

Abstract
The gamma interferon (IFN-γ) response, mediated by the STAT1 transcription factor, is crucial for host defense against the intracellular pathogen Toxoplasma gondii, but prior infection with Toxoplasma can inhibit this response. Recently, it was reported that the Toxoplasma type II NTE strain prevents the recruitment of chromatin remodeling complexes containing Brahma-related gene 1 (BRG-1) to promoters of IFN-γ-induced secondary response genes such as Ciita and major histocompatibility complex class II genes in murine macrophages, thereby inhibiting their expression. We report here that a type I strain of Toxoplasma inhibits the expression of primary IFN-γ response genes such as IRF1 through a distinct mechanism not dependent on the activity of histone deacetylases. Instead, infection with a type I, II, or III strain of Toxoplasma inhibits the dissociation of STAT1 from DNA, preventing its recycling and further rounds of STAT1-mediated transcriptional activation. This leads to increased IFN-γ-induced binding of STAT1 at the IRF1 promoter in host cells and increased global IFN-γ-induced association of STAT1 with chromatin. Toxoplasma type I infection also inhibits IFN-β-induced interferon-stimulated gene factor 3-mediated gene expression, and this inhibition is also linked to increased association of STAT1 with chromatin. The secretion of proteins into the host cell by a type I strain of Toxoplasma without complete parasite invasion is not sufficient to block STAT1-mediated expression, suggesting that the effector protein responsible for this inhibition is not derived from the rhoptries.
INTRODUCTION
Gamma interferon (IFN-γ) is a critical cytokine in both innate and adaptive immune responses to infection (1, 2). The cellular response to IFN-γ leads to the induction of many effector mechanisms that inhibit the growth and survival of intracellular pathogens. These include the p47 immunity-related GTPases (IRGs), p65 guanylate binding proteins (GBPs), iNOS/Nos2, indoleamine 2,3-dioxygenase 1 (IDO1), and major histocompatibility complex (MHC) genes (2–7). Mice deficient in various components of the IFN-γ pathway are acutely susceptible to many pathogens, including the parasite Toxoplasma gondii (8–12). Toxoplasma is an obligate intracellular protozoan parasite that infects virtually all warm-blooded animals, including mice and humans (13).
IFN-γ stimulation activates the signal transducer and activator of transcription 1 (STAT1) transcription factor and induces a broad transcriptional program (14). When IFN-γ binds to its receptors, IFNGR1 and IFNGR2, the receptors oligomerize and cause constitutively associated Janus activated kinase 1 (JAK1) and JAK2 to be activated (15, 16). Activated JAKs tyrosine-phosphorylate the IFN-γ receptor, creating a docking site for STAT1, which is subsequently phosphorylated by the JAKs at tyrosine 701, leading to its homodimerization and nuclear translocation. In the nucleus, STAT1 binds to gamma-activated sequence (GAS) sites in the DNA, leading to its serine phosphorylation at residue 727 (17). This additional serine phosphorylation is required for maximal STAT1 activity (18).
During IFN-γ stimulation, STAT1 undergoes nuclear-cytoplasmic cycling in order to constantly monitor the activity of the IFN-γ receptor and the JAKs (19, 20). After DNA binding and transcriptional activation, STAT1 dissociates from the DNA, is dephosphorylated at tyrosine 701, and is exported back into the cytoplasm. This STAT1 can then be reactivated to begin the cycle anew. These steps must occur in this order since DNA-bound STAT1 is protected from tyrosine phosphatases, and only unphosphorylated STAT1 can be exported back into the cytoplasm (19). This cycling is required for full STAT1 transcriptional activity; STAT1 mutants that have a decreased dissociation rate from DNA (20) or that are defective in nuclear export (21) have decreased transcriptional output. Two other STAT family members, STAT6 (22) and STAT3 (23), also need to undergo this cycling on and off DNA to produce their full transcriptional output.
Preinfection of cells with Toxoplasma globally inhibits the IFN-γ-induced, STAT1-mediated gene expression program in multiple cell types of multiple species, including human foreskin fibroblasts (HFFs) (24), murine bone-marrow derived macrophages (25), and RAW264.7 murine macrophages (26). It is thought that this inhibition is required for survival of the parasite and conversion to the chronic cyst stage but how this inhibition occurs remains a matter of contention. A recent study showed that in murine macrophages Toxoplasma infection inhibits the expression of IFN-γ-induced secondary response genes, such as class II transactivator (Ciita) and MHC class II genes, by imparing Brahma-related gene 1 (BRG-1)-mediated chromatin remodeling of their promoters (25). Treatment of these cells with histone deacetylase (HDAC) inhibitors decreased the inhibition of secondary response genes by the Toxoplasma type II NTE strain (25). However, the IFN-γ-induced expression of primary response genes such as IRF1 does not require BRG-1-mediated remodeling (27), suggesting that Toxoplasma might utilize a different mechanism to inhibit the expression of primary STAT1-induced genes.
In the present study, we further characterized the Toxoplasma-mediated inhibition of STAT1 transcriptional activity and expression of primary IFN-γ response genes by measuring each step of IFN-γ-induced STAT1 activation to determine where in the pathway Toxoplasma acts. We find that Toxoplasma infection inhibits STAT1 transcriptional activity by preventing STAT1 nuclear-cytoplasmic cycling. A Toxoplasma effector likely inhibits the dissociation of STAT1 from DNA since infection leads to increased association of IFN-γ-activated STAT1 with chromatin and the IRF1 promoter and prevents the dephosphorylation and nuclear export of IFN-γ-activated STAT1. Toxoplasma infection can also inhibit IFN-β-induced gene expression through a similar mechanism involving increased association of STAT1 with chromatin and decreased STAT1 nuclear-cytoplasmic cycling. We find that Toxoplasma can inhibit the expression of IFN-γ primary response genes in the presence of various HDAC inhibitors, which contrasts with the role of HDACs in the Toxoplasma-mediated inhibition of IFN-γ secondary response genes (25). This suggests that the mechanism of inhibition of IFN-γ-induced primary response genes is distinct from the mechanism by which secondary response genes are inhibited in murine macrophages. The Toxoplasma factor responsible for this inhibition is unknown; however, our results indicate that it is unlikely to be secreted into the host cell from the rhoptry secretory organelle and that it likely acts directly on DNA-bound tyrosine-phosphorylated STAT1.
MATERIALS AND METHODS
Parasites and cells.
Parasites were maintained in vitro by serial passage on monolayers of HFFs, as described previously (28). An RH strain engineered to express clickbeetle luciferase and green fluorescent protein (GFP) (RH 1-1) (29), an RHΔrop16 strain (provided by John Boothroyd, Stanford University) (30), an RHΔrop16 strain expressing firefly luciferase and GFP (31), a Pru strain engineered to express firefly luciferase and GFP (Pru Δhxgprt A7) (32), and a CEP strain engineered to express clickbeetle luciferase and GFP (CEP hxgprt− C22) (29) have been described previously. HFFs were cultured as previously described (28). 293FT and HEK293 cells were cultured with additional 10 mM HEPES. A HEK293-pGreenFire1-GAS IFN-γ responsive reporter cell line has been previously described (26). All parasite strains and cell lines were routinely checked for Mycoplasma contamination, and it was never detected.
Reagents.
Antibodies against total STAT1α p91 (C-24; Santa Cruz, catalog no. 345), phospho-STAT1Tyr701 (58D6; Cell Signaling, catalog no. 9167), phospho-STAT1Ser727 (Cell Signaling, catalog no. 9177), STAT2 (H-190; Santa Cruz, catalog no. 22816), IRF1 (BD Biosciences, catalog no. 612046), IRF9/ISGF-3γ p48 (C-20; Santa Cruz, catalog no. 496), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; 6C5; Santa Cruz, catalog no. 32233), Toxoplasma surface antigen 1 (SAG-1; kindly provided by John Boothroyd, Stanford University), Toxoplasma GRA7 (33), histone H3 (Abcam, catalog no. 1791), acetyl-histone H4Lys12 (Cell Signaling, catalog no. 2591), and phospho-STAT6Tyr641 (Santa Cruz, catalog no. 11762-R) were used in immunofluorescence and Western blot assays. Secondary antibodies coupled with either Alexa Fluor 488 or Alexa Fluor 594 (Molecular Probes) for immunofluorescence assays or conjugated to peroxidase (Kirkegaard & Perry Laboratories) for Western blots were used. Recombinant human IFN-γ (AbD Serotec), IFN-β (Peprotech), IL-4 (Peprotech), and TNF-α (Gibco/Life Technologies) were used to stimulate cells. Cycloheximide (CHX; 50 μg/ml, Sigma), cytochalasin D (1 μM; Enzo), mycalolide B (3 μM; Wako), trichostatin A (3 to 9 μM, Sigma), MS-275 (2 to 10 μM, Selleck), MC1568 (2 to 10 μM; Selleck), sodium butyrate (2 to 10 μM; Sigma), and MG132 (0.5 to 2.5 μM; Sigma) were also used to treat cells.
Immunofluorescence assay.
Immunofluorescence assays were performed as described previously (28). Quantification of nuclear signal was performed by randomly selecting at least 12 cells per condition and measuring the average signal intensity per nucleus using the NIS-Elements software and Hoechst dye to define nuclei.
Reporter cell line construction.
The construction of a HEK293-pGreenFire1-GAS IFN-γ responsive reporter cell line has been previously described (26). pGreenFire1-ISRE and STAT1 cell lines were constructed by the same method from ISRE (TR016PA-1, 5′-CAGTTTCACTTTCCCTTT-3′) and STAT1 (TR015PA-1, 5′-GATTTCCGGGAAATGGGGAAGG-3′) vectors purchased from System Biosciences. Briefly, lentivirus containing the vector was produced in 293FT cells and added to HEK293 cells (American Type Culture Collection). Cells containing the construct were selected with 750 μg of Geneticin (Invitrogen)/ml, cloned by limiting dilution, and assayed for responsiveness to IFN-γ, IFN-β, tumor necrosis factor alpha (TNF-α), and interleukin-4 (IL-4) (see Fig. S1 in the supplemental material).
Luciferase assay.
Luciferase assays of HEK293 pGF1-GAS, STAT1, or ISRE cells were performed as previously described using the Promega luciferase assay system (26). For all experiments, 3.5 × 104 to 4 × 104 cells were plated in 96-well plates for at least 4 h before any treatment or infection. Exact treatment and infection times varied slightly between experiments, but we obtained similar results for all time points and present averaged data with the standard errors. For ISRE experiments, cells were infected with RH parasites at a multiplicity of infection (MOI) of ∼1.5 for 3 to 5 h and subsequently stimulated with 100 U of either IFN-γ or IFN-β/ml for 14 to 17 h before lysis. For HDAC inhibitor experiments, cells were pretreated with HDAC inhibitors for 1 h, infected with RH parasites at an MOI of ∼4 for 1 to 3 h, and subsequently stimulated with 100 U of IFN-γ/ml for 14 to 20 h before lysis. For MG132 experiments, cells were pretreated with MG132 for 40 min, infected with RH parasites at an MOI of ∼1.5 for 3 to 5 h, and subsequently stimulated with 100 U of IFN-γ/ml for 15 h before lysis. HDAC inhibitors and MG132 were kept on the cells for the entire experiment. For cytochalasin D experiments, parasites were pretreated with 1 μM cytochalasin D for 15 min, and cytochalasin D was kept on the parasites for the entire experiment. For mycalolide B experiments, parasites were pretreated with 3 μM mycalolide B for 10 min and then pelleted and resuspended in normal medium. Pretreated parasites were added to cells for 1.5 h, and the cells were subsequently stimulated with 100 U of IFN-γ/ml for 18 h before lysis.
Reverse transcription-quantitative PCR (RT-qPCR).
For IFN-β infections, ∼9 × 105 HFFs were grown in six-well plates, infected with RH 1-1 parasites for 4 h, and subsequently stimulated with 100 U of IFN-γ or IFN-β/ml for 15 to 20 h. The exact treatment was between 15 and 20 h, varying slightly between experiments, but we obtained similar results for all time points and present averaged data with the standard errors. For CHX infections, 1.5 × 105 to 2 × 105 HFFs were grown in 12-well plates, pretreated with CHX for 40 min, infected with RH 1-1 for 1 h, and stimulated with human IFN-γ for 1 h. Cells were also left untreated and uninfected. RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Samples from the CHX experiments were cleaned and concentrated using an RNeasy MinElute kit (Qiagen). RNA was determined to be intact and of good quality by gel electrophoresis. Genomic DNA was removed from RNA preparations by DNase I treatment (Invitrogen), and first-strand cDNA was synthesized with SuperScript II or III RT (Invitrogen) and oligo(dT) (Ambion), according to the manufacturer's protocol. Quantitative PCR was performed using SYBR green reagent (Kapa Biosciences) and a LightCycler 480 II real-time PCR machine (Roche) according to the manufacturer's instructions. For IFN-β experiments, genes specifically induced by IFN-β and not by IFN-γ in human fibroblasts (RSAD2, MX2, and OASL) were chosen from published microarray results (34). The primer efficiencies were calculated using the Real-time PCR Miner program (35) and are listed with primer sequences in Table S1 in the supplemental material. The fold change was calculated using the ΔΔCT method (36), comparing the expression to two different control genes, ACTB and NFE2L1, that were not affected by Toxoplasma infection in previous gene expression analyses (28). Similar results were obtained from both normalizations.
Native PAGE and Western blotting.
HFFs were infected with either RHΔhxgprt or RHΔrop16 parasites at two different MOIs (actual MOIs of 5 and 7 for RHΔhxgprt and MOIs of 7 and 9 for RHΔrop16) for 3 h, or left uninfected, and subsequently stimulated with 100 U of human IFN-γ/ml for 1 h or left untreated. Cells were then lysed in nondenaturing buffer containing 1% sodium deoxycholate, and lysates were run on 7.5% PAGE gels in Tris-glycine buffer with 1% sodium deoxycholate in the cathode chamber at 4°C. Western transfer and blotting were performed as described previously (28). Blots were stained with Ponceau S to visualize protein standard (NativeMark; Life Technologies). After immunoblotting, the membranes were stripped with 2% sodium dodecyl sulfate (SDS) and 0.7% β-mercaptoethanol and reprobed.
ChIP and qPCR.
Chromatin immunoprecipitation (ChIP) experiments were performed according to the protocol of Lee et al. (37) with several modifications. HFFs were grown in 15-cm dishes to ∼90% confluence (∼107 cells). Coverslips were placed in dishes to measure nuclear phospho-STAT1Tyr and IRF1 as controls and processed after fixation according to the immunofluorescence assay methods described above. HFFs were infected with RHΔhxgprt or RHΔrop16 parasites for 4 h, or left uninfected, subsequently stimulated with 100 U of human IFN-γ/ml for 1 h, or left unstimulated, and fixed with 1% formaldehyde. A total of ∼5 × 106 cells were used for each immunoprecipitation. After cell lysis, DNA was sheared with a Bioruptor (Diagenode). Immunoprecipitation was performed using an IP-Star (Diagenode) and 3 μg of antibody (total STAT1α p91 [C-24; Santa Cruz, catalog no. 345]). After DNA purification, qPCR was performed using SYBR green reagent (Kapa Biosciences) and a LightCycler 480 II real-time PCR machine (Roche). The primers were designed using published STAT1 ChIP-seq data to amplify STAT1 binding sites in the promoters of IFN-γ-induced genes as well as negative-control regions in the promoters of genes unaffected by IFN-γ that STAT1 does not bind (38). Primer efficiencies were calculated using Real-time PCR Miner (35) and are listed with primer sequences in Table S2 in the supplemental material. The percentage of total DNA bound by STAT1 was calculated by comparing the qPCR results from immunoprecipitated and input samples.
Cell fractionation, STAT1 immunoprecipitation, and mass spectrometry.
Cells were fractionated into cytoplasmic, nuclear extract, and chromatin fractions using a Qiagen Qproteome nuclear protein kit according to the manufacturer's instructions. For Western blot analysis, samples were diluted in 2× reducing sample buffer and boiled before SDS-PAGE analysis and Western blotting as described previously (28). For STAT1 immunoprecipitations, all fractions were diluted to have a final concentration of 150 mM NaCl. Rabbit α-total STAT1α p91 (C-24; ∼1 μg/106 cells) was cross-linked to protein A-Dynabead slurry (Life Technologies; ∼20 μl/μg antibody) with 5 mM bis(sulfosuccinimidyl)suberate (BS3; Pierce) as described previously (39). The samples were incubated with the bead-antibody slurry for 1.5 h at 4°C, while rotating. Beads were then washed three times with immunoprecipitation wash buffer (10 mM HEPES-KOH [pH 7.5], 150 mM NaCl, 0.5% NP-40/IGEPAL, 2.5 mM EGTA-KOH, 20 mM β-glycerophosphate), washed twice with HEPES-buffered saline, and boiled in 2× reducing sample buffer. For mass spectrometry analysis, proteins were excised from each lane of a Coomassie blue-stained SDS-PAGE gel in pieces encompassing the entire molecular weight range. Trypsin-digested extracts were analyzed by using reversed-phase high-pressure liquid chromatography and a Thermo Fisher LTQ linear ion trap mass spectrometer. Peptides were identified from the mass spectrometry data using SEQUEST algorithms44 that searched a species-specific database generated from NCBI's nonredundant (nr.fasta) database. A summary of the number of peptides found and percent coverage for the STAT1 protein in each immunoprecipitation is shown in Table S3 in the supplemental material.
Plaque assay.
For native PAGE, ChIP, cell fractionation and immunoprecipitation, luciferase reporter, and RT-qPCR experiments, a plaque assay was performed to determine parasite viability and the actual MOI. One hundred parasites per well were added to confluent HFFs in a 24-well plate and were incubated undisturbed for 5 to 7 days at 37°C, and the number of plaques was counted.
Statistical analyses.
Two sample t tests, either paired or unpaired as applicable, were performed to assess statistical significance for ChIP-qPCR, luciferase reporter, and RT-qPCR assays.
RESULTS
HDAC activity is not required for Toxoplasma inhibition of IRF1 expression or STAT1 transcriptional activity.
The recruitment of histone acetyltransferases and increased histone H4 and H3 acetylation is associated with the formation of euchromatin and accessibility of DNA to transcription factors and RNA polymerase II (40), and actively transcribed STAT1 target genes have increased H3 and H4 acetylation upon IFN-γ treatment (25). It was recently reported that Toxoplasma infection inhibits the expression of IFN-γ-induced secondary response genes such as Ciita and MHC class II genes by activating HDACs and preventing the recruitment of chromatin remodeling complexes to gene promoters (25). However, many stimulus-induced primary response genes do not require chromatin remodeling for their expression (41). For example, the IFN-γ-induced primary response gene IRF1 does not require expression of the chromatin remodeler BRG-1 (27), which was previously implicated in Toxoplasma-mediated inhibition of the IFN-γ response (25). It is therefore unclear whether HDAC activity is required for the inhibition of primary IFN-γ-induced gene expression by Toxoplasma, and we decided to test this using a variety of HDAC inhibitors.
Since three clonal lineages of Toxoplasma—types I, II, and III—all equally inhibit STAT1 transcriptional activity (26), it is likely that all of these strains utilize a similar mechanism of inhibition, and we have focused our study on just one of these strains, the type I RH strain. Toxoplasma infection also equally inhibits STAT1 activity and IFN-γ-induced primary (IRF1) gene expression in a variety of cell types, including HFFs, HEK293 cells, murine macrophages, and murine dendritic cells (26, 42), and we have therefore focused our study on two readouts of primary STAT1-induced gene expression: the expression of IRF1 in HFFs and stable HEK293 STAT1 luciferase reporter cell lines.
First, we pretreated HFFs on coverslips for 1 h with trichostatin A (TSA), a class I/II HDAC inhibitor, subsequently infected the cells with RH parasites for 1 h, and then stimulated the cells with IFN-γ for 2 h. TSA was kept on the cells for the entire experiment. Cells were then fixed and stained for IRF1 expression and acetylated-histone H4. TSA treatment under all conditions increased the intensity of acetylated-histone H4 staining in the host nucleus, indicating that under these conditions it potently inhibits host HDACs (Fig. 1A). However, infection with RH parasites either in the presence or absence of TSA strongly inhibited the IFN-γ-induced expression of IRF1, indicating that the activity of class I and class II HDACs is not required for Toxoplasma's inhibition of IRF1 expression (Fig. 1A).
FIG 1.

HDAC activity is not required for Toxoplasma to inhibit IFN-γ-induced IRF1 expression or induction of IFN-γ-responsive reporter cell lines. (A) HFFs were plated on coverslips, pretreated with 3 μM trichostatin A (TSA), an HDAC inhibitor, for 1 h, infected with RH parasites for 1 h, and subsequently stimulated with 100 U of IFN-γ/ml for 2 h. Control cells were also left unstimulated (US) and/or uninfected (UI). Cells were fixed, permeabilized, and stained with α-acetyl-histone H4 (green), α-IRF1 (red), and with Hoechst dye (nucleus, blue). A representative cell from each condition is shown. Scale bar, 10 μm. This experiment was performed twice with similar results. (B) HEK293 GAS (top) or STAT1 (bottom) reporter cell lines were pretreated with a variety of HDAC inhibitors (TSA, MC1568, MS-275, and sodium butyrate) or left untreated (DMSO, vehicle-only control) for 1 h. Cells were left uninfected (UI) or infected with RH parasites for 1 to 3 h, subsequently stimulated with 100 U of IFN-γ/ml for 14 to 20 h or left unstimulated (US), and lysed, and the luciferase activity was measured. The data were normalized within each experiment to the sample with the maximum luciferase activity, and the data shown are averages and the standard errors of the mean (SEM) from three independent experiments. Asterisks (*) indicate P < 0.05, or the P values are shown above bars.
However, the chromatin environments of IFN-γ-induced genes may not all be regulated in the same manner. Therefore, we also tested Toxoplasma-mediated inhibition of STAT1 activity in two different stable HEK293 reporter cell lines: a “GAS” line and a “STAT1” line. These cell lines contain slightly different consensus STAT1 binding sites, both driving the expression of luciferase. Treatment of either of these reporters with IFN-γ, but not IFN-β, TNF-α, or IL-4, results in the induction of luciferase activity (26) (see Fig. S1B in the supplemental material). We pretreated both of these cell lines with a variety of HDAC inhibitors: TSA, MC1568, MS-275, or sodium butyrate, or with dimethyl sulfoxide (DMSO) as a control, using concentrations that were previously shown to inhibit Toxoplasma-mediated inhibition of Ciita and MHC class II gene expression (25). We then infected the lines with RH parasites, subsequently stimulated the cells with IFN-γ, and measured the induction of luciferase activity. In the DMSO control, prior infection with RH parasites significantly inhibited the IFN-γ-stimulated induction of luciferase activity in both cell lines (Fig. 1B), in agreement with previous results (26). In the GAS reporter line, pretreatment with MC1568, MS-275, or sodium butyrate did not affect the ability of RH infection to inhibit this induction (Fig. 1B). Treatment with TSA by itself inhibited the induction of luciferase after IFN-γ treatment in this cell line, although prior infection with RH still lowered the IFN-γ-induced luciferase activity further (Fig. 1B). Conversely, in the STAT1 reporter line treatment with TSA or MS-275 strongly induced luciferase activity even in the absence of IFN-γ treatment (Fig. 1B). Prior infection with RH still inhibited IFN-γ-induced luciferase activity under all conditions (Fig. 1B).
Together, these results suggest that the mechanism by which a type I strain of Toxoplasma inhibits the expression of STAT1-induced primary response genes is distinct from the mechanism of inhibition of secondary response genes, and does not involve the activation of HDACs. In addition, we find that inhibition of HDAC activity has both positive and negative effects on basal and IFN-γ-induced STAT1 transcriptional activity, depending on the exact promoter and the different HDACs that are targeted.
New host cell protein synthesis is dispensable for the inhibition of IFN-γ-induced primary response gene expression by Toxoplasma.
Since the mechanism of inhibition of primary and secondary response genes appears to be different, we sought to further characterize the HDAC-independent mechanism of inhibition of IFN-γ primary response genes. One possibility is that Toxoplasma induces host negative regulatory proteins that target STAT1 activity, such as suppressor of cytokine signaling (SOCS) family proteins, protein tyrosine phosphatases (PTPs), and protein inhibitor of activated STAT1 (PIAS) family proteins (43). To determine whether host proteins whose expression is induced by type I Toxoplasma infection play a role in inhibiting STAT1 activity, we pretreated HFFs with the protein translation inhibitor cycloheximide (CHX) for 40 min prior to infecting the cells with an RH strain for 1 h and subsequently stimulating with IFN-γ for 1 h, keeping CHX on the cells for the entire experiment. Under these conditions, infection-induced and IFN-γ-induced protein expression is prevented and cell viability is not affected (see Fig. S2 in the supplemental material). We then determined IFN-γ-induced IRF1 mRNA levels by RT-qPCR. IFN-γ treatment increased IRF1 mRNA levels ∼13-fold, and this induction was decreased in samples preinfected with RH parasites (Fig. 2A). Preinfection with RH parasites also inhibited IFN-γ-responsive IRF1 mRNA accumulation in the presence of CHX (Fig. 2A). However, conclusions from the IRF1 qPCR are complicated by the fact that both CHX treatment (∼5-fold) and RH infection combined with CHX treatment (∼12-fold) induce IRF1 mRNA transcription in the absence of IFN-γ (Fig. 2A). We therefore calculated the fold inhibition of IRF1 expression by RH preinfection in each of these conditions. The presence of CHX did not significantly alter the ability of RH to inhibit IRF1 gene expression (Fig. 2B), suggesting that new host protein synthesis is not required for the inhibition of primary response genes by type I Toxoplasma. Similar results were obtained when IRF1 qPCR data were normalized to a different control gene (see Fig. S3 in the supplemental material).
FIG 2.

Toxoplasma can inhibit IFN-γ-responsive gene expression in the presence of both cycloheximide and MG132. (A and B) HFFs were pretreated with 50 μg of CHX/ml for 40 min, infected with an RH strain for 1 h, and stimulated with 100 U of IFN-γ/ml for 1 h. CHX was left on the treated cells for the entire experiment. Cells were also left untreated (UT) and/or uninfected (UI). Induction of IRF1 mRNA was determined by RT-qPCR analysis and normalized to ACTB transcript levels. (A) The averages of two experiments are shown; error bars represent the SEM. (B) The fold inhibition by RH infection in each of the conditions was calculated for each experiment, and averages of two experiments are shown. Error bars represent the SEM. (C) HEK293 GAS or STAT1 reporter cell lines were pretreated with MG132 or left untreated for 40 min. Cells were then infected with RH parasites for 3 to 5 h, subsequently stimulated with 100 U of IFN-γ/ml for 15 h, and lysed, and the luciferase activity was measured. MG132 was left on the treated cells for the entire experiment. The data were normalized within each experiment to the uninfected, unstimulated (US) sample, and the data shown are the average fold induction and the SEM from three independent experiments. Asterisks (*) indicate P < 0.05.
Inhibition of STAT1 activity does not depend on the proteasome.
Toxoplasma infection could also cause the proteasomal degradation of a coactivator that is necessary for STAT1 to recruit general transcription machinery and RNA polymerase II to the promoters of IFN-γ primary response genes. To determine whether the ability of type I Toxoplasma to inhibit STAT1 transcriptional activity depends on the proteasome, we treated our HEK293 STAT1 and GAS reporter cell lines with MG132, which inhibits the proteolytic activity of the 26S proteasome. We then infected the cell lines with RH parasites, subsequently stimulated the cells with IFN-γ, and measured the induction of luciferase activity, keeping MG132 on the cells for the entire experiment. In both the GAS and STAT1 reporter cell lines, treatment with increasing concentrations of MG132 inhibited the induction of luciferase activity even in the absence of infection (Fig. 2C). However, in the STAT1 reporter line this inhibition was only partial, and preinfection with RH parasites significantly inhibited IFN-γ-induced luciferase activity further (Fig. 2C). This result indicates that type I Toxoplasma-induced inhibition of STAT1-mediated gene expression does not require proteolytic activity of the proteasome.
Toxoplasma infection does not interfere with IFN-γ-induced STAT1 dimerization.
Since Toxoplasma infection does not induce the expression or degradation of host proteins in order to inhibit STAT1 activity, we hypothesized that a Toxoplasma effector acts directly on STAT1 to inhibit its activity. We therefore decided to measure more proximal steps in IFN-γ/STAT1 signaling in infected cells. Previous results have shown that Toxoplasma infection does not interfere with STAT1 tyrosine phosphorylation, serine phosphorylation, or nuclear translocation (24, 26, 44). These data have led to the conclusion that STAT1 homodimerization is also not inhibited by infection. However, it is also possible that a Toxoplasma protein containing a nuclear localization sequence directly binds to single tyrosine phosphorylated STAT1 proteins and carries them into the nucleus. To determine the predominant complex(es) within which STAT1 is found in infected cells and therefore distinguish between these two possibilities, we visualized STAT1-containing complexes in nondenaturing conditions by native PAGE and Western blotting. We infected HFFs with the Toxoplasma RH strain for 3 h, subsequently stimulated the cells with IFN-γ for 1 h, lysed the cells in nondenaturing conditions, ran the lysates on native PAGE, and blotted for STAT1. In uninfected, unstimulated cells, STAT1α runs at a size between 66 and 146 kDa, but upon IFN-γ treatment the majority of STAT1α protein shifts into complex that runs at a size between 146 and 242 kDa (Fig. 3). These sizes are consistent with STAT1α monomers (91 kDa) and STAT1 homodimers, respectively. Additional blotting for the phosphotyrosine form of STAT1 also demonstrated that the majority of phosphorylated STAT1 is found in the slower-migrating band, consistent with this band representing the dimer since tyrosine phosphorylation is required for STAT1 dimerization (Fig. 3). In cells preinfected with RH parasites, STAT1α again runs at two different bands of exactly the same size as in uninfected cells, indicating that type I Toxoplasma does not inhibit STAT1 homodimerization and suggesting that a putative Toxoplasma effector does not strongly bind STAT1 under these conditions (Fig. 3). In type I strains the rhoptry kinase ROP16 can also induce the phosphorylation and nuclear translocation of STAT1 (26), and our results show that this ROP16-activated STAT1 also dimerizes (Fig. 3). We therefore additionally performed this experiment with RH parasites deficient for ROP16 to specifically measure IFN-γ-induced STAT1 dimerization and find that RHΔrop16 parasites also do not inhibit the dimerization of STAT1 (Fig. 3).
FIG 3.

Toxoplasma infection does not inhibit IFN-γ-induced STAT1 dimerization. HFFs were infected with RH or RHΔrop16 parasites, or left uninfected, for 4 h. Cells were then stimulated with 100 U of human IFN-γ/ml for the last hour of infection (+IFN-γ) or left unstimulated (US). Cell lysates were collected in nondenaturing buffer and analyzed by native PAGE, followed by Western blotting. Cells were infected at two different MOIs, and the actual MOIs for each sample calculated after plaque assay are indicated. Blots were probed for phospho-STAT1Tyr, stripped, and reprobed for total STAT1α. This experiment has been performed three times with similar results.
IFN-γ-induced STAT1 DNA association is increased upon Toxoplasma infection.
We next wondered whether the nuclear STAT1 in infected, IFN-γ-stimulated host cells is able to bind DNA, and specifically to the GAS sites it normally targets. Previous electrophoretic mobility shift assays (EMSAs) indicated that nuclear STAT1 is still able to bind GAS sites in vitro (25, 42, 45). In one study the binding was slightly weaker in extracts from infected cells (45), whereas another study found that binding was both increased and prolonged in infected extracts (42). In two of these studies, STAT1 from infected cells was able to bind to GAS oligonucleotides but was present in a different, more slowly migrating, aberrant complex compared to STAT1 from uninfected extracts (25, 42). Conversely, Toxoplasma was recently reported to inhibit STAT1 binding to the Irf1 promoter in murine bone marrow-derived dendritic cells (BMDCs) (42). To determine whether STAT1 binds GAS sites in the promoters of IFN-γ-responsive genes in HFFs, we performed STAT1 ChIP experiments. We infected HFFs with RH parasites for 4 h, or left cells uninfected, and subsequently stimulated the cells with IFN-γ for 1 h or left the cells unstimulated. Coverslips were included in sample plates to measure STAT1 phosphorylation and the inhibition of IRF1 expression by preinfection as controls (see Fig. S4 in the supplemental material). In uninfected cells, we detected a significant increase in STAT1 binding after IFN-γ treatment at all loci, except for a negative-control locus, CCND2 (Fig. 4A). Infection with RH parasites, in the absence of IFN-γ treatment, also resulted in a significant increase in STAT1 binding at all except one of these loci (Fig. 4A). We hypothesized that the STAT1 binding to DNA after RH infection was due to ROP16-activated STAT1, and we therefore also infected cells with an RHΔrop16 strain. In cells infected with RHΔrop16 parasites, STAT1 binding at these loci is not significantly higher than in uninfected cells, suggesting that ROP16-activated STAT1 is not only tyrosine phosphorylated, dimerized, and nuclear but that it is also able to bind to GAS sites in vivo (Fig. 4A). In cells preinfected with either RH or RHΔrop16 parasites, STAT1 binding upon IFN-γ stimulation was not inhibited at six of the seven IFN-γ induced loci we tested (Fig. 4A). At the IRF1 promoter, IFN-γ-induced STAT1 binding was significantly higher in infected samples compared to uninfected samples (Fig. 4A).
FIG 4.

IFN-γ-induced STAT1 DNA association is increased upon infection with Toxoplasma. HFFs were infected with RH or RHΔrop16 parasites, or left uninfected (UI), for 4 to 5 h. Cells were stimulated with 100 U of human IFN-γ/ml for the last hour of infection or left unstimulated (US). (A) Samples were fixed with 1% formaldehyde and collected for chromatin immunoprecipitation. qPCR of STAT1-binding regions of the promoters of IFN-γ-responsive genes was performed on both the immunoprecipitated STAT1-bound DNA and total input DNA. The percentage of the total DNA bound by STAT1 was calculated. A promoter region where STAT1 is not known to bind (CCND2) was also included as a negative control. The averages and SEM of three experiments are shown. The average MOI in the three experiments was 8. Asterisks (*) indicate P < 0.05 versus the uninfected, unstimulated sample, or the P value is indicated above the bars. (B to D) Samples were fractionated in cytoplasmic, nuclear extract, and chromatin fractions. Then, 1/4 to 1/6 of each fraction was diluted in 2× reducing sample buffer and boiled, and the protein levels were analyzed by SDS-PAGE and Western blotting (B and C). GAPDH and H3 are markers for cytoplasmic and chromatin fractions, respectively. The H3 antibody cross-reacts with Toxoplasma H3 and is therefore also found in the cytoplasmic fraction. STAT1 was then immunoprecipitated from the remaining portion of all three fractions, and mass spectrometry was performed. From each sample, the percentage of the total STAT1 peptides that were found in the chromatin fraction was calculated (D). The data are from three experiments performed independently at different times: HFFs infected with RH at a plaque assay-calculated MOI of ∼8 (B and D), RH at a plaque assay-calculated MOI of ∼1.5 (D), and RHΔrop16 at a plaque assay-calculated MOI of ∼5 (C and D).
To confirm that Toxoplasma does not inhibit STAT1's association with DNA and chromatin and to determine whether infection actually increases STAT1 global DNA binding and chromatin association as we observed at the IRF1 locus, we again infected HFFs with RH parasites for 3 to 4 h, or left the cells uninfected, and subsequently stimulated the cells with IFN-γ for 1 h. We then isolated cytoplasmic, nuclear, and chromatin fractions from these cells and analyzed the protein levels in these fractions by SDS-PAGE and Western blotting. As expected, in unstimulated, uninfected cells, STAT1 is present exclusively in the cytoplasmic fraction and, upon IFN-γ treatment, STAT1 is both tyrosine phosphorylated and present in the nuclear extract (Fig. 4B). Only a very small amount of this STAT1 is stably associated with the chromatin (Fig. 4B), a finding consistent with a model of STAT1 cycling on and off DNA. In cells preinfected with RH and stimulated with IFN-γ, the levels of STAT1 in both the nuclear and the chromatin fractions are significantly higher than in uninfected cells (Fig. 4B), suggesting that not only does type I Toxoplasma infection not inhibit STAT1 DNA binding but it actually increases STAT1 chromatin association. This result mirrors what we observed at the IRF1, SOCS3, and ICAM1 promoter regions in the ChIP assay. We obtained similar results from cells preinfected with an RHΔrop16 strain (Fig. 4C), indicating that this increased association is not simply due to ROP16-activated STAT1.
To quantitatively measure the relative amount of STAT1 in the chromatin fraction in each of these samples, we immunoprecipitated STAT1 from the cytoplasmic, nuclear, and chromatin fractions and performed mass spectrometry on the pulled-down protein. Complete data on the number of STAT1 peptides and the percent coverage of STAT1 found in each immunoprecipitation is shown in Table S3 in the supplemental material. We then calculated the percentage of STAT1 peptides present in the chromatin fraction compared to the total STAT1 peptides detected in all fractions of each sample. Consistent with our Western blot results, preinfection with either RH or RHΔrop16 resulted in substantially more IFN-γ-induced STAT1 in the chromatin fraction (Fig. 4D). Thus, the mechanism by which type I Toxoplasma inhibits STAT1 transcriptional activity likely involves the increased association of STAT1 with chromatin and DNA. Under the conditions of these immunoprecipitations, we did not find any Toxoplasma proteins consistently pulled down with STAT1 in any of the fractions (data not shown).
Toxoplasma prevents recycling of STAT1.
After IFN-γ-activated STAT1 binds to DNA and initiates transcription, it falls off the DNA, is dephosphorylated, and translocates back to the cytoplasm (19). Our data indicate that infection increases the association of STAT1 with DNA, and we therefore hypothesized that infection might inhibit STAT1 nuclear-cytoplasmic cycling. Because STAT1 cannot be dephosphorylated until it has fallen off DNA (19), our previous observation that in cells preinfected with Toxoplasma IFN-γ-induced phospho-STAT1Tyr levels were significantly higher than in uninfected cells (26) is consistent with this hypothesis.
If IFN-γ is still present and the receptors and JAKs are still active when STAT1 is exported back into the cytoplasm, STAT1 will be reactivated and cycle back to the nucleus to induce further transcription (19, 20). However, when IFN-γ is removed its signaling pathway is downregulated through dephosphorylation of the JAKs and internalization of the IFN-γ receptor (43, 46). To determine how quickly the IFN-γ pathway is deactivated after IFN-γ is removed, we stimulated HFFs on coverslips with IFN-γ for 30 min, washed the IFN-γ away, fixed the cells after either 3.5 or 21.5 h, and quantified the nuclear accumulation of phospho-STAT1Tyr by immunofluorescence (Fig. 5). After 3.5 h, we observed nuclear levels of phospho-STAT1Tyr that were ∼2.5-fold higher than unstimulated cells (Fig. 5), indicating that the IFN-γ pathway is still active at this time point. This level of activation is similar to that of cells that have been continuously stimulated with IFN-γ for 2 h (26). Later, 21.5 h after IFN-γ was removed, the nuclear levels of phospho-STAT1Tyr had decreased to be ∼1.8-fold higher than unstimulated cells (Fig. 5).
FIG 5.
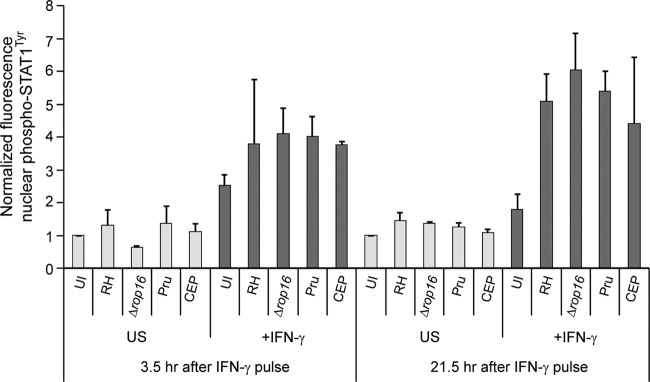
Type I, II, and III Toxoplasma strains prevent dephosphorylation and nuclear export of STAT1. HFFs were plated on coverslips, preinfected with RH, RHΔrop16, Pru, or CEP Toxoplasma parasites for 2 h at an MOI of ∼2, or left uninfected (UI), and stimulated with a pulse of 100 U of IFN-γ/ml for 30 min before the cells were washed and the medium was changed, or left unstimulated (US). Cells were fixed after 3.5 or 21.5 additional hours, permeabilized, and stained with α-phospho-STAT1Tyr and with Hoechst dye (nucleus). Nuclear accumulation of phospho-STAT1Tyr was quantified in at least 12 randomly selected cells per condition and normalized to the UI, US condition. Averaged data from at least two experiments per condition are shown; error bars represent standard deviations.
To determine whether Toxoplasma inhibits STAT1 cycling back to the cytoplasm, we preinfected HFFs with RH parasites for 2 h, subsequently stimulated the cells with a 30-min pulse of IFN-γ, and measured nuclear levels of phospho-STAT1Tyr after both 3.5 and 21.5 additional hours. At 3.5 h after the IFN-γ pulse, the levels of phospho-STAT1Tyr were slightly higher in RH- and RHΔrop16-infected cells, ∼3.8 to 4-fold higher than in unstimulated cells (Fig. 5). At 21.5 h after the IFN-γ pulse, when phospho-STAT1Tyr has returned to near baseline levels in uninfected cells, in RH- and RHΔrop16-infected cells, levels of phospho-STAT1Tyr were even higher than at 3.5 h (~5- to 6-fold over unstimulated cells), indicating that infection with a type I strain prevents the dephosphorylation and nuclear export of IFN-γ-activated STAT1 (Fig. 5). This effect was independent of ROP16 since in RHΔrop16 preinfected cells levels of nuclear phospho-STAT1Tyr were even higher than in RH-infected cells at both time points. We obtained similar results after infection with a type II (Pru) or type III (CEP) strain (Fig. 5), suggesting that all three strains inhibit STAT1 nuclear-cytoplasmic cycling.
Toxoplasma infection also inhibits IFN-β-induced STAT1 activity.
Our data indicate that Toxoplasma inhibits the dissociation of STAT1 from DNA, thereby blocking its dephosphorylation and export back into the cytoplasm. STAT1 can also be present in a complex with STAT2 and IRF9, which is primarily activated by type I IFNs, i.e., IFN-α and IFN-β (14). This complex is called IFN-stimulated gene factor 3 (ISGF3) and binds to IFN-stimulated response elements (ISREs) in DNA to induce the expression of a subset of genes that partially overlaps with the set of genes induced by IFN-γ (14). To test whether Toxoplasma infection can inhibit the activity of type I IFN-activated STAT1, we developed a stable ISRE reporter cell line in HEK293 cells. We infected this cell line with RH parasites or left cells uninfected, stimulated the cells with IFN-β, or left the cells unstimulated, and we measured the induction of luciferase activity. Treatment with IFN-β led to the induction of luciferase by ∼3.5-fold, and preinfection with RH parasites significantly inhibited this induction (Fig. 6A), suggesting that RH parasites can inhibit IFN-β-induced STAT1 activity. However, this reporter cell line also responds to IFN-γ treatment (Fig. 6A; see Fig. S1A in the supplemental material), and IFN-β treatment can induce STAT1 homodimers in addition to ISGF3, making it unclear what complex is being inhibited by infection.
FIG 6.

Toxoplasma also inhibits IFN-β-induced gene expression through a similar mechanism. (A) A HEK293 ISRE reporter cell line was infected with RH parasites for 3 to 5 h or left uninfected (UI), subsequently stimulated with 100 U of IFN-γ/ml or 100 U of IFN-β/ml, or left unstimulated (US) for 14 to 17 h and lysed, and the luciferase activity was measured. The data were normalized within each experiment to the uninfected, unstimulated sample, and the data shown are the average fold inductions and the SEM from three experiments. Asterisks (*) indicate P < 0.05. (B) HFFs were plated in six-well plates, infected with RH parasites for 4 h, and subsequently stimulated with 100 U of IFN-γ/ml or 100 U of IFN-β/ml for 15 to 20 h. Cells were also left uninfected (UI) and unstimulated (US). Transcript levels of three ISGF3-induced (ISRE promoter) genes and one STAT1 homodimer-induced (GAS promoter) gene were analyzed by RT-qPCR and normalized to ACTB levels. Averages of three independent experiments are shown; error bars represent the SEM. Asterisks (*) indicate P < 0.05 versus uninfected samples with the same stimulation. (C and D) HFFs were plated in 60-cm dishes, infected with RH parasites for 3 h, and subsequently stimulated with 100 U of IFN-γ/ml or 100 U of IFN-β/ml for 1 h. Cells were also left uninfected (UI) and unstimulated (US). A portion of the sample was lysed, boiled, separated by SDS-PAGE, and subjected to Western blotting (C). The rest of the samples were fractionated into cytoplasmic, nuclear extract, and chromatin fractions, diluted in 2× reducing SDS sample buffer, and boiled, and protein levels were analyzed by SDS-PAGE and Western blotting (D). GAPDH and H3 are markers for cytoplasmic and chromatin fractions, respectively. These experiments (C and D) were performed twice from two independent infections with similar results. (E) HFFs were plated on coverslips, preinfected with RH or RHΔrop16 parasites for 2 h at an MOI of ∼2 or left uninfected (UI), and stimulated with a pulse of 100 U of IFN-β/ml for 30 min before the cells were washed and the medium was changed or left unstimulated (US). Cells were fixed after 3.5 or 21.5 additional hours, permeabilized, and stained with α-phospho-STAT1Tyr and with Hoechst dye (nucleus). Nuclear accumulation of phospho-STAT1Tyr was quantified in at least 18 randomly selected cells per condition and normalized to the UI, US condition. Averaged data from two experiments are shown; error bars represent standard deviations.
To directly test the ability of Toxoplasma to inhibit ISGF3-mediated gene expression, we analyzed the expression of genes specifically induced by IFN-β and not by IFN-γ. We infected HFFs with RH parasites, or left cells uninfected, subsequently stimulated the cells with either IFN-γ or IFN-β, or left cells unstimulated, isolated RNA from cells, and analyzed transcript levels by RT-qPCR. IRF1 expression was induced strongly by IFN-γ treatment but also slightly induced by IFN-β treatment (Fig. 6B), likely via STAT1 homodimers in both conditions. Preinfection with RH significantly decreased this expression in both conditions (Fig. 6B). The expression of genes specifically induced by IFN-β—RSAD2, MX2, and OASL (34)—was also significantly inhibited by preinfection with RH parasites (Fig. 6B), indicating that infection also inhibits the activity of ISGF3 complexes. Normalization of qPCR data to a different control gene yielded virtually identical results (see Fig. S5 in the supplemental material).
To assess whether Toxoplasma infection inhibits STAT1 homodimer and ISGF3 transcriptional activity by similar mechanisms, we first determined whether Toxoplasma inhibits IFN-β-induced phosphorylation of STAT1. We infected HFFs with RH parasites, or left cells uninfected, for 3 h and subsequently stimulated cells with IFN-γ or IFN-β for 1 h or left the cells unstimulated. As measured by SDS-PAGE and Western blotting, infection with RH parasites does not inhibit IFN-β-induced STAT1 tyrosine or serine phosphorylation, even at an MOI that inhibits IFN-γ-induced IRF1 induction (Fig. 6C). Next, we measured the association of the ISGF3 complex with chromatin by isolating cytoplasmic, nuclear, and chromatin fractions from these cells and analyzing protein levels by SDS-PAGE and Western blot. The nuclear translocation and chromatin association of IFN-β-induced STAT1 and STAT2 was not inhibited by prior infection with Toxoplasma, and in fact we observed an increase in IFN-β-induced STAT1 in the chromatin fraction after infection (Fig. 6D), similar to what we observed for IFN-γ-induced STAT1. IRF9 was found in the nuclear extract fraction in all samples, but we observed a slight increase in chromatin association after IFN-γ or IFN-β stimulation or RH infection, and the combination of RH infection and IFN-β treatment together led to a strong increase in the chromatin association of IRF9 (Fig. 6D). These results indicate that, as with IFN-γ-induced STAT1, type I Toxoplasma infection does not inhibit the association of IFN-β-induced STAT1, STAT2, or IRF9 with host cell chromatin and in fact increases the chromatin association of STAT1 and IRF9.
We then tested whether a type I Toxoplasma strain inhibits nuclear-cytoplasmic cycling of STAT1 after stimulation with a pulse of IFN-β, as it does after stimulation with a pulse of IFN-γ. We preinfected HFFs with RH parasites for 2 h and subsequently stimulated the cells with a 30-min pulse of IFN-β. After either 3.5 or 21.5 additional hours, we fixed the cells and measured the nuclear levels of phospho-STAT1Tyr. Similar to our results after an IFN-γ pulse (Fig. 5), phospho-STAT1Tyr levels were 2- to 6-fold higher in RH- or RHΔrop16-infected cells compared to uninfected cells after the IFN-β was washed away (Fig. 6E), suggesting that type I infection also prevents the dephosphorylation and nuclear export of IFN-β-activated STAT1.
Toxoplasma rhoptry secretion is not sufficient for STAT1 inhibition.
The Toxoplasma effector(s) responsible for the inhibition of IFN-γ-induced, STAT1-mediated primary response gene expression remains unknown. It was previously reported that UV-treated parasites, which are unable to replicate, can still inhibit the IFN-γ-induced upregulation of MHC class II molecules (44). Similarly, we have found that with just 3 h of infection, before the parasites have replicated, Toxoplasma consistently inhibits the IFN-γ-induced expression of IRF1 (26). We therefore wondered whether the Toxoplasma effector(s) that modulates STAT1 transcriptional activity is a secreted factor that the parasite injects into the host cell upon invasion (47). To test this hypothesis, RH parasites were pretreated with cytochalasin D, an inhibitor of actin polymerization that allows parasites to attach to a host cell and secrete rhoptry contents but inhibits active invasion, which requires Toxoplasma actin polymerization (48). Pretreated parasites were added to HFFs and allowed to attach for 1.5 h, after which the cells were stimulated with IFN-γ for 18 h and the expression of IRF1 was measured by immunofluorescence. Cytochalasin D-treated parasites did not invade the HFF host cells but still attached and secreted rhoptry proteins, including ROP16, as demonstrated by the presence of phospho-STAT6 in host cell nuclei (49) (Fig. 7A). However, the injection of rhoptry contents into a cell was not sufficient to inhibit IFN-γ-induced IRF1 expression (Fig. 7A).
FIG 7.

Invasion is required for Toxoplasma's ability to inhibit IFN-γ-induced gene expression. RH parasites were pretreated with 1 μM cytochalasin D (cytoD) or 3 μM mycalolide B (mycaB) or left untreated (UT) and added to host cells for 1.5 h. Cells were then stimulated with 100 U of IFN-γ/ml or left unstimulated (US) for 18 h. (A) HFFs were fixed and stained for IRF1 (green), phospho-STAT6 (red), and with Hoechst dye (nucleus, blue). Scale bar, 10 μm. Arrows indicate infected cells, and arrowheads indicate uninfected cell with parasites attached and rhoptry proteins secreted. This experiment was performed three times with similar results. (B) A HEK293 GAS luciferase reporter cell line was then lysed, and the luciferase activity was measured. The results from two experiments per condition, except for the uninfected mycalolide B-treated condition for which only one experiment was done, were normalized to the maximum luciferase activity within the experiment and then averaged. In these experiments, 100% maximum induction represents an average of 10-fold induction over uninfected, unstimulated samples. Error bars represent the SEM. Asterisks (*) indicate P < 0.05 compared to uninfected control.
Similarly, parasites pretreated with cytochalasin D were unable to prevent IFN-γ-induced luciferase activity in our GAS reporter cell line (Fig. 7B). Cytochalasin D is a reversible inhibitor and must be kept on the cells for the entire experiment, also inhibiting host actin polymerization. Because it was recently shown that host actin plays a role in chromatin remodeling at IFN-γ-induced promoters (25), we also pretreated parasites with mycalolide B, an irreversible actin-depolymerizing agent, which was washed away before parasites were added to host cells. Parasites pretreated with mycalolide B also were unable to inhibit IFN-γ-induced STAT1 transcriptional activity in the GAS reporter cell line (Fig. 7B). Thus, our results suggest that secretion of rhoptry proteins by a type I parasite into an uninvaded host cell is not sufficient to inhibit IFN-γ-induced STAT1 transcriptional activity.
DISCUSSION
In this study, we have further elucidated the mechanism by which type I Toxoplasma parasites inhibit STAT1 activity at primary IFN-γ response genes. STAT1 undergoes multiple steps of activation at which an effector could act, including its phosphorylation, dimerization, nuclear translocation, DNA binding, and recycling (Fig. 8). It was previously demonstrated that Toxoplasma infection does not inhibit STAT1 tyrosine phosphorylation or nuclear translocation (24–26, 44). We report that STAT1 dimerization (Fig. 3) and STAT1 DNA binding in vivo (Fig. 4A) are also not inhibited by type I Toxoplasma preinfection. Instead, our results support a model where Toxoplasma infection inhibits STAT1 recycling by inhibiting its dissociation from the DNA, thereby inhibiting further and repeated rounds of STAT1 activation to prevent the full activation of the STAT1-mediated transcriptional program (Fig. 8).
FIG 8.

Intersection of IFN-STAT1 pathways and Toxoplasma. IFN-β and IFN-γ activate the expression of downstream target genes through ISGF3 and STAT1 homodimer complexes, respectively. The activation pathways of these cytokines are outlined. In a cell preinfected with Toxoplasma, the STAT1-mediated expression of both IFN-γ- and IFN-β-induced target genes is inhibited. We have measured multiple steps of these pathways and indicate here whether each step is inhibited by Toxoplasma infection or still occurs in a Toxoplasma-infected cell. Arrows indicate activation, inhibitory arrows indicate negative regulation, an “X” indicates steps which do not occur in Toxoplasma-infected cells, and a check mark indicates steps that do still occur in Toxoplasma-infected cells. We find that the Toxoplasma effector responsible for the inhibition of STAT1 activity and the expression of IFN-γ primary response genes is unlikely to be a protein secreted from the Toxoplasma rhoptry organelle prior to invasion. Although Toxoplasma infection induces the expression of SOCS family proteins, which negatively regulate JAK/STAT activation, this induction is not necessary for Toxoplasma to inhibit STAT1-mediated gene expression. We find that Toxoplasma infection inhibits the release of STAT1 from DNA, inhibiting downstream STAT1 recycling and further rounds of transcriptional activation.
Other data from previous studies are consistent with this model. STAT1 serine phosphorylation can only occur when STAT1 is chromatin associated (17), and it is known that Toxoplasma infection does not inhibit this phosphorylation (24, 26). Several EMSAs have shown that Toxoplasma does not inhibit STAT1 DNA-binding activity in vitro by EMSA (25, 42, 45), and one of these studies also observed increased and prolonged STAT1 DNA binding upon infection (42). However, our results differ from a ChIP experiment recently reported in murine BMDCs, where it was found that preinfection with type I Toxoplasma parasites did inhibit STAT1 binding at the Irf1 promoter (42). This could be due to differences in the species and cell type tested, although Toxoplasma infection can equally inhibit STAT1-mediated transcription in both human fibroblasts and murine macrophages (26).
This mechanism appears to be distinct from how Toxoplasma inhibits the expression of secondary IFN-γ response genes, such as Ciita, H2-Eβ, and Gbp2 (25), since it does not require the activity of HDACs (Fig. 1). In fact, treatment of two different STAT1 reporter cell lines with various HDAC inhibitors illustrated that altering histone acetylation can affect both basal and IFN-γ-induced STAT1-mediated gene expression, both positively and negatively (Fig. 1B). Although Toxoplasma targets histone acetylation to inhibit the expression of IFN-γ-induced secondary response genes, our results suggest that it is unlikely that Toxoplasma activates HDACs to inhibit IFN-γ-induction of primary response genes such as IRF1. In our ChIP experiments we did test the DNA-binding activity of STAT1 at several secondary response genes, including Ciita, GBP1, and IDO1. At the Ciita and GBP1 promoters, we observed IFN-γ induced STAT1 DNA binding in infected cells equivalent to that in uninfected cells (Fig. 4A), while at the IDO1 promoter Toxoplasma preinfection inhibited IFN-γ-induced STAT1 binding (Fig. 4A). Many secondary IFN-γ response genes, including IDO1, require the IRF1 transcription factor in addition to STAT1 for maximal induction (50, 51), and IRF1 can directly contact and recruit RNA polymerase II to promoters (52). At the IDO1 promoter, STAT1 may not stably bind in the absence of IRF1.
To target STAT1 and inhibit its DNA dissociation, a Toxoplasma effector could directly bind STAT1, or it could activate or inhibit a host protein. However, experiments with a proteasomal inhibitor (MG132) and a protein translation inhibitor (CHX) exclude several possible mechanisms of inhibition via modulation of a host factor. The ability of Toxoplasma to inhibit STAT1 activity in the presence of MG132 rules out the possibility that Toxoplasma infection induces the proteasomal degradation of a necessary STAT1 coactivator. IFN-γ-induced activation of both of our HEK293 STAT1 reporter cell lines was inhibited by MG132 treatment alone (Fig. 2C), which has been observed before (53), actually arguing for a role of the proteasome in STAT1 activation. Degradation of transcriptional activators has been linked to increased target gene expression (54, 55), and STAT1 may undergo similar turnover. Toxoplasma also does not require new host protein synthesis to inhibit STAT1-mediated primary response gene expression (Fig. 2A and B), ruling out the transcriptional or translational activation of a host negative regulatory protein or transcriptional repressor as a possible mechanism. Toxoplasma infection does induce the expression of SOCS proteins, which regulate STAT activity (56), but these proteins target the phosphorylation of the JAK and STAT proteins (57), while infection inhibits STAT1 activity downstream of these steps (Fig. 8). In this CHX experiment, CHX treatment and CHX treatment in combination with infection resulted in the expression of IRF1 independent of IFN-γ treatment (Fig. 2A). This expression likely occurs via a different transcription factor, since IRF1 can also be induced by NF-κB (2, 26, 58), and CHX treatment leads to the activation of NF-κB by preventing synthesis of inhibitory IκB proteins (59, 60). The activation of another transcription factor such as NF-κB could also explain the synergistic induction of IRF1 transcript by the combination of CHX and IFN-γ.
A Toxoplasma effector could also directly target STAT1. In previous EMSAs a more slowly migrating aberrant STAT1 complex bound to GAS oligonucleotides was observed specifically in infected cell extracts (25, 42); however, we did not detect any proteins stably bound to STAT1 in either native PAGE experiments or immunoprecipitations.
We report for the first time that type I Toxoplasma infection also inhibits the expression of IFN-β-induced genes (Fig. 6A and B). IFN-β signals through a transcription factor complex, ISGF3, consisting of STAT1, STAT2, and IRF9. Type I Toxoplasma infection does not prevent the IFN-β-induced tyrosine or serine phosphorylation of STAT1 (Fig. 6C) or the chromatin association of STAT1, STAT2, or IRF9 (Fig. 6D). Infection does prevent the dephosphorylation and nuclear export of IFN-β-activated STAT1 (Fig. 6E), suggesting that type I Toxoplasma infection inhibits STAT1 homodimer and ISGF3 activity by similar mechanisms.
The Toxoplasma effector responsible for this inhibition is still unknown. Our results suggest that this factor is not secreted into the host cell upon invasion, since cytochalasin D- or mycalolide B-treated parasites cannot inhibit IFN-γ-induced gene expression (Fig. 7). Three major possibilities remain for the identity of the Toxoplasma effector: (i) a rhoptry or dense granule protein that is secreted into the host cell upon invasion but must traffic back to the parasitophorous vacuole (PV) to be modified or to act, (ii) a small molecule or metabolite that can diffuse or be transported into the host cell from the PV that activates a host cell protein such as a nuclear receptor, or (iii) a protein that is secreted into the host cell postinvasion. Future experiments may distinguish between these possibilities.
Supplementary Material
ACKNOWLEDGMENTS
J.S. was supported by National Institutes of Health R01-AI080621. E.R. was supported by a predoctoral grant in the Biological Sciences (5-T32-GM007287-33) and the Cleo and Paul Schimmel Fund. A.C. was supported by a postdoctoral fellowship from the American Heart Association.
We thank members of the Saeij lab for useful comments and discussion, and we thank J. Wamstad, L. Surface, S. Thornton, and V. Subramanian of Laurie Boyer's lab at MIT for indispensable advice on ChIP assays. We also thank K. Thai and the MIT BioMicro Center for technical assistance with ChIP experiments.
Footnotes
Published ahead of print 25 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01291-13.
REFERENCES
- 1.Boehm U, Klamp T, Groot M, Howard JC. 1997. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15:749–795. 10.1146/annurev.immunol.15.1.749 [DOI] [PubMed] [Google Scholar]
- 2.Saha B, Jyothi Prasanna S, Chandrasekar B, Nandi D. 2010. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine 50:1–14. 10.1016/j.cyto.2009.11.021 [DOI] [PubMed] [Google Scholar]
- 3.Hunn JP, Feng CG, Sher A, Howard JC. 2011. The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mammalian Genome 22:43–54. 10.1007/s00335-010-9293-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, Huang DCS, Soldati-Favre D, Horie K, Takeda J, Takeda K. 2012. A cluster of interferon-γ-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37:302–313. 10.1016/j.immuni.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 5.Scharton-Kersten TM, Yap G, Magram J, Sher A. 1997. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185:1261–1273. 10.1084/jem.185.7.1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfefferkorn ER, Eckel M, Rebhun S. 1986. Interferon-gamma suppresses the growth of Toxoplasma gondii in human fibroblasts through starvation for tryptophan. Mol. Biochem. Parasitol. 20:215–224. 10.1016/0166-6851(86)90101-5 [DOI] [PubMed] [Google Scholar]
- 7.Degrandi D, Kravets E, Konermann C, Beuter-Gunia C, Klümpers V, Lahme S, Wischmann E, Mausberg AK, Beer-Hammer S, Pfeffer K. 2013. Murine guanylate binding protein 2 (mGBP2) controls Toxoplasma gondii replication. Proc. Natl. Acad. Sci. U. S. A. 110:294–299. 10.1073/pnas.1205635110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Grunvald E, Hieny S, Gazzinelli RT, Sher A. 1996. In the absence of endogenous IFN-γ, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 157:4045. [PubMed] [Google Scholar]
- 9.Yap GS, Sher A. 1999. Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-gamma- and tumor necrosis factor (TNF)-alpha-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J. Exp. Med. 189:1083–1091. 10.1084/jem.189.7.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman LA, Banica M, Reiner SL, Hunter CA. 2004. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. J. Immunol. 172:457–463 [DOI] [PubMed] [Google Scholar]
- 11.Khan IA, Matsuura T, Fonseka S, Kasper LH. 1996. Production of nitric oxide (NO) is not essential for protection against acute Toxoplasma gondii infection in IRF-1−/−mice. J. Immunol. 156:636. [PubMed] [Google Scholar]
- 12.Yap GS, Pesin M, Sher A. 2000. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 165:1–4 [DOI] [PubMed] [Google Scholar]
- 13.Sibley LD, Khan A, Ajioka JW, Rosenthal BM. 2009. Genetic diversity of Toxoplasma gondii in animals and humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364:2749–2761. 10.1098/rstb.2009.0087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat. Rev. Immunol. 5:375–386. 10.1038/nri1604 [DOI] [PubMed] [Google Scholar]
- 15.Bach EA, Aguet M, Schreiber RD. 1997. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 15:563–591. 10.1146/annurev.immunol.15.1.563 [DOI] [PubMed] [Google Scholar]
- 16.Darnell JE, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. 10.1126/science.8197455 [DOI] [PubMed] [Google Scholar]
- 17.Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmüller A, Yang E, Schaljo B, Kovarik P. 2008. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. U. S. A. 105:8944. 10.1073/pnas.0801794105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Müller M, Decker T. 2003. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-γ-dependent innate immunity. Immunity 19:793–802. 10.1016/S1074-7613(03)00322-4 [DOI] [PubMed] [Google Scholar]
- 19.Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. 2003. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev. 17:1992–2005. 10.1101/gad.268003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch V, Staab J, Ruppert V, Meyer T. 2012. Two glutamic acid residues in the DNA-binding domain are engaged in the release of STAT1 dimers from DNA. BMC Cell Biol. 13:22. 10.1186/1471-2121-13-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Begitt A, Meyer T, van Rossum M, Vinkemeier U. 2000. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc. Natl. Acad. Sci. U. S. A. 97:10418–10423. 10.1073/pnas.190318397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andrews RP, Ericksen MB, Cunningham CM, Daines MO, Hershey GKK. 2002. Analysis of the life cycle of Stat6: continuous cycling of Stat6 is required for IL-4 signaling. J. Biol. Chem. 277:36563–36569. 10.1074/jbc.M200986200 [DOI] [PubMed] [Google Scholar]
- 23.Lerner L, Henriksen MA, Zhang X, Darnell JE. 2003. STAT3-dependent enhanceosome assembly and disassembly: synergy with GR for full transcriptional increase of the α2-macroglobulin gene. Genes Dev. 17:2564–2577. 10.1101/gad.1135003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SK, Fouts AE, Boothroyd JC. 2007. Toxoplasma gondii dysregulates IFN-γ-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. J. Immunol. 178:51–54 [DOI] [PubMed] [Google Scholar]
- 25.Lang C, Hildebrandt A, Brand F, Opitz L, Dihazi H, Lüder CGK 2012. Impaired chromatin remodeling at STAT1-regulated promoters leads to global unresponsiveness of Toxoplasma gondii-infected macrophages to IFN-γ. PLoS Pathog. 8:e1002483. 10.1371/journal.ppat.1002483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosowski EE, Saeij JPJ. 2012. Toxoplasma gondii clonal strains all inhibit STAT1 transcriptional activity but polymorphic effectors differentially modulate IFNγ induced gene expression and STAT1 phosphorylation. PLoS One 7:e51448. 10.1371/journal.pone.0051448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Gao B, Xu W, Xiong S. 2011. BRG1 is indispensable for IFN-γ-induced TRIM22 expression, which is dependent on the recruitment of IRF-1. Biochem. Biophys. Res. Commun. 410:549–554. 10.1016/j.bbrc.2011.06.022 [DOI] [PubMed] [Google Scholar]
- 28.Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KDC, Saeij JPJ. 2011. Strain-specific activation of the NF-κB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208:195–212. 10.1084/jem.20100717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle JP, Saeij JPJ, Boothroyd JC. 2007. Toxoplasma gondii: inconsistent dissemination patterns following oral infection in mice. Exp. Parasitol. 116:302–305. 10.1016/j.exppara.2007.01.010 [DOI] [PubMed] [Google Scholar]
- 30.Ong Y-C, Reese ML, Boothroyd JC. 2010. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 285:28731–28740. 10.1074/jbc.M110.112359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen KDC, Wang Y, Wojno EDT, Shastri A, Hu K, Cornel L, Boedec E, Ong Y-C, Chien Y-H, Hunter C a, Boothroyd JC, Saeij JPJ. 2011. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 9:472–483. 10.1016/j.chom.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S-K, Karasov A, Boothroyd JC. 2007. Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect. Immun. 75:1626–1634. 10.1128/IAI.01862-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunn JD, Ravindran S, Kim S-K, Boothroyd JC. 2008. The Toxoplasma gondii dense granule protein GRA7 is phosphorylated upon invasion and forms an unexpected association with the rhoptry proteins ROP2 and ROP4. Infect. Immun. 76:5853–5861. 10.1128/IAI.01667-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Indraccolo S, Pfeffer U, Minuzzo S, Esposito G, Roni V, Mandruzzato S, Ferrari N, Anfosso L, Dell'Eva R, Noonan DM, Chieco-Bianchi L, Albini A, Amadori A. 2007. Identification of genes selectively regulated by IFNs in endothelial cells. J. Immunol. 178:1122–1135 [DOI] [PubMed] [Google Scholar]
- 35.Zhao S, Fernald RD. 2005. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 12:1047–1064. 10.1089/cmb.2005.12.1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 37.Lee T, Johnstone S, Young R. 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1:729–748. 10.1038/nprot.2006.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4:651–657. 10.1038/nmeth1068 [DOI] [PubMed] [Google Scholar]
- 39.Niedelman W, Gold DA, Rosowski EE, Sprokholt JK, Lim D, Farid Arenas A, Melo MB, Spooner E, Yaffe MB, Saeij JPJ. 2012. The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog. 8:e1002784. 10.1371/journal.ppat.1002784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 41.Medzhitov R, Horng T. 2009. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9:692–703. 10.1038/nri2634 [DOI] [PubMed] [Google Scholar]
- 42.Schneider AG, Abi Abdallah DS, Butcher BA, Denkers EY. 2013. Toxoplasma gondii triggers phosphorylation and nuclear translocation of dendritic cell STAT1 while simultaneously blocking IFN-γ-induced STAT1 transcriptional activity. PLoS One 8:e60215. 10.1371/journal.pone.0060215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shuai K, Liu B. 2003. Regulation of JAK-STAT signaling in the immune system. Nat. Rev. Immunol. 3:900–911. 10.1038/nri1226 [DOI] [PubMed] [Google Scholar]
- 44.Lang C, Algner M, Beinert N, Gross U, Lüder CGK. 2006. Diverse mechanisms employed by Toxoplasma gondii to inhibit IFN-γ-induced major histocompatibility complex class II gene expression. Microbes Infect. 8:1994–2005. 10.1016/j.micinf.2006.02.031 [DOI] [PubMed] [Google Scholar]
- 45.Lüder CGK, Walter W, Beuerle B, Maeurer MJ, Gross U. 2001. Toxoplasma gondii downregulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1α. Eur. J. Immunol. 31:1475–1484. [DOI] [PubMed] [Google Scholar]
- 46.Blouin CM, Lamaze C. 2013. Interferon gamma receptor: the beginning of the journey. Frontiers Immunol. 4:267. 10.3389/fimmu.2013.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boothroyd JC, Dubremetz J-F. 2008. Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat. Rev. Microbiol. 6:79–88. 10.1038/nrmicro1800 [DOI] [PubMed] [Google Scholar]
- 48.Håkansson S, Charron AJ, Sibley LD. 2001. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J. 20:3132–3144. 10.1093/emboj/20.12.3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327. 10.1038/nature05395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hobart M, Ramassar V, Goes N, Urmson J, Halloran P. 1997. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J. Immunol. 158:4260–4269 [PubMed] [Google Scholar]
- 51.Silva NM, Rodrigues CV, Santoro MM, Reis LFL, Alvarez-Leite JI, Gazzinelli RT. 2002. Expression of indoleamine 2,3-dioxygenase, tryptophan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect. Immun. 70:859. 10.1128/IAI.70.2.859-868.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramsauer K, Farlik M, Zupkovitz G, Seiser C, Kroger A, Hauser H, Decker T. 2007. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-γ-inducible gbp2 gene. Proc. Natl. Acad. Sci. U. S. A. 104:2849–2854. 10.1073/pnas.0610944104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li XL, Hassel BA. 2001. Involvement of proteasomes in gene induction by interferon and double-stranded RNA. Cytokine 14:247–252. 10.1006/cyto.2001.0887 [DOI] [PubMed] [Google Scholar]
- 54.Geng F, Wenzel S, Tansey WP. 2012. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 81:177–201. 10.1146/annurev-biochem-052110-120012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipford J, Smith G, Chi Y, Deshaies R. 2005. A putative stimulatory role for activator turnover in gene expression. Nature 438:8–11. 10.1038/nature04098 [DOI] [PubMed] [Google Scholar]
- 56.Zimmermann S, Murray PJ, Heeg K, Dalpke AH. 2006. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-γ signaling. J. Immunol. 176:1840–1847 [DOI] [PubMed] [Google Scholar]
- 57.Fujimoto M, Naka T. 2003. Regulation of cytokine signaling by SOCS family molecules. Trends Immunol. 24:659–666. 10.1016/j.it.2003.10.008 [DOI] [PubMed] [Google Scholar]
- 58.Robinson CM, Hale PT, Carlin JM. 2006. NF-κB activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-gamma and tumor necrosis factor-alpha. Cytokine 35:53–61. 10.1016/j.cyto.2006.07.007 [DOI] [PubMed] [Google Scholar]
- 59.Casado M, Díaz-Guerra M. 1997. Differential regulation of nitric oxide synthase mRNA expression by lipopolysaccharide and proinflammatory cytokines in fetal hepatocytes treated with cycloheximide. Biochem. J. 823:819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hershko DD, Robb BW, Wray CJ, Luo G-J, Hasselgren P-O. 2004. Superinduction of IL-6 by cycloheximide is associated with mRNA stabilization and sustained activation of p38 map kinase and NF-κB in cultured caco-2 cells. J. Cell. Biochem. 91:951–961. 10.1002/jcb.20014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.