

Abstract
Enterotoxigenic Escherichia coli (ETEC) is a major cause of morbidity and mortality due to infectious diarrhea in developing countries for which there is presently no effective vaccine. A central challenge in ETEC vaccinology has been the identification of conserved surface antigens to formulate a broadly protective vaccine. Here, we demonstrate that EatA, an immunogenic secreted serine protease of ETEC, contributes to virulence by degrading MUC2, the major protein present in the small intestinal mucous layer, and that removal of this barrier in vitro accelerates toxin access to the enterocyte surface. In addition, we demonstrate that vaccination with the recombinant secreted passenger domain of EatA (rEatAp) elicits high titers of antibody and is protective against intestinal infection with ETEC. These findings may have significant implications for development of both subunit and live-attenuated vaccines against ETEC and other enteric pathogens, including Shigella flexneri, that express similar proteins.
INTRODUCTION
Infectious diarrhea continues to cause substantial morbidity and remains a leading cause of death among children in areas where clean water and sanitation are limited (1, 2). Enterotoxigenic Escherichia coli (ETEC) infections range in severity from asymptomatic colonization to severe, cholera-like diarrhea (3, 4) and death that ensues from rapid dehydration. Currently, it is estimated that these infections are responsible for millions of infections and hundreds of thousands of deaths, particularly among children under the age of 2 years (5–7). While in general the death rate from diarrheal illness has declined (1, 8), these infections are still commonly associated with severe dehydrating forms of illness similar to Vibrio cholerae (9, 10). Recent data from the large Global Enteric Multicenter Study reaffirm the important role that these infections play particularly in young children (12). ETEC infections have also been shown to play a significant part in the complex association between malnutrition and repeated bouts of diarrheal illness among young children in developing countries (13, 14).
ETEC bacteria are defined by the production heat-stable toxin (ST) and/or heat-labile toxin (LT) enterotoxins that elicit increases in cyclic nucleotides cyclic GMP (cGMP) and cyclic AMP (cAMP), respectively, in target intestinal epithelial cells. Activation of cyclic-nucleotide-dependent cellular kinases results in activation of the cystic fibrosis transmembrane regulatory (CFTR) channel leading to chloride efflux into the intestinal lumen with commensurate decreased absorption of sodium and water, ultimately resulting in profuse watery diarrhea (15).
Currently, there are no vaccines to protect against these important pathogens (16, 17). Most vaccines thus far have specifically targeted a relatively small group of antigens, namely, plasmid-encoded surface antigens known as colonization factors (CFs), or the heat-labile toxin. While prior infections with ETEC do appear to offer protection against subsequent infection (6, 18, 19), recent vaccine studies would suggest that the precise nature of this protection and the protective antigens involved are still being defined (20). Moreover, recent immunoproteomic studies (21) suggest that the immune response to ETEC infections is complex and involves recognition of multiple antigens, including both classical antigens, such as LT and the CF, and a number of novel pathotype-specific antigens.
One of the antigens identified in these studies, EatA, appears to be relatively conserved in the ETEC pathovar (22) and is represented by a geographically diverse collection of ETEC strains (23). Earlier studies demonstrated that EatA is a member of a family of molecules referred to as serine protease autotransporters of the Enterobacteriaceae (SPATE) (24). These molecules serve a variety of virulence functions in different enteric pathogens. EatA appears to modulate both adherence to epithelial cells and intestinal colonization in part by digesting EtpA, a novel exoprotein adhesin molecule that is secreted by ETEC (25).
However, a number of factors would suggest that this picture is incomplete and that EatA could have additional functions. First, while many strains that possess the eatA gene also encode the etpBAC two-partner secretion locus (26) responsible for production and secretion of EtpA, some do not. Furthermore, EatA homologues are present in other bacteria which do not make EtpA including Shigella (SepA) (27), strains of enteroaggregative E. coli (EAEC) associated with more severe diarrheal illness (28), and Shiga toxin-producing E. coli serotype O104:H4 strains that recently emerged in outbreaks of hemolytic-uremic syndrome (HUS) (29). Collectively, these data would suggest that EatA and similar molecules play an important common role in the pathogenesis of these enteric pathogens.
Here we demonstrate that EatA degrades MUC2, the major mucin secreted by intestinal epithelium. These data potentially provide an explanation for the widespread distribution of EatA and its homologues in a variety of mucosal pathogens and suggest that targeting of intestinal mucins may be critical to their virulence.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in these studies are outlined in Table 1. ETEC strain H10407, originally isolated from a patient with severe cholera-like diarrheal illness in Bangladesh (30), was kindly supplied from stocks of this strain maintained at the Walter Reed Army Institute of Research according to good manufacturing practices by Stephen Savarino.
TABLE 1.
Bacterial strains and plasmids used in these studies
| Strain or plasmid | Descriptiona | Reference(s) or source |
|---|---|---|
| Strains | ||
| H10407 | Wild-type ETEC strain O78:H11; CFA/I; LT+/ST+; EtpA+; EatA+ | 30, 52 |
| TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu)7697 galU galK rpsL (Strr) | Invitrogen |
| jf904 | H10407 eatA::Cmr mutant | 24 |
| jf876 | H10407 ΔlacZYA::Kmr | 39 |
| Plasmids | ||
| pBAD/Myc-HisA | Arabinose-inducible expression plasmid | Invitrogen |
| pSB001 | 7,064-bp BamHIΔlac subclone from pBAD-TOPO/lacZ/V5-His | 25 |
| pSP014 | 4,185-bp eatA amplicon cloned into pBAD-TOPO | 24 |
| pSP019 | pSP014 altered by SDM for His-134→Arg-134 substitution of EatA | 24 |
Strr, streptomycin resistance; Cmr, chloramphenicol resistance; Kmr, kanamycin resistance; SDM, site-directed mutagenesis.
Maintenance and propagation of human cell lines.
LS174T intestinal epithelial cells (31) were propagated in Eagle's minimum essential medium (E-MEM) supplemented with 10% (final concentration) fetal bovine serum (FBS). Jurkat cells were propagated in RPMI 1640 medium supplemented with 10% FBS. All cells were cultivated at 37°C in a 5% CO2 atmosphere.
Purification of recombinant EatA passenger domain.
Recombinant EatA passenger domain (rEatAp) was purified from E. coli LMG194ΔfliC(pSP014) (25) culture supernatants by ultrafiltration, followed by the addition of ammonium sulfate to 80% saturation and centrifugation. Pellets containing rEatAp were redissolved in 50 mM sodium phosphate and 200 mM NaCl and further purified by gel filtration chromatography (HiLoad 16/60 Superdex 200 prep grade; GE Life Sciences) in the same buffer. The passenger domain of the rEatApH134R mutant (rEatAp with a passenger domain mutation within the known catalytic site at histidine residue 134 [H134R]) was purified in an identical manner from E. coli LMG194ΔfliC(pSP019). When required, additional purification was performed by anion exchange. rEatAp in 25 mM imidazole (pH 7.0) was applied to a 5-ml column (HiTrap Q HP), and bound protein was then eluted over a linear 0 to 1 M gradient of NaCl in the same buffer. Protein purity was assessed by SDS-PAGE, and purified protein was buffer exchanged against phosphate-buffered saline (PBS) (pH 7.4) containing 10% glycerol before storage at −80°C.
Antibody purification.
Polyclonal rabbit antisera raised against a fragment of the EatA passenger domain (24) was cross absorbed against an immobilized E. coli lysate column (Thermo Scientific) and then affinity purified using the passenger protein immobilized on nitrocellulose as previously described (25, 32).
Isolation of mucin.
To isolate intestinal gel-forming mucin MUC2, medium supernatants from tissue culture of LS174T cells (ATCC CL-188) that produce abundant MUC2 (33, 34), were first concentrated by ultrafiltration using a 100-kDa-molecular-weight cutoff (MWCO) filter. Following buffer exchange with 10 mM Tris-HCl and 250 mM NaCl (pH 7.4), size exclusion chromatography was performed using Sepharose CL-2B (35). Fractions containing intact MUC2 as confirmed by immunoblotting using anti-MUC2 antibody (described below) were pooled and saved at −80°C.
Mucin degradation.
Degradation of purified MUC2 was examined by adding various amounts of rEatAp to 0.2 μg of purified MUC2 in 20-μl final reaction mixture volume in PBS (pH 7.4) and incubating at 37°C for the indicated time. The reaction digest was resolved on 3 to 8% Tris-acetate gradient gel (NuPAGE; Invitrogen) and transferred to nitrocellulose, and MUC2 degradation products were visualized by immunoblotting using anti-MUC2 rabbit polyclonal (IgG) antibody (H-300 [catalog no. sc-15334; Santa Cruz]) (1:2,000) that recognizes an epitope corresponding to amino acids 4880 to 5179 mapping at the C terminus of mucin 2 of human origin (gene identification [ID] 4583). To inhibit serine protease activity of rEatAp, 4-amidino-phenylmethanesulfonyl fluoride hydrochloride (APMSF) was used at a final concentration of 100 μM. A previously constructed active site mutant rEatApH134R (24) was also tested for its protease activity as described above. Degradation of cell-associated MUC2 was carried out by treating confluent monolayers of LS174T cells grown in 96-well plates with rEatAp (50 μg/ml final concentration in MEM without FBS) for 3 h or overnight at 37°C. Cells were lysed by boiling in SDS-PAGE sample buffer, and immunoblotting was performed as described above.
Substrate specificity of EatA.
In order to study substrate specificity of rEatAp, we used fluorescein isothiocyanate (FITC)-labeled substrates, including casein, IgG, gelatin, and bovine submaxillary mucin (BSM) in a kinetic assay based on the release of fluorescence from quenched substrates due to proteolysis as previously described (36).
We also tested other glycoproteins as potential substrates for rEatAp, including IgA, lactoferrin, and heavily glycosylated mucin-like protein CD43 expressed at the surfaces of human T lymphocytes (37). Briefly, bovine lactoferrin (Sigma) or human IgA (Sigma) was dissolved at 0.5 mg/ml in 100 μl of 100 mM morpholinepropanesulfonic acid (MOPS) and 200 mM NaCl (pH 7.3). Digests were initiated by the addition of either 5 μg rEatAp or 1 μg proteinase K and then incubated at 37°C. Samples were removed at intervals, the reaction was stopped by boiling in SDS-PAGE buffer, and the products were separated by SDS-PAGE and visualized by Coomassie blue staining.
To test CD43 degradation, 1 × 106 Jurkat cells were incubated for 3 h at 37°C and 5% CO2 with 5 μg of rEatAp diluted in a total volume of 100 μl of RPMI 1640 medium without FBS, a protocol similar to that outlined by Szabady et al. (38). The cells were pelleted at 400 × g for 5 min, washed with PBS, and blocked with 1% BSA in PBS, and cell surface CD43 was then labeled with phycoerythrin-conjugated anti-CD43 (mouse anti-human CD43 monoclonal clone L10; Invitrogen) and analyzed by flow cytometry.
In vitro MUC2 degradation.
LS174T cells were treated with rEatAp or rEatApH134R (at a final concentration of 50 μg/ml) or vehicle control (PBS) for 3 h and examined by immunofluorescence confocal microscopy with anti-MUC2 rabbit polyclonal antibody (IgG) (H-300 [catalog no. sc-15334; Santa Cruz]). Images were saved as z-stacks, and image data were then analyzed using Volocity three-dimensional (3D) image analysis software (version 6.2; PerkinElmer, Inc.).
Binding of labeled cholera toxin binding to epithelial cells.
LS174T cells were treated with rEatAp (at a final concentration of 5 μg/ml) or vehicle control (PBS) for 3 h as described above and examined by immunofluorescence confocal microscopy using cholera toxin B subunit conjugated with Alexa Fluor 488 (Life Technologies). Images were saved as z-stacks, and image data were then analyzed using Volocity 3D image analysis software (version 6.2; PerkinElmer, Inc.).
Cellular cytotoxicity assay.
To examine rEatAp-treated cells for potential cytotoxicity, LS174T cells were grown to confluent monolayers in 96-well plates. After the tissue culture medium was removed, the cells were treated with prewarmed MEM alone or supplemented with rEatAp (25 or 50 μg/ml) for 3 h at 37°C and 5% CO2. Triton X-100 (at final concentration of 0.1%) was used as a positive control. After the cells were treated, the medium was aspirated, and the cells were treated with 0.5 mg/ml MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) in MEM with 10% FBS at 37°C and 5% CO2 for 30 min. The medium was aspirated immediately, and the cells were washed with 200 μl of PBS. Cells were lysed by adding 200 μl dimethyl sulfoxide (DMSO), and optical density at 630 nm (OD630) was recorded with DMSO alone as a blank. Data represent percentage of the mock-treated cells from triplicate experiments.
EatA induction studies.
In order to test whether intestinal mucins can induce secretion of EatA by ETEC, we grew overnight cultures of ETEC H10407 in M9 medium containing 0.2% glucose for 3 h, and then the medium was supplemented with purified MUC2 (final concentration of 2 μg/ml). Samples of culture supernatant were collected at indicated time points, precipitated with trichloroacetic acid (TCA), and used to detect secreted EatA by immunoblotting using affinity-purified EatA antibody.
Immunization and intestinal colonization of mice.
Groups of 10 CD-1 mice were immunized intranasally with either PBS, 1 μg of heat-labile toxin (LT), or 1 μg of LT plus 30 μg of rEatAp on days 0, 14, and 28. On day 42, mice were pretreated with streptomycin; the mice were given streptomycin (5 g per liter) in drinking water for 24 h, followed by drinking water alone for 12 h. Fecal samples (6 pellets/mouse) were collected on day 42, resuspended in buffer (10 mM Tris, 100 mM NaCl, 0.05% Tween 20, 5 mM sodium azide [pH 7.4]) overnight at 4°C, and then centrifuged the next day to pellet insoluble material and recover supernatant to test fecal antibody response (see below). After administration of famotidine to reduce gastric acidity, mice were challenged with ∼105 CFU of the lacZYA::Kmr jf876 (39) strain by oral gavage. Twenty-four hours after infection, the mice were sacrificed, their sera were collected, and dilutions of saponin intestinal lysates (40) were plated onto Luria agar plates containing kanamycin (25 μg/ml).
Immune responses to EatA and LT were determined using previously described kinetic enzyme-linked immunosorbent assay (ELISA) (41, 42). Briefly, ELISA wells were coated with 1 μg/ml of rEatAp or LT in 0.1 M NaHCO3 buffer (pH 8.6) overnight at 4°C. The next day, the wells were washed three times with Tris-buffered saline containing 0.005% Tween 20 (TBS-T), blocked with 1% bovine serum albumin (BSA) in TBS-T for 1 h at 37°C, and 100 μl of fecal suspensions (undiluted) or sera (diluted 1:100 in TBS-T with 1% BSA) was added per ELISA well and incubated at 37°C for 1 h. Horseradish peroxidase-conjugated secondary antibodies were used, and signal was detected with TMB (3,3′,5,5′-tetramethylbenzidine)-peroxidase substrate (KPL) substrate.
To examine any differences in clearance of wild-type (WT) and eatA mutant strains, we monitored fecal shedding of bacteria by infected mice. Briefly, mice were challenged with either the lacZYA::Kmr strain or eatA::Cmr mutant bacteria at 105 CFU/dose, and stools (6 fecal pellets/mouse) were collected at days 1, 2, 3, 4, and 5 postinfection, resuspended in phosphate-buffered saline, and plated onto agar containing kanamycin (25 μg/ml) or chloramphenicol (15 μg/ml).
RESULTS
EatA preferentially degrades MUC2 intestinal mucin.
Previous studies of EatA demonstrated that this protease was associated with enhanced intestinal fluid accumulation and epithelial cell destruction in the rabbit ileal loop model (24). One possible explanation for these findings is that EatA could enhance access to enterocytes by degradation of the mucin barrier in the small intestine. To test this hypothesis, we first examined the ability of purified rEatAp (Fig. 1a) to degrade the major mucin present in the small intestinal lumen, MUC2. Treatment of LS174T cell monolayers with rEatAp significantly reduced the amount of MUC2 associated with these cells (Fig. 1b to d) as detected by immunoblotting of cellular lysates (Fig. 1b) and by confocal microscopy (Fig. 1c and d).
FIG 1.

EatA degrades the major intestinal mucin MUC2. (a) EatA is a serine protease autotransporter protein with an N-terminal signal peptide (SP) passenger domain containing the predicted HDS catalytic triad and C-terminal β-domain required for extracellular secretion of passenger domain. (b) Immunoblot showing the degradation of MUC2 (black arrow) in lysates of LS174T cell monolayers treated with recombinant EatA passenger domain (rEatAp) compared to mock (ø) treatment. The positions of molecular mass markers (in kilodaltons) are indicated to the left of the blot. (c and d) Abundant cell-associated MUC2 (green) on the surfaces of mock-treated LS174T cells (c) is depleted following the addition of recombinant rEatAp passenger domain (d). Shown at the left of each panel are individual z-stack images, while composite yz and xz reconstructions are shown to the right and at the bottom of each panel, respectively. Nuclei are stained blue, and cell membranes are shown in red.
Deletion of eatA in ETEC H10407 resulted in significant loss of its MUC2-degrading activity. Concentrated culture supernatants from the H10407 strain degraded MUC2, while those obtained from the eatA mutant grown under identical conditions had no appreciable activity (Fig. 2a). Likewise, eatA mutant complementation with pSP014 expressing rEatAp regained the ability to degrade MUC2, while complementation with the plasmid (pSP019) expressing mutant rEatApH134R with a passenger domain mutation within the known catalytic site at histidine residue 134 (H134R) or the vector alone (pSB001) failed to rescue this phenotype. Similarly, purified rEatAp from a recombinant E. coli LMG194ΔfliC strain expressing pSP014 showed activity against mucin, while rEatApH134R purified from LMG194ΔfliC(pSP019) displayed no activity (Fig. 2b).
FIG 2.
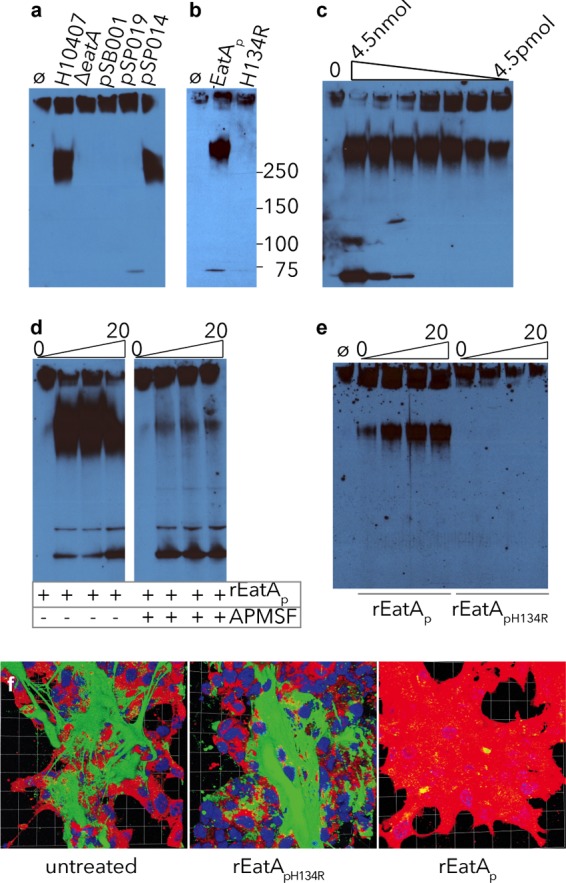
Mucin degradation by EatA requires serine protease activity. Using concentrated culture supernatant from the eatA mutant, we observed no MUC2 degradation compared to that from WT ETEC H10407. (a) Complementation of the eatA mutant with WT EatA (pSP014), but not H134R mutant EatA (pSP019) or vector control (pSB001), restored activity. (b) H134R mutant EatA (pSP019) purified from E. coli LMG194ΔfliC was unable to degrade MUC2 compared to WT EatA (pSP014). (c) rEatAp degrades purified MUC2 in a dose-dependent fashion. Untreated MUC2 is shown in the leftmost lane (lane 0) followed by serial dilutions of rEatAp passenger domain starting at 4.5 nmol/ml of protein. (d) Protease activity of rEatAp was inhibited by APMSF. (e) MUC2 is degraded in a time-dependent manner (samples obtained at 0, 5, 10, and 20 minutes) by the purified passenger domain of recombinant wild-type EatA (rEatAp) but not by the purified active-site mutant protein (rEatApH134R). (f) EatA degrades cell-associated MUC2. Abundant MUC2 (green) on the surfaces of untreated LS174T cells and monolayers treated with recombinant H134R mutant EatA passenger protein (rEatApH134R) compared to cells treated with recombinant wild-type EatA passenger domain (rEatAp). The images shown in panel f are Volocity-generated 3D projections from z-stacks.
Purified rEatAp degraded MUC2 in a dose-dependent fashion (Fig. 2c). Mucin degradation by rEatAp was inhibited by the addition of the serine protease inhibitor APMSF (Fig. 2d). Consistent with the previously described proteolytic activity of EatA, rEatApH134R was completely deficient in the ability to degrade MUC2 over time (Fig. 2e and f).
Recombinant rEatAp passenger domain exhibited its greatest proteolytic activity against MUC2 at neutral to slightly acidic pH (Fig. 3), consistent with the environment of the small intestine (43). However, with this pH range, we were not able to demonstrate activity against other substrates, including IgA, IgG, casein, gelatin, lactoferrin, mucin-like glycoprotein CD43, or commercially available bovine submaxillary mucin (Fig. 4), indicating a high specificity of rEatAp for human intestinal mucin MUC2.
FIG 3.

Mucin-degrading activity of EatA is pH dependent. Anti-MUC2 immunoblot demonstrates cleavage of MUC2 (top band) in the presence of recombinant EatA (rEatAp). Control lanes at each pH are shown (no rEatAp added [− lanes]).
FIG 4.

Specificity of rEatAp activity. (a and b) EatA lacks the ability to degrade lactoferrin (a) or IgG (b). Proteinase K (pr.K) activity is shown as a positive control for each of these substrates in Coomassie blue-stained gel images. Small black arrows to the right of the gels indicate the respective substrate bands, while small white arrows in the leftmost lane show the migration of the rEatAp passenger domain. (c) Degradation of FITC-labeled substrates casein, BSM, gelatin, and IgA by the bacterial protease subtilisin (as evident by an increase in fluorescence) but not rEatAp. PMSF, phenylmethylsulfonyl fluoride. (d) Fluorescence-activated cell sorting (FACS) data demonstrating loss of mucin-like glycoprotein CD43 from the surfaces of Jurkat cells following proteinase K treatment but not rEatAp.
We also examined the effect of mucin on growth of wild-type and eatA mutant strains of ETEC. We saw no evidence that supplementation of media with purified MUC2 enhanced growth of either strain (data not shown). Likewise, the addition of MUC2 to growth media failed to stimulate the secretion or production of EatA by ETEC (data not shown).
EatA enhances toxin access to epithelial cells.
Both heat-labile toxin (LT) and the closely related cholera toxin (CT) must engage GM1 ganglioside receptors on the surfaces of intestinal epithelial cells for their cellular activity. We reasoned that degradation of MUC2 on the surfaces of epithelial cells would unmask these receptors and promote enterotoxin binding. Indeed, treatment of epithelial cells with rEatAp significantly enhanced access of labeled cholera toxin B subunit to cognate receptors on the cell surface (Fig. 5a and b). In contrast, as might be predicted from its lack of homology to known cytotoxins in this class of proteins (44), rEatAp had no effect on the integrity of target epithelial cells, suggesting that it lacks any direct cytopathic effect (Fig. 5c).
FIG 5.

rEatA treatment enhances cholera toxin binding to epithelial cells. (a) rEatAp treatment enhanced cholera toxin subunit B-Alexa Fluor 488 (AF488) conjugate (green) binding to LS174T cells (bottom row) compared to untreated cells (top row). (b) Quantification of signal intensity by Volocity software shows a significant increase in cholera toxin subunit B (CTB) binding to rEatAp-treated LS174T cells compared to untreated cells (P less than 0.0001). The dotted horizontal line shows the mean value for the group of mice. Each symbol represents the value for an individual mouse. (c) MTT cytotoxicity assay. rEatAp treatment of LS174T cells does not cause cellular toxicity (Triton X-100 [TX100] is shown as a positive control).
EatA is highly immunogenic and a protective antigen.
ETEC virulence is thought to require bacterial transit to the mucosal surface where these organisms directly engage enterocytes to effectively deliver LT and ST to receptors on the surfaces of these cells. In theory, the ability to degrade mucin would accelerate this process by promoting direct access of the bacteria to the enterocyte surface. Theoretically, bacteria that are incapable of degrading mucin would be quickly eliminated from the intestine as they are propelled along the intestinal lumen by peristaltic flow (45). However, we found that in a murine ETEC infection model, the eatA mutant appeared in only slightly greater numbers in stool (Fig. 6a) and while the mutant was eliminated somewhat faster than the wild-type ETEC parent (Fig. 6b), these differences in fecal shedding were not statistically different.
FIG 6.

Impact of eatA on fecal shedding and clearance. (a) CFU/ml of fecal suspensions from 10 mice following oral (gavage) challenge with equal numbers of either wild-type (wt) ETEC H10407 strain or eatA mutant strain. The dotted horizontal lines represent the mean values (CFU/ml) for the groups of mice. Each symbol represents the value for an individual mouse. Mice with no organisms detected are shown at the theoretical lower limit of detection (1 or 100 CFU/ml). There was no statistically significant difference (ns) between the values for the groups by Mann-Whitney two-tailed testing. (b) Percentage of mice shedding measurable amount of ETEC in stool over 5 days after challenge.
Nevertheless, we also questioned whether neutralization of EatA activity could serve as an effective complement to existing vaccine strategies, which so far have primarily targeted either the toxins themselves or fimbrial adhesin molecules (16, 17). Because we have recently shown that EatA is recognized by antibody present in convalescent-phase sera from patients following ETEC infection (21), we examined whether vaccination with rEatAp would offer protection against ETEC infection of the small intestinal mucosa in the murine model.
As predicted by earlier studies, immunization with rEatAp generated robust serum and mucosal antibody responses (Fig. 7a to d). More importantly, we found that mice vaccinated with rEatAp demonstrated significantly reduced small intestinal colonization following challenge with ETEC (Fig. 7e). Collectively, these data provide additional evidence suggesting that EatA plays a significant role in the pathogenesis of ETEC and potentially other important enteric pathogens that make very similar molecules.
FIG 7.

Immunogenicity and protective efficacy of recombinant EatA passenger (rEatAp) domain. (a to d) Kinetic ELISA data (expressed as Vmax in milliunits/min) demonstrate antibody in samples from mice vaccinated with either LT alone (1 μg/dose), LT plus rEatAp (1 μg LT plus 30 μg rEatAp/dose), or PBS. The immunogen used is shown on the x axis of each graph. Serum (IgG, IgM, and IgA) responses (at 1:100 dilution of sera) to LT (a) and rEatAp (b) and fecal (IgA) responses (undiluted samples) to rEatAp (c) and LT (d) are shown. αLT, anti-LT antibody. (e) LT/rEatAp-immunized mice were protected against colonization by ETEC following intestinal challenge with jf876 (data obtained 24 h after challenge; P = 0.0063 by Mann-Whitney two-tailed nonparametric analysis).
DISCUSSION
Intestinal mucin represents a major host defense mechanism that significantly limits interaction of both commensal and pathogenic organisms with enterocytes to maintain mucosal integrity (46, 47). MUC2, the predominant gel-forming mucin present in the lumen of the small intestine (46, 48, 49) forms a complex multimeric glycoprotein network that bacterial pathogens must navigate if they are to effectively engage epithelial cells. Enteric pathogens have evolved a number of mechanisms to counter this barrier (46). These mechanisms include glycosidases to degrade oligosaccharide residues and peptidases that degrade the peptide backbone of these complex glycoproteins.
MUC2 is the major gel-forming mucin in both the small and large intestine. The core region of this molecule is heavily glycosylated and very resistant to the action of proteases, while interactions of the C-terminal and N-terminal ends contribute to the formation of dimers and polymers of MUC2, respectively (49). Additional study will be needed to determine whether EatA cleaves MUC2 at either the unprotected amino or carboxy termini, similar to a cysteine protease of Entamoeba histolytica (49).
Our studies demonstrate that vaccination with the passenger domain of EatA, which possesses MUC2-degrading activity, afforded significant protection against infection of the small intestinal mucosa in the murine model. However, we should point out that it is not clear from these studies that this relates directly to neutralization of EatA enzymatic activity. Earlier studies of the ability of EatA to modulate adhesion, toxin delivery, and intestinal colonization by degrading the EtpA adhesin (25) would suggest that this enzyme potentially plays a complex role in the pathogenesis of ETEC. Moreover, despite the robust activity of EatA in degradation of human MUC2 in vitro, the anticipated impact on fecal shedding in mice proved to be modest, potentially relating to structural differences in the major gel-forming mucins of mice and humans or differential enzymatic activity in the complex environment of the intestine. Additional effort will likely be required to address the impact of EatA and related proteases on these very complex phenotypes.
EatA is a member of the serine protease autotransporters of the Enterobacteriaceae (SPATE) family of proteins. Another SPATE protein that is shared by both enteroaggregative E. coli and Shigella flexneri, Pic, has been shown to degrade a number of substrates, including mucins (50). However, unlike Pic, we were unable to demonstrate that EatA had any activity against bovine submaxillary mucin. These results are consistent with earlier studies of the EatA homologue SepA (44). Although several SPATE molecules, including Pic, have been shown to target a number of glycoproteins rich in O-linked glycans involved in the leukocyte trafficking and inflammation (37), this property is not shared by SepA, suggesting that these proteins may target a different class of molecules. Indeed, EatA was incapable of degrading the mucin-like CD43 molecule in our studies. Collectively, these findings suggest that enteric pathogens have evolved divergent mechanisms to address the barrier imposed by intestinal mucins.
EatA shares approximately 80% homology to SepA, a serine protease of Shigella flexneri. Similar to EatA of ETEC, the SepA Shigella homologue appeared to accelerate fluid accumulation in the ileal loop model of infection (24, 27). Although the exact function of SepA in Shigella pathogenesis is still unknown, our studies could suggest that SepA may be involved in degradation of MUC2 covering the colonic mucosa, the preferential site for Shigella colonization.
Additional molecular epidemiology studies may attest to the importance of EatA as a virulence determinant in ETEC and other enteric pathogens. In otherwise heterogeneous collections of ETEC from Chile (23) and Guinea Bissau (22), eatA genes were present in more than 70% of ETEC strains. Recently, EatA homologues have been identified in enteroaggregative E. coli associated with more-severe infections (28) and in serotype O104 Shiga toxin-producing E. coli that emerged suddenly in food-borne outbreaks in Europe (29). These organisms likely share in their need to penetrate mucin to cause severe infections. While ETEC infections may range from mild clinical illness to severe cholera-like diarrhea (3) associated with rapid dehydration, the contribution of eatA to the more severe manifestations is not clear at present. Nevertheless, the clear association of these organisms with death due to severe diarrheal illnesses in young children (12) provides a strong impetus to understand which virulence factors might contribute to these more life-threatening phenotypes.
As noted in our accompanying paper (53), ETEC strains also encode the type II secretion system effector protein YghJ, a metalloprotease which also degrades intestinal mucins. Collectively, these studies provide further corroborative evidence that the ability to breach the physical barrier imposed by mucins is an important virulence attribute for enteric pathogens, including enterotoxigenic E. coli.
Four decades after the discovery of ETEC (51), a broadly protective vaccine for ETEC has yet to be developed (16). The identification of novel immunogens and elucidation of the contribution of EatA and other antigens to virulence and protective immunity should inform rational approaches to ETEC vaccinology.
ACKNOWLEDGMENTS
This work was supported by funding from the Department of Veterans Affairs, grant R01AI89894 from the National Institutes of Allergy and Infectious Diseases (NIH) (J.M.F.), and the Washington University School of Medicine Digestive Disease Research Core Center was supported by grant P30DK052574 from the National Institute of Diabetes and Digestive and Kidney Disease.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIAID, NIH, or VA.
We thank Robin Klein for the Jurkat cell line used in these studies.
Footnotes
Published ahead of print 11 November 2013
REFERENCES
- 1.Levine MM, Kotloff KL, Nataro JP, Muhsen K. 2012. The Global Enteric Multicenter Study (GEMS): impetus, rationale, and genesis. Clin. Infect. Dis. 55(Suppl 4):S215–S224. 10.1093/cid/cis761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black RE, Morris SS, Bryce J. 2003. Where and why are 10 million children dying every year? Lancet 361:2226–2234. 10.1016/S0140-6736(03)13779-8 [DOI] [PubMed] [Google Scholar]
- 3.Sack RB, Gorbach SL, Banwell JG, Jacobs B, Chatterjee BD, Mitra RC. 1971. Enterotoxigenic Escherichia coli isolated from patients with severe cholera-like disease. J. Infect. Dis. 123:378–385. 10.1093/infdis/123.4.378 [DOI] [PubMed] [Google Scholar]
- 4.Sack DA, McLaughlin JC, Sack RB, Orskov F, Orskov I. 1977. Enterotoxigenic Escherichia coli isolated from patients at a hospital in Dacca. J. Infect. Dis. 135:275–280. 10.1093/infdis/135.2.275 [DOI] [PubMed] [Google Scholar]
- 5.Gupta SK, Keck J, Ram PK, Crump JA, Miller MA, Mintz ED. 2008. Part III. Analysis of data gaps pertaining to enterotoxigenic Escherichia coli infections in low and medium human development index countries, 1984–2005. Epidemiol. Infect. 136:721–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qadri F, Saha A, Ahmed T, Al Tarique A, Begum YA, Svennerholm AM. 2007. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect. Immun. 75:3961–3968. 10.1128/IAI.00459-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qadri F, Das SK, Faruque ASG, Fuchs GJ, Albert MJ, Sack RB, Svennerholm A-M. 2000. Prevalence of toxin types and colonization factors in enterotoxigenic Escherichia coli isolated during a 2-year period from diarrheal patients in Bangladesh. J. Clin. Microbiol. 38:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, Rudan I, Campbell H, Cibulskis R, Li M, Mathers C, Black RE. 2012. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet 379:2151–2161. 10.1016/S0140-6736(12)60560-1 [DOI] [PubMed] [Google Scholar]
- 9.Fischer Walker CL, Sack D, Black RE. 2010. Etiology of diarrhea in older children, adolescents and adults: a systematic review. PLoS Negl. Trop. Dis. 4:e768. 10.1371/journal.pntd.0000768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury F, Rahman MA, Begum YA, Khan AI, Faruque AS, Saha NC, Baby NI, Malek MA, Kumar AR, Svennerholm AM, Pietroni M, Cravioto A, Qadri F. 2011. Impact of rapid urbanization on the rates of infection by Vibrio cholerae O1 and enterotoxigenic Escherichia coli in Dhaka, Bangladesh. PLoS Negl. Trop. Dis. 5:e999. 10.1371/journal.pntd.0000999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reference deleted.
- 12.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2 [DOI] [PubMed] [Google Scholar]
- 13.Mondal D, Haque R, Sack RB, Kirkpatrick BD, Petri WA., Jr 2009. Attribution of malnutrition to cause-specific diarrheal illness: evidence from a prospective study of preschool children in Mirpur, Dhaka, Bangladesh. Am. J. Trop. Med. Hyg. 80:824–826 [PMC free article] [PubMed] [Google Scholar]
- 14.Mondal D, Minak J, Alam M, Liu Y, Dai J, Korpe P, Liu L, Haque R, Petri WA., Jr 2012. Contribution of enteric infection, altered intestinal barrier function, and maternal malnutrition to infant malnutrition in Bangladesh. Clin. Infect. Dis. 54:185–192. 10.1093/cid/cir807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. 2010. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 12:89–98. 10.1016/j.micinf.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svennerholm AM, Lundgren A. 2012. Recent progress toward an enterotoxigenic Escherichia coli vaccine. Expert Rev. Vaccines 11:495–507. 10.1586/erv.12.12 [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Sack DA. 2012. Progress and hurdles in the development of vaccines against enterotoxigenic Escherichia coli in humans. Expert Rev. Vaccines 11:677–694. 10.1586/erv.12.37 [DOI] [PubMed] [Google Scholar]
- 18.Steinsland H, Valentiner-Branth P, Gjessing HK, Aaby P, Molbak K, Sommerfelt H. 2003. Protection from natural infections with enterotoxigenic Escherichia coli: longitudinal study. Lancet 362:286–291. 10.1016/S0140-6736(03)13971-2 [DOI] [PubMed] [Google Scholar]
- 19.Cravioto A, Reyes RE, Trujillo F, Uribe F, Navarro A, De La Roca JM, Hernandez JM, Perez G, Vazquez V. 1990. Risk of diarrhea during the first year of life associated with initial and subsequent colonization by specific enteropathogens. Am. J. Epidemiol. 131:886–904 [DOI] [PubMed] [Google Scholar]
- 20.Darsley MJ, Chakraborty S, DeNearing B, Sack DA, Feller A, Buchwaldt C, Bourgeois AL, Walker R, Harro CD. 3 October 2012. The oral, live attenuated enterotoxigenic Escherichia coli vaccine ACE527 reduces the incidence and severity of diarrhea in a human challenge model of diarrheal disease. Clin. Vaccine Immunol. 19:1921–1931 10.1128/CVI.00364-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy K, Bartels S, Qadri F, Fleckenstein JM. 2010. Enterotoxigenic Escherichia coli elicits immune responses to multiple surface proteins. Infect. Immun. 78:3027–3035. 10.1128/IAI.00264-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahl JW, Steinsland H, Redman JC, Angiuoli SV, Nataro JP, Sommerfelt H, Rasko DA. 2011. A comparative genomic analysis of diverse clonal types of enterotoxigenic Escherichia coli reveals pathovar-specific conservation. Infect. Immun. 79:950–960. 10.1128/IAI.00932-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Del Canto F, Valenzuela P, Cantero L, Bronstein J, Blanco JE, Blanco J, Prado V, Levine M, Nataro J, Sommerfelt H, Vidal R. 2011. Distribution of classical and nonclassical virulence genes in enterotoxigenic Escherichia coli isolates from Chilean children and tRNA gene screening for putative insertion sites for genomic islands. J. Clin. Microbiol. 49:3198–3203. 10.1128/JCM.02473-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel SK, Dotson J, Allen KP, Fleckenstein JM. 2004. Identification and molecular characterization of EatA, an autotransporter protein of enterotoxigenic Escherichia coli. Infect. Immun. 72:1786–1794. 10.1128/IAI.72.3.1786-1794.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roy K, Kansal R, Bartels SR, Hamilton DJ, Shaaban S, Fleckenstein JM. 2011. Adhesin degradation accelerates delivery of heat-labile toxin by enterotoxigenic Escherichia coli. J. Biol. Chem. 286:29771–29779. 10.1074/jbc.M111.251546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleckenstein JM, Roy K, Fischer JF, Burkitt M. 2006. Identification of a two-partner secretion locus of enterotoxigenic Escherichia coli. Infect. Immun. 74:2245–2258. 10.1128/IAI.74.4.2245-2258.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjelloun-Touimi Z, Sansonetti PJ, Parsot C. 1995. SepA, the major extracellular protein of Shigella flexneri: autonomous secretion and involvement in tissue invasion. Mol. Microbiol. 17:123–135. 10.1111/j.1365-2958.1995.mmi_17010123.x [DOI] [PubMed] [Google Scholar]
- 28.Boisen N, Scheutz F, Rasko DA, Redman JC, Persson S, Simon J, Kotloff KL, Levine MM, Sow S, Tamboura B, Toure A, Malle D, Panchalingam S, Krogfelt KA, Nataro JP. 2012. Genomic characterization of enteroaggregative Escherichia coli from children in Mali. J. Infect. Dis. 205:431–444. 10.1093/infdis/jir757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Moller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365:709–717. 10.1056/NEJMoa1106920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans DJ, Jr, Evans DG. 1973. Three characteristics associated with enterotoxigenic Escherichia coli isolated from man. Infect. Immun. 8:322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tom BH, Rutzky LP, Jakstys MM, Oyasu R, Kaye CI, Kahan BD. 1976. Human colonic adenocarcinoma cells. I. Establishment and description of a new line. In Vitro 12:180–191 [DOI] [PubMed] [Google Scholar]
- 32.Harlow E, Lane D, Harlow E. 1999. Using antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 33.van Klinken BJ, Oussoren E, Weenink JJ, Strous GJ, Buller HA, Dekker J, Einerhand AW. 1996. The human intestinal cell lines Caco-2 and LS174T as models to study cell-type specific mucin expression. Glycoconj. J. 13:757–768. 10.1007/BF00702340 [DOI] [PubMed] [Google Scholar]
- 34.Bu XD, Li N, Tian XQ, Huang PL. 2011. Caco-2 and LS174T cell lines provide different models for studying mucin expression in colon cancer. Tissue Cell 43:201–206. 10.1016/j.tice.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 35.Davies J, Carlstedt I. 2000. Isolation of large gel-forming mucins. Methods Mol. Biol. 125:3–13 [DOI] [PubMed] [Google Scholar]
- 36.Lewis WG, Robinson LS, Perry J, Bick JL, Peipert JF, Allsworth JE, Lewis AL. 2012. Hydrolysis of secreted sialoglycoprotein immunoglobulin A (IgA) in ex vivo and biochemical models of bacterial vaginosis. J. Biol. Chem. 287:2079–2089. 10.1074/jbc.M111.278135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruiz-Perez F, Wahid R, Faherty CS, Kolappaswamy K, Rodriguez L, Santiago A, Murphy E, Cross A, Sztein MB, Nataro JP. 2011. Serine protease autotransporters from Shigella flexneri and pathogenic Escherichia coli target a broad range of leukocyte glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 108:12881–12886. 10.1073/pnas.1101006108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szabady RL, Yanta JH, Halladin DK, Schofield MJ, Welch RA. 2011. TagA is a secreted protease of Vibrio cholerae that specifically cleaves mucin glycoproteins. Microbiology 157:516–525. 10.1099/mic.0.044529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorsey FC, Fischer JF, Fleckenstein JM. 2006. Directed delivery of heat-labile enterotoxin by enterotoxigenic Escherichia coli. Cell. Microbiol. 8:1516–1527. 10.1111/j.1462-5822.2006.00736.x [DOI] [PubMed] [Google Scholar]
- 40.Allen KP, Randolph MM, Fleckenstein JM. 2006. Importance of heat-labile enterotoxin in colonization of the adult mouse small intestine by human enterotoxigenic Escherichia coli strains. Infect. Immun. 74:869–875. 10.1128/IAI.74.2.869-875.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsang VC, Wilson BC, Maddison SE. 1980. Kinetic studies of a quantitative single-tube enzyme-linked immunosorbent assay. Clin. Chem. 26:1255–1260 [PubMed] [Google Scholar]
- 42.Roy K, Hamilton DJ, Fleckenstein JM. 2012. Cooperative role of antibodies against heat-labile toxin and the EtpA adhesin in preventing toxin delivery and intestinal colonization by enterotoxigenic Escherichia coli. Clin. Vaccine Immunol. 19:1603–1608. 10.1128/CVI.00351-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fallingborg J. 1999. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 46:183–196 [PubMed] [Google Scholar]
- 44.Dutta PR, Cappello R, Navarro-Garcia F, Nataro JP. 2002. Functional comparison of serine protease autotransporters of Enterobacteriaceae. Infect. Immun. 70:7105–7113. 10.1128/IAI.70.12.7105-7113.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. 2010. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 6:e1000902. 10.1371/journal.ppat.1000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGuckin MA, Linden SK, Sutton P, Florin TH. 2011. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9:265–278. 10.1038/nrmicro2538 [DOI] [PubMed] [Google Scholar]
- 47.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. 2008. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:15064–15069. 10.1073/pnas.0803124105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J. 2010. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 1:254–268. 10.4161/gmic.1.4.12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansson ME, Sjovall H, Hansson GC. 2013. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 10:352–361. 10.1038/nrgastro.2013.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson IR, Czeczulin J, Eslava C, Noriega F, Nataro JP. 1999. Characterization of Pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect. Immun. 67:5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sack RB. 2011. The discovery of cholera-like enterotoxins produced by Escherichia coli causing secretory diarrhoea in humans. Indian J. Med. Res. 133:171–180 [PMC free article] [PubMed] [Google Scholar]
- 52.Evans DG, Silver RP, Evans DJ, Jr, Chase DG, Gorbach SL. 1975. Plasmid-controlled colonization factor associated with virulence in Escherichia coli enterotoxigenic for humans. Infect. Immun. 12:656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo Q, Kumar P, Vickers T, Sheikh A, Lewis WG, Rasko DA, Sistrunk J, Fleckenstein JM. 2014. Enterotoxigenic Escherichia coli secretes a highly conserved mucin-degrading metalloprotease to effectively engage intestinal epithelial cells. Infect. Immun. 82:509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]