

Abstract
A mutant that exhibited less cytotoxic activity toward INT-407 human intestinal epithelial cells than the wild type was screened from a random transposon mutant library of Vibrio vulnificus, and an open reading frame encoding an Fe-S cluster regulator, IscR, was identified using a transposon-tagging method. A mutational analysis demonstrated that IscR contributes to mouse mortality as well as cytotoxicity toward the INT-407 cells, indicating that IscR is essential for the pathogenesis of V. vulnificus. A whole-genome microarray analysis revealed that IscR influenced the expression of 67 genes, of which 52 were upregulated and 15 were downregulated. Among these, 12 genes most likely involved in motility and adhesion to host cells, hemolytic activity, and survival under oxidative stress of the pathogen during infection were selected and experimentally verified to be upregulated by IscR. Accordingly, the disruption of iscR resulted in a significant reduction in motility and adhesion to INT-407 cells, in hemolytic activity, and in resistance to reactive oxygen species (ROS) such as H2O2 and tert-butyl hydroperoxide (t-BOOH). Furthermore, the present study demonstrated that iscR expression was induced by exposure of V. vulnificus to the INT-407 cells, and the induction appeared to be mediated by ROS generated by the host cells during infection. Consequently, the combined results indicated that IscR is a global regulator that contributes to the overall success in the pathogenesis of V. vulnificus by regulating the expression of various virulence and survival genes in addition to Fe-S cluster genes.
INTRODUCTION
Most of virulence factors of pathogenic bacteria act cooperatively to obtain maximum effectiveness during pathogenesis while their expression is coordinately regulated by common global regulators in response to environmental conditions, and this coordinated regulation facilitates the cooperation of virulence factors and is crucial to the overall success of the pathogens during infection (1, 2). Vibrio vulnificus is an opportunistic Gram-negative pathogen that frequently contaminates oysters. It has been proposed that numerous virulence factors account for the fulminating and destructive nature of V. vulnificus infections and contribute to not only disease development but also the survival and multiplication on or within the host (for a recent review, see reference 3). However, studies about global regulators involved in the regulation of V. vulnificus virulence factors are still very limited.
Iron-sulfur proteins containing the Fe-S cluster as a cofactor carry out multiple important cellular processes such as electron transfer, metabolic reactions, and gene regulation and are widely distributed (for recent reviews, see references 4 and 5). A highly conserved isc operon, iscRSUA-hscBA-fdx, was discovered to encode all of the proteins required for the biogenesis of the majority, if not all, of the Fe-S cluster proteins in Escherichia coli (for recent reviews, see references 5 and 6). Expression of the isc operon is autoregulated by IscR, a [2Fe-2S] cluster-containing transcription factor (7). IscR functions as a sensor of the cellular Fe-S cluster status and homeostatically regulates Fe-S cluster biogenesis (7, 8). When the cellular Fe-S cluster is sufficient to occupy IscR, the resulting [2Fe-2S]-IscR represses the isc operon. The repression is relieved by decreased [2Fe-2S] occupancy of IscR under iron starvation or oxidative stress conditions (8–12).
IscR is proposed to be a member of the Rrf2 family of transcription factors, consisting of a winged helix-turn-helix (HTH) DNA-binding domain in the N-terminal region and a motif with three cysteines and one histidine (CCCH) required for Fe-S cluster ligation in the C-terminal region (13, 14). It has been reported that IscR controls the expression of more than 40 genes, including the genes encoding additional proteins involved in Fe-S cluster biogenesis, such as the suf operon (sufABCDSE), and anaerobic respiratory enzymes containing the Fe-S cluster (15). IscR also regulates genes involved in biofilm formation in response to changes in cellular Fe-S cluster levels in E. coli (16). These findings suggested that IscR is a global regulator having broader roles beyond modulating Fe-S cluster homeostasis in bacteria.
In the present study, an open reading frame (ORF) encoding an E. coli IscR (EcIscR) homologue was identified in an effort to screen for V. vulnificus virulence factors by a transposon-tagging method. A V. vulnificus null mutant, in which the iscR gene was inactivated, was constructed by allelic exchanges, and the possible roles of the IscR protein during infection of V. vulnificus were explored. As a result, it was discovered that IscR is essential for the virulence of V. vulnificus in mice and in tissue culture. A transcriptome analysis newly identified several genes involved in motility and adhesion to host cells, hemolytic activity, and survival under oxidative stress as components of the IscR regulon. Furthermore, IscR expression was found to be induced by exposure of the pathogen to host cells. Therefore, the results indicated that IscR is a global regulator and plays an essential role in bacterial pathogenesis by being induced during infection.
MATERIALS AND METHODS
Strains, plasmids, and culture conditions.
The strains and plasmids used in this study are listed in Table 1. Unless noted otherwise, the V. vulnificus strains were grown in LB medium supplemented with 2.0% (wt/vol) NaCl (LBS) at 30°C.
TABLE 1.
Plasmids and bacterial strains used in this study
| Strain or plasmid | Relevant characteristic(s)a | Reference or source |
|---|---|---|
| Bacterial strains | ||
| V. vulnificus | ||
| M06-24/O | Clinical isolate; virulent | Laboratory collection |
| JK093 | M06-24/O with ΔiscR | This study |
| E. coli | ||
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1; plasmid replication | Laboratory collection |
| S17-λpir | λ-pir lysogen; thi pro hsdR hsdM+ recA RP4-2 Tc::Mu-Km::Tn7;Tpr Smr; host for π-requiring plasmids, conjugal donor | 17 |
| BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | Laboratory collection |
| Plasmids | ||
| pRL27 | Tn5-RL27; oriR6K; Kmr | 18 |
| pGEM-T Easy | PCR product cloning vector; Apr | Promega |
| pDM4 | R6K γ ori sacB; suicide vector; oriT of RP4; Cmr | 22 |
| pJK0909 | pDM4 with ΔiscR; Cmr | This study |
| pRK415 | IncP ori; broad-host-range vector, oriT of RP4; Tcr | 23 |
| pJK1016 | pRK415 with iscR; Tcr | This study |
| pET22b(+) | His6 tag fusion expression vector; Apr | Novagen |
| pJK0928 | pET22b(+) with iscR; Apr | This study |
Tpr, trimethoprim resistant; Smr, streptomycin resistant; Kmr, kanamycin resistant; Apr, ampicillin resistant; Cmr, chloramphenicol resistant; Tcr, tetracycline resistant.
Identification of V. vulnificus iscR and generation of an iscR mutant.
To generate a random transposon mutant library of V. vulnificus, the E. coli S17-1 λpir tra strain (17) containing pRL27, a suicide vector carrying the hypertransposable mini-Tn5 (Kmr) element (18), was used as a conjugal donor to V. vulnificus MO6-24/O. Transposon mutants of V. vulnificus grown on the LBS agar containing polymyxin B (100 U/ml; to exclude E. coli) (19) and kanamycin (100 μg/ml) were selected and grown with 200 μl of LBS medium in 96-well culture dishes (Nunc, Roskilde, Denmark). From the transposon mutants, a mutant exhibiting decreased cytotoxic activity against INT-407 (ATCC CCL-6) human intestinal epithelial cells was screened. A DNA segment flanking the transposon insertion was amplified by PCR as previously described (20) and a search of the V. vulnificus MO6-24/O genome sequence (GenBank accession numbers CP002469 and CP002470 [www.ncbi.nlm.nih.gov]) for homology to the sequence of the resulting PCR product singled out iscR, which encodes a putative Fe-S cluster regulator.
The iscR gene was inactivated in vitro by deletion of the iscR ORF (300 bp of 507 bp) using a PCR-mediated linker-scanning mutation method as described previously (21). The primer pairs ISCR001F and ISCR001R (for amplification of the 5′ amplicon) and ISCR002F and ISCR002R (for amplification of the 3′ amplicon) were designed (see Table S1 in the supplemental material). The iscR gene with the 300-bp deletion was amplified by PCR using a mixture of both amplicons as the template and ISCR001F and ISCR002R as primers. The resulting 1,702-bp DNA fragment containing the deleted iscR was ligated with SpeI-SphI-digested pDM4 (22) to generate pJK0909 (Table 1). The E. coli S17-1 λpir tra strain (containing pJK0909) was used as a conjugal donor to V. vulnificus MO6-24/O. The conjugation and isolation of the transconjugants were conducted as previously described (21), and the V. vulnificus iscR mutant chosen for further analysis was named JK093 (Table 1).
Complementation of the iscR mutant.
To complement the iscR mutation, the iscR coding region was amplified from the genomic DNA of V. vulnificus M06-24/O by PCR with the primer pair ISCR004F and ISCR004R (Table S1) and then digested with HindIII and XbaI. The amplified iscR coding region was subcloned into the broad-host-range vector pRK415 (23) linearized with the same enzymes (Table 1) to result in pJK1016. The plasmid pJK1016 was delivered into the iscR mutant JK093 by conjugation as described above.
Cytotoxicity and mouse mortality.
Cytotoxicity was evaluated by measuring cytoplasmic lactate dehydrogenase (LDH) activity that is released from the INT-407 cells by damage of plasma membranes (24). The INT-407 cells were grown in minimum essential medium containing 1% (vol/vol) fetal bovine serum (MEMF) (Gibco-BRL, Gaithersburg, MD) in 96-well culture dishes (Nunc) as described previously (25). Each well with 2 × 104 INT-407 cells was infected with the V. vulnificus strains at a multiplicity of infection (MOI) of 10 for various incubation times. The LDH activity released into the supernatant was determined using a cytotoxicity detection kit (Roche, Mannheim, Germany).
Mortalities of mice infected with the wild-type and iscR mutant strains were compared as described elsewhere (26). Groups of (n = 20) 7-week-old ICR female mice (specific-pathogen-free; Seoul National University) were starved without food and water for 12 h until infection. Then the mice, without iron-dextran pretreatment, were intragastrically administered with 50 μl of 8.5% (wt/vol) sodium bicarbonate solution, followed immediately with 50 μl of the inoculum, representing approximately 109 cells of either the wild-type or iscR mutant strain. Mouse mortalities were recorded for 24 h. All manipulations of mice were approved by the Animal Care and Use Committee of Seoul National University.
Transcriptome analysis.
A transcriptome analysis was performed using a V. vulnificus Whole-Genome Twin-Chip as described previously (27). Total RNAs from the V. vulnificus strains grown to an A600 of 0.5 were isolated with an RNeasy Mini Kit (Qiagen, Valencia, CA). Then aminoallyl-cDNA was synthesized using an Amino Allyl-cDNA Labeling Kit (Ambion, Austin, TX) according to the manufacturer's procedures. The aminoallyl-cDNAs from the iscR mutant and wild-type strains were, respectively, labeled with Cy5 and Cy3, and equal amounts of the labeled cDNAs were combined and used to hybridize the microarray slides at 42°C for 16 h. The arrays were washed, dried, scanned, and analyzed by GenePix Pro, version 3.0, software (Axon Instruments, Union City, CA). ORF spots that showed a 2-fold or greater difference in expression with a P value of ≤0.05 were considered to be regulated by IscR.
Quantitative real-time PCR (qRT-PCR).
Total RNA from the V. vulnificus strains was isolated as described above, and cDNA was synthesized using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time PCR amplification of the cDNA was performed by using a Chromo 4 real-time PCR detection system (Bio-Rad) with a pair of specific primers (listed in Table S2 in the supplemental material), as described previously (21). Relative expression levels of the specific transcripts were calculated by using the 16S rRNA expression level as the internal reference for normalization.
When necessary, the wild-type V. vulnificus grown to A600 0.5 was exposed to MEMF (control), hydrogen peroxide (H2O2), INT-407 cells, or INT-407 cells preincubated with 25 mM N-acetyl-l-cysteine (NAC) for 1 h to scavenge reactive oxygen species (ROS) (28). For H2O2 exposure experiments, V. vulnificus was grown anaerobically as described elsewhere (29). The mixture of the INT-407 and V. vulnificus cells was centrifuged at 250 × g for 10 min to precipitate INT-407 cells, and then the V. vulnificus cells were harvested from the supernatant and used to isolate total RNAs as described previously (20).
Purification of V. vulnificus IscR and electrophoretic mobility shift assays (EMSAs).
The coding region of the iscR gene was amplified by PCR using V. vulnificus MO6-24/O chromosomal DNA and primers ISCR003F and ISCR003R (see Table S1 in the supplemental material). The 513-bp PCR product was subcloned into a His6 tag expression vector, pET22b(+) (Novagen, Madison, WI), resulting in pJK0928 (Table 1). His-tagged IscR was then expressed in E. coli BL21(DE3) and purified aerobically by affinity chromatography according to the manufacturer's procedure (Qiagen).
For EMSAs, four genes were randomly chosen from the pool of the 12 predicted IscR-regulated genes. The putative regulatory regions (about 300 bp) of the genes were amplified and radioactively labeled by PCR using the primer pairs FLGE01F and FLGE01R for flgE, GBPA01F and GBPA01R for gbpA, VVHBA01F and VVHBA01R for vvhB, and PRX01F and PRX01R for prx (see Table S1 in the supplemental material). For the negative control, a 250-bp DNA fragment of the iscR coding region was amplified and radioactively labeled by PCR using the primer pair ISCR01F and ISCR01R (see Table S1). The labeled DNA (2.5 nM) fragments were incubated with the purified IscR (10 nM) for 30 min at 30°C in a 20-μl reaction mixture containing 1× binding buffer (15) and 0.1 μg of poly(dI-dC). Electrophoretic analyses of the DNA-protein complexes were performed as described previously (21).
Motility and adhesion assays.
For motility assays, V. vulnificus strains were grown to an A600 of 0.5 and subsequently stabbed into LBS semisolid medium solidified with 0.3% agar (21). The plates were incubated at 30°C for 24 h, and migration through the agar was photographed using a digital camera (Canon PowerShot SX220 HS; Japan).
For the adhesion assay, 2 × 105 INT-407 cells per well in 12-well culture dishes (Nunc) were infected with the V. vulnificus strains at an MOI of 10 for 30 min, and then plates were washed two times to remove nonadherent bacteria as described previously (25). Following the last wash, the INT-407 cells were broken with 0.1% Triton X-100 treatment for 20 min, and the recovered bacterial cells were enumerated as CFU counts per well (25). Adhesion of V. vulnificus to INT-407 cells was also examined microscopically. For this purpose, INT-407 cells seeded onto glass coverslips were infected with the V. vulnificus strains at an MOI of 10 for 30 min and then fixed in methanol and stained with 0.4% Giemsa (25).
Hemolysis assay and survival under oxidative stress.
For a hemolysis assay, an aliquot of the culture supernatants of V. vulnificus strains grown to an A600 of 0.9 was mixed with an equal volume of human red blood cell (hRBC) suspension (1.0% in phosphate-buffered saline) (Innovative Research, Novi, MI) and incubated at 37°C for 20 min. The level of hemolysis was determined as described elsewhere (30) and expressed using the complete hemolysis by 1% Triton X-100 as 100%.
Survival of the V. vulnificus strains under oxidative stress was determined by measuring the growth on the LBS agar medium containing either 250 μM H2O2 or 60 μM tert-butyl hydroperoxide (t-BOOH) (31). Equal volumes of the strains grown to an A600 of 0.5 were serially diluted and then spotted onto the agar medium. Growth was then photographed after 16 h as described above for the motility assay.
Western blot analysis.
The purified IscR was used to raise rabbit anti-IscR polyclonal antibodies (AB Frontier, Seoul, South Korea). The wild-type V. vulnificus was exposed to MEMF (control), H2O2, INT-407 cells, or INT-407 cells preincubated with NAC and then harvested as described above for qRT-PCR. V. vulnificus cells were lysed using complete lysis B buffer (Roche) for 1 min, and residual cell debris was removed by centrifugation (21). Protein samples from the cell lysates, equivalent to 10 μg of total protein, were resolved by using SDS-PAGE. The resolved proteins were then transferred to a nitrocellulose membrane (Bio-Rad) and probed with a 1:5,000 dilution of the anti-IscR polyclonal antibodies. The bound antibodies were detected using goat anti-rabbit IgG conjugated with alkaline phosphatase (Sigma) and visualized by incubation with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (Sigma) (29).
Data analyses and statistics.
Averages and standard errors of the means (SEM) were calculated from at least three independent experiments. Mouse mortality was evaluated using a log rank test program (http://bioinf.wehi.edu.au/software/russell/logrank/). All other data were analyzed by Student's t tests with the SAS program (SAS software, SAS Institute, Inc.). Significance of differences between experimental groups was accepted at a P value of <0.05.
Microarray data accession number.
All primary microarray data were deposited in the Gene Expression Omnibus (GEO [http://www.ncbi.nlm.nih.gov/projects/geo/]) database under accession number GSE44679.
RESULTS
Identification of V. vulnificus IscR.
Using a transposon-tagging method as described in Materials and Methods, an ORF encoding an E. coli IscR homologue was identified from a mutant of V. vulnificus that exhibited less cytotoxicity to INT-407 cells than the wild type. The amino acid sequence deduced from the putative V. vulnificus iscR nucleotide sequence revealed a protein, the putative IscR (VvIscR), composed of 168 amino acids having a theoretical molecular mass of 18,199 Da and a pI of 6.82. VvIscR exhibits a high level of identity (77% in amino acid sequences) with E. coli IscR (EcIscR) (see Fig. S1 in the supplemental material) (7). Furthermore, VvIscR contains a winged HTH DNA-binding domain and the highly conserved CCCH motif at positions corresponding to those of EcIscR. The predicted hydrophobicity profile (http://web.expasy.org/protscale/) was similar to that of EcIscR and consistent with the fact that IscR is a cytosolic soluble protein (data not shown). The results indicated that VvIscR is also an Fe-S cluster-containing transcription factor as observed in EcIscR (7, 14).
IscR is important for virulence.
In an effort to understand the role of IscR in V. vulnificus pathogenesis, an isogenic mutant JK093 (Table 1) which lacked functional iscR gene was constructed by allelic exchanges, and its virulence in tissue culture and in mice was evaluated. For this purpose, the monolayers of INT-407 cells were infected with the wild-type and iscR mutant strains, and the activity of LDH released from the INT-407 cells was compared at various incubation times (Fig. 1A). The iscR mutant exhibited significantly lower LDH-releasing activity than the wild-type strain for as long as 2 h, indicating that IscR is important for V. vulnificus to infect and injure host cells (Fig. 1A). Complementation of the iscR gene in JK093 with a functional iscR gene (pJK1016) restored the LDH-releasing activity to levels comparable to the wild-type level (Fig. 1A). Therefore, the decreased cytotoxicity of JK093 resulted from inactivation of iscR rather than reduced expression of any genes downstream of iscR.
FIG 1.
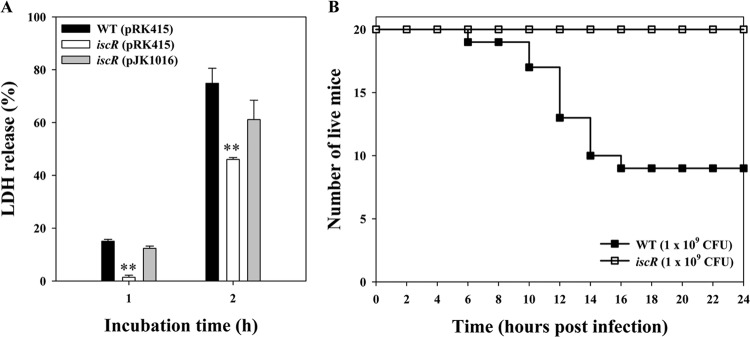
Cytotoxicity and mouse mortality of V. vulnificus. (A) INT-407 cells were infected with the V. vulnificus strains at an MOI of 10. The cytotoxicity was determined by an LDH release assay and expressed using the total LDH released from the cells completely lysed by 1% Triton X-100 as 100%. Error bars represent the SEM. **, P < 0.005 relative to groups infected with the wild type at each incubation time. (B) Seven-week-old specific-pathogen-free female ICR mice were intragastrically infected with the wild type (WT) or the iscR mutant at doses of 109 CFU. WT (pRK415), wild type; iscR (pRK415), iscR mutant; iscR (pJK1016), complemented strain.
To examine the role of IscR in mouse mortality, mice were infected intragastrically with the wild-type and iscR mutant strains, and the numbers of dead mice were counted. As shown in Fig. 1B, the death of mice infected with the iscR mutant was significantly inhibited by 24 h of postinfection, while more than 50% of mice infected with the parental wild-type strain were dead by this time point (P = 2.88 × 10−6, log rank test). The combined results suggested that IscR is essential for the virulence of V. vulnificus in mice and in tissue culture. However, in contrast to results in the cell culture experiments, the complemented strain JK093(pJK1016) was not able to restore the reduced virulence of JK093, as determined by measuring mouse mortality (data not shown). One possible explanation for this lack of complementation is that the plasmid pJK1016 was not well maintained in bacteria used to infect mice, where no antibiotics were added.
IscR-regulated genes involved in pathogenesis.
A whole-genome microarray analysis was used to compare the transcriptional profiles of the wild-type and iscR mutant strains. The microarray analysis predicted 67 genes potentially regulated by IscR, of which 52 genes were upregulated and 15 were downregulated (see Tables S3 and S4 in the supplemental material). Among them, 12 genes that most likely participate in the pathogenesis of V. vulnificus (3) were upregulated by IscR (Fig. 2). Among the products of the 12 genes are the flagellar protein FlgE contributing to motility (32), methyl-accepting chemotaxis proteins (MCPs), and signal transduction proteins contributing to chemotaxis (33). A gene encoding GbpA known to participate in adhesion (34), two genes (vvhBA) involved in the production of a cytolysin/hemolysin (35), and two genes encoding the antioxidant protein peroxiredoxin (Prx) (36) and glutaredoxin 2 (Grx2) (37) are also upregulated by IscR (Fig. 2).
FIG 2.
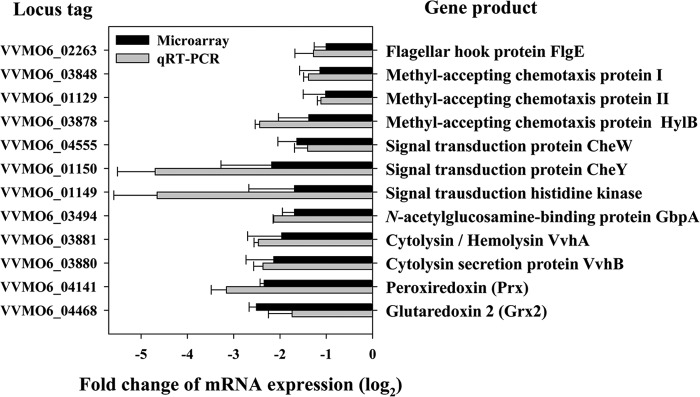
IscR-regulated genes possibly involved in the pathogenesis of V. vulnificus. Twelve genes possibly involved in the pathogenesis of V. vulnificus were chosen from the pool of the IscR regulon members predicted by microarray analysis. Regulation of their transcription by IscR was confirmed by qRT-PCR. Each column represents the mRNA expression level in the iscR mutant relative to that of the parental wild-type strain. Error bars represent the SEM. Locus tags are based on the database of the V. vulnificus MO6-24/O genome sequence, which was retrieved from GenBank (accession numbers CP002469 and CP002470), and the products of the 12 genes are listed on the right.
Since many of the genes that we predicted as potential members of the IscR regulon were not previously reported to be IscR regulated in other bacteria, IscR regulation of the selected 12 genes was experimentally verified. qRT-PCR revealed that IscR indeed regulates transcription of all of the genes predicted, implying that IscR controls not only Fe-S cluster biogenesis but also the virulence and survival of V. vulnificus (Fig. 2). To determine whether IscR regulates the genes directly or indirectly, four genes, flgE, gbpA, vvhB, and prx, were randomly chosen from the 12 genes, and the binding of IscR to their upstream regulatory regions was examined. EMSAs revealed that IscR binds to the upstream regions of all of the genes that were tested. IscR binding was specific because assays were performed in the presence of 0.1 μg of poly(dI-dC) as a nonspecific competitor. Furthermore, IscR was not able to bind to the iscR coding region used as a negative control (Fig. 3A). These results suggested that the genes are under direct control of IscR. Furthermore, the regulatory-region sequences of the four genes were analyzed by comparing them with a 25-bp consensus sequence that was previously known for binding of E. coli IscR (13, 38). The analysis predicted the potential sequences for binding of V. vulnificus IscR from the upstream regions, and alignment of the sequences revealed substantial levels of matches (over 20 of 25) with the consensus E. coli IscR-binding sequence (Fig. 3B).
FIG 3.
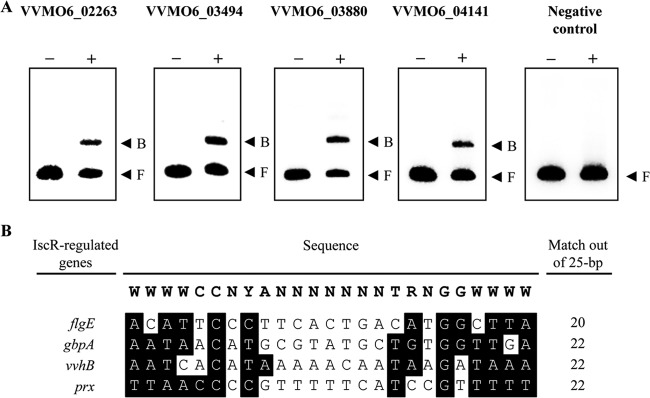
Verification of IscR binding to the regulatory region of the newly identified IscR regulon. (A) The regulatory regions of VVMO6_02263 (flgE), VVMO6_03494 (gbpA), VVMO6_03880 (vvhB), or VVMO6_04141 (prx) and a part of the iscR coding region (negative control) were radioactively labeled and then used as probe DNAs. The same locus tag numbers that appear in Fig. 2 are at the top of each panel. −, without IscR; +, with IscR; B, bound DNA; F, free DNA. (B) Comparison of the consensus E. coli IscR-binding sequence and the potential V. vulnificus IscR-binding sequences. The consensus E. coli IscR-binding sequence (type 2) (38) is shown at the top. Sequences from the regulatory regions of flgE, gbpA, vvhB, and prx, retrieved from GenBank (accession numbers CP002469 and CP002470), are aligned below. Individual bases identical to those conserved in the consensus E. coli IscR-binding sequence are highlighted. Numbers of bases that match those of the consensus E. coli IscR-binding sequence are indicated on the right. R, A or G; Y, C or T; W, A or T; N, any base.
Effects of the iscR mutation on the virulence-related phenotypes of V. vulnificus.
To extend our understanding of the role of IscR in V. vulnificus pathogenesis, the effect of the iscR mutation on several phenotypes related to the virulence of many enteropathogenic bacteria were examined. The iscR mutant JK093 was less motile, as determined by its ability to migrate on a semisolid plate surface, than the wild-type strain (Fig. 4A). The diameter of the swimming area of the mutant was significantly reduced, to approximately 50% of that of the wild type (Fig. 4B). Complementation of the iscR mutant by introduction of pJK1016 substantially restored the reduced motility (Fig. 4). Since motility has been known to be essential for the adhesion of enteropathogens to host cells (39), INT-407 cells were infected with the V. vulnificus strains, and the numbers of bacterial cells adhered to INT-407 cells were compared. The number of the iscR mutant cells adhering to INT-407 cells was about 3-fold lower than the numbers of cells of the wild-type and the complemented strains (Fig. 5A). The wild-type and the complemented strains revealed the formation of small clusters of aggregated bacteria on the INT-407 cell surface (Fig. 5B). In contrast, when INT-407 cells were infected with the iscR mutant, a much smaller area of the cell surfaces was covered with the pathogen, and no clusters of aggregated bacteria were observed (Fig. 5B). These results indicated that IscR is required for the optimum adhesion to host cells as well as for the motility of V. vulnificus. These findings were consistent with the previous observation that IscR activates the expression of the genes encoding proteins involved in motility, chemotaxis, and adhesion (Fig. 2).
FIG 4.
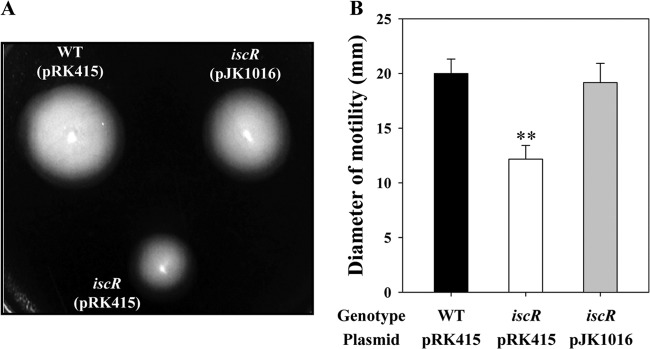
Motility of the V. vulnificus strains. (A) The areas of motility of the strains grown at 30°C for 24 h on plates with LBS and 0.3% agar were photographed. (B) The diameters of motility areas are the means plus SEM of results from three independent experiments. **, P < 0.005 relative to the wild type. WT (pRK415), wild type; iscR (pRK415), iscR mutant; iscR (pJK1016), complemented strain.
FIG 5.
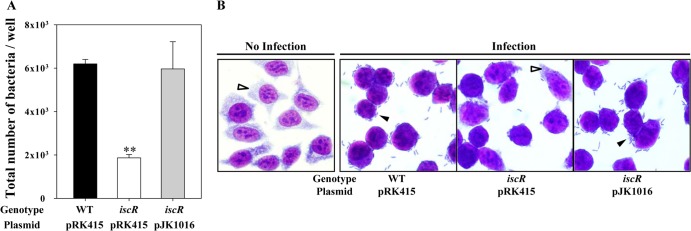
Adhesion of the V. vulnificus strains. (A) INT-407 cells were infected at an MOI of 10 with the V. vulnificus strains as indicated. After 30 min, adherent bacteria were enumerated, and results are presented as the numbers of bacteria per well of the tissue culture dishes. Error bars represent the SEM. **, P < 0.005 relative to the wild type. (B) INT-407 cells were infected with the V. vulnificus strains at an MOI of 10 for 30 min and morphologically observed using a light microscope after Giemsa staining (original magnification, ×1,200). The adhered V. vulnificus cells (filled arrowheads) and the cytoplasm of the INT-407 cells (open arrowheads) are indicated. WT (pRK415), wild type; iscR (pRK415), iscR mutant; iscR (pJK1016), complemented strain.
The effects of the iscR mutation on hemolytic activity and survival under oxidative stress were also assessed. When the hemolytic activities in the culture supernatant were quantitated, the hemolytic activity of the iscR mutant was about 4-fold lower than that of the wild-type and the complemented strains (Fig. 6). These results indicated that IscR is required for full hemolytic activity of V. vulnificus. The decreased expression of the vvhBA gene encoding cytolysin in the absence of IscR could be an important, if not the sole, reason for the lower hemolytic activity of the iscR mutant (Fig. 2).
FIG 6.
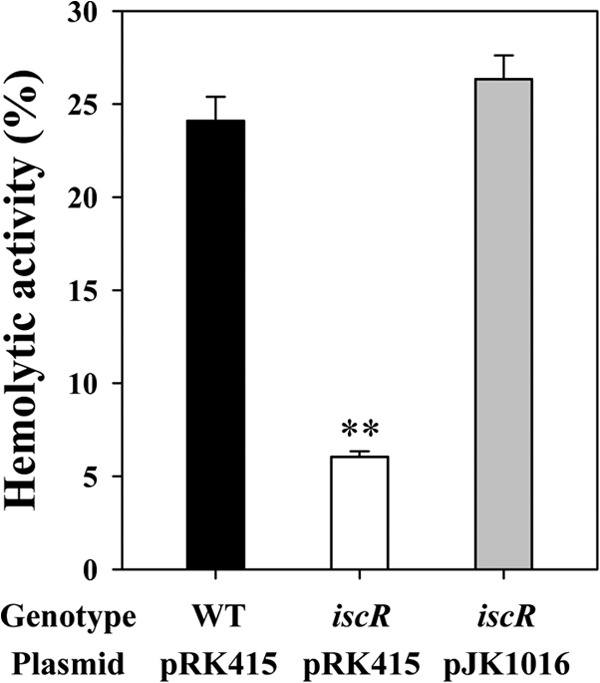
Hemolytic activities of V. vulnificus. An aliquot of the culture supernatants of V. vulnificus strains was mixed with an equal volume of hRBCs and then incubated at 37°C for 20 min. The lysis of hRBCs was determined, and results are presented using the complete lysis of hRBC by 1% Triton X-100 as 100%. Error bars represent the SEM. **, P < 0.005 relative to the wild type. WT (pRK415), wild type; iscR (pRK415), iscR mutant; iscR (pJK1016), complemented strain.
Compared to growth of the wild-type strain, the growth of the iscR mutant was substantially impaired on LBS medium containing 250 μM H2O2 (Fig. 7B) or 60 μM t-BOOH (Fig. 7C). However, neither defective nor advantageous growth was observed for the strains on LBS plates without oxidative stress (Fig. 7A). These results suggested that the iscR mutant is more sensitive to H2O2- and t-BOOH-induced oxidative stress than its parental wild type. The decreased growth of the iscR mutant under oxidative stress was partially recovered by the reintroduction of pJK1016 (Fig. 7B and C). These results indicated that IscR is essential for survival of V. vulnificus under oxidative stress. The susceptibility of the iscR mutant to oxidative stress could possibly be attributed to decreased expression of the antioxidant Prx and Grx2 (Fig. 2). Taken together, the results suggested that IscR might contribute to V. vulnificus pathogenesis by indeed regulating the newly identified genes of the IscR regulon to ensure motility and adhesion to host cells, hemolytic activity, and survival under oxidative stress during infection.
FIG 7.

Growth of V. vulnificus under oxidative stress. The V. vulnificus strains were compared for their ability to grow on LBS plates supplemented without oxidants (LBS) or with 250 μM H2O2 or 60 μM t-BOOH. Serial 10-fold dilutions of each culture were spotted on the plates, and plates were photographed after 16 h of growth. WT (pRK415), wild type; iscR (pRK415), iscR mutant; iscR (pJK1016), complemented strain.
Effects of host cells on IscR expression.
Expression of iscR in the presence of INT-407 cells was analyzed by qRT-PCR and Western blotting (Fig. 8). The qRT-PCR analyses were performed with RNA isolated from wild-type V. vulnificus cultures exposed to different numbers of INT-407 cells. The results revealed that the iscR mRNA level increased along with the increase in the numbers of INT-407 cells (Fig. 8A). Expression of iscR was almost 3-fold greater in the V. vulnificus cells exposed to 4 × 106 cells of INT-407 than in cells exposed to MEMF alone (control). Cellular levels of the IscR protein were also higher in V. vulnificus cells exposed to INT-407 cells (Fig. 8B). These results suggested that the expression of V. vulnificus iscR is induced by the host cells.
FIG 8.
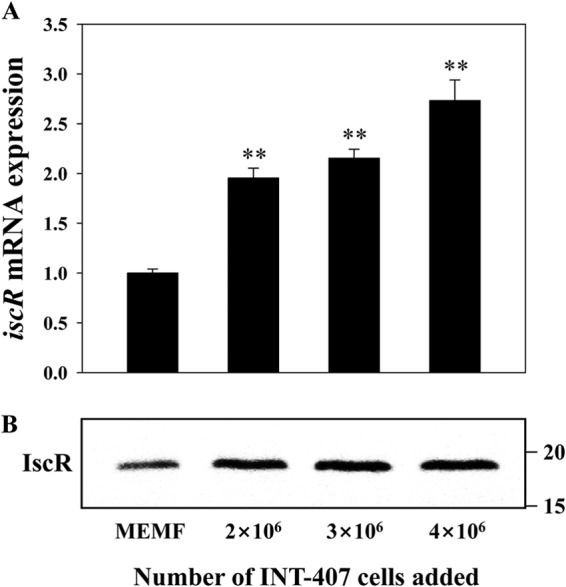
Induction of iscR expression by INT-407 host cells. Wild-type V. vulnificus was exposed to various numbers of INT-407 cells for 30 min as indicated and then used to isolate total RNAs and proteins as described in Materials and Methods. (A) The iscR mRNA levels were determined by qRT-PCR analyses, and the iscR mRNA level in the bacteria exposed to MEMF alone (control) was set as 1. Error bars represent the SEM. **, P < 0.005 relative to the bacteria exposed to MEMF alone. (B) Protein samples were resolved by SDS-PAGE, and IscR was detected by Western blotting using a rabbit anti-IscR antiserum. The positions of protein size markers (in kDa; Bio-Rad) are shown on the right of the gel.
It has been reported that ROS induce the expression of E. coli iscR (9, 10, 12), and the host epithelial cells infected with V. vulnificus generate ROS (28). Therefore, we hypothesized that the induction of iscR expression upon exposure to INT-407 cells could result from the increased level of ROS generated from the host cells. To examine this hypothesis, wild-type V. vulnificus cultures grown anaerobically were exposed to different levels of H2O2, and qRT-PCR and Western blot analyses were performed with the total RNAs and proteins isolated from the cultures. Both iscR mRNA and IscR protein levels were elevated by exposure of V. vulnificus to H2O2 (Fig. 9A and B). In V. vulnificus cells exposed to H2O2, the iscR mRNA level was more than 3.5-fold greater than that of the cells not exposed to H2O2 (control), demonstrating that ROS also induce the expression of V. vulnificus iscR. To examine whether induction of iscR expression by INT-407 cells is indeed due to the ROS generated by the host cells, the levels of iscR expression were determined in V. vulnificus cells exposed to either INT-407 cells or INT-407 cells that were preincubated with NAC to scavenge ROS (Fig. 10). The results revealed that the increased expression level of iscR in V. vulnificus cells exposed to INT-407 cells decreased to the level of the pathogen exposed to MEMF alone (control). The combined results suggested that the iscR expression induced by the INT-407 cells might be mediated by the oxidative stress imposed by the host cells during infection.
FIG 9.
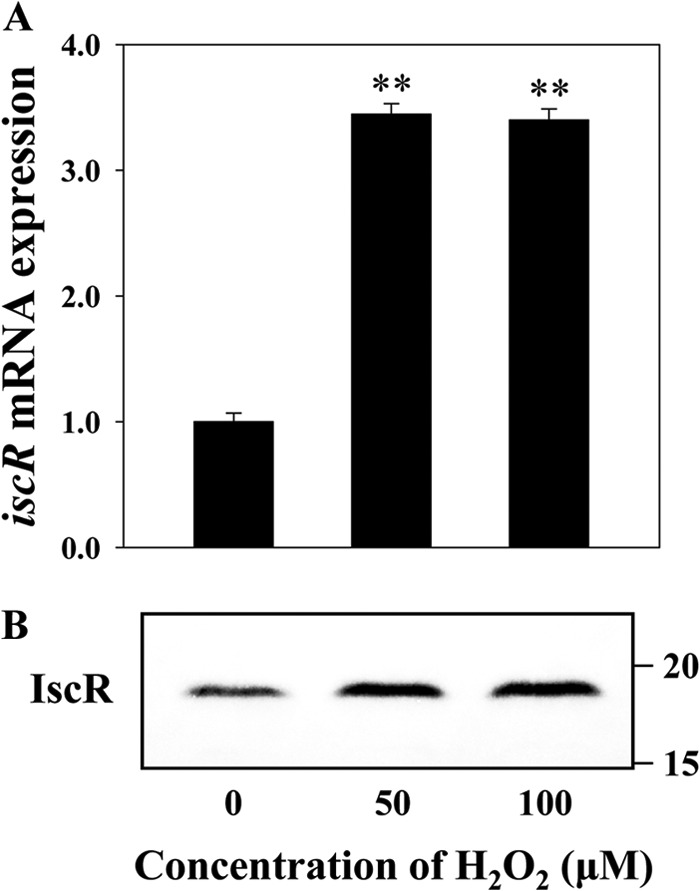
Induction of iscR expression by H2O2. Total RNAs and proteins were isolated from wild-type V. vulnificus, grown anaerobically to an A600 of 0.5, and then exposed to various levels of H2O2 for 10 min as indicated. (A) The iscR mRNA levels were determined by qRT-PCR analyses, and the iscR mRNA level in the bacteria not exposed to H2O2 (control) was set as 1. Error bars represent the SEM. **, P < 0.005 relative to the bacteria unexposed to H2O2. (B) Protein samples were resolved by SDS-PAGE; IscR was detected by Western blotting, and results are presented as described in the legend of Fig. 8.
FIG 10.
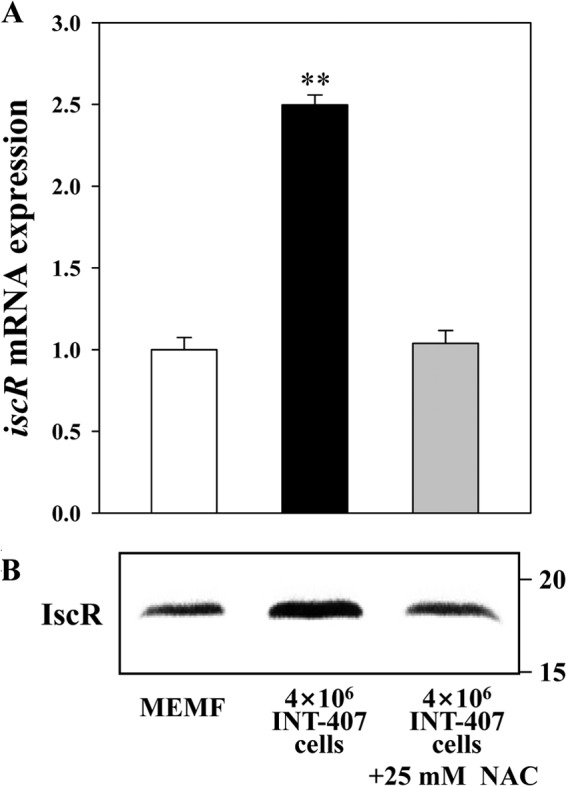
Effects of scavenging ROS on host cell induction of iscR expression. (A and B) Wild-type V. vulnificus grown to an A600 of 0.5 was exposed to MEMF (control), 4 × 106 INT-407 cells, or 4 × 106 INT-407 cells preincubated with NAC for 30 min at an MOI of 30 and then used to isolate total RNAs and proteins as described in Materials and Methods. The iscR mRNA and IscR protein levels in V. vulnificus cells were determined by qRT-PCR and Western blot analyses, respectively, and results are presented as described in the legend of Fig. 8. Error bars represent the SEM. **, P < 0.005 relative to the bacteria exposed to MEMF alone.
DISCUSSION
The iscRSUA-hscBA-fdx operon of E. coli is well characterized at the molecular level (5, 6). IscS is a cysteine desulfurase that removes the sulfur from cysteine and provides the sulfur to build a transient Fe-S cluster on the scaffold protein IscU. IscA, an A-type carrier, facilitates the cluster transfer from the scaffold protein to target apoproteins. HscB and HscA interact with IscU to stabilize the conformation for transfer of the cluster from IscU to a subset of apoproteins. The ferredoxin (Fdx) protein might be involved in electron transfer. The V. vulnificus MO6-24/O genome sequence (GenBank accession numbers CP002469 and CP002470) is predicted to possess the isc operon (iscRSUA-hscBA-fdx; VVMO6_02440-VVMO6_02434) (see Table S4 in the supplemental material) as a sole Fe-S biogenesis system. The amino acid sequence of each coding region of the isc operon is about 47 to 85% identical to the respective sequence of the E. coli isc operon (data not shown). The combined findings mentioned above suggested that the products of the V. vulnificus isc operon are indeed involved in Fe-S cluster biogenesis, as are the products of the E. coli isc operon, and that expression of the V. vulnificus isc operon is also regulated by IscR (as confirmed by microarray analysis) (see Table S4).
Iron starvation and oxidative stress, frequently encountered by most bacterial pathogens upon infection (40, 41), are two conditions that are highly detrimental for maintaining Fe-S cluster homeostasis (9–12, 42). Accordingly, Fe-S cluster biogenesis and its regulation are crucial for the pathogenesis of Erwinia chrysanthemi and Shigella flexneri, allowing adaptation to hostile host environments (43, 44). It is noteworthy that the Pseudomonas aeruginosa IscR also plays a role in survival under H2O2- and paraquat-induced oxidative stress and in virulence (45). In the present study, mutation of iscR significantly reduced the virulence of V. vulnificus in mice and in tissue culture, indicating that regulation of Fe-S cluster biogenesis is also essential for the pathogenesis of the bacteria (Fig. 1).
Furthermore, this study demonstrated that V. vulnificus IscR regulates the 12 genes that are potentially involved in pathogenesis in addition to Fe-S cluster biogenesis (Fig. 2 and 3; see also Tables S3 and S4 in the supplemental material). The flagellar hook protein FlgE is required for motility, adhesion to host epithelial cells, and virulence in mice (32). Chemotaxis signal transduction proteins such as MCPs, CheW, CheY, and histidine kinase (33) are essential for the pathogenesis of many pathogenic Vibrio spp. (46–48). GbpA is a homologue of Vibrio cholerae N-acetylglucosamine-binding protein A, which is an adhesion factor required for initial adherence to intestinal mucin (34, 49). Although RtxA1 is a major hemolytic toxin of V. vulnificus (50), VvhA is also a secreted cytolysin/hemolysin pore-forming toxin and causes intestinal tissue damage and inflammation (51). VvhA promotes dissemination of V. vulnificus to the bloodstream in an intragastric infection of mice (51). VvhB protein is a chaperone-like protein required for synthesis of active VvhA (52). Prx, homologous to human Prx5 (36), is presumably an antioxidant that protects V. vulnificus from oxidative stress during infection. Grx2 is homologous to E. coli Grx2 required for the reduction of cytosolic protein disulfides and survival under oxidative stress (37). Consistent with this, the iscR mutant showed decreased motility and adhesion to host cells (Fig. 4 and 5), hemolytic activity (Fig. 6), and survival under oxidative stress (Fig. 7).
In addition to these 12 genes, the transcriptome analysis revealed that many genes primarily involved in transport, metabolism, and energy production were also regulated by IscR (see Tables S3 and S4 in the supplemental material). When enteropathogens invade the host, they compete with the host cells and endogenous bacterial flora for the specific nutrients. Therefore, the ability to acquire and metabolize nutrients under these adverse environments is often crucial for the bacteria to survive and multiply in the host (for a recent review, see reference 53). It is thus possible that mutation of the iscR gene might impair the coordinated expression of the genes involved in metabolism and energy production in the host, leading to a reduction in virulence. It is not difficult to imagine that through IscR, V. vulnificus recognizes the host environment by sensing iron bioavailability and/or ROS levels and, in turn, coordinately regulates the metabolism and energy production genes. Consistent with this, the expression of IscR was induced upon exposure to host epithelial cells (Fig. 8), and the induction appeared to be mediated by ROS generated by the host cells during infection (Fig. 9 and 10).
In summary, our data presented here extended our understanding of the role of IscR in V. vulnificus pathogenesis by demonstrating that iscR expression is induced by host cells, and, in turn, that the induced IscR activates the expression of many genes primarily required for motility, adhesion, hemolytic activity, and survival under oxidative stress. Although comprehending the exact mechanism of IscR in the regulation of these genes needs additional work, the combined results suggest that IscR is a global regulator contributing to the overall success in the pathogenesis of V. vulnificus.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants to S.H.C from the Mid-Career Researcher Program ( 2012R1A2A1A03009679) and the Public Welfare and Safety Research Program (2012M3A2A1051679) through the National Research Foundation funded by the Ministry of Science, ICT, and Future Planning and the R&D Convergence Center Support Program of the Ministry of Agriculture, Food and Rural Affairs, Republic of Korea.
Footnotes
Published ahead of print 18 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01141-13.
REFERENCES
- 1.Cotter PA, DiRita VJ. 2000. Bacterial virulence regulation: an evolutionary perspective. Annu. Rev. Microbiol. 54:519–565. 10.1146/annurev.micro.54.1.519 [DOI] [PubMed] [Google Scholar]
- 2.Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 75:5542–5549. 10.1128/IAI.01094-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones MK, Oliver JD. 2009. Minireview Vibrio vulnificus: disease and pathogenesis. Infect. Immun. 77:1723–1733. 10.1128/IAI.01046-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fontecave M. 2006. Iron-sulfur clusters: ever-expanding roles. Nat. Chem. Biol. 2:171–174. 10.1038/nchembio0406-171 [DOI] [PubMed] [Google Scholar]
- 5.Py B, Barras F. 2010. Building Fe-S proteins: bacterial strategies. Nat. Rev. Microbiol. 8:436–446. 10.1038/nrmicro2356 [DOI] [PubMed] [Google Scholar]
- 6.Johnson DC, Dean DR, Smith AD, Johnson MK. 2005. Structure, function, and formation of biological Fe-S clusters. Annu. Rev. Biochem. 74:247–281. 10.1146/annurev.biochem.74.082803.133518 [DOI] [PubMed] [Google Scholar]
- 7.Schwartz CJ, Giel JL, Patschkowski T, Luther C, Ruzicka FJ, Beinert H, Kiley PJ. 2001. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc. Natl. Acad. Sci. U. S. A. 98:14895–14900. 10.1073/pnas.251550898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giel JL, Nesbit AD, Mettert EL, Fleischhacker AS, Wanta BT, Kiley PJ. 2013. Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe-2S]-IscR in Escherichia coli. Mol. Microbiol. 87:478–492. 10.1111/mmi.12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. 2001. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 183:4562–4570. 10.1128/JB.183.15.4562-4570.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Outten FW, Djaman O, Storz G. 2004. A suf operon requirement for Fe-S cluster assembly during iron starvation in Escherichia coli. Mol. Microbiol. 52:861–872. 10.1111/j.1365-2958.2004.04025.x [DOI] [PubMed] [Google Scholar]
- 11.Imlay JA. 2006. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 59:1073–1082. 10.1111/j.1365-2958.2006.05028.x [DOI] [PubMed] [Google Scholar]
- 12.Yeo WS, Lee JH, Lee KC, Roe JH. 2006. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol. Microbiol. 61:206–218. 10.1111/j.1365-2958.2006.05220.x [DOI] [PubMed] [Google Scholar]
- 13.Nesbit AD, Giel JL, Rose JC, Kiley PJ. 2009. Sequence-specific binding to a subset of IscR-regulated promoters does not require IscR Fe-S cluster ligation. J. Mol. Biol. 387:28–41. 10.1016/j.jmb.2009.01.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleischhacker AS, Stubna A, Hsueh KL, Guo Y, Teter SJ, Rose JC, Brunold TC, Markley JL, Münck E, Kiley PJ. 2012. Characterization of the [2Fe-2S] cluster of Escherichia coli transcription factor IscR. Biochemistry 51:4453–4462. 10.1021/bi3003204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. 2006. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol. Microbiol. 60:1058–1075. 10.1111/j.1365-2958.2006.05160.x [DOI] [PubMed] [Google Scholar]
- 16.Wu Y, Outten FW. 2009. IscR controls iron-dependent biofilm formation in Escherichia coli by regulating type I fimbria expression. J. Bacteriol. 191:1248–1257. 10.1128/JB.01086-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 18.Larsen RA, Wilson MM, Guss AM, Metcalf WW. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178:193–201. 10.1007/s00203-002-0442-2 [DOI] [PubMed] [Google Scholar]
- 19.Cerdà-Cuéllar M, Jofre J, Blanch AR. 2000. A selective medium and a specific probe for detection of Vibrio vulnificus. Appl. Environ. Microbiol. 66:855–859. 10.1128/AEM.66.2.855-859.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee BC, Lee JH, Kim MW, Kim BS, Oh MH, Kim KS, Kim TS, Choi SH. 2008. Vibrio vulnificus rtxE is important for virulence, and its expression is induced by exposure to host cells. Infect. Immun. 76:1509–1517. 10.1128/IAI.01503-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SM, Lee DH, Choi SH. 2012. Evidence that the Vibrio vulnificus flagellar regulator FlhF is regulated by a quorum sensing master regulator SmcR. Microbiology 158:2017–2025. 10.1099/mic.0.059071-0 [DOI] [PubMed] [Google Scholar]
- 22.Milton DL, O'Toole R, Horstedt P, Wolf-Watz H. 1996. Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 178:1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keen NT, Tamaki S, Kobayashi D, Trollinger D. 1988. Improved broad-host-range plasmids for DNA cloning in Gram-negative bacteria. Gene 70:191–197. 10.1016/0378-1119(88)90117-5 [DOI] [PubMed] [Google Scholar]
- 24.Sepp A, Binns RM, Lechler RI. 1996. Improved protocol for colorimetric detection of complement-mediated cytotoxicity based on the measurement of cytoplasmic lactate dehydrogenase activity. J. Immunol. Methods 196:175–180. 10.1016/0022-1759(96)00112-3 [DOI] [PubMed] [Google Scholar]
- 25.Jeong HG, Oh MH, Kim BS, Lee MY, Han HJ, Choi SH. 2009. The capability of catabolic utilization of N-acetylneuraminic acid, a sialic acid, is essential for Vibrio vulnificus pathogenesis. Infect. Immun. 77:3209–3217. 10.1128/IAI.00109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang J, Kim BS, Jang SY, Lim JG, You DJ, Jung HS, Oh TK, Lee JO, Choi SH, Kim MH. 2013. Structural insights into the regulation of sialic acid catabolism by the Vibrio vulnificus transcriptional repressor NanR. Proc. Natl. Acad. Sci. U. S. A. 110:e2829–e2837. 10.1073/pnas.1302859110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong HG, Choi SH. 2008. Evidence that AphB essential for the virulence of Vibrio vulnificus is a global regulator. J. Bacteriol. 190:3768–3773. 10.1128/JB.00058-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung KJ, Cho EJ, Kim MK, Kim YR, Kim SH, Yang HY, Chung KC, Lee SE, Rhee JH, Choy HE, Lee TH. 2010. RtxA1-induced expression of the small GTPase Rac2 plays a key role in the pathogenicity of Vibrio vulnificus. J. Infect. Dis. 201:97–105. 10.1086/648612 [DOI] [PubMed] [Google Scholar]
- 29.Bang YJ, Oh MH, Choi SH. 2012. Distinct characteristics of two 2-Cys peroxiredoxins of Vibrio vulnificus suggesting differential roles in detoxifying oxidative stress. J. Biol. Chem. 287:42516–42524. 10.1074/jbc.M112.421214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HJ, Kim JA, Lee MA, Park SJ, Lee KH. 2013. Regulation of haemolysin (VvhA) production by ferric uptake regulator (Fur) in Vibrio vulnificus: repression of vvhA transcription by Fur and proteolysis of VvhA by Fur-repressive exoproteases. Mol. Microbiol. 88:813–826. 10.1111/mmi.12224 [DOI] [PubMed] [Google Scholar]
- 31.Baek WK, Lee HS, Oh MH, Koh MJ, Kim KS, Choi SH. 2009. Identification of the Vibrio vulnificus ahpC1 gene and its influence on survival under oxidative stress and virulence. J. Microbiol. 47:624–632. 10.1007/s12275-009-0130-x [DOI] [PubMed] [Google Scholar]
- 32.Lee JH, Rho JB, Park KJ, Kim CB, Han YS, Choi SH, Lee KH, Park SJ. 2004. Role of flagellum and motility in pathogenesis of Vibrio vulnificus. Infect. Immun. 72:4905–4910. 10.1128/IAI.72.8.4905-4910.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker MD, Wolanin PM, Stock JB. 2006. Signal transduction in bacterial chemotaxis. Bioessays 28:9–22. 10.1002/bies.20343 [DOI] [PubMed] [Google Scholar]
- 34.Kirn TJ, Jude BA, Taylor RK. 2005. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438:863–866. 10.1038/nature04249 [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto K, Wright AC, Kaper JB, Morris JG., Jr 1990. The cytolysin gene of Vibrio vulnificus: sequence and relationship to the Vibrio cholerae E1 Tor hemolysin gene. Infect. Immun. 58:2706–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knoops B, Loumaye E, Van Der Eecken V. 2007. Evolution of the peroxiredoxins: taxonomy, homology and characterization, p 27–40 In Flohé L, Harris JR. (ed), Peroxiredoxin systems. Springer, New York, NY: [DOI] [PubMed] [Google Scholar]
- 37.Vlamis-Gardikas A, Potamitou A, Zarivach R, Hochman A, Holmgren A. 2002. Characterization of Escherichia coli null mutants for glutaredoxin 2. J. Biol. Chem. 277:10861–10868. 10.1074/jbc.M111024200 [DOI] [PubMed] [Google Scholar]
- 38.Rajagopalan S, Teter SJ, Zwart PH, Brennan RG, Phillips KJ, Kiley PJ. 2013. Studies of IscR reveal a unique mechanism for metal-dependent regulation of DNA binding specificity. Nat. Struct. Mol. Biol. 20:740–747. 10.1038/nsmb.2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ottemann KM, Miller JF. 1997. Role for motility in bacterial-host interactions. Mol. Microbiol. 24:1109–1117. 10.1046/j.1365-2958.1997.4281787.x [DOI] [PubMed] [Google Scholar]
- 40.Wilks A, Burkhard KA. 2007. Heme and virulence: how bacterial pathogens regulate, transport and utilize heme. Nat. Prod. Rep. 24:511–522. 10.1039/b604193k [DOI] [PubMed] [Google Scholar]
- 41.Miller RA, Britigan BE. 1997. Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 10:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Py B, Moreau PL, Barras F. 2011. Fe-S clusters, fragile sentinels of the cell. Curr. Opin. Microbiol. 14:218–223. 10.1016/j.mib.2011.01.004 [DOI] [PubMed] [Google Scholar]
- 43.Rincon-Enriquez G, Crété P, Barras F, Py B. 2008. Biogenesis of Fe/S proteins and pathogenicity: IscR plays a key role in allowing Erwinia chrysanthemi to adapt to hostile conditions. Mol. Microbiol. 67:1257–1273. 10.1111/j.1365-2958.2008.06118.x [DOI] [PubMed] [Google Scholar]
- 44.Runyen-Janecky L, Daugherty A, Lloyd B, Wellington C, Eskandarian H, Sagransky M. 2008. Role and regulation of iron-sulfur cluster biosynthesis genes in Shigella flexneri virulence. Infect. Immun. 76:1083–1092. 10.1128/IAI.01211-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim SH, Lee BY, Lau GW, Cho YH. 2009. IscR modulates catalase A (KatA) activity, peroxide resistance and full virulence of Pseudomonas aeruginosa PA14. J. Microbiol. Biotechnol. 19:1520–1526. 10.4014/jmb.0906.06028 [DOI] [PubMed] [Google Scholar]
- 46.O'Toole R, Milton DL, Wolf-Watz H. 1996. Chemotactic motility is required for invasion of the host by the fish pathogen Vibrio anguillarum. Mol. Microbiol. 19:625–637. 10.1046/j.1365-2958.1996.412927.x [DOI] [PubMed] [Google Scholar]
- 47.Kim YR, Lee SE, Kim CM, Kim SY, Shin EK, Shin DH, Chung SS, Choy HE, Progulske-Fox A, Hillman JD, Handfield M, Rhee JH. 2003. Characterization and pathogenic significance of Vibrio vulnificus antigens preferentially expressed in septicemic patients. Infect. Immun. 71:5461–5471. 10.1128/IAI.71.10.5461-5471.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butler SM, Camilli A. 2005. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat. Rev. Microbiol. 3:611–620. 10.1038/nrmicro1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhowmick R, Ghosal A, Das B, Koley H, Saha DR, Ganguly S, Nandy RK, Bhadra RK, Chatterjee NS. 2008. Intestinal adherence of Vibrio cholerae involves a coordinated interaction between colonization factor GbpA and mucin. Infect. Immun. 76:4968–4977. 10.1128/IAI.01615-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim YR, Lee SE, Kook H, Yeom JA, Na HS, Kim SY, Chung SS, Choy HE, Rhee JH. 2008. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell Microbiol. 10:848–862. 10.1111/j.1462-5822.2007.01088.x [DOI] [PubMed] [Google Scholar]
- 51.Jeong HG, Satchell KJ. 2012. Additive function of Vibrio vulnificus MARTXVv and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog. 8:e1002581. 10.1371/journal.ppat.1002581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senoh M, Okita Y, Shinoda S, Miyoshi S. 2008. The crucial amino acid residue related to inactivation of Vibrio vulnificus hemolysin. Microb. Pathog. 44:78–83. 10.1016/j.micpath.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 53.Brown SA, Palmer KL, Whiteley M. 2008. Revisiting the host as a growth medium. Nat. Rev. Microbiol. 6:657–666. 10.1038/nrmicro1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.