

Abstract
Malaria is a widespread infectious disease caused by the parasite Plasmodium. During pregnancy, malaria infection leads to a range of complications that can affect both the mother and fetus, including stillbirth, infant mortality, and low birth weight. In this study, we utilized a mouse model of placental malaria (PM) infection to determine the importance of the protein MyD88 in the host immune response to Plasmodium during pregnancy. Initially, we demonstrated that Plasmodium berghei NK65GFP adhered to placental tissue via chondroitin sulfate A and induced PM in mice with a C57BL/6 genetic background. To evaluate the involvement of MyD88 in the pathology of PM, we performed a histopathological analysis of placentas obtained from MyD88−/− and wild-type (WT) mice following infection on the 19th gestational day. Our data demonstrated that the detrimental placental alterations observed in the infected mice were correlated with the expression of MyD88. Moreover, in the absence of this protein, production of interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α) was significantly reduced in the infected mice. More importantly, in contrast to fetuses from infected WT mice, which exhibited a reduction in body weight, the fetuses from infected MyD88−/− mice did not display significant weight loss compared to their noninfected littermates. In addition, we observed a decrement of maternal care associated with malaria infection, which was attenuated in the MyD88-deficient mice. Collectively, the results of this study illustrate the pivotal importance of the MyD88 signaling pathway in the pathogenesis of placental malaria, thus presenting new possibilities for targeting MyD88 in therapeutic interventions.
INTRODUCTION
Malaria is still the world's most threatening infectious disease found in tropical and subtropical areas and is transmitted via the bites of mosquitoes that are infected with the parasite Plasmodium. Following infection, the Plasmodium parasites multiply in the liver and then infect red blood cells. The symptoms of malaria often include fever, headache, and vomiting and typically appear 10 to 15 days after the mosquito bite. If left untreated, malaria can develop into a severe form of the disease (1). Plasmodium infection during pregnancy has been implicated in a number of pregnancy-related complications, such as spontaneous abortion, intrauterine growth retardation, and low birth weight, which are known risk factors for neonatal mortality (2, 3).
Many of the complications of pregnancy caused by Plasmodium infection are related to the accumulation of infected red blood cells (iRBCs) in the placenta (2–4). During Plasmodium falciparum infection, VAR2CSA, a member of the Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) family (5–8), which interacts with chondroitin sulfate A (CSA) and hyaluronic acid (HA) (9, 10), facilitates the sequestration of iRBCs in the placenta. The increased expression of CSA and HA in placental tissue promotes iRBC accumulation and interaction between the parasite and host cells.
The placenta is a unique transient organ responsible for maternal-fetal exchange and functions as an immunologic barrier against microbial infection, recognizing and responding to pathogens via innate pattern recognition receptors (PRRs) and ultimately preventing harmful or detrimental effects on fetal development (11, 12). In malaria, it has been demonstrated that members of the Toll-like receptor (TLR) family recognize components of Plasmodium sp. and stimulate an immune response. For example, studies have shown that glycosylphosphatidylinositol (GPI) is recognized by TLR2 and TLR4 (13), while the parasite's DNA in association with hemozoin, a metabolite derived from the heme detoxification mechanism in the parasite, has been implicated in the stimulation of TLR9 (14–16). TLR activation is mediated by a range of adaptor proteins, such as MyD88, Tirap (also known as Mal), Trif, and Tram, and culminates in the expression of proinflammatory genes (17). In acute malaria, TLR9 association with MyD88 is essential for the initiation of an interleukin 12 (IL-12)/gamma interferon (IFN-γ)-mediated immune response and the subsequent increased expression of proinflammatory cytokines (18).
Polymorphisms in TLRs have been correlated with the severity of malaria (19–21). More specifically, in pregnant women, these polymorphisms increase the susceptibility to placental malaria (19, 21). Innate immune activation during pregnancy has been associated with a range of complications for the mother and fetus, such as intrauterine growth restriction, low birth weight, and abortion. Several studies have demonstrated that TLRs are pivotal for the host immune responses to Plasmodium and related pathogen-associated molecular patterns (12–15, 22). However, the roles of these TLRs in Plasmodium-induced pregnancy complications have not yet been examined. Accordingly, the mechanism that triggers immune activation following malaria infection during pregnancy is not well understood. In the present study, using a mouse model of placental malaria (3) and validating it in mice with a C57BL/6 genetic background, we analyzed the involvement of MyD88, an adapter molecule for all TLRs except TLR3 (23), in the detrimental effects of the severe inflammatory host immune response to Plasmodium during pregnancy.
MATERIALS AND METHODS
Mice and parasites.
C57BL/6 (wild-type [WT]) and MyD88−/− mice from the animal breeding facility of the Institute of Biomedical Sciences at the University of São Paulo (ICB/USP), aged 8 to 10 weeks, were bred and maintained with a constant light-dark cycle (12 h/12 h) in conventional housing at the ICB/USP Department of Parasitology's animal facility. The mice received water and were fed with commercial NUVILAB CR-1 feed (Nuvital, Brazil) ad libitum. Erythrocytes infected with Plasmodium berghei NK65, constitutively expressing green fluorescent protein (GFP) (24), were used to induce placental malaria and to conduct in vitro experiments. Infected red blood cells (iRBCs) to be used in experimental infections were obtained through in vivo passaging in C57BL/6 mice when the percentage of iRBCs reached approximately 10%. Infection was monitored daily, and parasitemia was assessed in blood smears stained with Giemsa, followed by counting under a microscope. All procedures were performed in accordance with the national guidelines on animal welfare and were authorized by the ethics committee at ICB/USP under protocol number 065fls104livro2.
Synchronization and enrichment of parasitized erythrocytes.
To obtain mature forms of the parasite (i.e., trophozoites and schizonts), iRBCs were synchronized as described previously (25). Briefly, iRBCs were collected from infected WT mice exhibiting 10 to 20% parasitemia through cardiac puncture and transferred to RPMI 1640 culture medium (Gibco, Life Technologies, Paisley, Scotland) supplemented with 25% fetal bovine serum (FBS). The iRBCs were subsequently maintained in vitro at 37°C for 14 h in an atmosphere containing 5% CO2, 85% N2, and 10% O2. The parasitized erythrocytes were then enriched using a magnetic separation column (Miltenyi Biotec, Bergisch Gladbach, Germany) to generate cell populations consisting of approximately 95% iRBCs, as assessed by thick blood smears.
Cytoadherence assays.
To confirm the ability of P. berghei NK65GFP-infected erythrocytes to bind to specific receptors, the iRBCs were enriched, synchronized, and coincubated with CHO cells stably expressing chondroitin sulfate A (CSA) (CHO-K1 cells) for 1 h at 37°C in an incubator with 5% CO2. For inhibition assays, following the first incubation period, nonadherent cells were removed by washing 3 times, and the cells were then incubated for 20 min in the presence of increasing doses of soluble CSA, chondroitin sulfate B (CSB), or colominic acid (CA) (3). Following a final wash, the cultures were fixed and stained using the Instant-Prov kit for differential staining (New Prov, Brazil). Cytoadherence was determined by counting the number of iRBCs attached per CHO cell.
Ex vivo adherence of the iRBCs to placental tissue was achieved using placentas from uninfected females, obtained at G19 and treated as described previously (26). Briefly, the placentas were fixed in 2% formalin and 0.5% glutaraldehyde for 10 min, heated in a microwave oven prior to paraffin embedment, cut into 5-μm sections, and placed on glass slides. This fixation protocol preserves the binding capacity of the glycosylaminoglycans (GAG) in the placental intervillous spaces. Following deparaffinization and rehydration, the boundaries of tissue sections were delimited using a Dako pen. For iRBC ligand blocking experiments, synchronized iRBCs were preincubated with the indicated concentrations of CSA, CSB, or colominic acid from bovine trachea (Sigma) at 37°C for 30 min with moderate agitation. After being washed, the iRBCs were used to overlay the placental sections as described above. The slides were mounted using Vectashield (Vector Laboratories, Inc., Burlingame, CA) and examined by fluorescence microscopy. The number of iRBCs (green dots) adhering to the placental sections under each experimental condition was determined in a blinded fashion, using the cell count plugin from Image J Software, and 50 fields were counted in each of 3 independent experiments.
Pregnancy monitoring and experimental infection.
Detection of a vaginal plug and measurement of body weight were used in combination to determine the gestation time, as described previously (27). Females (2 or 3) were placed with one male for 24 h and then examined for the presence of a vaginal plug. The day the vaginal plug was detected was considered the first day of gestation (G1). The progression of pregnancy was monitored every other day based on weight gain. Successful fertilization was confirmed between G8 and G10 when the females exhibited an average increase of 3 to 4 g in body weight. Therefore, weight gain was taken as an indication of pregnancy, whereas abrupt weight loss was interpreted as an indicator of pregnancy complications or interruption. Pregnant mice were infected intravenously (i.v.) on G13 with 1 × 105 iRBCs, and parasitemia was assessed daily. G13 was determined as the optimal time point for infection to allow analysis of the pathological features of malaria during the course of pregnancy and in the developing fetus, because infection at an earlier stage did not allow the pregnancy to reach term, which is consistent with previous reports (28–30). The pregnant mice underwent cesarean sections on G19, and their placentas were collected for histopathological analysis and RNA extraction. Nonpregnant infected females or noninfected pregnant females were used as controls when appropriate. To monitor the offspring, foster mothers were employed in postnatal follow-up analyses of the newborns to avoid weight bias caused by differential maternal nourishment. To evaluate maternal care in relation to infection, we monitored the offspring for 30 days after birth. To construct the survival rate, we did not distinguish the cause of death of the pups (as infanticide or cannibalism).
Collection of placentas.
The placentas from the infected and noninfected females were treated in a similar manner. The placentas were divided into two parts: one half was fixed in 1.6% paraformaldehyde with 20% sucrose for further processing, and the other half was collected for RNA extraction. Paraffin-embedded, nonconsecutive placental sections were stained with hematoxylin-eosin (H&E), periodic acid-Schiff (PAS), or picrosirius red and then examined under a conventional Zeiss microscope (Axio imager M2) coupled with a Zeiss camera (Axio Cam HRc). For histological and morphometric analyses, the placental sections were coded and examined by two different individuals in a blinded fashion.
Morphometric analysis.
Morphometric analyses of the placentas were performed as described previously (3). Briefly, the vascular space was quantified through analysis of hematoxylin-eosin (H&E)-stained sections of the placentas. For each section, three areas of the intervillous space were randomly selected for image acquisition (magnification, ×200) using a Zeiss color camera (Axio Cam HRc) connected to a Zeiss light microscope (Axio Imager M2). The images were analyzed using Image J software (http://rsbweb.nih.gov/ij/). Briefly, the images were subjected to an automated light analysis procedure in which noise removal was applied to ensure color and image quality standardization across the sections and specimens. A color threshold was applied to the images to cover the area corresponding to the blood space lumen. The percentage of coverage was calculated as the ratio between the number of pixels covered by the area defined by the threshold and the total number of pixels in the image. The blood vascular area in each placenta was estimated based on the analysis of three nonconsecutive sections.
Gene expression analysis via qRT-PCR.
RNA was extracted from the placenta obtained at G19 (6th day postinfection) samples using the RNeasy minikit (Qiagen) in accordance with the manufacturer's protocol for animal tissue (Animal Cell 1). Total RNA (1 μg) was converted into cDNA (First Strand cDNA Synthesis Transcriptor kit; Roche, Penzberg, Germany) using random hexamer primers. Relative gene expression was quantified via quantitative reverse transcription-PCR (qRT-PCR) using TaqMan assays (Applied Biosystems) with probes specific for IL-6 (Mm00446190_m1), gamma interferon-induced protein 10 (IP-10) (CXCL10; Mm00445235_m1), and tumor necrosis factor alpha (TNF-α) (Mm00443258_m1). Briefly, 1.25 μl of 20× target assay and 1.25 μl of 20× mouse GAPD (Applied Biosystems) mix was added to 12.5 μl of MasterMix (Applied Biosystems), 5 μl of deionized distilled water, and 100 ng of cDNA. All experiments were carried out in duplicate. The thermal cycler conditions were 50°C for 2 min, 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. The data were normalized according to the constitutive expression of the GAPDH gene, and the 2ΔΔCT method was applied for quantification (31).
Cytokine measurements.
IL-6, TNF-α, IFN-γ, and IL-10 production was evaluated in serum samples from the pregnant mice obtained at G19 (6th day postinfection) using cytometric bead arrays. This procedure was performed in accordance with the manufacturer's instructions (cytometric bead arrays [CBA]; BD Biosciences). The serum samples were analyzed in a FACS Canto II flow cytometer (BD Biosciences) using the supplied cytometer setup beads and FCAP software (BD Biosciences).
Statistical analysis.
Significant differences between the groups were evaluated via analysis of variance (ANOVA) following Bonferroni's posttest. Survival rates were analyzed using the log rank test. The statistical tests were performed with GraphPad Prism version 5.0 software (GraphPad Software, San Diego, CA). P values of <0.05, <0.01, and <0.001 were considered statistically significant.
RESULTS
P. berghei NK65GFP-iRBCs adhere to placental tissue via CSA and could be related to the placental malaria.
Adhesion of iRBCs to placental tissue may be detrimental to pregnancy and fetal development. In P. falciparum infections, parasite adhesion to placental tissue is mediated by CSA (9, 32). Similar results were obtained in a murine model using P. berghei ANKAGFP in BALB/c mice (3). P. berghei ANKAGFP infection normally kills C57BL/6 mice after 1 week of infection due to neurological complications and is therefore characterized as an experimental model of cerebral malaria. Thus, to allow the use of knockout mice together with C57BL/6 background mice, we established a new mouse model of placental malaria using a parasite strain (P. berghei NK65GFP) that does not cause cerebral malaria (33). In our model, pregnant mice were infected on gestational day 13 (G13) and killed at G19 to allow us to obtain intact placentas (see Fig. S1 in the supplemental material).
Initially, to validate our model, we evaluated the alterations induced in the placental tissue by parasite infection. Our results showed that at G19, placentas from C57BL/6 mice infected with P. berghei NK65GFP exhibited increased tissue disorganization compared to placentas from noninfected mice (Fig. 1A versus B), collagen deposition in the labyrinth zone (Fig. 1C and D), and necrotic areas between the spongiotrophoblast and decidua (Fig. 1E). Moreover, these events appear to be related to the presence of iRBCs in the syncytiotrophoblast (Fig. 1F).
FIG 1.
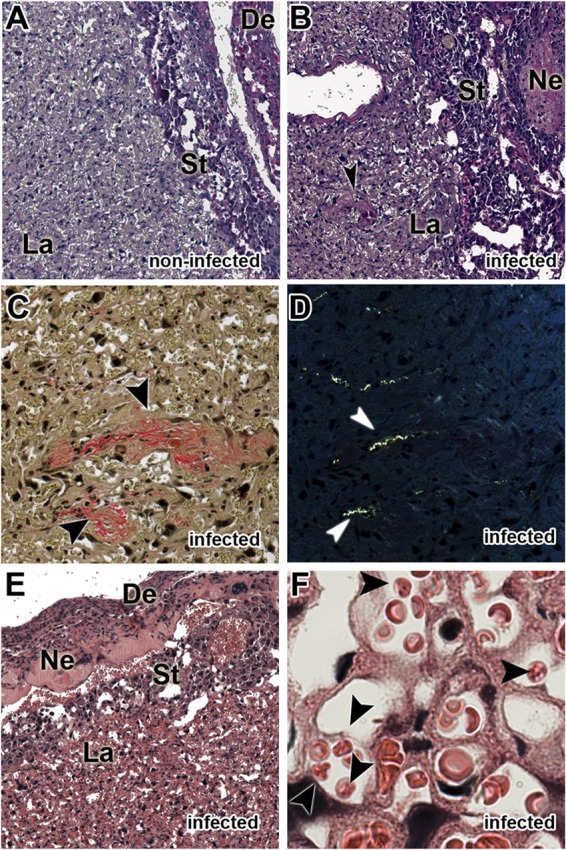
P. berghei NK65GFP induces placental malaria. Pregnant WT females were infected i.v. with 1 × 105 P. berghei NK65GFP iRBCs on the 13th gestational day. (A and B) Photomicrographs of the sinusoidal areas of placentas from noninfected (A) and infected (B) mice stained with PAS, showing the tissue organization and vascular space. (C and D) Collagen fibers were stained with picrosirius red and visualized via conventional light microscopy (C) and polarization light microscopy (D). (E) Necrotic areas were stained with H&E. (F) iRBCs are present in the syncytiotrophoblast. The arrows indicate necrosis in panel B, collagen in panels C and D, and iRBCs in panel F. De, decidua; St, spongiotrophoblast; Ne, necrosis; La, labyrinth zone. Magnification, ×400 (A to E) and ×1,000 (F).
Next, to determine whether P. berghei NK65GFP-iRBCs have the capacity to adhere to CSA, we evaluated the effect of CSA on the adhesion of synchronized mature iRBCs to CHO-K1 cells exhibiting a high level of surface expression of CSA. Figure 2A shows that CSA, but not CSB or CA, competitively inhibited iRBC adhesion in a dose-dependent manner. Finally, through a series of ex vivo adhesion assays, we demonstrated that P. berghei NK65GFP-iRBCs adhered to the placental tissue (Fig. 2B and C). Together, these results validate our model of placental malaria.
FIG 2.

P. berghei NK65GFP adheres to placental tissue via CSA, inducing placental malaria. (A) Competition assay of iRBCs binding to CHO-K1 cells preincubated with increasing concentrations of CSA, CSB, and CA. The numbers of iRBCs in 10 randomly selected fields were counted, and the values presented are mean percentages of binding compared to the control. (B) Ex vivo adhesion assay in placental tissue from noninfected mice. iRBCs were preincubated with increasing concentrations of CSA and used in the binding assays as described in Materials and Methods. The data are the proportion of bound iRBCs, expressed as a percentage of the control. (C) Typical microscopic image obtained from adherence assays, showing iRBCs adhering to the syncytiotrophoblast cell layer (left). Representative images from adherence-blocking assays, in which the iRBCs were preincubated with 100 μg/ml (middle) or 1,000 μg/ml (right) of CSA. All data are the means ± SE and are representative of three independent experiments (n = 3). **, P < 0.01; ***, P < 0.001.
The increased parasitemia resulting from pregnancy is not dependent on MyD88 activation.
Next, to determine the role of MyD88 in the increased susceptibility to malaria observed during pregnancy, a phenomenon previously observed in P. berghei ANKAGFP-infected BALB/c mice (32, 34), we analyzed parasitemia on the 6th day postinfection. Our results showed that both MyD88−/− and WT (C57BL/6 background) pregnant mice exhibited higher parasitemia levels than nonpregnant mice (Fig. 3).
FIG 3.
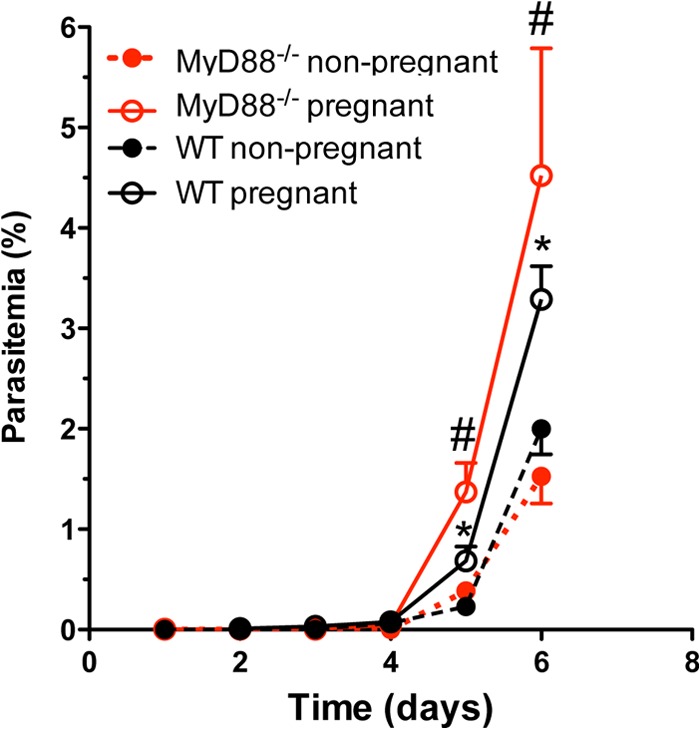
Pregnancy influences parasitemia levels. Pregnant WT and MyD88−/− mice were infected intravenously on G13 with 1 × 105 P. berghei NK65GFP iRBCs. As a control, nonpregnant mice were infected in parallel. The animals were killed 6 days postinfection. Data are means ± SE (10 mice/group). *, P < 0.05 compared with nonpregnant WT mice; #, P < 0.05 compared with nonpregnant MyD88−/− mice.
Placental pathology is associated with the activation of the MyD88 adaptor protein pathway.
The characteristics of placental malaria include changes in tissue organization and a reduction of the vascular space (35). As described previously for other strains (34), placentas from infected C57BL/6 mice exhibited a reduction in the vascular space due to malaria infection compared to placentas from noninfected mice (Fig. 4A). Using a morphometric method to quantify the area of blood sinusoids in cross sections of the placentas, we observed a reduction in the area of blood sinusoids in the infected WT pregnant mice compared to the noninfected WT pregnant mice (48.13% ± 0.88% versus 39.44% ± 0.64%; mean ± standard error [SE]) of the vascular space in the noninfected and infected mice, respectively; P < 0.001). However, when the analysis was conducted in placentas from MyD88−/− pregnant mice, no differences were observed between the infected and noninfected groups (Fig. 4B).
FIG 4.

MyD88 signaling is associated with a diminished vascular placental space and reduced growth rate in the progeny of infected mothers. Pregnant WT and MyD88−/− females were infected i.v. with 1 × 105 P. berghei NK65GFP iRBCs on the 13th gestational day. (A) Representative images of the sinusoidal areas of placentas from noninfected (left) and infected (right) mice. (B) The sinusoidal areas of the placentas were quantified relative to the total area of the placenta using an automated method of morphometric analysis. (C) Images of mice at 21 days of age from noninfected (left) and infected (right) mothers. (D) Fetal body weights in noninfected and infected mothers at G19 at the time of cesarean section. Data are presented as a scatter plot, with the medians indicated (15 to 40 mice/group). ***, P < 0.001.
Next, to evaluate the influence of the reduction of vascular space on fetal development, we analyzed fetus weight on day G19. Figure 4C illustrates the sizes of the 21-day-old mice born to a noninfected mother (left) and an infected mother (right). Consistent with previous studies, the fetuses from infected WT mice displayed a 30% reduction in body weight compared to fetuses from noninfected WT mice (Fig. 4C). However, the fetuses from infected MyD88−/− did not exhibit significant differences in body weight compared to those from their noninfected counterparts (Fig. 4D).
Because infection with P. berghei NK65GFP did not result in placental pathology in MyD88−/− mice, we hypothesized that the MyD88-mediated inflammatory response might be involved in this process. Therefore, we investigated the cytokine profiles in the serum and the placenta tissue of WT and MyD88−/− pregnant mice following P. berghei NK65GFP infection. When the infected groups were compared, the WT mice showed higher serum levels of IL-6, IFN-γ, and TNF-α (Fig. 5A, B, and C). In addition, IL-10 serum levels were significantly reduced in MyD88−/− mice compared to WT mice, in both the infected and noninfected groups (Fig. 5D). Regarding the proinflammatory markers examined in the placental tissue, we observed increased expression of TNF-α, IP-10, and IL-6 only in infected WT mice compared to noninfected WT (Fig. 6). In addition, we did not observe differences between infected and noninfected MyD88−/− mice.
FIG 5.

MyD88 is involved in the induction of systemic proinflammatory cytokines in pregnant mice. (A to D) Serum cytokine production of IL-6, TNF-α, IFN-γ, and IL-10 measured via cytometric bead arrays (CBA). Serum samples were obtained at G19 (6th day postinfection). Data are means ± SE (5 mice/group). *, P < 0.05, ***, P < 0.001.
FIG 6.
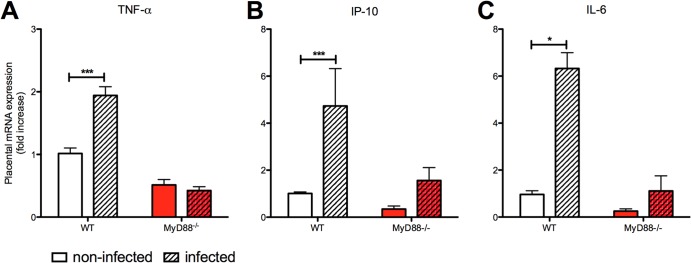
Placental pathology is associated with altered gene expression of inflammation markers. Placental gene expression was quantified for TNF-α (A), gamma interferon-induced protein 10 (IP-10) (B), and IL-6 (C). mRNA expression was measured in placentas from WT and MyD88−/− mice that were either infected with P. berghei NK65GFP (hatched bars) or not infected (open bars). Relative quantification was performed via normalization to GAPDH expression levels. Samples were obtained at G19 (6th day postinfection). The results are plotted as the fold change over noninfected WT mice, and each bar represents the mean ± SE (5 placentas/group from different donors). *, P < 0.01; ***, P < 0.001.
Finally, to evaluate the effects of maternal infection in the progeny, we followed the pups' survival for 30 days after parturition. Analysis of the survival rates of the pups indicated an interesting phenomenon regarding maternal care. The results presented in Fig. 7 show that infected WT mothers displayed decrements in maternal care that influenced the pup survival rate. Surprisingly, however, pups from infected MyD88-deficient mothers exhibited better survival rates than pups from infected WT mothers.
FIG 7.
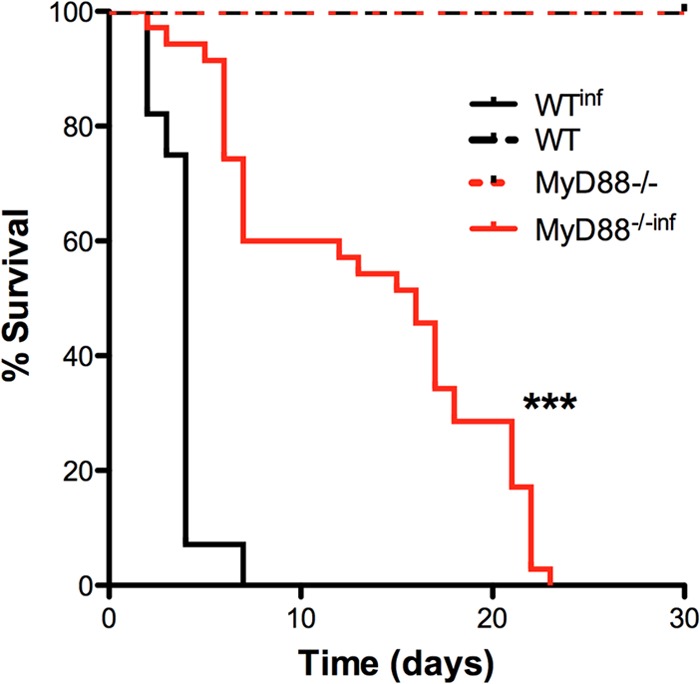
Attenuation of the maternal care decrement is associated with MyD88 expression in placental malaria. The survival curve for pups from infected WT mothers (n = 28) and MyD88−/− mothers (n = 35) up to 20 days after birth is shown. ***, P < 0.001.
DISCUSSION
Placental malaria is a major issue in many regions where malaria is endemic, and the immunological mechanisms that play a role in the evolution of this outcome are still not completely understood. It is well established that in humans, accumulation of P. falciparum-iRBCs in the placenta occurs through the adhesion of PfEMP-1 to CSA, leading to malaria pathogenesis during pregnancy (2, 5, 36). However, this characteristic is not exclusive to P. falciparum. Using a murine model of placental malaria, our group and others have demonstrated the ability of P. berghei ANKA to adhere to placental tissue (37). In addition, Plasmodium chabaudi- and Plasmodium yoelii-iRBCs have been reported to accumulate in the placenta (4, 38, 39). To evaluate the involvement of the MyD88 adapter protein in placental malaria, we first validated the P. berghei variant NK65GFP as a potential model for this complication, inasmuch as this parasite, in contrast to P. berghei ANKA, does not induce cerebral malaria in mice with a C57BL/6 background. Through in vitro and ex vivo experiments, we demonstrated that the membrane components of the P. berghei NK65GFP iRBCs adhered to CSA on the surface of the chondroitin sulfate A-expressing CHO-K1 cell line and to placental tissue.
Using this infection model, we showed that P. berghei NK65GFP-induced activation of the innate immune system via the MyD88 pathway is clearly involved in the development of PM. Moreover, because P. berghei NK65GFP-infected MyD88−/− pregnant mice do not show an increase in proinflammatory cytokines in the serum and placenta that correlates with the reduced placental vascular space observed in infected WT pregnant mice, our results indicate that PM development occurs because of excessive TLR-induced local inflammation.
These findings show potential significance in relation to malaria during pregnancy and directional cytokine release by the placenta, suggesting that TLRs may act as a placental tissue sensor of Plasmodium components that, through the MyD88 pathway, trigger the release of inflammatory cytokines into the maternal blood.
Inflammation as a result of innate immune system activation via TLRs has a negative effect on the outcome of pregnancy (40). In rats, intraperitoneal administration of LPS, the best-described TLR4 agonist, during pregnancy increases the concentrations of TNF-α and IL-6 in maternal serum as well as the placental expression of TNF-α, IL-6, and IL-1β (40). Moreover, LPS from Gram-negative bacteria has been implicated in preterm delivery (41). TLR3 activation via poly(I·C), a synthetic double-stranded RNA that mimics viral RNA, during late pregnancy also has a negative effect on pregnancy (42). However, TLR2 activation does not appear to be related to inflammatory responses in placental tissue. TLR stimulation is also known to induce fetal reabsorption (43) and preeclampsia (44). In addition, clinical studies have linked TLR activation to pregnancy disorders such as preterm labor, preeclampsia, and intrauterine growth restriction (IUGR) (12).
A consequence of TLR activation is the production of proinflammatory cytokines. In infected pregnant women, it has been shown that the soluble levels of TNF receptors are correlated with P. falciparum parasitemia (45). Moreover, systemic production of TNF-α has been observed following malaria infection during pregnancy and has been found to be associated with maternal anemia and low birth weight. In addition to TNF-α, other proinflammatory cytokines, such as IFN-γ, have been implicated in spontaneous abortion in humans (46). Similarly, murine models developed in an attempt to induce pregnancy failure through malaria infection point to systemic increases in TNF-α and IFN-γ production as a cause of abortions (47, 48). In our model system, we did not observe an increase in abortion or reabsorption due to systemic TNF-α or IFN-γ production. However, our results indicated that the production of these cytokines in WT infected mice was clearly associated with the observed placental impairment (i.e., inflammation, tissue disorganization, and reduction of vascular space). A possible explanation for the absence of abortions in this study may be the time of infection, which, in contrast to previous studies (47), was chosen to allow the pregnancies to reach term in our murine model (3). In addition to the systemic maternal levels of TNF-α, the placentas from the infected WT mice showed increased expression of TNF-α mRNA. Interestingly, both systemically and locally, the increase in proinflammatory cytokines was dependent on MyD88 expression, suggesting that the absence of this adaptor protein may have a negative effect on the immune response. In this regard, Franklin and collaborators showed that MyD88 is essential for eliciting TNF-α and IFN-γ production during infection with P. chabaudi (49). In addition, those authors showed that in the absence of MyD88 expression, mice exhibited an attenuated pathology.
Malaria infection during pregnancy is associated with alterations in placental structure that result in insufficient gas exchange and reduced nutrient availability, leading to impaired fetal growth and development (35, 50, 51). In this regard, murine models have been useful for investigating the causes and consequences of placental modifications. Working with BALB/c mice, we previously demonstrated that pregnant mice are more susceptible to malaria infection than nonpregnant mice (52). In this study, we showed that infected C57BL/6 mice also exhibited increased susceptibility to malaria infection by P. berghei. Another primary finding of this study was that fetuses from the infected WT mice had lower body weights than the controls, which is consistent with the symptoms of infection observed in pregnant women. Additionally, the experiments in knockout mice demonstrated a clear correlation between reduced weight and the expression of MyD88. In the absence of this protein, a reduction of placental inflammation was observed, and fetal weight was therefore not affected. Consistent with this observation, using a model of preterm delivery induced by Escherichia coli infection, Filipovich and collaborators demonstrated that MyD88-deficient mice were protected from preterm delivery induced by bacterial infection compared to WT mice (53). Notably, although the MyD88-deficient mice were able to produce TNF-α in response to bacterial infection, the production of this cytokine was lower than in WT mice and was correlated with delayed TRIF signaling.
Infected animals display a range of characteristics, including anorexia, adipsia, lethargy, and reduced social interaction (54), which are referred to as sickness behavior and represent an adaptive strategy to aid the organism in recovery from infection (55). Using a model of infection, Weil and collaborators showed that mice injected with LPS exhibited a decrement of maternal care, and their litters were therefore not able to maintain normal growth (55). In the present work, we observed that infected WT mothers also exhibited impaired maternal care, showing increased infanticide and cannibalism events. In eutherian mammals, there is a major expenditure of energy during the reproductive cycle due to lactation. For this reason, infanticide and cannibalism in rodents could be explained as a mechanism to prevent the waste of additional investments in infants (56). This behavior could be the result of stress induced by infection. In this regard, similar behavior has been observed in rodents deprived of food (57). Surprisingly however, the MyD88-deficient mice exhibited delayed and attenuated infanticide and cannibalism events.
Studies characterizing the pathogenesis of PM are useful for the development of therapeutic strategies. As a continuation of our previous findings, in this study, we identified a key pathway stimulated by the parasites that triggers the innate immune system, leading to a local inflammatory response. This process culminated in the placental pathology observed in the infected mice and, ultimately, reduced fetal body weight. In conclusion, this study illustrates the importance of a severe local inflammatory response in the pathogenesis of PM via MyD88 signaling. Consequently, blocking this pathway may become a critical approach for the treatment of placental inflammation induced by Plasmodium. The combination of blocking innate immune receptors or proteins related to the activation of these receptors and concomitant use of antiplasmodial drugs is the keystone of our conceptual model for investigating novel interventional strategies for treating malaria in pregnancy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bernardo Paulo Albe for his technical assistance in preparing the histological sections. F.T.M.C. is enrolled by the Programa Estratégico de Ciência, Tecnologia & Inovação nas Fundações Estaduais de Saúde (PECTI/AM Saúde) of FAPEAM.
This work was supported by São Paulo Research Foundation (FAPESP) (grants 2009/53889-0, 2009/53256-7, 2011/17880-8, and 2012/02270-2), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES (grants AUX-PE-PNPD 2751/2010 and 258/2010), and Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (grants 475771/2009-5 and 404213/2012).
Footnotes
Published ahead of print 9 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01288-13.
REFERENCES
- 1.van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. 2006. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22:503–508. 10.1016/j.pt.2006.09.002 [DOI] [PubMed] [Google Scholar]
- 2.Rogerson SJ, Hviid L, Duffy PE, Leke RF, Taylor DW. 2007. Malaria in pregnancy: pathogenesis and immunity. Lancet Infect. Dis. 7:105–117. 10.1016/S1473-3099(07)70022-1 [DOI] [PubMed] [Google Scholar]
- 3.Neres R, Marinho CRF, Gonçalves LA, Catarino MB, Penha-Gonçalves C. 2008. Pregnancy outcome and placenta pathology in Plasmodium berghei ANKA infected mice reproduce the pathogenesis of severe malaria in pregnant women. PLoS One 3:e1608. 10.1371/journal.pone.0001608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poovassery J, Moore JM. 2006. Murine malaria infection induces fetal loss associated with accumulation of Plasmodium chabaudi AS-infected erythrocytes in the placenta. Infect. Immun. 74:2839–2848. 10.1128/IAI.74.5.2839-2848.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy MF, Caragounis A, Noviyanti R, Kyriacou HM, Choong EK, Boysen K, Healer J, Rowe JA, Molyneux ME, Brown GV, Rogerson SJ. 2006. Transcribed var genes associated with placental malaria in Malawian women. Infect. Immun. 74:4875–4883. 10.1128/IAI.01978-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuikue Ndam NG, Salanti A, Bertin G, Dahlbäck M, Fievet N, Turner L, Gaye A, Theander T, Deloron P. 2005. High level of var2csa transcription by Plasmodium falciparum isolated from the placenta. J. Infect. Dis. 192:331–335. 10.1086/430933 [DOI] [PubMed] [Google Scholar]
- 7.Salanti A, Dahlback M, Turner L, Nielsen MA, Barfod L, Magistrado P, Jensen AT, Lavstsen T, Ofori MF, Marsh K, Hviid L, Theander TG. 2004. Evidence for the involvement of VAR2CSA in pregnancy-associated malaria. J. Exp. Med. 200:1197–1203. 10.1084/jem.20041579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricke CH, Staalsoe T, Koram K, Akanmori BD, Riley EM, Theander TG, Hviid L. 2000. Plasma antibodies from malaria-exposed pregnant women recognize variant surface antigens on Plasmodium falciparum-infected erythrocytes in a parity-dependent manner and block parasite adhesion to chondroitin sulfate A. J. Immunol. 165:3309–3316 [DOI] [PubMed] [Google Scholar]
- 9.Fried M, Duffy PE. 1996. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science 272:1502–1504. 10.1126/science.272.5267.1502 [DOI] [PubMed] [Google Scholar]
- 10.Beeson JG, Rogerson SJ, Cooke BM, Reeder JC, Chai W, Lawson AM, Molyneux ME, Brown GV. 2000. Adhesion of Plasmodium falciparum-infected erythrocytes to hyaluronic acid in placental malaria. Nat. Med. 6:86–90. 10.1038/71582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abrahams VM, Mor G. 2005. Toll-like receptors and their role in the trophoblast. Placenta 26:540–547. 10.1016/j.placenta.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 12.Abrahams VM. 2008. Pattern recognition at the maternal-fetal interface. Immunol. Invest. 37:427–447. 10.1080/08820130802191599 [DOI] [PubMed] [Google Scholar]
- 13.Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC. 2005. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J. Biol. Chem. 280:8606–8616. 10.1074/jbc.M413541200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coban C, Igari Y, Yagi M, Reimer T, Koyama S, Aoshi T, Ohata K, Tsukui T, Takeshita F, Sakurai K, Ikegami T, Nakagawa A, Horii T, Nuñez G, Ishii KJ, Akira S. 2010. Immunogenicity of whole-parasite vaccines against Plasmodium falciparum involves malarial hemozoin and host TLR9. Cell Host Microbe 7:50–61. 10.1016/j.chom.2009.12.003 [DOI] [PubMed] [Google Scholar]
- 15.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. 2007. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. U. S. A. 104:1919–1924. 10.1073/pnas.0608745104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, Heppner DG, Stewart VA, Hasegawa H, Looareesuwan S, Shanks GD, Miller RS. 2004. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J. Immunol. 172:4926–4933 http://www.jimmunol.org/content/172/8/4926.long [DOI] [PubMed] [Google Scholar]
- 17.Pasare C, Medzhitov R. 2005. Toll-like receptors: linking innate and adaptive immunity. Adv. Exp. Med. Biol. 560:11–18. 10.1007/0-387-24180-9_2 [DOI] [PubMed] [Google Scholar]
- 18.Franklin BS, Parroche P, Ataíde MA, Lauw F, Ropert C, de Oliveira RB, Pereira D, Tada MS, Nogueira P, da Silva LHP, Bjorkbacka H, Golenbock DT, Gazzinelli RT. 2009. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc. Natl. Acad. Sci. U. S. A. 106:5789–5794. 10.1073/pnas.0809742106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh N-R, Otchwemah RN, Dietz E, Ehrhardt S, Schröder NWJ, Bienzle U, Schumann RR. 2006. Toll-like receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. Proc. Natl. Acad. Sci. U. S. A. 103:177–182. 10.1073/pnas.0506803102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leoratti FMS, Farias L, Alves FP, Suarez-Mútis MC, Coura JR, Kalil J, Camargo EP, Moraes SL, Ramasawmy R. 2008. Variants in the Toll-like receptor signaling pathway and clinical outcomes of malaria. J. Infect. Dis. 198:772–780. 10.1086/590440 [DOI] [PubMed] [Google Scholar]
- 21.Hamann L, Bedu-Addo G, Eggelte TA, Schumann RR, Mockenhaupt FP. 2010. The toll-like receptor 1 variant S248N influences placental malaria. Infect. Genet. Evol. 10:785–789. 10.1016/j.meegid.2010.05.005 [DOI] [PubMed] [Google Scholar]
- 22.Meuris S, Piko BB, Eerens P, Vanbellinghen AM, Dramaix M, Hennart P. 1993. Gestational malaria: assessment of its consequences on fetal growth. Am. J. Trop. Med. Hyg. 48:603–609 [DOI] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 24.Sultan AA, Thathy V, Nussenzweig V, Ménard R. 1999. Green fluorescent protein as a marker in Plasmodium berghei transformation. Infect. Immun. 67:2602–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janse CJ, Franke-Fayard B, Mair GR, Ramesar J, Thiel C, Engelmann S, Matuschewski K, van Gemert GJ, Sauerwein RW, Waters AP. 2006. High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol. Biochem. Parasitol. 145:60–70. 10.1016/j.molbiopara.2005.09.007 [DOI] [PubMed] [Google Scholar]
- 26.Muthusamy A, Achur RN, Bhavanandan VP, Fouda GG, Taylor DW, Gowda DC. 2004. Plasmodium falciparum-infected erythrocytes adhere both in the intervillous space and on the villous surface of human placenta by binding to the low-sulfated chondroitin sulfate proteoglycan receptor. Am. J. Pathol. 164:2013–2025. 10.1016/S0002-9440(10)63761-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freyre A, Falcón J, Méndez J, Rodriguez A, Correa L, González M. 2006. Refinement of the mouse model of congenital toxoplasmosis. Exp. Parasitol. 113:154–160. 10.1016/j.exppara.2005.12.019 [DOI] [PubMed] [Google Scholar]
- 28.Oduola AM, Holbrook TW, Galbraith RM, Bank H, Spicer SS. 1982. Effects of malaria (Plasmodium berghei) on the maternal-fetal relationship in mice. J. Protozool. 29:77–81. 10.1111/j.1550-7408.1982.tb02883.x [DOI] [PubMed] [Google Scholar]
- 29.Hioki A, Hioki Y, Ohtomo H. 1990. Influence of pregnancy on the course of malaria in mice infected with Plasmodium berghei. J. Protozool. 37:163–167. 10.1111/j.1550-7408.1990.tb01121.x [DOI] [PubMed] [Google Scholar]
- 30.Desowitz RS, Shida KK, Pang L, Buchbinder G. 1989. Characterization of a model of malaria in the pregnant host: Plasmodium berghei in the white rat. Am. J. Trop. Med. Hyg. 41:630–634 [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 32.Marinho CRF, Neres R, Epiphanio S, Gonçalves LA, Catarino MB, Penha-Gonçalves C. 2009. Recrudescent Plasmodium berghei from pregnant mice displays enhanced binding to the placenta and induces protection in multigravida. PLoS One 4:e5630. 10.1371/journal.pone.0005630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodrigues-Duarte L, de Moraes LV, Barboza R, Marinho CR, Franke-Fayard B, Janse CJ, Penha-Goncalves C. 2012. Distinct placental malaria pathology caused by different Plasmodium berghei lines that fail to induce cerebral malaria in the C57BL/6 mouse. Malar. J. 11:231. 10.1186/1475-2875-11-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodrigues-Duarte L, Vieira de Moraes L, Barboza R, Marinho CR, Franke-Fayard B, Janse CJ, Penha-Goncalves C. 2012. Distinct placental malaria pathology caused by different Plasmodium berghei lines that fail to induce cerebral malaria in the C57Bl/6 mouse. Malar. J. 11:231. 10.1186/1475-2875-11-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brabin BJ, Romagosa C, Abdelgalil S, Menendez C, Verhoeff FH, McGready R, Fletcher KA, Owens S, D'Alessandro U, Nosten F, Fischer PR, Ordi J. 2004. The sick placenta—the role of malaria. Placenta 25:359–378. 10.1016/j.placenta.2003.10.019 [DOI] [PubMed] [Google Scholar]
- 36.Rogerson SJ. 1995. Chondroitin sulfate A is a cell surface receptor for Plasmodium falciparum-infected erythrocytes. J. Exp. Med. 182:15–20. 10.1084/jem.182.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hviid L, Marinho CRF, Staalsoe T, Penha-Gonçalves C. 2010. Of mice and women: rodent models of placental malaria. Trends Parasitol. 26:412–419. 10.1016/j.pt.2010.04.010 [DOI] [PubMed] [Google Scholar]
- 38.Cox J, Semoff S, Hommel M. 1987. Plasmodium chabaudi: a rodent malaria model for in-vivo and in-vitro cytoadherence of malaria parasites in the absence of knobs. Parasite Immunol. 9:543–561. 10.1111/j.1365-3024.1987.tb00529.x [DOI] [PubMed] [Google Scholar]
- 39.Pavia CS, Niederbuhl CJ. 1991. Immunization and protection against malaria during murine pregnancy. Am. J. Trop. Med. Hyg. 44:176–182 [DOI] [PubMed] [Google Scholar]
- 40.Gayle DA, Beloosesky R, Desai M, Amidi F, Nuñez SE, Ross MG. 2004. Maternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286:R1024–R1029. 10.1152/ajpregu.00664.2003 [DOI] [PubMed] [Google Scholar]
- 41.Hirsch E, Wang H. 2005. The molecular pathophysiology of bacterially induced preterm labor: insights from the murine model. J. Soc. Gynecol. Invest. 12:145–155. 10.1016/j.jsgi.2005.01.007 [DOI] [PubMed] [Google Scholar]
- 42.Ilievski V, Lu S-J, Hirsch E. 2007. Activation of toll-like receptors 2 or 3 and preterm delivery in the mouse. Reprod. Sci. 14:315–320. 10.1177/1933719107302959 [DOI] [PubMed] [Google Scholar]
- 43.Li L, Kang J, Lei W. 2010. Role of Toll-like receptor 4 in inflammation-induced preterm delivery. Mol. Hum. Reprod. 16:267–272. 10.1093/molehr/gap106 [DOI] [PubMed] [Google Scholar]
- 44.Faas MM, Schuiling GA, Baller JF, Visscher CA, Bakker WW. 1994. A new animal model for human preeclampsia: ultra-low-dose endotoxin infusion in pregnant rats. Am. J. Obstet. Gynecol. 171:158–164. 10.1016/0002-9378(94)90463-4 [DOI] [PubMed] [Google Scholar]
- 45.Thévenon AD, Zhou JA, Megnekou R, Ako S, Leke RGF, Taylor DW. 2010. Elevated levels of soluble TNF receptors 1 and 2 correlate with Plasmodium falciparum parasitemia in pregnant women: potential markers for malaria-associated inflammation. J. Immunol. 185:7115–7122. 10.4049/jimmunol.1002293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marzi M, Vigano A, Trabattoni D, Villa ML, Salvaggio A, Clerici E, Clerici M. 1996. Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin. Exp. Immunol. 106:127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poovassery J, Moore JM. 2009. Association of malaria-induced murine pregnancy failure with robust peripheral and placental cytokine responses. Infect. Immun. 77:4998–5006. 10.1128/IAI.00617-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poovassery JS, Sarr D, Smith G, Nagy T, Moore JM. 2009. Malaria-induced murine pregnancy failure: distinct roles for IFN-gamma and TNF. J. Immunol. 183:5342–5349. 10.4049/jimmunol.0901669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franklin BS, Rodrigues SO, Antonelli LR, Oliveira RV, Goncalves AM, Sales-Junior PA, Valente EP, Alvarez-Leite JI, Ropert C, Golenbock DT, Gazzinelli RT. 2007. MyD88-dependent activation of dendritic cells and CD4+ T lymphocytes mediates symptoms, but is not required for the immunological control of parasites during rodent malaria. Microbes Infect. 9:881–890. 10.1016/j.micinf.2007.03.007 [DOI] [PubMed] [Google Scholar]
- 50.Le Cessie S. 2002. Changes in haemoglobin levels in infants in Malawi: effect of low birth weight and fetal anaemia. Arch. Dis. Child. Fetal Neonatal 86:182F–187F. 10.1136/fn.86.3.F182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brabin BJ, Kalanda BF, Verhoeff FH, Chimsuku LH, Broadhead RL. 2004. Risk factors for fetal anaemia in a malarious area of Malawi. Ann. Trop. Paediatr. 24:311–321. 10.1179/027249304225019136 [DOI] [PubMed] [Google Scholar]
- 52.Neres R, Marinho CRF, Gonçalves LA, Catarino MB, Penha-Gonçalves C. 2008. Pregnancy outcome and placenta pathology in Plasmodium berghei ANKA infected mice reproduce the pathogenesis of severe malaria in pregnant women. PLoS One 3:e1608. 10.1371/journal.pone.0001608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filipovich Y, Lu J, Akira S, Hirsch E. 2009. The adaptor protein MyD88 is essential for E coli-induced preterm delivery in mice. Am. J. Obstet. Gynecol. 200:93.e1–93.e8. 10.1016/j.ajog.2008.08.038 [DOI] [PubMed] [Google Scholar]
- 54.Hart BL. 1988. Biological basis of the behavior of sick animals. Neurosci. Biobehav. Rev. 12:123–137. 10.1016/S0149-7634(88)80004-6 [DOI] [PubMed] [Google Scholar]
- 55.Weil ZM, Bowers SL, Dow ER, Nelson RJ. 2006. Maternal aggression persists following lipopolysaccharide-induced activation of the immune system. Physiol. Behav. 87:694–699. 10.1016/j.physbeh.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 56.Ebensperger LA. 1998. Strategies and counterstrategies to infanticide in mammals. Biol. Rev. 73:321–346. 10.1111/j.1469-185X.1998.tb00034.x [DOI] [Google Scholar]
- 57.Elwood RW. 1980. The development, inhibition, and disinhibition of pup-cannibalism in the Mongolian gerbil. Anim. Behav. 28:1188–1194. 10.1016/S0003-3472(80)80107-2 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.