

Abstract
We developed a multilocus sequence typing (MLST) scheme and used it to study the population structure and evolutionary relationships of three pathogenic Yersinia species. MLST of these three Yersinia species showed a complex of two clusters, one composed of Yersinia pseudotuberculosis and Yersinia pestis and the other composed of Yersinia enterocolitica. Within the first cluster, the predominant Y. pestis sequence type 90 (ST90) was linked to Y. pseudotuberculosis ST43 by one locus difference, and 81.25% of the ST43 strains were from serotype O:1b, supporting the hypothesis that Y. pestis descended from the O:1b serotype of Y. pseudotuberculosis. We also found that the worldwide-prevalent serotypes O:1a, O:1b, and O:3 were predominated by specific STs. The second cluster consisted of pathogenic and nonpathogenic Y. enterocolitica strains, two of which may not have identical STs. The pathogenic Y. enterocolitica strains formed a relatively conserved group; most strains clustered within ST186 and ST187. Serotypes O:3, O:8, and O:9 were separated into three distinct blocks. Nonpathogenic Y. enterocolitica STs were more heterogeneous, reflecting genetic diversity through evolution. By providing a better and effective MLST procedure for use with the Yersinia community, valuable information and insights into the genetic evolutionary differences of these pathogens were obtained.
INTRODUCTION
Recent reports concerning the taxonomy of the genus Yersinia show it consists of 17 species (1), among which only three of 11 currently recognized species (2) are human pathogens: Yersinia enterocolitica, Yersinia pseudotuberculosis, and Yersinia pestis. The three pathogenic Yersinia species differ radically in their pathogenicities. Y. pestis is the deadliest bacterium known in human history; it is primarily a rodent pathogen transmitted via the bite of an infected flea. Y. pseudotuberculosis and Y. enterocolitica are zoonotic food-borne pathogens that spread through the fecal-oral route; they have a broad host range, infecting animals, including swine, dogs, rodents, birds, and wild animals (3, 4). Many studies show that swine and dogs are the most common sources of Y. enterocolitica infections in humans (5, 6). These enteropathogens cause human enteric diseases, both sporadically and in epidemics worldwide. Y. enterocolitica infections are primarily reported in northern Europe (7), but outbreaks have occurred in Finland, Japan, the United States, and Brazil (8–11). In China, two outbreaks in the 1980s caused >500 infections (12). Y. pseudotuberculosis outbreaks have been reported in the Northern Hemisphere, including in Canada, Japan, and Russia (13). Most infections reported in FoodNet (The Food-borne Diseases Active Surveillance Network [see http://www.cdc.gov/foodnet/] launched by the U.S. government in 1996) appear to be severe and invasive (14). Plague, known as the Black Death, has claimed millions of human lives through multiple pandemics (15). Human epidemics occur each year in China along with animal epidemics. Recently, a dog-associated outbreak involving 12 persons was reported in 2009 in Qinghai Province (16). To date, several MLST analyses were reported for pathogenic Yersinia species (17–19); however, neither the Y. pestis nor Y. pseudotuberculosis analyses turned out to be satisfactory. Thus, an MLST analysis was developed in this study based on a method by Laukkanen-Ninios et al. (19) to integrate the three pathogenic Yersinia species and reveal further their genetic similarities and evolutionary relationships.
MATERIALS AND METHODS
Sources of strains.
One thousand fifteen strains of three pathogenic Yersinia species were used in this study (Table 1). One hundred eighty-seven Y. enterocolitica strains were chosen from nearly four thousand strains isolated by our laboratory from 19 provinces in China, from 1985 to 2013, collected from various sources, including a patient with diarrhea, animals (swine, dogs, rats, hens, sheep, and fish), and the environment. The pathogenicities, serotypes, and host distributions of the isolated and reference strains are shown in Table 2, and the isolation locations and biotype distributions are shown in Table 3. Surveillance of the patient with diarrhea was aimed at the whole population; all specimens were collected at an intestine outpatient clinic, regardless of patient symptoms, to minimize any sampling biases. Seventy-six Y. pseudotuberculosis strains were isolated from rats, dogs, and swine from seven provinces in China (Table 4; see isolation locations and serotypes for the isolated and reference strains). Among 35 Y. pestis strains isolated from different natural plague foci in China, 15 were isolated from patients (seven from Yunnan Province, six from Qinghai Province, one from Gansu Province, and one from Inner Mongolia), and the rest were from rats, Marmota spp., Xenopsylla cheopis, and Suncus murinus. The sample collection and detection protocols were approved by the ethics review committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention. Informed consents were obtained from all patients. The reference strain Y. enterocolitica WA, provided by Enshu Yu of the Fujian Provincial Center for Disease Control and Prevention, was donated by an American scholar from the Food Research Institute, University of Wisconsin, Madison. Other reference strains were purchased from the Institute Pasteur by the Institute of Chinese Biomedicine or were provided by H. Fukushima at the Shimane Prefectural Institute of Public Health, Matsue, Japan. The genome sequence data were obtained from GenBank (see http://ncbi.nlm.nih.gov). Six hundred sixty-three sequence data, five of which were from Y. pestis, were downloaded from the ERI-UCC (Environmental Research Institute, University College Cork) database (http://mlst.ucc.ie/mlst/dbs/Ypseudotuberculosis). The provenances of all 1,015 strains are listed in Table S2 in the supplemental material.
TABLE 1.
Summary of 1,015 strains of pathogenic Yersinia in this study
| Strain or sequence source | No. of strains from: |
||
|---|---|---|---|
| Y. enterocolitica | Y. pseudotuberculosis | Y. pestis | |
| Strains | |||
| Isolate | 187 | 76 | 35 |
| Reference | 11 | 24 | 0 |
| Sequences | |||
| Genome sequence | 3 | 4 | 12 |
| UCC database | 0 | 658 | 5 |
| Total | 201 | 762 | 52 |
TABLE 2.
Pathogenicity, serotype, and host distribution of Y. enterocolitica strains
| Strain source | Host | No. of pathogenic strains |
No. of nonpathogenic strains |
Total no. of strains by source | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O:3 | O:5,27 | O:8 | O:9 | Total | O:3 | O:5 | O:6,30 | O:7,8 | O:8 | O:9 | UNa | Total | |||
| Isolate strains | Patient | 15 | 7 | 22 | 5 | 3 | 1 | 1 | 7 | 17 | 39 | ||||
| Swine | 41 | 8 | 49 | 3 | 1 | 5 | 2 | 10 | 21 | 70 | |||||
| Dog | 18 | 3 | 21 | 2 | 3 | 5 | 26 | ||||||||
| Rat | 2 | 10 | 12 | 3 | 3 | 6 | 18 | ||||||||
| Hen | 1 | 2 | 3 | 1 | 2 | 4 | 7 | 10 | |||||||
| Cow | 1 | 1 | 1 | 2 | 1 | 3 | 7 | 8 | |||||||
| Sheep | 3 | 3 | 1 | 3 | 1 | 5 | 8 | ||||||||
| Raw meat | 2 | 1 | 3 | 3 | |||||||||||
| Milk | 2 | 2 | 2 | ||||||||||||
| Rabbit | 1 | 1 | 1 | ||||||||||||
| Fish | 1 | 1 | 1 | ||||||||||||
| Refrigerator | 1 | 1 | 1 | ||||||||||||
| Reference strains | 6 | 1 | 5 | 2 | 14 | 14 | |||||||||
| Total no. of strains | 87 | 1 | 5 | 37 | 130 | 1 | 10 | 3 | 2 | 19 | 4 | 32 | 71 | 201 | |
UN, undetermined serotype.
TABLE 3.
Isolation location and biotype distribution of Y. enterocolitica strains
| Strain type/origin | No. of strains of Y. enterocolitica biotype: |
Total no. of strains per location | ||||
|---|---|---|---|---|---|---|
| 1A | 1B | 2 | 3 | 4 | ||
| Isolated strains | ||||||
| Inland provinces | ||||||
| Anhui | 2 | 1 | 3 | |||
| Guizhou | 1 | 1 | ||||
| Henan | 9 | 8 | 15 | 32 | ||
| Heilongjia | 2 | 2 | ||||
| Jilin | 1 | 7 | 10 | 18 | ||
| Inner Mongolia | 2 | 2 | ||||
| Ningxia | 4 | 13 | 15 | 32 | ||
| Qinghai | 1 | 1 | ||||
| Sichuan | 1 | 2 | 3 | |||
| Yunnan | 4 | 1 | 5 | |||
| Total | 27 | 29 | 43 | 99 | ||
| Coastal provinces | ||||||
| Beijing | 1 | 3 | 4 | |||
| Fujian | 1 | 8 | 1 | 10 | ||
| Guangxi | 2 | 2 | ||||
| Jiangsu | 18 | 20 | 38 | |||
| Shandong | 17 | 2 | 19 | |||
| Shanghai | 2 | 2 | ||||
| Shenzhen | 1 | 1 | ||||
| Tianjin | 5 | 5 | ||||
| Zhejiang | 4 | 3 | 7 | |||
| Total | 44 | 41 | 3 | 88 | ||
| Reference strains | 5 | 1 | 5 | 3 | 14 | |
| Total no. of strains | 71 | 5 | 30 | 89 | 6 | 201 |
TABLE 4.
Isolation locations and serotypes of Y. pseudotuberculosis strains
| Strain type | Province of isolation | No. of strains from serotype: |
Total no. of strains | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O:1 | O:1a | O:1b | O:2a | O:2b | O:3 | O:4b | O:5a | O:6 | O:8 | O:9 | O:10 | O:11 | O:14 | O:15 | NTa | |||
| Isolated | Guangxi | 1 | 2 | 1 | 2 | 3 | 9 | |||||||||||
| Guizhou | 1 | 1 | 1 | 1 | 4 | |||||||||||||
| Jiangxi | 2 | 2 | ||||||||||||||||
| Ningxia | 1 | 2 | 2 | 5 | ||||||||||||||
| Sichuan | 1 | 1 | 2 | |||||||||||||||
| Yunnan | 1 | 1 | ||||||||||||||||
| Zhejiang | 2 | 13 | 25 | 9 | 4 | 53 | ||||||||||||
| Total isolated strains | 5 | 14 | 32 | 1 | 13 | 1 | 3 | 7 | 76 | |||||||||
| Reference | 1 | 3 | 4 | 3 | 1 | 2 | 2 | 1 | 2 | 1 | 3 | 1 | 1 | 3 | 28 | |||
| Total no. of strains | 1 | 3 | 9 | 3 | 1 | 16 | 34 | 1 | 14 | 3 | 1 | 3 | 1 | 4 | 3 | 7 | 104 | |
NT, nontypeable serotype.
Culture and identification.
Y. enterocolitica and Y. pseudotuberculosis enrichment was performed using peptone sorbitol bile broth (Sigma-Aldrich, USA [pH, 7.6 ± 0.2]) at 4°C for 21 days. The presumptive Yersinia strains with colonies having a typical bull's-eye appearance (deep-red centers surrounded by an outer transparent zone) on Yersinia selective agar (CIN agar; Oxoid, Basingstoke, United Kingdom) were inoculated onto brain heart infusion (BHI) agar (Beijing Land Bridge Technology Co., Ltd., China) plates incubated at 25°C for 24 to ∼48 h to obtain pure cultures (4, 12). The inoculation of Y. pestis was performed using the method described by Achtman et al. (20). The whole genome was extracted using the DNeasy blood and tissue kit (Qiagen, USA) and a DNA nucleic acid extraction kit (Tiangen, China), according to each step in the handbook, and the elution volume was 100 μl. Biochemical identification tests used the bacterial identification system API 20E test strips (bioMérieux). Commercial serotype identification test kits for Y. enterocolitica were purchased from Denka Seiken Co., Ltd., Japan, and the Institute of Chinese Biomedicine. The biotypes of Y. enterocolitica strains were identified using the scheme described by Bottone (7). The virulence genes (ail, ystA, ystB, virF, and yadA) of the Y. enterocolitica isolates were amplified. Pathogenic Y. enterocolitica strains were positive for all (ail+, ystA+, virF+, and yadA+) virulence genes; however, some pathogenic strains lost virulence genes that were carried on a plasmid (ail+, ystA+, virF-negative, and yadA-negative strains) (3, 4). Serotype identification of the Y. pseudotuberculosis strains was conducted using multiplex PCR as described by Bogdanovich et al. (21). The detection of genes ypm and yadA was described by Fukushima et al. (22) and Thoerner et al. (3), respectively. The identification of Yersinia similis was described by Sprague et al. (23).
MLST genes, primer design, amplification, and sequencing.
The Y. enterocolitica housekeeping genes were selected based on an ERI-UCC-constructed MLST scheme designed for Y. pseudotuberculosis: adk (adenylate kinase), argA (amino acid acetyltransferase), aroA (3-phosphoshikimate-1-carboxyvinyltransferase), glnA (glutamine synthase), thrA (aspartokinase-homoserine dehydrogenase 1), tmk (thymidylate kinase), and trpE (anthranilate synthase component 1). A BLAST search was performed with the 28 amplification and sequencing primers for Y. enterocolitica strain 8081 to see if there were mismatches and to modify four primers: tmk-p4, aroA-s3, tmk-s2, and trpE-s4. Twenty-eight primers for Y. enterocolitica are shown in Table 5 (synthetized by Sangon Biotech, China). Each PCR included 2 μl of DNA template (approximately 20 ng), 25 μl Premix Taq version 2.0 (TaKaRa), 2 μl (25 pmol/μl) of each forward and reverse primer, and Milli-Q water, resulting in a total volume of 50 μl. The thermal cycling conditions performed using an MJ PTC200 (MJ, USA) were initial DNA denaturation for 5 min at 94°C, followed by 30 cycles of DNA denaturation for 15 s at 94°C, primer annealing for 30 s at 53°C, and polymerization for 30 s at 72°C, with a final extension of 5 min at 72°C. The reaction products were detected using gel electrophoresis (2,000-bp DNA ladder [TaKaRa, Japan]), and the gel image was captured using a Gel Doc 2000 (Bio-Rad, USA). The amplicon was purified using a gel extraction kit (Qiagen, USA) and sequenced with an ABI Prism BigDye Terminator cycle sequencing ready reaction kit using AmpliTaq DNA polymerase, according to the instructions of the manufacturer, and an ABI Prism 377xl DNA sequencer (Applied Biosystems, Foster City, CA, USA) at TaKaRa Biotechnology (Dalian)/Tsingke BioTech Co., Ltd., to sequence the amplicons in both directions.
TABLE 5.
Amplification and sequencing primers for Y. enterocolitica
| Target gene | Data by primer type: |
||||
|---|---|---|---|---|---|
| Amplification |
Sequencing |
||||
| Name | Sequence | Length (bp) | Name | Sequence | |
| adk | adk-p1 | ATGCGTATCATTCTGCTGGG | 641 | adk-s1 | TGGAGAAATACGGTATTCCG |
| adk-p2 | CCGAGAATAGTCGCCAGTTC | adk-s2 | ACTTTACGGGTTCCGTCCAG | ||
| argA | argA-p1 | GGATTTCGCCACTCAGTTCC | 615 | argA-s3 | CAAGACATTTGTTGTCATGC |
| argA-p2 | ATCCGTCACCCCTTGTGATG | argA-s4 | ATAGCTAATTGAGTTGCAAC | ||
| aroA | aroA-p1 | AGCGGCCAATTGGTCATTTG | 802 | aroA-s3a | AGCACAGATTGATTATCTGG |
| aroA-p2 | CACATCGCCATGCGGTGGTC | aroA-s2 | ATGGTCATTGCAGCATCAGG | ||
| glnA | glnA-p1 | GCTGACTTCTTCGAAGAAGG | 701 | glnA-s1 | TTTGATGGCTCCTCGATTGGTG |
| glnA-p2 | GACATATGGCAGTGCATACC | glnA-s2 | TTGGTCATGGTATTGAAGCG | ||
| thrA | thrA-p3 | CGTCTTTGCGGTGATGTCG | 823 | thrA-s1 | GATGTGATGGAACATCTGGC |
| thrA-p2 | GTTGGTGTCATACAAGAATTTACG | thrA-s2 | GTCACAACATGGAAGCCATC | ||
| tmk | tmk-p3 | TATTGAAGGGCTTGAAGGGG | 606 | tmk-s5 | CGCCCAAGGGATTAACGATAT |
| tmk-p4a | CGGCTGGTCAGCCATTGCTT | tmk-s2a | AAGCGGTTGAGAAGCATCAAT | ||
| trpE | trpE-p3 | CACCAATTGCAACAAGCGCC | 743 | trpE-s1 | CCAGAGATGGCGTTACAGTG |
| trpE-p4 | GTATCCAAATCACCATGAGC | trpE-s4a | TAGCCGACAGCACCGCCGTA | ||
tmk-p4, aroA-s3, tmk-s2, and trpE-s4 were redesigned according to Y. enterocolitica 8081. The four primers of Y. pseudotuberculosis are TGATTGGTCAGCCACTGAGC (tmk-p4), TGCACAGATCGATTATCTGG (aroA-s3), AATGGGTTGGGAGGCATCAAT (tmk-s2), and TAGCCCACTGCACCGCCGTA (trpE-s4).
MLST data analysis.
The reading of trace files and assembly of contigs were performed using Chromas and DNAStar. Next, the sequence reads were trimmed by removing low-quality nucleotide sequences from the ends and were reamplified and sequenced if necessary. The sequences were aligned with the reference sequence from the UCC database using MEGA (version 5.0). BioEdit (version 7.0.1) was used to determine the allele assignments of the housekeeping genes before composing a profile of each strain. We submitted the new sequence patterns to the UCC database for their allele and ST assignments. New profiles of published-pattern alleles were submitted for ST assignments. Unrooted neighbor-joining trees (maximum likelihood criteria) based on concatenated sequences were performed using MEGA (version 5.0). The minimum spanning tree and shortest spanning path were constructed using BioNumerics 5.10 (Applied Maths) (maximum neighbor distance, 1 change; minimum size, 1 type). The latter approach demonstrated the shortest spanning path between STs that differed at one locus and provided more specific information than the minimum spanning tree. eBURST (24) was utilized to draw a snapshot, including three pathogenic Yersinia species (minimum number of identical loci for group definition, 0; minimum single-locus variant [SLV] count for subgroup definition, 0; number of resamplings for bootstrap, 1,000; diagram group number, 1).
RESULTS
Housekeeping gene variations and sequence type distribution.
MLST analysis of 1,015 strains identified 188 sequence types (see Table S1 in the supplemental material for the profile and number of strains from each ST). The fragments thrA and tmk diverged with the most sequence patterns (Table 6; see sequence pattern numbers for each locus). Y. pestis and Y. pseudotuberculosis shared one sequence pattern for each housekeeping gene except trpE. Within Y. enterocolitica, the pathogenic and nonpathogenic strains shared two sequence patterns for each housekeeping gene.
TABLE 6.
Sequence pattern number of each housekeeping gene for each species
| Species and pathogenicity | Data (n) by housekeeping gene: |
No. in ST/total no. of strainsb | ||||||
|---|---|---|---|---|---|---|---|---|
| adk | argA | aroA | glnA | thrA | tmk | trpE | ||
| Y. enterocolitica | ||||||||
| Pathogenic | 6a | 7a | 6a | 5a | 5a | 6a | 8a | 17/130 |
| Nonpathogenic | 14a | 16a | 12a | 13a | 22a | 24a | 16a | 46/71 |
| Y. pseudotuberculosis | 7 | 6 | 17 | 18 | 17 | 11 | 11 | 121/762 |
| Y. pestis | 1a | 2a | 1a | 1a | 3a | 1a | 1 | 4/52 |
| Total Yersinia speciesa | 25 | 28 | 33 | 34 | 44 | 39 | 32 | 188/1,015 |
The total number of each allele was not directly added, as Y. pestis and Y. pseudotuberculosis share one sequence pattern for each locus except trpE. Pathogenic and nonpathogenic Y. enterocolitica strains share two sequence patterns for each locus.
The ratio of the number of STs/number of strains showed that Y. pestis was the least diverse species, and Y. enterocolitica had proportionately more STs than the other two species; however, pathogenic Y. enterocolitica strains were nearly as conserved as Y. pseudotuberculosis strains.
The Y. pseudotuberculosis strains collected by our laboratory formed 45 STs, 19 of which were previously published in the UCC database; the new STs were submitted for ST assignment (ST97 to ST122). The ST assignment numbers of Y. pestis and Y. enterocolitica were positioned after those of Y. pseudotuberculosis. The ratio of the number of STs to the number of strains showed that Y. pestis is the least diverse species of the three; there were proportionately more Y. enterocolitica STs than for the other two species; however, pathogenic Y. enterocolitica strains were nearly as conserved as those of Y. pseudotuberculosis (Table 6).
Therefore, all Y. pseudotuberculosis strains showed 121 STs (ST1 to ST89 and ST91 to ST122), Y. pestis strains showed four STs (ST90 and ST123 to ST125), and Y. enterocolitica strains showed 63 STs (see Table S1 in the supplemental material). The STs were not shared among the species. Within Y. enterocolitica, the only sequence type shared by the pathogenic and nonpathogenic strains was ST187.
Cluster analysis of sequence types.
The clustering analysis data of three pathogenic Yersinia species were obtained using neighbor-joining tree, minimum spanning tree, and eBURST (see Fig. S1 in the supplemental material) analyses.
Neighbor-joining tree.
Generally, two blocks were demonstrated based on the neighbor-joining tree of the housekeeping gene concatenated sequences (Fig. 1): one included Y. pseudotuberculosis and Y. pestis, and the other was composed of Y. enterocolitica. In the Y. enterocolitica block, the only sequence type shared by the pathogenic and nonpathogenic strains was ST187.
FIG 1.

Neighbor-joining tree of 1,015 isolates, housekeeping genes, and concatenated sequences (topology graph). Besides three pathogenic Yersinia species, Y. similis and the Korean group are colored according to the description by Laukkanen-Ninios et al. (19). **, ST187 was the only sequence type shared by pathogenic and nonpathogenic Y. enterocolitica strains. The nonpathogenic strain was a biotype 1A ail+ strain. *, Two particular nonpathogenic, ystB+ Y. enterocolitica strains, ST184, clustered next to ystB-negative strain STs (ST181 to ST183), and ST185, a biotype 1A ail+ strain, clustering next to pathogenic Y. enterocolitica STs, indicating its potential pathogenicity. s.s., sensu stricto.
Minimum spanning tree.
To add genetic diversity and compare the genetic differences and evolutionary relationships between the strains isolated in and outside China, we introduced the UCC database data in our analysis. Two trees were obtained before and after importing the UCC sequences to the strains collected by our laboratory. As seen in Fig. 2, the genetic diversity of three species from China was generally consistent with that around the world. See Fig. S2 in the supplemental material for the shortest spanning path between the STs that differed at one locus. Three pathogenic species were separated into two distinct blocks, similar to the results obtained with the neighbor-joining tree analysis. No obvious association was found between the isolation background (isolation year, location, or host) and sequence types. Y. pseudotuberculosis STs were numerous and scattering, Y. enterocolitica STs were more heterogeneous but presented low genetic diversity within the pathogenic strains, and Y. pestis was the least diverse.
FIG 2.

Minimum spanning tree, colored by species, of three pathogenic Yersinia species before (A) and after (B) importing the UCC database sequences and combining them with 352 strains collected by our laboratory. Pathogenic Yersinia species fell into two distinct clusters; one included Y. pseudotuberculosis and Y. pestis, and the other involved pathogenic and nonpathogenic Y. enterocolitica strains. Y. pseudotuberculosis STs were numerous and scattering, Y. enterocolitica was relatively conserved, especially within pathogenic strains, and Y. pestis was the least diverse. The Arabic numerals show ST assignments. The circle sizes are directly proportional to the numbers of isolates. The numbers of locus differences are represented by bold lines (1 locus), plain lines (2 loci), black dotted lines (3 loci), dark-gray dotted lines (4 loci), and light-gray dotted lines (>5 loci). The colored shaded areas around the circles represent single-locus variants (SLVs).
Within the pathogenic Y. enterocolitica strains, a strong specificity pattern for O serotypes was observed. ST187 was the predominant sequence type of the pathogenic O:3 clonal group. ST186 was the predominant sequence type of the pathogenic O:9 clonal group, which differed from ST187 at one locus. Within each clonal group, most STs diverged from ST186 or ST187 by one locus. The pathogenic O:3 and O:9 clonal groups were found to have a close genetic relationship. ST131 contains a pathogenic O:5,27 strain, and with one locus difference from ST186, it can be regarded as a member of the pathogenic O:9 clonal group. The pathogenic O:8 clonal group was composed of reference strains isolated from countries beyond China, as it is genetically more distant from the other pathogenic strains. Within ST187, 80 of 81 strains were pathogenic, and the last strain was 1A/O:3, harboring the same-sequence pattern ail as the rest of pathogenic strains assigned to ST187, which indicated its potential pathogenicity. The nonpathogenic strains were genetically distant from the pathogenic strains and presented with more genetic variability. The pathogenic and nonpathogenic strains that were assigned the same serotypes were genetically distant from each other. Most nonpathogenic STs contained a single strain for each ST, and these are linked by a dotted line on the neighbor-joining tree. The nonpathogenic ystB-negative strains (ST181 to ST183) had seven radically different loci compared to the ystB+ strains. Two particular ystB+ strains (ST184 and ST185) had no identical loci with either ystB-negative strains or other ystB+ strains (Fig. 3).
FIG 3.
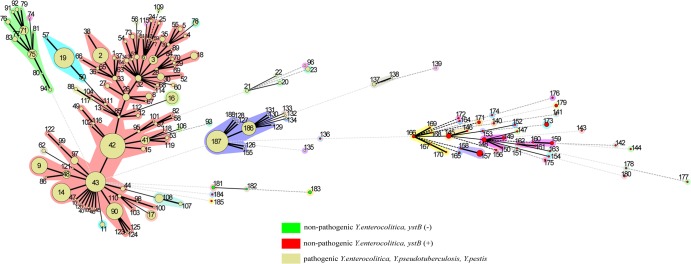
ystB distribution in nonpathogenic Y. enterocolitica strains. Within the nonpathogenic Y. enterocolitica strains, ystB-negative strains (ST181 to ST183) had seven radically different loci compared to ystB+ strains. Two particular ystB+ strains (ST184 and ST185) had no identical loci with either ystB-negative strains or other ystB+ strains.
Y. pseudotuberculosis STs were numerous and scattering. STs containing no more than 10 strains accounted for 87.7% of the strains. From the minimum spanning tree based on definitive serotype (the 21 serotypes reported by T. Bogdanovich et al. [21]) data, a specificity pattern for the O serotypes was observed (Fig. 4). ST42, ST43, and ST19, which contained most of the strains, each had a predominant serotype (Table 7).
FIG 4.

Y. pseudotuberculosis minimum spanning tree based on definitive serotype (the 21 serotypes reported by Bogdanovich et al. [21]) data. A specificity pattern for O serotypes was detected. The three STs containing the most strains (ST42, ST43, and ST19) each had a particular predominant serotype.
TABLE 7.
Three STs containing the most Y. pseudotuberculosis strains had predominant serotype
| ST | No. of strains: |
Predominant serotypes: |
||
|---|---|---|---|---|
| In serotype | With definitive serotypea | Nameb | % of serotype for ST | |
| 42 | 108 | 40 | O:1a | 80.00 |
| 43 | 70 | 32 | O:1b | 81.25 |
| 19 | 69 | 51 | O:3 | 98.04 |
The definitive serotypes are the 21 serotypes reported by Bogdanovich et al. (21).
The predominant serotype is the serotype that contained the most strains, among the definitive serotypes.
Three Y. pestis genome sequencing strains diverged from ST90 at argA or thrA, whereas the rest of the strains were uniformly from ST90. Y. pestis strain Pestoides F varied in 66 bp of the argA 5′-ends, including a 37-bp deletion. Y. pestis strain D106004 had a deleted G at base 282 of thrA. Y. pestis strain D182038 was found to have a G-to-A transversion at base 232 of thrA (25).
DISCUSSION
We developed a multilocus sequence typing (MLST) scheme for three pathogenic Yersinia species based on a method by Laukkanen-Ninios et al. (19) and used it to study the heterogeneity of housekeeping genes and better understand the population structure and evolutionary relationships of the three strains. An MLST-based analysis showed that pathogenic Yersinia species fell into two distinct clusters (Fig. 2), one that included Y. pseudotuberculosis and Y. pestis, and the other that included pathogenic and nonpathogenic Y. enterocolitica strains. Population genetics studies have shown that Y. pestis recently evolved from Y. pseudotuberculosis O:1b about 1,500 to 20,000 years ago. DNA hybridization analyses demonstrated that Y. pseudotuberculosis and Y. pestis were genetically close (20, 26, 27). In our study, the predominant Y. pestis sequence type ST90 was linked to Y. pseudotuberculosis ST43 (O:1b comprised 81.25% of its serotypes) by one locus difference (Fig. 4). This again supported the conclusion above, and it is possible that this highly virulent pathogen evolved in a short time from the relatively mildly pathogenic progenitor, Y. pseudotuberculosis (20, 28). Although Y. pestis and Y. pseudotuberculosis were indistinguishable using multiple molecular typing methods (29), our analysis showed that Y. pestis and Y. pseudotuberculosis have no shared STs. This reconfirmed the intrinsic genetic differences between Y. pestis and Y. pseudotuberculosis that cause different diseases (30). In contrast, Y. enterocolitica were found to have a much more distant genetic relationship with Y. pseudotuberculosis. Phylogenetic studies have suggested that Y. enterocolitica and Y. pseudotuberculosis diverged within the last 200 million years (31). Sequence alignment showed that Y. enterocolitica strains have no identical sequence patterns with Y. pseudotuberculosis for any locus.
A recently described species, Y. similis, showed a minimum distance of six loci compared with Y. pseudotuberculosis ST42 (Fig. 2). These strains are biochemically similar to Y. pseudotuberculosis and cannot be distinguished using API 20E (23). However, Y. similis is not thought to be pathogenic for humans, as it has been isolated from small mammals and the environment. It lacks pYV and carries YPMb, and it is ultimately distinguished from Y. pseudotuberculosis on the basis of 16S rRNA sequencing (19, 23). Hence, STs linked by ≥5 locus differences in the minimum spanning tree do not necessarily share many genetic similarities. A comparable case was the “Korean group,” named by Laukkanen-Ninios et al. (19) (Fig. 2, purple segments). Five loci differentiated its ST21 and Y. pseudotuberculosis ST58. This group had not been defined, and its 16S rRNA sequences clustered between Y. similis and Y. pseudotuberculosis. Laukkanen-Ninios et al. inferred that the Korean group is genetically somewhat distinct from Y. pseudotuberculosis and may be in the process of becoming a distinct species. Within the nonpathogenic Y. enterocolitica strains in our study, the ystB+ strains evidently differed from the ystB-negative strains (ST181 to ST183) (Fig. 3). The biotype 1A strains are generally regarded as avirulent, as they lack the pYV plasmid and major chromosomal virulence genes. However, some biovar 1A strains produce disease symptoms that are indistinguishable from those produced by known pathogenic biovars (1B, 2, 3, 4, and 5). In particular, strains harboring ystB may be responsible for sporadic cases of diarrhea (32). Seven different housekeeping genes of ystB+ and ystB-negative strains added genetic evidence to support these distinct phenotypes. Two particular ystB+ strains (Fig. 3, ST184 and ST185) should also be noted; they had no identical loci with either ystB− strains or other ystB+ strains. In the neighbor-joining tree, ST184 clusters with ystB-negative strain STs (Fig. 1), and ST185 is a biotype 1A ail+ strain, clustering between other ystB+ strain STs and pathogenic Y. enterocolitica STs, indicating its potential pathogenicity (33).
Comparing the number of sequence patterns for each allele, it was obvious that Y. pestis is the least diverse, followed by Y. pseudotuberculosis and Y. enterocolitica. However, the pathogenic Y. enterocolitica strains were nearly as conserved as those of Y. pseudotuberculosis (Table 6). Within pathogenic Y. enterocolitica strains, a strong specificity pattern for O serotypes was detected. The serotype classifications were used to identify a strain as pathogenic or nonpathogenic. With the progress of molecular biology, virulence genes detected on the chromosome and pYV proved to be more accurate and coincided with the assigned serotype to some degree; e.g., the worldwide serotype O:3 and O:9 Y. enterocolitica strains are mostly pathogenic. In America and Japan, 1B/O:8 strains are historically considered to be the most common and highly pathogenic bio-serotype. Within the pathogenic Y. enterocolitica strains in our study, close genetic relationships between the serotypes and specificity patterns for the serotypes were demonstrated (Fig. 6). However, the pathogenicity classifications being dependent on serotype had limitations. All serotype O:8 strains isolated from China lacked virulence factors and were classified as being from biovar 1A (12). In MLST analysis, the pathogenic and nonpathogenic O:8 strains were distributed in separate branches of the minimum spanning tree (Fig. 6). In addition, we found that the allele primers had multiple or even no binding sites on the genomes of nonpathogenic Y. enterocolitica strains, so the target fragments could be amplified or sequenced. In contrast, all amplification genes in the pathogenic Y. enterocolitica strains were a single band and were sequenced. Being genetically heterogeneous, nonpathogenic Y. enterocolitica strains appeared to be much more adapted in response to host and environmental changes (26). In Y. pseudotuberculosis, the specificity pattern for O serotypes was also detected, which differed from the findings from a previous study (19): the worldwide prevalent serotypes O:1a, O:1b, and O:3 each were predominant in particular STs (Fig. 5).
FIG 6.
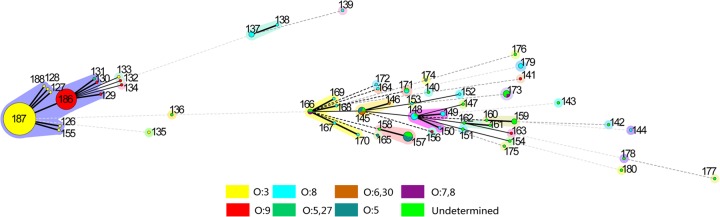
Minimum spanning tree of pathogenic Y. enterocolitica strains, colored by serotype. A strong specificity pattern for O serotypes within the pathogenic strains was demonstrated. ST187 and ST186 include most pathogenic strains. Nonpathogenic Y. enterocolitica STs are more heterogeneous, presenting genetic diversity through evolution. Same-serotype strains of pathogenic and nonpathogenic Y. enterocolitica strains were distributed in separate branches of the tree.
FIG 5.
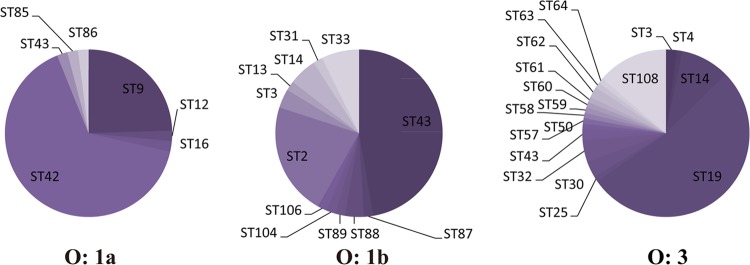
ST composition of the three worldwide-prevalent Y. pseudotuberculosis serotypes. The sequence types of O:1a, O:1b, and O:3 were dominated by ST42, ST43, and ST19, respectively.
This new modulation of the MLST method for three pathogenic Yersinia species was critical in our research, as it furnished a better and effective MLST procedure for use in this field. Though not particularly groundbreaking either in its methods or in the generation of new insights into the evolution of the Yersinia group, we found parts of novel sequence types and alleles and submitted them to public MLST databases, providing valuable information to the Yersinia research community. In addition, the typing results revealed close relationships with either the pathogenicities or serotypes of the bacteria, indicating the different evolutionary pathways of these pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (general project no. 31100101) and the National Sci-Tech Key Project (grant no. 2012ZX10004201 and 2013ZX10004203-002). The UCC sequence data are publicly available at http://mlst.ucc.ie, which is currently supported by a grant from the Science Foundation of Ireland (grant no. 05/FE1/B882).
We thank Liuying Tang and Jim Nelson for the critical reading of and helpful comments on our manuscript.
Footnotes
Published ahead of print 16 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02185-13.
REFERENCES
- 1.Hurst MR, Becher SA, Young SD, Nelson TL, Glare TR. 2011. Yersinia entomophaga sp. nov., isolated from the New Zealand grass grub Costelytra zealandica. Int. J. Syst. Evol. Microbiol. 61:844–849. 10.1099/ijs.0.024406-0 [DOI] [PubMed] [Google Scholar]
- 2.Ewing WH, Ross AJ, Brenner DJ. 1978. Yersinia ruckeri sp. nov., the Redmouth (RM) bacterium. Int. J. Syst. Bacteriol. 28:37–44 [Google Scholar]
- 3.Thoerner P, Bin Kingombe C, Bögli-Stuber K, Bissig-Choisat B, Wassenaar T, Frey J, Jemmi T. 2003. PCR detection of virulence genes in Yersinia enterocolitica and Yersinia pseudotuberculosis and investigation of virulence gene distribution. Appl. Environ. Microbiol. 69:1810–1816. 10.1128/AEM.69.3.1810-1816.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Cui Z, Jin D, Tang L, Xia S, Wang H, Xiao Y, Qiu H, Hao Q, Kan B, Xu J, Jing H. 2009. Distribution of pathogenic Yersinia enterocolitica in China. Eur. J. Clin. Microbiol. Infect. Dis. 28:1237–1244. 10.1007/s10096-009-0773-x [DOI] [PubMed] [Google Scholar]
- 5.Liang J, Wang X, Xiao Y, Cui Z, Xia S, Hao Q, Yang J, Luo L, Wang S, Li K, Yang H, Gu W, Xu J, Kan B, Jing H. 2012. Prevalence of Yersinia enterocolitica in pigs slaughtered in Chinese abattoirs. Appl. Environ. Microbiol. 78:2949–2956. 10.1128/AEM.07893-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Cui Z, Wang H, Tang L, Yang J, Gu L, Jin D, Luo L, Qiu H, Xiao Y, Xiong H, Kan B, Xu J, Jing H. 2010. Pathogenic strains of Yersinia enterocolitica isolated from domestic dogs (Canis familiaris) belonging to farmers are of the same subtype as pathogenic Y. enterocolitica strains isolated from humans and may be a source of human infection in Jiangsu Province, China. J. Clin. Microbiol. 48:1604–1610. 10.1128/JCM.01789-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bottone EJ. 1997. Yersinia enterocolitica: the charisma continues. Clin. Microbiol. Rev. 10:257–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.WHO Scientific Working Group 1980. Cholera and other Vibrio-associated diarrhoeas. Bull. World Health Organ. 58:353–374 [PMC free article] [PubMed] [Google Scholar]
- 9.Gutman L, Ottesen E, Quan TJ, Noce P, Katz SL. 1973. An inter-familial outbreak of Yersinia enterocolitica enteritis. N. Engl. J. Med. 288:1372–1377 [DOI] [PubMed] [Google Scholar]
- 10.Toivanen P, Olkkonen L, Toivanen A, Aantaa S. 1973. Hospital outbreak of Yersinia enterocolitica infection. Lancet 301:801–803 [DOI] [PubMed] [Google Scholar]
- 11.Toma S, Lafleur L. 1974. Survey on the incidence of Yersinia enterocolitica infection in Canada. Appl. Microbiol. 28:469–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Qiu H, Jin D, Cui Z, Kan B, Xiao Y, Xu Y, Xia S, Wang H, Yang J, Wang X, Hu W, Xu J, Jing H. 2008. O:8 serotype Yersinia enterocolitica strains in China. Int. J. Food Microbiol. 125:259–266. 10.1016/j.ijfoodmicro.2008.04.016 [DOI] [PubMed] [Google Scholar]
- 13.Niskanen T. 2010. Diagnostics and epidemiology of Yersinia pseudotuberculosis. Department of Food and Environmental Hygiene, University of Helsinki, Helsinski, Finland [Google Scholar]
- 14.Long C, Jones TF, Vugia DJ, Scheftel J, Strockbine N, Ryan P, Shiferaw B, Tauxe RV, Gould LH. 2010. Yersinia pseudotuberculosis and Y. enterocolitica infections, FoodNet, 1996–2007. Emerg. Infect. Dis. 16:566. 10.3201/eid1603.091106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morelli G, Song Y, Mazzoni CJ, Eppinger M, Roumagnac P, Wagner DM, Feldkamp M, Kusecek B, Vogler AJ, Li Y, Cui Y, Thomson NR, Jombart T, Leblois R, Lichtner P, Rahalison L, Petersen JM, Balloux F, Keim P, Wirth T, Ravel J, Yang R, Carniel E, Achtman M. 2010. Phylogenetic diversity and historical patterns of pandemic spread of Yersinia pestis. Nat. Genet. 42:1140. 10.1038/ng.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Cui Y, Wang Z, Wang X, Guo Z, Yan Y, Li C, Cui B, Xiao X, Yang Y, Qi Z, Wang G, Wei B, Yu S, He D, Chen H, Chen G, Song Y, Yang R. 2011. A dog-associated primary pneumonic plague in Qinghai Province, China. Clin. Infect. Dis. 52:185–190. 10.1093/cid/ciq107 [DOI] [PubMed] [Google Scholar]
- 17.Ch'ng SL, Octavia S, Xia Q, Duong A, Tanaka MM, Fukushima H, Lan R. 2011. Population structure and evolution of pathogenicity of Yersinia pseudotuberculosis. Appl. Environ. Microbiol. 77:768–775. 10.1128/AEM.01993-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotetishvili M, Kreger A, Wauters G, Morris JG, Jr, Sulakvelidze A, Stine OC. 2005. Multilocus sequence typing for studying genetic relationships among Yersinia species. J. Clin. Microbiol. 43:2674–2684. 10.1128/JCM.43.6.2674-2684.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laukkanen-Ninios R, Didelot X, Jolley KA, Morelli G, Sangal V, Kristo P, Brehony C, Imori PF, Fukushima H, Siitonen A, Tseneva G, Voskressenskaya E, Falcao JP, Korkeala H, Maiden MC, Mazzoni C, Carniel E, Skurnik M, Achtman M. 2011. Population structure of the Yersinia pseudotuberculosis complex according to multilocus sequence typing. Environ. Microbiol. 13:3114–3127. 10.1111/j.1462-2920.2011.02588.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96:14043–14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogdanovich T, Carniel E, Fukushima H, Skurnik M. 2003. Use of O-antigen gene cluster-specific PCRs for the identification and O-genotyping of Yersinia pseudotuberculosis and Yersinia pestis. J. Clin. Microbiol. 41:5103–5112. 10.1128/JCM.41.11.5103-5112.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukushima H, Matsuda Y, Seki R, Tsubokura M, Takeda N, Shubin FN, Paik IK, Zheng XB. 2001. Geographical heterogeneity between far eastern and western countries in prevalence of the virulence plasmid, the superantigen Yersinia pseudotuberculosis-derived mitogen, and the high-pathogenicity island among Yersinia pseudotuberculosis strains. J. Clin. Microbiol. 39:3541–3547. 10.1128/JCM.39.10.3541-3547.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sprague LD, Scholz HC, Amann S, Busse HJ, Neubauer H. 2008. Yersinia similis sp. nov. Int. J. Syst. Evol. Microbiol. 58:952–958. 10.1099/ijs.0.65417-0 [DOI] [PubMed] [Google Scholar]
- 24.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518–1530. 10.1128/JB.186.5.1518-1530.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Hai R, Song Z, Xia L, Liang Y, Cai H, Liang Y, Shen X, Zhang E, Xu J, Yu D, Yu XJ. 2009. Spatial variation of Yersinia pestis from Yunnan Province of China. Am. J. Trop. Med. Hyg. 81:714. 10.4269/ajtmh.2009.09-0174 [DOI] [PubMed] [Google Scholar]
- 26.Garzetti D, Bouabe H, Heesemann J, Rakin A. 2012. Tracing genomic variations in two highly virulent Yersinia enterocolitica strains with unequal ability to compete for host colonization. BMC Genomics 13:467. 10.1186/1471-2164-13-467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bercovier H, Mollaret H, Alonso JM, Brault J, Fanning GR, Steigerwalt AG, Brenner DJ. 1980. Intra- and interspecies relatedness of Yersinia pestis by DNA hybridization and its relationship to Yersinia pseudotuberculosis. Curr. Microbiol. 4:225–229 [Google Scholar]
- 28.Wang X, Zhou D, Qin L, Dai E, Zhang J, Han Y, Guo Z, Song Y, Du Z, Wang J, Wang J, Yang R. 2006. Genomic comparison of Yersinia pestis and Yersinia pseudotuberculosis by combination of suppression subtractive hybridization and DNA microarray. Arch. Microbiol. 186:151–159. 10.1007/s00203-006-0129-1 [DOI] [PubMed] [Google Scholar]
- 29.Trebesius K, Harmsen D, Rakin A, Schmelz J, Heesemann J. 1998. Development of rRNA-targeted PCR and in situ hybridization with fluorescently labelled oligonucleotides for detection of Yersinia species. J. Clin. Microbiol. 36:2557–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wren BW. 2003. The yersiniae–a model genus to study the rapid evolution of bacterial pathogens. Nat. Rev. Microbiol. 1:55–64. 10.1038/nrmicro730 [DOI] [PubMed] [Google Scholar]
- 31.Thomson NR, Howard S, Wren BW, Holden MT, Crossman L, Challis GL, Churcher C, Mungall K, Brooks K, Chillingworth T, Feltwell T, Abdellah Z, Hauser H, Jagels K, Maddison M, Moule S, Sanders M, Whitehead S, Quail MA, Dougan G, Parkhill J, Prentice MB. 2006. The complete genome sequence and comparative genome analysis of the high pathogenicity Yersinia enterocolitica strain 8081. PLoS Genet. 2:e206. 10.1371/journal.pgen.0020206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramamurthy T, Yoshino KI, Huang X, Balakrish Nair G, Carniel E, Maruyama T, Fukushima H, Takeda T. 1997. The novel heat-stable enterotoxin subtype gene (ystB) of Yersinia enterocolitica: nucleotide sequence and distribution of the yst genes. Microb. Pathog. 23:189–200 [DOI] [PubMed] [Google Scholar]
- 33.Tennant SM, Grant TH, Robins-Browne RM. 2003. Pathogenicity of Yersinia enterocolitica biotype 1A. FEMS Immunol. Med. Microbiol. 38:127–137. 10.1016/S0928-8244(03)00180-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.