
- 31.Debierre-Grockiego F, Rabi K, Schmidt J, Geyer H, Geyer R, Schwarz RT. 2007. Fatty acids isolated from Toxoplasma gondii reduce glycosylphosphatidylinositol-induced tumor necrosis factor alpha production through inhibition of the NF-kappaB signaling pathway. Infect. Immun. 75:2886–2893. 10.1128/IAI.01431-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seeber F. 1997. Consensus sequence of translational initiation sites from Toxoplasma gondii genes. Parasitol. Res. 83:309–311. 10.1007/s004360050254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.