

Abstract
Periodontitis is a disease of polymicrobial etiology characterized by inflammation, degradation of host tissue, and bone that irreversibly destroys the supporting apparatus of teeth. Porphyromonas gingivalis contains lipid A with structural heterogeneity that has been postulated to contribute to the initiation of dysbiosis in oral communities by modulating the host response, thereby creating a permissive environment for its growth. We examined two P. gingivalis lipid A phosphatase mutants which contain different “locked” lipid A structures that induce different host cellular responses for their ability to induce dysbiosis and periodontitis in rabbits. Lipopolysaccharide (LPS) preparations obtained from these strains were also examined. After repeated applications of all strains and their respective LPS preparations, P. gingivalis wild type, but not the lipid A mutants, had a significant impact on both the oral commensal microbial load and composition. In contrast, in rabbits exposed to the mutant strains or the LPS preparations, the microbial load did not increase, and yet significant changes in the oral microbial composition were observed. All strains and their respective LPS preparations induced periodontitis. Therefore, the ability to alter the lipid A composition in response to environmental conditions by lipid A phosphatases is required for both colonization of the rabbit and increases in the microbial load. Furthermore, the data demonstrate that multiple dysbiotic oral microbial communities can elicit periodontitis.
INTRODUCTION
Porphyromonas gingivalis is a Gram-negative, anaerobic bacterium that is associated with periodontitis. Periodontitis is a complex disease that is characterized by a shift from aerobic Gram-positive bacteria to anaerobic Gram-negative bacteria. The complex Gram-negative biofilm induces a significant increase in inflammation and eventually a complex immune lesion that results in degradation of host tissues, alveolar bone reduction, chronic inflammatory disease and ultimately tooth loss. Over 600 bacterial taxa have been identified in the oral cavity; yet, only a relatively small percentage has been associated with disease (1). P. gingivalis has been shown to cause disease in several different animal models (2). The mechanism by which P. gingivalis transforms healthy microbial/host homeostasis to destructive periodontitis is not clear.
To date, two in vivo animal models have implicated commensal bacteria as having a role in P. gingivalis induced periodontal disease. The first study, utilizing a rabbit model of periodontal disease showed that inoculation of P. gingivalis via ligature leads to an outgrowth of oral bacteria, as well as a shift in commensal profile and bone loss (3). There was no direct implication that commensal bacteria contributed to disease; however, it was clear that introduction of P. gingivalis caused the outgrowth of the oral bacterial community. More recently, P. gingivalis infection by oral gavage in mice was followed by an increase of oral bacteria that resulted in bone loss, while P. gingivalis-infected germfree mice lacking oral bacteria were protected from disease (4). The latter study clearly demonstrated that oral commensal bacteria were required for disease in the mouse gavage model and that P. gingivalis orchestrated the shift from a healthy bacterial community to a periopathogenic and dysbiotic community. Due to its significant contribution to the remodeling of commensal bacterial community resulting in disease, P. gingivalis was termed a keystone pathogen (5–8). P. gingivalis contains several virulence factors that may contribute to its ability to modulate the oral microbial composition (9). One of the virulence factors, the lipopolysaccharide (LPS), has been proposed to contribute changes to the oral microbial community (6). For example, P. gingivalis can alter its lipid A phosphate composition in response to different environmental conditions resulting in lipid A structures that are either agonists or antagonists for inflammatory activation at Toll-like receptor 4 (TLR4) (10–12). Alterations in the host environment by modulation of host TLR4 activity can have global effects on the microbial community by enhancing or suppressing the growth of different members of the oral community (7).
Here, we show that two P. gingivalis mutants that are unable to modulate their lipid A structural composition display significantly different phenotypes with respect to interactions with innate host components TLR4 and antimicrobial peptides. One mutant expresses a lipid A that is a TLR4 agonist, whereas the other mutant expresses a lipid A that is capable of antagonizing TLR4. One hypothesis we tested is that the antagonist structure disrupts host cell functions and causes overgrowth of the oral microbiota. Therefore, the two mutants were examined for their ability to induce periodontitis. LPS isolated from these two mutant strains was also examined. It was found that in the rabbit model of periodontal disease the wild-type (WT) strain, but not the mutant strains, of P. gingivalis was able to colonize the rabbit periodontium, significantly increase the oral commensal bacterial load, and result in periodontitis. Conversely, although the mutant strains did not significantly colonize rabbit periodontal tissue, both they and the isolated LPS preparations were able to create dysbiotic oral communities that were also associated with periodontitis. These data demonstrate that in the rabbit ligature model of periodontitis P. gingivalis lipid A phosphatases are required for colonization and that multiple different oral dysbiotic microbial communities can disrupt host homeostasis and result in disease.
MATERIALS AND METHODS
Bacterial growth conditions.
P. gingivalis strain ATCC 33277 was obtained from our stock collection. Bacteria were grown in TYHK medium consisting of Trypticase soy broth (30 g/liter; Becton Dickinson, Sparks, MD), yeast extract (5 g/liter; Becton Dickinson), and vitamin K3 (menadione; Sigma-Aldrich, St. Louis, MO). The basal TYHK medium was sterilized by autoclaving, followed by the addition of filter-sterilized hemin (Sigma-Aldrich) to the desired final concentration of either (1 μg/ml) or (10 μg/ml) as indicated in the text and figure legends. Cultures were grown in an anaerobic growth chamber (5% H2, 5% CO2, 90% N2) and maintained at 37°C on TYHK-agar plates.
Gene deletions in P. gingivalis 33277.
The genomic nucleotide sequences encoding the putative lipid A 1-phosphatase, PG1773, and the putative lipid A 4′-phosphatase, PG1587, were obtained from searches of the annotated P. gingivalis W83 genome at The Comprehensive Microbial Resource (http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi). Gene deletions were created by introducing either a tetracycline resistance cassette (tetQ) in place of the coding region for PG1773 or an erythromycin resistance cassette (ermF/AM) in place of the coding region for PG1587. PCR amplification of genomic DNA from P. gingivalis A7436 was performed using primer sets designed against the W83 sequence to amplify 1,000 bp upstream and 1,000 bp downstream from the regions adjacent to the PG1773 and PG1587 coding regions, respectively. The amplified 5′ and 3′ flanking regions for PG1773 and PG1587, respectively, were coligated with the tetQ and ermF/AM cassettes, respectively, into pcDNA3.1(−) to generate the gene disruption plasmids, p1773 5′flank:tetQ:3′flank and p1587 5′flank:erm:3′flank. P. gingivalis 33277 deficient in either PG1587 (1587KO) or PG1773 (1773KO) was generated by introducing either p1587 5′flank:erm:3′flank or p1773 5′flank:tetQ:3′flank into P. gingivalis 33277 by electroporation in a GenePulser Xcell (Bio-Rad, Hercules, CA). Bacteria were plated on TYHK/agar plates containing the appropriate selective medium, which included either erythromycin (5 μg ml−1) or tetracycline (1 μg ml−1) and incubated anaerobically. One week later, colonies were selected for characterization. Loss of the PG1587 and PG1773 coding sequences were confirmed in all clones by PCR analyses using primers designed to detect the coding sequences (11).
Isolation of LPS and lipid A.
Bacteria were cultured for 48 h in TYHK medium containing hemin at a concentration of either 1 or 10 μg/ml. LPS was isolated using a modified version of the Tri-Reagent protocol for LPS isolation as previously described (10). To generate lipid A, dried LPS samples were resuspended in 10 mM sodium acetate (pH 4.5) containing 1% (wt/vol) sodium dodecyl sulfate. The solution was heated 100°C for 1 h, followed by lyophilization overnight. The resulting lipid A pellets washed once in ice-cold 95% ethanol containing 0.02 N HCl and three times in 95% ethanol, followed by a final extraction with 1,160 μl of chloroform-methanol-water (1:1:0.9 [vol/vol/vol]) to remove residual carbohydrate contaminants. The chloroform layer containing the lipid A was dried and used for matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) or MALDI-TOF/TOF tandem MS analyses.
MALDI-TOF MS analyses.
For MALDI-TOF MS analyses, lipid A samples were dissolved in 10 μl of a mixture of 5-chloro-2-mercaptobenzothiazole (20 mg/ml) in chloroform-methanol at 1:1 (vol/vol), and 0.5 μl of each sample was analyzed in both positive- and negative-ion modes on an AutoFlex Analyzer (Bruker Daltonics). The data were acquired with a 50 Hz repletion rate and up to 3,000 shots were accumulated for each spectrum. Instrument calibration and all other tuning parameters were optimized using HP Calmix (Sigma-Aldrich). The data were acquired and processed using flexAnalysis software (Bruker Daltonics) (11).
HEK293 TLR4 activation assays.
HEK293 cells were plated in 96-well plates at a density of 4 × 104 cells per well and transfected the following day with plasmids bearing firefly luciferase, Renilla luciferase, human TLR4, and MD-2 by a standard calcium phosphate precipitation method as described previously (13) The test wells were stimulated in triplicate for 4 h at 37°C with the indicated doses of LPS or intact bacteria that had been suspended by vortexing in DMEM containing 10% human serum. After stimulation, the HEK293 cells were rinsed with phosphate-buffered saline and lysed with 50 μl of passive lysis buffer (Promega, Madison, WI). Luciferase activity was measured using the dual luciferase assay reporter system (Promega). The data are expressed as the fold increase of NF-κB-activity, which represents the ratio of NF-κB-dependent firefly luciferase activity to β-actin promoter-dependent Renilla luciferase activity (11).
Polymyxin B sensitivity assays.
Overnight cultures of wild-type P. gingivalis A7436 and its isogenic putative phosphatase mutants were grown in THYK medium containing hemin (1 μg/ml) or (10 μg/ml) as indicated above. Liquid cultures that had been grown for 24 h in an anaerobic growth chamber were diluted to a starting optical density at 600 nm (OD600) of 1.0, which represents ∼109 CFU/ml for all of the strains examined (data not shown). Subsequently, 10−3 to 10−8 dilutions of each strain were plated on TYHK-agar plates containing PMB (0, 5, and 200 μg/ml). After 8 to 10 days of incubation in an anaerobic chamber, the resulting colonies were counted to determine the viability at the different PMB treatments. The results for each strain were plotted as percent survival, which was derived from the ratio of the number of colonies detected on the experimental plates containing polymyxin B (5 or 200 μg/ml) to the number of colonies detected on the control plates that did not contain polymyxin B (11).
P. gingivalis wild type (WT).
P. gingivalis (strain A7436 [14]) was grown using standard procedures as described previously (15–18). Briefly, bacteria were cultured on agar plates containing Trypticase soy agar supplemented with 0.5% (wt/vol) yeast extract (Invitrogen Life Technologies), 5% defibrinated sheep red blood cells, 5 μg of hemin, and 1 μg of vitamin K (Sigma-Aldrich)/ml. Plates were incubated for 3 days at 37°C in an anaerobic chamber maintained by hydrogen gas mixture (hydrogen/nitrogen) that is circulated through a heated palladium catalyst. Colonies were randomly selected and anaerobically cultured overnight at 37°C in Wilkins-Chalgren anaerobe broth (3).
Slurry preparations.
Bacterial numbers with P. gingivalis strains and mutants were spectrophotometrically determined at 600 nm, adjusted to 109 CFU (i.e., an OD600 of 0.8), while 35 ng of LPS was used for LPS preparations. Bacterial strains and LPS preparations were mixed with carboxymethyl cellulose (CMC) to form a thick slurry.
Animal model.
The study was approved by the Forsyth Institute Institutional Animal Care and Use Committee (IACUC). Specific-pathogen-free New Zealand White rabbits (24 males, 3.5 to 4.0 kg) were purchased from Covance Research Products, Inc. (Denver, PA), equilibrated for 7 days prior to experiments. The rabbits were, kept in individual cages, received water ad libitum, and fed standard rabbit chow (Purina LabDiet 5321; Purina Mills, LLC, St. Louis, MO) throughout the experiment. Periodontitis was induced and established in all animals using a previously established protocol (3). A 3-0 silk suture (ligature) was placed around the second premolar teeth of both mandibular quadrants under general anesthesia using injections of 40 mg of ketamine (Ketaset; Fort Dodge Animal Health)/kg and 5 mg of xylazine (Anased; Lloyd Laboratories, Inc.)/kg administered intramuscularly. The CMC slurry containing P. gingivalis, phosphatase mutants or purified LPS (35 ng) was topically applied to the ligatures three times a week (Monday, Wednesday, and Friday) over a 6-week period to induce periodontal disease. The sutures were checked at every application, and lost or loose sutures were replaced (3). At the end of the 6-week period, animals were euthanized using an overdose of sodium pentobarbital (Euthasol [Virbac Animal Health], 120 mg/kg) according to the approved protocol by the Forsyth IACUC. Upon harvest, each animal was numbered arbitrarily so that the examiner was blinded during all subsequent measurements and analyses. Due to the large number of animals required for this study, historical data were used for the ligated control. This control has been tested many times in our laboratory with consistent results (3).
Morphometric analysis.
Immediately after euthanasia, the mandibles were dissected free of muscle and soft tissue, keeping the attached gingiva intact. Hemimandibles were obtained by splitting each mandible from the midline between the central incisors. The left hemimandible was processed for morphometric analysis of bone loss, while the other half was utilized for histological evaluation. For morphometric analysis, the hemimandible was defleshed by immersion in 10% hydrogen peroxide, followed by careful soft tissue removal, washed with distilled water, air dried, and stained with 1% methylene blue for visual distinction between the tooth and bone. The bone level around the second premolar was measured directly by a 0.5-mm calibrated periodontal probe. Measurements were made at three points each, at buccal and lingual sides, for crestal bone level. The mean crestal bone level around the tooth was calculated for statistical analysis. Similarly, for the proximal (intrabony) bone level, measurements were made at mesial and distal aspects of the tooth. The measurements were taken from both the buccal and the lingual sides on both proximal aspects of the second premolar, and the mean proximal bone level was calculated (3). The tip of the tooth at the measured site was used as the reference point for these measurements.
Qualitative histological evaluations.
Half of the mandible was immersed in 10 volumes of 10% EDTA with continuous agitation. The solution was replaced every 24 h for 4 weeks. Demineralization was confirmed by serial radiographs. After a series of hydration and drying, the specimens were embedded in paraffin. About 30 sections (5 μm) were cut for each paraffin block. Selected section for each specimen were either stained with hematoxylin-eosin for descriptive histology and histomorphometry or with tartrate-resistant acid phosphatase (TRAP) to examine the osteoclastic activity using light microscopy (3).
Quantitative histomorphometry.
To quantify the changes in bone, the mean value (± the standard deviation [SD]) of the linear distance and the area of bone loss were calculated for each group. A previously developed measurement technique (3) was used to calculate the bone changes at three different sections of the root using the ProImage software. The linear measurements were made at three levels, each corresponding to one-third of the root and alveolar bone interface: crestal, middle, and apical. Linear distance is reported as the distance from the base of the epithelium to the alveolar crest border at the three chosen levels, the apical, middle, and the coronal third of the root and is expressed as the difference between ligated and unligated sites. Osteoclast counts were performed to evaluate the osteoclastogenic activity in TRAP-stained sections by calculating the osteoclasts in affected areas. The total numbers of osteoclasts at the surface of the bone were compared between the groups.
Microbial sampling.
Microbial dental plaque was sampled at baseline, at 3 and 6 weeks using paper points. The area was isolated to prevent saliva contamination, and 30-s samples were collected using sterile paper points according to previously reported methods (3). Each sample was placed in an individual Eppendorf tube containing 0.15 ml of TE (10 mM Tris-HCl, 1 mM EDTA [pH 7.6]) and 0.5 M NaOH was added for long-term stabilization and stored at −80°C until analysis. Twenty-eight species representing periodontal organisms, including P. gingivalis, Aggregatibacter actinomycetemcomitans, Actinomyces odontolyticus, Actinomyces viscosus, Actinomyces israelii, Peptostreptococcus micros, Prevotella intermedia, Prevotella nigrescens, Capnocytophaga curva, Capnocytophaga rectus, Streptococcus oralis, Streptococcus intermedius, Treponema denticola, Eikenella corrodens, Fusobacterium nucleatum subsp. vincenti, Escherichia coli, Campylobacter concisus, Capnocytophaga sputigena, Prevotella bivia, Selenomonad noxia, Veillonella parvula, Capnocytophaga ochracea, Filifactor alocis, Actinomyces naeslundii, Lactobacillus acidophilus, Eubacterium saphenum, and Streptococcus sanguis were investigated in each plaque sample using the checkerboard DNA-DNA hybridization technique (3). Evaluation of the chemiluminescent signals is performed by radiographic detection, comparing the obtained signals with the signals generated by pooled standard comparisons of 0, 103, and 106 bacteria of each of the 28 species.
Statistical analyses.
Mean values for histomorphometric measurements were used to determine the changes in bone level. In addition, TRAP-positive cell counts were calculated to detect the osteoclastogenesis. The data were analyzed by two-tailed unpaired Student t tests (GraphPad Prism) where indicated. P < 0.05 was considered indicative of statistical significance.
RESULTS
Phenotypic characterization of the P. gingivalis lipid A mutants used in this study.
Two phosphatase mutants that result in P. gingivalis bacteria that have different lipid A profiles were created to test lipid A phenotypes in the rabbit model of periodontal disease. The initial identification and characterization of P. gingivalis lipid A mutants that are unable to modify their lipid A structural composition and display significantly different lipid A structural profiles was performed in strain 33277 (11). However, since previous work in the rabbit ligature model was performed with P. gingivalis strain A7436, in order to prevent potential undefined strain variability effects in the rabbit model, the lipid A mutants were constructed in strain A7436. The mutant strains, designated PG1587 and PG1773, contain deletion mutations in the lipid A 4′-phosphatase (PG1587) and 1-phosphatase (PG1773) genes, respectively (11). Characterization of the lipid A structural composition confirmed that similar to strain 33277, strain A7436 containing these mutations displayed an altered lipid A structural composition in that PG1587 accumulated the diphosphate lipid A structural peak designated m/z 1,770, whereas strain PG1773 accumulated peak m/z 1,449 (Fig. 1). These data confirm that the genes PG1587 and PG1773 result in the same alterations in the lipid A structural profile in both P. gingivalis strains 33277 and A7436.
FIG 1.

P. gingivalis expresses both lipid A 4′- and 1-phosphatase activities. Lipid A isolated from wild-type P. gingivalis A7436 or mutant bacteria bearing deletions in PG1587 and PG1773 gene loci were examined by MALDI-TOF MS to elucidate the position of the lipid A phosphates. Representative structures of lipid A corresponding to peak are shown. Lipid A samples were examined in the negative-ion mode or the positive-ion mode for each sample.
Next, the mutant strains and their respective LPS preparations were examined for their TLR4 and TLR2 responses. Similar to the results previously shown in strain 33277 (11), TLR4 displayed differing responses to PG1587 and PG1773 in A7436. PG1587 bacteria, as well as their isolated LPS, demonstrated strong TLR4 agonist activity (Fig. 2A). Likewise, LPS obtained from PG1773, but not from PG1587, displayed TLR4 antagonism (Fig. 2B). Interestingly, and different from that observed in strain 33277, both PG1587 and PG1773 strains in A7436 displayed increased TLR2 activation compared to the WT (Fig. 2B). We recently reported contaminating lipoproteins in the P. gingivalis LPS preparations that could explain this TLR2 activity (8).
FIG 2.

Lipid A phosphatases allow P. gingivalis to evade host innate immune differences. Phosphatase mutants have different phenotypic host innate effects through TLRs and antimicrobial susceptibility. HEK293 cells expressing either human TLR4 and MD-2 (A and C) or TLR2 and TLR1 (B) were exposed to the indicated doses of LPS or whole bacteria for 4 h. The fold activation of NF-κB over the medium control was determined by measuring inducible firefly luciferase activity. The positive control for TLR4 is E. coli and TLR2 is PamC3K4. The results shown are means ± the SD of triplicate samples from one of three independent experiments. Asterisks indicate statistically significant differences (P < 0.001 [two-tailed unpaired t tests]) in the potency of TLR4 antagonism by PGWT or PG1773. (D) Lipid A phosphatases confer P. gingivalis resistance to killing by antimicrobial cationic peptides. The indicated strains of P. gingivalis were plated on TYHK-agar plates containing polymyxin B (PMB; 0, 5, and 200 mg ml−1) and measured by spectrophotometry (OD600).
Finally, it has been previously reported that PG1587 and PG1773 in strain 33277 display significantly different susceptibilities to polymyxin B, a cationic antimicrobial peptide antibiotic (11). Examination of these mutations in A7436 yielded similar results in that PG1587 was exquisitely susceptible to polymyxin B, whereas the wild-type strain and PG1773 were completely resistant (Fig. 2D). These data confirm that the lipid A phosphatase mutations previously described in strain 33277 display nearly identical phenotypes when contrasted in A7436.
P. gingivalis requires lipid A phosphatase modulation to colonize and cause overgrowth of commensal bacteria.
We used a rabbit model of periodontal disease to examine P. gingivalis colonization and changes to total bacteria. Samples of plaque from the ligature area were taken at three time points, including baseline (time zero), 3 weeks, and 6 weeks (Fig. 3). Chemiluminescence units from DNA-checkerboard were plotted for plaque samples that were collected from each treatment group shown in Fig. 4. Remarkably, P. gingivalis WT was the only group to cause robust P. gingivalis colonization (Fig. 4A). PG1587 also displayed a significant increase in P. gingivalis colonization at 6 weeks, although much lower than the WT (Fig. 4A). It is possible that the low chemiluminescence from the baseline-uninfected rabbits is indicative of an endogenous strain of P. gingivalis that may have increased over the time course of infection with the PG1587. The test group LPS1587 shows a similar increase, and since no bacteria were added, this increase, although not significant, strongly implicates an endogenous strain that could be affected by the mutant strain or its LPS.
FIG 3.
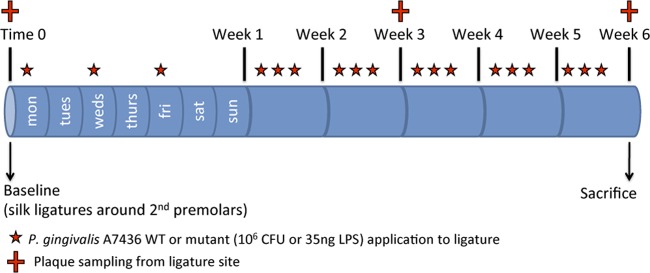
Timeline of experimental design. 3-0 silk ligatures were tied to second premolars in mandibular quadrants at baseline in all groups, and P. gingivalis, phosphatase mutants or purified LPS was applied in methylcellulose slurry three times per week (Monday-Wednesday-Friday) for 6 weeks. Plaque samples were taken for microbial analysis at 0, 3, and 6 weeks during treatment. At 6 weeks, all were sacrificed, and the extent of disease was determined.
FIG 4.

P. gingivalis phosphate modulation is required for colonization and commensal overgrowth. Chemiluminescence units of DNA-DNA checkerboard hybridization assay specific to P. gingivalis (A) or the summation of all bacteria analyzed (B) (see Materials and Methods; P < 0.01 [Mann-Whitney test]).
Next, the relative biomass of the dental plaque was examined for the different experimental groups (Fig. 4B). Biomass was determined by the total chemiluminescence activity found in the dental plaque samples, and it was found that plaque samples from each experimental group (except the plaque LPS 1773ko group) showed a drastic reduction in total bacteria at 3 weeks from the baseline uninfected point (Fig. 4B). At 6 weeks, the wild type was also the only bacterial strain to cause significant increase in commensal biomass (Fig. 4B). This result was not found in any other experimental group, demonstrating that both lipid A phosphate mutants were unable to significantly colonize the rabbit periodontium or significantly increase the total dental plaque biomass.
Phosphatase mutants exert distinct affects on oral bacterial communities compared to the wild type, whereas LPS preparations yielded a less complex but discrete bacterial profile.
To examine major increases and/or decreases to specific bacteria, the changes in abundance of bacterial species that experienced a significant change (P ≥ 0.01 [Mann-Whitney test]) after 6 weeks of applications were plotted in Table 1. Although wild-type P. gingivalis was the only group to generate an increase in oral bacteria at 6 weeks, all experimental groups had significant effects on the composition of the oral bacterial community. The increase in total oral bacteria is markedly increased and significant without the inclusion of P. gingivalis WT. Of the three species of Campylobacter that were identified at baseline; two species (C. concisus and C. curva) showed significant reductions in all experimental groups. The changes to oral bacteria induced by wild-type P. gingivalis affected 16 species of the panel of 28 (59% Gram negative and 41% Gram positive). The P. gingivalis wild type altered the abundance of many more bacterial species than any of the other mutant strains and LPS preparations. Conversely, the phosphatase mutants induced unique profiles predominantly in the Gram-negative communities. The changes to commensal bacteria by LPS preparations were very few and virtually identical, indicating a phenotype common to all LPS types that caused the discrete changes to the bacterial communities.
TABLE 1.
Significant changes to the chemiluminescence signal from specific oral bacteria at 6 weeks compared to uninfected controlsa
| Group | Gram-negative bacteria |
Gram-positive bacteria |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P. bivia | C. curva | C. rectus | E. coli | S. noxia | C. ochracea | C. sputigena | C. concisus | V. parvula | F. nucleatum subsp. vincetii | P. gingivalis | T. denticola | P. micros | S. sanguis | A. viscosus | F. alocis | L. acidophilus | |
| WT bacteria | + | – | + | + | + | + | – | + | + | + | + | + | + | + | + | + | |
| 1587ko bacteria | – | + | + | – | – | + | + | + | + | + | |||||||
| 1773ko bacteria | + | – | + | + | + | – | + | + | |||||||||
| WT LPS | – | + | – | ||||||||||||||
| 1587ko LPS | – | + | – | ||||||||||||||
| 1773ko LPS | – | + | + | – | |||||||||||||
Significance (P < 0.01) was determined by using the Mann-Whitney test. Bacterial complexes were grouped into “color” complexes based on previously published data (18). Red complex, T. denticola and P. gingivalis; orange complex, F. nucleatum subsp. vincetii and P. micros; yellow complex, V. parvula; purple complex, S. sanguis; green complex, C. ochracea, C. sputigena, and C. concisus.
In order to assess the changes to Gram-negative and Gram-positive communities; the relative abundance was plotted in a percentage plot (Fig. 5A). To better visualize prominent Gram-negative bacteria, P. gingivalis, C. curva, and C. sputigena were separated by pattern. The first major change observed was the reduction in the Gram-negative community at 3 weeks in all treatment groups, which was principally due to the marked decline of C. curva. For example, the Gram-negative community, which includes C. curva, makes up ca. 50% of the bacterial community at baseline in animals that received P. gingivalis WT; however, 3 weeks after P. gingivalis WT application, there was a reduction of 30% in the Gram-negative bacteria, and this seemed to be due to the disappearance of C. curva. At the end of 6 weeks, P. gingivalis wild type became the predominant strain among the Gram-negative bacteria; this can be attributed largely to the colonization of P. gingivalis.
FIG 5.

Distinct changes to bacterial communities result from the application of P. gingivalis, phosphatase mutants, or LPS. (A) Relative biomass of Gram-negative bacteria, including P. gingivalis, C. curva, and C. sputigena. (B) Gram-positive bacterial profiles and chemiluminescence units of C. sputigena plotted for each treatment group over time. Statistics were scored by using the Mann-Whitney test.
A low abundance of C. sputigena was seen at baseline and at 3 weeks; however, at 6 weeks, the LPS preparations showed a remarkable increase in C. sputigena, while there were decreases in the wild type and the mutant PG1587. The LPS groups showed no significant change in the percentages of Gram-negative bacteria from baseline to 6 weeks. In these groups, the dramatic loss of C. curva was replaced by the increase in C. sputigena, masking overall changes to Gram-negative bacteria, while the phosphatase mutant bacteria and wild type had reduced amounts of C. sputigena in addition to the reduction of C. curva.
To examine the impact of bacterial strains and LPS preparations on C. sputigena, chemiluminescence units were plotted in Fig. 5B. All three LPS treatment groups yielded remarkable increases in C. sputigena, whereas the bacterial treatment groups showed marked reductions (Fig. 5B).
Clinical parameters of disease show similar bone loss for all groups.
In order to evaluate amount of periodontal disease caused by P. gingivalis or LPS preparations, clinical disease parameters, including crestal bone level (the distance between the crest of the bone and the tip of the tooth), intrabony defect depth (the distance between the crestal bone and the base of the bone defect-vertical bone loss), were evaluated (Fig. 6). Histological bone levels and osteoclast activity were assessed by histological staining of tissue samples. Osteoclasts were identified by positive staining for their tartrate-resistant acid phosphatases (TRAPs). Strikingly, all experimental groups showed similar levels of clinical crestal and histological bone loss. The only difference was in the intrabony defect assessment; rabbits receiving PGWT, PG1773, and LPS1773 showed significant bone loss over ligature alone (historical data [3]), while other differences were not statistically significant. It is interesting that this difference was common to all strains that had predominantly the 1-phosphorylated, tetra-acylated lipid A structure capable of inhibiting host responses. However, all other assessments of disease showed similar bone loss and osteoclast activity between treatment groups. Together, these findings indicate that, regardless of the etiology, changes to lipid A, or bacterial communities, all of the tested bacterial strains and LPS preparations cause similar levels of periodontal disease.
FIG 6.

Analyses of periodontal disease from P. gingivalis, phosphatase, or LPS-treated rabbit lesions. Alveolar bone loss for all animals was directly measured on defleshed jaws (see Materials and Methods) for characteristics of human periodontitis, including soft (A; intrabony defect) and hard (B; crestal bone loss) tissue destruction. Rabbit mandibles were harvested and prepared for histologic analysis (see Materials and Methods). (C) Histologic analysis and quantification of histomorphometric changes. (D) Osteoclastogenesis in the alveolar bone assessed by TRAP staining (see Materials and Methods). Asterisks indicate statistically significant differences (P < 0.01 [Mann-Whitney test]).
DISCUSSION
P. gingivalis has a number of virulence factors that contribute to disease, such as proteases (gingipains), lipopeptides, fimbriae, hemagglutinins, and LPS. Although some of these factors have been explored (9), the function of P. gingivalis' unique heterogeneous LPS in disease in vivo has not been examined until now. In the present study, the use of P. gingivalis lipid A phosphatase mutants that are unable to remodel their lipid A structural composition do not colonize well or induce overgrowth of the commensal oral microbiota. However, the P. gingivalis mutants and their respective isolated LPS preparations induced significant changes in the qualitative composition of the oral bacterial communities and caused similar disease profiles. This observation supports the idea that multiple different microbial compositions result in dysbiosis and disease (7).
Modifications to lipid A resulted in phenotypic changes to P. gingivalis. We have previously reported that P. gingivalis grown in high temperature or lacking the PG1587 phosphatase can result in bacteria susceptible to antimicrobial peptides (11, 12). A potential explanation for this antimicrobial peptide sensitivity may be revealed by a recent study of Helicobacter pylori lipid A phosphatases that showed the location of the phosphate group determines susceptibility to polymyxin B (19). Considering this study, the dominant lipid A structure possessing the phosphate at the 1 position in the PG1773 and the P. gingivalis wild type could confer resistance to polymyxin B, whereas the lipid A with the 4′-phosphate is susceptible. Since PG1587 was susceptible to polymyxin B, it was postulated that this mutant could be sensitive to antimicrobial peptides in vivo. Indeed, the PG1587 was unable to colonize. However, despite the antimicrobial peptide resistance of PG1773, it was also unable to colonize. It is likely that the phosphatase mutants could not colonize due to the inability to modify the lipid A moiety but for reasons not associated with antimicrobial peptide sensitivity. It has been shown in two other studies that alteration of phosphate position can affect colonization ability. Utilizing bacterial mutants with changes to lipid A phosphate position in either Salmonella enterica serovar Typhimurium or Helicobacter pylori resulted in a reduction or inability to colonize in a mouse model of disease (19, 20. The phosphate position on lipid A appears to be a determining factor for virulence; however, other pleiotropic effects from phosphate mutations have not thoroughly been examined. For example, we have demonstrated here that TLR2 activities are different between whole bacteria wild type and phosphatase mutants. At present, there is no explanation for this, and therefore more investigation is required. Regardless of the phosphatase phenotype or ability to colonize, all of the bacterial treatments caused changes to the bacterial communities.
DNA checkerboard hybridization was utilized to examine bacterial communities that have been shown to change with the application of P. gingivalis in a rabbit model of periodontal disease (3). Although the DNA checkerboard hybridization has limitations, such as high DNA requirement and species-specific probes, this particular method allows for the analysis of a large panel of known bacterial species. The checkerboard assay has been compared to the 16S ribosomal DNA (rDNA)-based PCR method and found to be comparable in sensitivity for prevalent species, although the sensitivity diminished with species at lower concentrations (21). The results from the checkerboard analysis revealed changes to bacterial communities were very different between the PGWT, PG1587, and PG1773. PGWT showed the most complex changes to oral microbiota. However, the bacterial changes seen from treatment with PG1587 and PG1773 were distinctly different from each other, but common to PGWT. For example, significant increases of bacteria associated with periodontal disease in humans were common to PGWT and PG1587, while a significant increase in unclassified bacteria was shared between PGWT and PG1773. It is interesting that the increase in bacteria associated with PG1587 and PTWT were predominantly part of the complexes of bacteria associated with periodontal disease originally described by Socransky et al. (22), although it is not known whether similar complexes exist in the rabbit periodontium. The PG1587 and PGWT also showed a decrease in two bacterial species that are described in the “green complex” of bacteria associated with healthy flora. PG1773 did not have any increases in bacteria associated with periodontal disease, except for one of the “yellow complex.” Perhaps a shared phenotype common to PGWT and PG1587 produced a suitable environment for the disease-associated bacterial complexes. For example, the agonist lipid A from PG1587, also produced by PGWT when environmental conditions allow (10, 11), could elicit an inflammatory host response favorable to the disease-associated bacteria. Alternately, the antagonist lipid A common to PGWT and PG1773 could inhibit the host response and allow an outgrowth of certain commensal bacteria. Interestingly, PGWT, PG1773, and LPS1773 treatment groups caused similar intrabony defects again indicating a shared phenotype such as the antagonist lipid A structure. It is possible that this particular lipid A structure may be directly involved in disease progression since there are similarities between these treatment groups' disease profiles but not their bacterial profiles. However, the abundance and species of oral bacteria that resulted from each treatment were distinctly different from each other and yet all resulted in disease.
Different LPS preparations do not appear to cause any specific alterations. Instead, similar changes were made to bacterial profiles, indicating a general environmental alteration. The striking feature common to all purified LPS was the robust TLR2 activity (8); this alteration of host environment could be the cause of the similar bacterial profiles observed. Further, the LPS preparations appear to alter the environment in a way that is beneficial to C. sputigena, which is not seen in the whole bacterium preparations.
One trend common to all treatment groups except for P. gingivalis WT was the disappearance of two species of Campylobacter, C. concisus, and C. curva. The presence of the P. gingivalis wild type did not reduce the C. concisus species, as seen in all other treatment groups. This suggests that the environment created by the modulation of lipid A in wild-type P. gingivalis may be required for C. concisus to maintain its niche; this niche may change to allow some bacteria to bind where others may lose their binding sites.
The present study demonstrates that lipid A phosphatases are required for P. gingivalis colonization and commensal overgrowth. Overgrowth of commensal bacteria due to P. gingivalis colonization has recently been shown to be responsible for periodontitis using a mouse model of periodontal disease by gavage (4). In this study, since all bacterial strains and their respective LPS preparations caused microbial dysbiosis that resulted in very similar disease, it is likely that disease in this model occurred through different etiologies that all resulted in a disruption of the normal oral flora. We could not distinguish microbial changes that occurred from an altered local inflammatory environment from those that may have induced the inflammatory response. Rather, the present study demonstrates that multiple different bacterial communities are associated with periodontal disease.
ACKNOWLEDGMENTS
This study was supported in part by U.S. Public Health Service grants DE012768 (R.P.D.), DE020906 (A.K.), DE19938, DE15566 (T.E.V.D.), and DE18917 (H.H.) from the National Institute of Dental and Craniofacial Research.
Footnotes
Published ahead of print 25 November 2013
REFERENCES
- 1.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J. Bacteriol. 192:5002–5017. 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oz HS, Puleo DA. 2011. Animal models for periodontal disease. J. Biomed. Biotechnol. 2011:754–857. 10.1155/2011/754857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, Van Dyke TE. 2007. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J. Immunol. 179:7021–7029 [DOI] [PubMed] [Google Scholar]
- 4.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. 10.1016/j.chom.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajishengallis G, Darveau RP, Curtis MA. 2012. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10:717–725. 10.1038/nrmicro2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darveau RP. 2009. The oral microbial consortium's interaction with the periodontal innate defense system. DNA Cell Biol. 28:389–395. 10.1089/dna.2009.0864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 8:481–490. 10.1038/nrmicro2337 [DOI] [PubMed] [Google Scholar]
- 8.Jain S, Coats SR, Chang AM, Darveau RP. 2013. A novel class of lipoprotein lipase-sensitive molecules mediates Toll-like receptor 2 activation by Porphyromonas gingivalis. Infect. Immun. 81:1277–1286. 10.1128/IAI.01036-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bostanci N, Belibasakis GN. 2012. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 333:1–9. 10.1111/j.1574-6968.2012.02579.x [DOI] [PubMed] [Google Scholar]
- 10.Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP. 2006. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect. Immun. 74:4474–4485. 10.1128/IAI.01924-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, To TT, Goodlett DR, Ernst RK, Darveau RP. 2009. Human Toll-like receptor 4 responses to Porphyromonas gingivalis are regulated by lipid A 1- and 4′-phosphatase activities. Cell Microbiol. 11:1587–1599. 10.1111/j.1462-5822.2009.01349.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, Rangarajan M, Tarelli E, Marsh PD. 2011. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect. Immun. 79:1187–1193. 10.1128/IAI.00900-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coats SR, Pham TT, Bainbridge BW, Reife RA, Darveau RP. 2005. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J. Immunol. 175:4490–4498 [DOI] [PubMed] [Google Scholar]
- 14.Shapira L, Champagne C, Van Dyke TE, Amar S. 1998. Strain-dependent activation of monocytes and inflammatory macrophages by lipopolysaccharide of Porphyromonas gingivalis. Infect. Immun. 66:2736–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsen I, Socransky SS. 1981. Ultrasonic dispersion of pure cultures of plaque bacteria and plaque. Scand. J. Dent. Res. 89:307–312 [DOI] [PubMed] [Google Scholar]
- 16.Olsen I, Socransky SS. 1981. Comparison of three anaerobic culture techniques and media for viable recovery of subgingival plaque bacteria. Scand. J. Dent. Res. 89:165–174 [DOI] [PubMed] [Google Scholar]
- 17.Doan N, Contreras A, Flynn J, Morrison J, Slots J. 1999. Proficiencies of three anaerobic culture systems for recovering periodontal pathogenic bacteria. J. Clin. Microbiol. 37:171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Socransky SS, Haffajee AD. 1994. Evidence of bacterial etiology: a historical perspective. Periodontology 2000 5:7–25. 10.1111/j.1600-0757.1994.tb00016.x [DOI] [PubMed] [Google Scholar]
- 19.Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. 2011. Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 7:e1002454. 10.1371/journal.ppat.1002454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong Q, Six DA, Liu Q, Gu L, Wang S, Alamuri P, Raetz CR, Curtiss R., III 2012. Phosphate groups of lipid A are essential for Salmonella enterica serovar Typhimurium virulence and affect innate and adaptive immunity. Infect. Immun. 80:3215–3224. 10.1128/IAI.00123-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siqueira JF, Rocas IN, De Uzeda M, Colombo AP, Santos KR. 2002. Comparison of 16S rDNA-based PCR and checkerboard DNA-DNA hybridisation for detection of selected endodontic pathogens. J. Med. Microbiol. 51:1090–1096 [DOI] [PubMed] [Google Scholar]
- 22.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144. 10.1111/j.1600-051X.1998.tb02419.x [DOI] [PubMed] [Google Scholar]