

Abstract
Downregulation of specific transcripts is one of the mechanisms utilized by eukaryotic checkpoint systems to prevent cell cycle progression. Here we identified and explored such a mechanism in the yeast Saccharomyces cerevisiae. It involves the Mec1-Rad53 kinase cascade, which attenuates G2/M-specific gene transcription upon genotoxic stress. This inhibition is achieved via multiple Rad53-dependent inhibitory phosphorylations on the transcriptional activator Ndd1 that prevent its chromatin recruitment via interactions with the forkhead factor Fkh2. Relevant modification sites on Ndd1 were identified by mass spectrometry, and corresponding alanine substitutions were able to suppress a methyl methanesulfonate-induced block in Ndd1 chromatin recruitment. Whereas effective suppression by these Ndd1 mutants is achieved for DNA damage, this is not the case under replication stress conditions, suggesting that additional mechanisms must operate under such conditions. We propose that budding yeast cells prevent the normal transcription of G2/M-specific genes upon genotoxic stress to precisely coordinate the timing of mitotic and postmitotic events with respect to S phase.
INTRODUCTION
Different key events in the cell cycle, such as DNA replication, mitosis, and cytokinesis, can be considered independent cycling processes which are orchestrated and synchronized by checkpoints to ensure that the genetic information from a mother cell is faithfully inherited by the daughter cells (1). Various checkpoints utilize different mechanisms to halt cell cycle progression when proper completion of the previous event is compromised. One such mechanism is to switch off the transcription of genes necessary to progress through later cell cycle stages. This is exemplified by the inhibition of G2/M phase-specific gene transcription via the morphogenesis checkpoint and the cell wall integrity pathway in the yeast Saccharomyces cerevisiae (2, 3). G2/M cluster genes include the mitotic cyclin genes CLB1 and CLB2, Polo-like kinase gene CDC5, the anaphase promoting complex (APC) factor gene CDC20, and several other candidates that are involved in the cell cycle functions through M and early G1 phases (4). Transcription of these genes is regulated by three transcription factors, Mcm1, Fkh2, and Ndd1, and a histone deacetylase complex, Sin3-Rpd3. Mcm1 and Fkh2 can be found at the promoters throughout the cell cycle, in which Fkh2 acts as an adaptor protein recruiting either the repressor Sin3-Rpd3 during G1 phase (5) or the activator Ndd1 (6, 7) during S, G2, and M phases. Removal of Sin3 from the promoters after Start leads to derepression of G2/M genes. It is noteworthy that the derepression step is sufficient to initiate low levels of transcription that can eventually lead to substantial levels of the respective mRNAs (5). However, full transcriptional activation is achieved only when Ndd1 is recruited (7). Both Fkh2 and Ndd1 are subjected to Cdk-mediated serine and threonine phosphorylations (S/T-Ps) (8, 9), which are functionally important. The interaction between the forkhead-associated (FHA) domain of Fkh2 and multiple phosphorylated residues on Ndd1 is essential for the latter to be recruited to G2/M promoters (10). Additionally, direct phosphorylation of Ndd1 by Cdc5 has been postulated to increase activation of G2/M promoters (11), whereas Pkc1-dependent phosphorylation was claimed to inhibit Ndd1 function in order to coordinate the timing of mitotic gene transcription with the cell cycle (12).
Here we investigated whether activation of genotoxic stress-dependent checkpoint signals during S phase prevent G2/M transcription as well. Our study was instigated by observations made in Schizosaccharomyces pombe and mammalian cells, where transcription of genes promoting mitosis is known to be downregulated upon genotoxic stress treatment (13, 14). Furthermore, global transcriptome analysis in the budding yeast revealed that methyl methanesulfonate (MMS)-treated mec1 and rad53 cells contain higher levels of G2/M-specific gene transcripts than wild-type cells (15, 16). Nevertheless, until very recently, no further experimental evidence that would confirm these observations as well as the role of Mec1 or its downstream kinases, such as Rad53/Chk1, in the regulation of G2/M transcription came forward.
Induction of genotoxic stress by hydroxyurea (HU) activates the replication checkpoint, whereas MMS is known to activate both the replication checkpoint and the DNA damage-induced G2/M checkpoint in a concentration-dependent manner. Once activated, these checkpoints block cell cycle progression in S phase or G2/M phase to provide sufficient time for successful completion of replication and DNA damage repair. Mec1, a phosphoinositide 3-kinase-like kinase family member and a homologue of mammalian ATM/ATR kinases, is the principal modulator of these checkpoints. When DNA damage or stalled replication forks are detected in a cell, Mec1 is activated by different mechanisms, depending on the cell cycle phase in which the signal is received (17). Two additional protein kinases, Rad53 and Chk1, serve as the most important downstream effectors of Mec1, as they are activated through a signaling mechanism mediated by Rad9 and Mrc1 (18, 19). These effector kinases regulate many downstream targets, such as the protein kinase Dun1, to facilitate DNA repair processes and inhibit cell cycle progression. Anaphase inhibitor Pds1/securin is perhaps the most important downstream target whose degradation is prevented by Rad53 and Chk1 through various mechanisms to inhibit anaphase entry (20, 21). Additionally, Mec1-Rad53 prevents premature chromosome segregation in HU-treated cells by regulating the spindle dynamics (22).
In this study, we have identified and characterized an additional mechanism by which the cell cycle progression is prevented in HU- or MMS-treated cells. In this case, activation of genotoxic stress-dependent checkpoint pathways suppresses G2/M-specific gene transcription by obstructing the recruitment of Ndd1 to G2/M promoters. This inhibition is primarily achieved by preventing the interaction of Ndd1 with the FHA domain of Fkh2. Our results strongly suggest that in MMS-treated cells, Mec1-Rad53 dependent modification of Ndd1 is mainly responsible for the inhibition of Ndd1 function. In HU-treated cells, however, additional Mec1-Rad53-independent pathways might contribute to the blockade of Ndd1 chromatin recruitment. Finally, we present data that hint at the physiological importance of this regulatory phenomenon.
MATERIALS AND METHODS
The S. cerevisiae strains used in this study, unless mentioned otherwise, were haploid and congenic to strain W303. A list of the strains used in this study is provided in Table 1. Original sml1, sml1 mec1, and sm11 rad53 strains were obtained from Maria Pia Longhese (23). rad9 mrc1AQ strains were provided by Stephen Elledge (24). Tagging of genes at an endogenous locus was carried out by transformation of PCR-amplified cassettes as described previously (5). A Rad53-tandem affinity purification (TAP)-tagged strain was taken from the yeast EUROSCARF collection. Integration of NDD1, wild type or mutant, at the LEU locus was done by transforming the respective plasmids after linearization with the ClaI restriction enzyme. An NDD1-HTBeaq strain was obtained by transforming the PCR-amplified HTBeaq cassette as described previously (25). In all cases, positive clones were confirmed by PCR-based analysis and/or Western blotting. Strains with multiple mutations were generated by standard genetic analysis. Standard molecular biology and molecular genetics techniques were used to construct plasmids. NDD1 point mutants were generated by using a QuikChange site-directed mutagenesis kit (Stratagene) or by whole-gene synthesis (Gene Art; Life Technologies). The circular minichromosome containing an ADE2 gene (pSKY393) used in chromosome loss studies was derived from p3 (26), a kind gift from Franz Klein. A 4.4-kb region containing the RAD50 gene in p3 was removed by ClaI digestion, and the residual 13-kb fragment was religated to generate pSKY393. A list of the plasmids used in this study is provided in Table 2.
TABLE 1.
Strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| MK155 | MATa NDD1-HA6::HIS3 | 6 |
| JV323 | MATa ndd1::KanMX p[GAL1-10 NDD1 CEN URA3] | 5 |
| JV717 | MATa FKH2-HA6::TRP1 | This study |
| JV753 | MATa sml1::kanMX mec1::HIS3 | 23 |
| JV754 | MATa sml1::kanMX | 23 |
| SKY101 | W303 MATa ade2-1 trp1-1 can1-100 leu2-3 his3-11ura3-52 ssd1 | 5 |
| SKY170 | MATa sml1::kanMX mec1::HIS3 NDD1-HTBeaq::hphMX | 23 and this study |
| SKY174 | MATa sml1::kanMX NDD1-HTBeaq::hphMX | 23 and this study |
| SKY260 | MATa LEU2::NDD1-HA6 | This study |
| SKY270 | MATa ndd1::KanMX p[GAL1-10 NDD1 CEN URA3] FKH2-MYC18::HIS3 | This study |
| SKY276 | MATa sml1::kanMX LEU2::NDD1-HA | 23 and this study |
| SKY277 | MATa sml1::kanMX mec1::HIS3 LEU2::NDD1-HA | 23 and this study |
| SKY293 | MATa LEU2::p[GAL1-10 NDD1-GFP] | This study |
| SKY301 | MATa rad9Δ::HIS3 mrc1Δ-2::HIS3 URA3-MRC1 GAP-RNR1-TRP1 LEU2::NDD1-HA | 24 and this study |
| SKY303 | MATa rad9Δ::HIS3 mrc1Δ-2::HIS3 URA3-mrc1AQ GAP-RNR1-TRP1 LEU2::NDD1-HA | 24 and this study |
| SKY324 | MATa sml1::kanMX rad53::HIS3 LEU2::NDD1-HA | 23 and this study |
| SKY325 | MATa sml1::kanMX rad53::HIS3 NDD1-HTBeaq::hphMX | 23 and this study |
| SKY361a | MATa RAD53-TAP::HIS | EUROSCARF collection |
| SKY362a | MATa dun1::kanMX | EUROSCARF collection |
| SKY363a | BY4741 MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | EUROSCARF collection |
| SKY364 | MATa ndd1::KanMX LEU2::NDD1-HA | This study |
| SKY365 | MATa ndd1::KanMX LEU2::NDD1-CD-10A-HA | This study |
Isogenic to BY4741.
TABLE 2.
Plasmids used in this study
| Plasmid | Details | Reference or source |
|---|---|---|
| pAS40 | YCplac33 NDD1-HA6 native promoter | 5 |
| pJV287 | pGBT9 GAL4DBD-fkh21-306 | 5 |
| pSKY285 | YIplac128 NDD1-GFP GAL1-10 promoter | This study |
| pSKY307 | YIplac128 NDD1-HA native promoter | This study |
| pSKY315 | YIplac128 NDD1-CD-8A–HA (S21/25/261/286/384/454/527/530A) native promoter | This study |
| pSKY324 | YIplac128 NDD1-CD-10A–HA (S21/25/261/286/384/437/440/454/527/530/525A) native promoter | This study |
| pSKY339 | YIplac128 NDD1-CD-S437/440A–HA native promoter | This study |
| pSKY379 | YIplac204 NDD1-HA GAL1-10 promoter | This study |
| pSKY381 | YIplac204 NDD1-CD-10A–HA GAL1-10 promoter | This study |
| pSKY393 | YCp50 ADE2 | 26 and this study |
Yeast media and reagents.
Cells were grown in standard yeast extract-peptone (YEP) or selective medium supplemented with 2% glucose or 1% raffinose–1% galactose (Raf-Gal). In all experiments, unless specifically mentioned, 104 mM hydroxyurea (Sigma) and 0.015% methyl methanesulfonate (Sigma) were used.
Cell synchronization, FACS, and fluorescence microscopy.
Cell synchronization in G1 using α-factor was performed as described previously (5). All conditional GAL1-10 NDD1 strains were synchronized in G1 for 2 h 5 min in YEP-Raf-Gal. When intended, 1% glucose was added to the culture and α-factor synchronization was prolonged for an additional 50 min in order to suppress NDD1 gene transcription. Subsequently, cells were washed and released in YEP-Raf-Gal or YEP-glucose. Cells for fluorescence-activated cell sorter (FACS) analysis were fixed in 70% ethanol, stained with propidium iodide, and analyzed with a BD FACSCalibur flow cytometer. For fluorescence microscope analysis, 1 ml of culture was fixed in ethanol, followed by nuclear staining with Hoechst 33342 (Molecular Probes) in 1× phosphate-buffered saline (PBS). Fluorescence microscopy studies were performed using a personal Delta Vision epifluorescence microscope from Applied Precision Inc. Data were analyzed using ImageJ software. More detailed protocols for FACS and fluorescence microscopy are provided in the procedures presented in the supplemental material.
ChIP and coimmunoprecipitation.
The protocols used for chromatin immunoprecipitation (ChIP) and multiplex PCR as well as the antibodies used for ChIP have been published previously (5). Quantitative PCR (qPCR) analysis was performed with primers amplifying the promoter regions of the CLB2, SWI5, ASE1, CDC5, and CDC20 genes and the GAL1-10 promoter region. Primers amplifying the ALD3 and FUS1 promoter regions as well as a telomeric region (TEL1) were used as internal immunoprecipitation (IP)-nonspecific DNA controls. (All sequences and detailed ChIP protocols are available upon request.) qPCR was done in triplicate for every ChIP sample, and individual experiments were repeated multiple times, as indicated in the appropriate figure legends. In all ChIP experiments, nonspecific signals were constant and evenly distributed throughout the experiment. Data are presented as the fold change over the value for the respective nonspecific DNA control. For coimmunoprecipitation, cells were washed twice in ice-cold Tris-buffered saline buffer and lysed in a 10 mM Tris, pH 8, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 10 mM Na pyrophosphate lysis buffer supplemented with protease inhibitors. The lysates were incubated overnight at 4°C with 40 μl pan-mouse IgG Dynabeads. Beads were preequilibrated at 4°C with 0.5% bovine serum albumin–PBS buffer for at least 1 h prior to use. After IP, beads were washed four times for 4 min each time with lysis buffer, suspended in 1× Laemmli buffer, and separated on SDS-polyacrylamide gels.
Northern blot and Western blot analyses.
Northern blot analysis was performed as described elsewhere (5). For Western blot analysis, standard SDS-PAGE blotting methods were used. Cell extracts were made in 8 M urea–20 mM Tris, pH 8–300 mM NaCl–20 mM Na-PO4–0.5% NP-40 buffer. To detect hemagglutinin (HA)-tagged Ndd1, anti-HA antibody (supernatant of 12CA5 hybridoma cells) was used (1:1,000 dilution). Anti-alpha-tubulin antibody (Ray Biotech Inc.) was used to detect Tub1. Anti-protein A antibody (Sigma-Aldrich) was used to detect TAP-tagged proteins.
Viability and chromosome loss assay.
HU and MMS sensitivity assays were performed in wild-type cells expressing NDD1-WT(pSKY379) or NDD1-CD-10A(pSKY381) under the control of the GAL1-10 promoter. Cells were grown to an optical density at 600 nm of 1 in yeast extract-peptone-dextrose (YEPD). Tenfold serial dilutions of these cultures were spotted on YEP-Raf-Gal plates containing either 50 mM HU or 0.005% MMS and on plain medium plates as a control. Plates were incubated at 30°C and photographed after 3 days. The basic methodology followed to perform the chromosome loss assay is described elsewhere (27). For the chromosome loss assay, ndd1 cells expressing either Ndd1-WT (SKY364) or Ndd1-CD-10A (SKY365) from an endogenous promoter were transformed with a centromeric minichromosome URA3 plasmid containing the ADE2 gene (pSKY393). Cells were grown for at least 5 generations in medium without uracil and plated on respective plates containing either 50 mM HU or 0.005% MMS and on YEPD plates as a control. Loss of the minichromosome leads to the formation of pink sectors in white colonies. At 5 days after incubation at 30°C, colonies with pink sectors were scored, and the percentage of minichromosome loss was calculated by the formula (number of colonies with pink sectors/total number of colonies) × 100.
Ndd1-HTBeaq purification and MS analysis.
For all the experiments involving mass spectrometry (MS), cultures were synchronized in G1 as mentioned above and released into the respective conditions. Cells were harvested by filtration and deep-frozen in liquid N2. The downstream protocol used to purify Ndd1-HTBeaq is described elsewhere (25). Proteins were digested with trypsin or chymotrypsin directly on the beads, and peptides were separated by reversed-phase chromatography and analyzed in a hybrid mass spectrometer (LTQ-Orbitrap Velos; Thermo Fisher Scientific). Details are described in the supplemental material. To identify the phosphorylation sites that are differentially regulated under certain conditions, we manually integrated the peak areas of the corresponding peptide signals over the elution time in the full scan and put them in relation to the peak area of the unphosphorylated counterpeptides.
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium database (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (28) with the data set identifier PXD000405.
RESULTS
Genotoxic stress affects transcription of G2/M-specific genes.
Our initial aim was to corroborate observations that implicate genotoxic stress in the regulation of G2/M-specific transcription. To reduce the significant noise and variability observed in asynchronous cultures, we used cells synchronized in G1 in almost all experiments. For synchronization, we exclusively used arrest by pheromone (α-factor), as this treatment leads to a short and highly synchronous release into S phase of both mother and daughter cells. From samples isolated at the time points indicated below, we analyzed the RNA levels of G2/M genes such as CLB2 and SWI5 by Northern blotting. Transcription of these genes was found to be downregulated upon HU or MMS treatment compared to the transcription in untreated cells (Fig. 1A; see Fig. S1A in the supplemental material). FACS analysis of the samples indicated that the cell cycle progression was inhibited in S phase upon HU treatment and G2/M transition upon MMS treatment (Fig. 1B). Significantly, the effect of MMS on G2/M transcription was dosage dependent; increasing the MMS concentration from 0.0075% to 0.015% increased the degree of suppression of CLB2 transcription (see Fig. S1B in the supplemental material). In the subsequent studies, we continued to use 0.015% MMS, which induced a G2/M checkpoint without causing G1 or intra-S-phase arrest. Taken together, our data confirmed the downregulation of G2/M-specific gene expression upon different genotoxic stress treatments.
FIG 1.

Genotoxic stress inhibits G2/M-specific gene transcription by preventing Ndd1 promoter recruitment. (A) Induction of genotoxic stress by HU or MMS downregulates G2/M-specific gene transcription. An NDD1-HA strain (MK155) was synchronized in G1 using α-factor and released into YEPD alone and YEPD with 0.1 M HU or 0.0075% MMS. CLB2 transcription was analyzed using Northern blot analysis from samples isolated at the indicated time points. Error bars indicate standard deviations in three independent experiments. fold to, fold expression of CLB2 relative to that of CMD1. (B) DNA content of cells from the experiment whose results are presented in panel A analyzed by FACS. (C) Recruitment of Ndd1 to G2/M promoters is prevented by genotoxic stress. Ndd1 recruitment to the CLB2 promoter was examined by ChIP, followed by qPCR analysis from samples for which the results are described in panel A. Error bars indicate standard deviations in three independent experiments. (D and E) The Ndd1 protein is abundant and localized to the nucleus in HU- and MMS-treated cells. (D) MK155 cells synchronized in G1 were released into YEPD with or without HU or MMS. The Ndd1-HA levels in samples isolated at the indicated time points were detected by Western blotting. Western blotting for alpha-tubulin was performed as a loading control. (E) A wild-type strain expressing NDD1-GFP from the GAL1-10 promoter (SKY293) was synchronized in G1 and released into YEP with or without HU or MMS. NDD1-GFP expression was induced by galactose addition 30 min prior to release. Cells were fixed at 60 min postrelease, and nuclei were stained with Hoechst. Ndd1-green fluorescent protein (GFP) localization was examined by fluorescence microscopy. DIC, differential inference contrast.
Ndd1 recruitment to G2/M promoters is prevented by genotoxic stress.
Under physiological conditions, recruitment of the transcriptional activator Ndd1 to G2/M gene promoters is known to upregulate their transcription. We therefore suspected that genotoxic stress might inhibit the function of Ndd1, either by regulating its recruitment to the chromatin or by affecting the protein stability and nuclear localization. To explore the mechanistic details, we initially examined the G2/M promoter occupation rates of Ndd1 using ChIP, followed by qPCR analysis. Our Ndd1 ChIP data confirmed that the recruitment of Ndd1 to various G2/M-specific gene promoters, such as CLB2 (Fig. 1C) and CDC5, CDC20, and ASE1 (see Fig. S1C to E in the supplemental material), was inhibited upon HU or MMS treatment compared to that for untreated cells. Moreover, speculating that the block in Ndd1 recruitment might be a general phenomenon associated with genotoxic stresses, we investigated whether shifting cdc13-1 cells to nonpermissive temperatures produces phenotypes similar to those produced by HU or MMS treatment. At high temperature, cdc13-1 cells are known to arrest in G2/M phase due to telomere damage. We found that G2/M transcription and Ndd1 recruitment to the G2/M promoters were inhibited when cdc13-1 cells were grown at a nonpermissive temperature (see Fig. S1F in the supplemental material). Ndd1-HA Western blot analysis showed that Ndd1 is maintained at high steady-state levels in both HU- and MMS-treated cells (Fig. 1D), even though NDD1 transcripts were slightly reduced in number under these conditions (see Fig. S2 in the supplemental material). Finally, Ndd1 localization studies performed using cells expressing GAL1-10 promoter-driven NDD1-GFP indicated that the intracellular distribution of Ndd1 remained unaffected by HU or MMS treatment (Fig. 1E). Taken together, our results suggest that genotoxic stress prevents the recruitment of Ndd1 to G2/M promoters by mechanisms that do not depend on either lower protein stability or increased nuclear export of Ndd1.
Genotoxic stress indirectly affects Fkh2 occupation at G2/M promoters.
Since promoter occupation of Ndd1 is dependent on Fkh2, we also tested the occupation rates of Fkh2 during genotoxic stress conditions. Although a Fkh2 binding signal could be observed at G2/M promoters throughout the cell cycle, one could measure a substantial increase during S-M phases at time points that coincided with Ndd1 binding (Fig. 2A and B; see Fig. S3B in the supplemental material). This increase in Fkh2 binding, however, could not be found in cells under genotoxic stress (Fig. 2A and B; see Fig. S3B in the supplemental material). To test whether this effect was due to loss of Ndd1 interaction or due to genotoxic signals, we examined Fkh2 abundance at the CLB2 promoter in Ndd1-depleted cells. An FKH2-MYC strain containing the only copy of NDD1 under the control of the GAL1-10 promoter was synchronized in G1 in galactose medium and released into galactose or glucose medium. Depletion of NDD1-mRNA was controlled by Northern blot analysis (see Fig. S3C in the supplemental material). ChIP data clearly indicated that Fkh2 binding to the CLB2 promoter was also significantly reduced in the cells in which NDD1 expression was switched off (Fig. 2C and D). Our results therefore rather support the notion that HU or MMS treatment only indirectly affects the Fkh2 recruitment signal by preventing cooperation between the two transcription factors.
FIG 2.

Genotoxic stress indirectly affects Fkh2 recruitment kinetics by preventing Ndd1 interaction with the FHA domain of Fkh2. (A to D) Fkh2 recruitment to G2/M promoters is indirectly affected by genotoxic stress. (A) An FKH2-HA strain (JV717) was synchronized in G1 and released into YEPD with or without HU or MMS. Fkh2 recruitment to the CLB2 promoter was examined by ChIP, followed by qPCR analysis. (B) Cell cycle analysis of the samples from the experiment whose results are presented in panel A by FACS. (C) A conditional GAL promoter-driven NDD1 strain (GALpr-NDD1) expressing FKH2-MYC (SKY270) was synchronized in G1 and released into YEP-Raf-Gal (Ndd1) or YEP-glucose (ndd1). Fkh2 recruitment to the CLB2 promoter was examined by ChIP, followed by qPCR analysis. (D) Cell cycle analysis of the samples whose results are presented in panel C by FACS. (E and F) Interaction of Ndd1 with the FHA domain of Fkh2 is inhibited by genotoxic stress. (E) Model describing important domains in full-length Fkh2 (i) and the GAL4DBD-Fkh2 FHA domain fusion construct (Gal4DBD-fkh21-306) (ii) and a model showing fusion protein-mediated recruitment of Ndd1 to the GAL1-10 promoter (iii). (F) An NDD1-HA strain transformed with GAL4DBD-fkh21-306 (pJV287) was synchronized in G1 and released into YEPD with or without HU or MMS. Ndd1 recruitment to the GAL1-10 promoter was examined by ChIP, followed by qPCR analysis. The results of cell cycle analysis of the samples by FACS are shown to the right. In all panels, error bars indicate standard deviations in at least two independent experiments.
The Sin3 deacetylase complex normally binds to the N terminus of Fkh2 during G1. A possible role of Sin3 in inhibiting the recruitment of Ndd1 by competing for interaction surfaces at Fkh2 was also excluded, as Sin3 was removed from the G2/M promoters in HU-treated cells with kinetics similar to those in untreated cells (see Fig. S2A in the supplemental material). To test whether promoter context and perhaps additional Mcm1/Fkh2 binding factors provided important parameters for Ndd1 regulation by genotoxic stresses, we used a chimera consisting of the Gal4 DNA-binding domain (Gal4DBD) and the N terminus of Fkh2 from amino acids 1 to 306, including the forkhead-associated (FHA) domain (Gal4DBD-fkh21-306), to monitor Ndd1 recruitment to the GAL1-10 locus. It has previously been observed that such a fusion construct consisting of Gal4DBD-fkh21-306 is sufficient to recruit Ndd1 to GAL1-10 promoters (Fig. 2E) (5, 10). NDD1-HA cells expressing Gal4DBD-fkh21-306 were synchronized in G1 and released into YEPD with or without HU or MMS. Interestingly, Ndd1 recruitment to the GAL1-10 promoter was still inhibited upon genotoxic stress (Fig. 2F). The results indicate that genotoxic stresses can inhibit the recruitment of Ndd1 to chromatin by preventing its interaction with the FHA domain of Fkh2 independently of a natural G2/M promoter context.
A Mec1-Rad53-dependent signal inhibits the recruitment of Ndd1 to G2/M promoters upon MMS treatment.
The Mec1 kinase cascade, including Rad9 plus Mrc1 as scaffolds and Rad53 plus Chk1 and Dun1 as downstream kinases, is known to regulate DNA repair processes and inhibition of cell cycle progression as the main conduit of the DNA damage-induced G2/M checkpoint. Each of these kinases was therefore an obvious candidate for orchestrating the inhibition of G2/M-specific gene transcription upon MMS treatment. If it is indeed important, deletion of these genes should relieve the inhibitory effect of genotoxic stress on G2/M-specific transcription. To test this assumption, we analyzed CLB2 mRNA levels and Ndd1 recruitment kinetics in the respective kinase mutant strains. Since a deletion of SML1 is essential for the viability of mec1 and rad53 cells, we used a sml1 strain as the wild-type equivalent. We observed that deletions of MEC1 or RAD53 in this background significantly relieved the inhibition of CLB2 transcription and Ndd1 recruitment to the CLB2 promoter upon MMS treatment (Fig. 3A and B; for the results of FACS analysis, see Fig. S4 in the supplemental material). A much less significant increase in transcription, however, was observed in a dun1 mutant (Fig. 3C), implying that the inhibition of transcription of G2/M-specific genes upon MMS treatment is largely carried out by a Mec1-Rad53-dependent mechanism.
FIG 3.

The inhibitory effect of MMS on G2/M gene transcription and Ndd1 recruitment is dependent on Mec1 and Rad53. (A) Deletion of MEC1 or RAD53 abrogates the inhibitory effect on mitotic gene transcription by MMS treatment. Northern blot analysis of CLB2 mRNA isolated from sml1 (JV754), sml1 mec1 (JV753), and sml1 rad53 (SKY324) cells released into YEPD with or without MMS after G1 arrest. (B) Ndd1 recruitment to G2/M promoters is reestablished in MMS-treated mec1 or rad53 mutants. Ndd1-HA ChIP followed by qPCR analysis targeted to the CLB2 promoter was performed with the lysates of sml1 (SKY276), sml1 mec1 (SKY277), and sml1 rad53 (SKY324) cells released into YEPD with or without MMS after G1 arrest. (C) DUN1 deletion has only a minor effect on G2/M gene transcription in MMS-treated cells. Northern blotting was used to analyze CLB2 mRNA isolated from wild-type (SKY363) and dun1 (SKY362) cells released into YEPD with or without MMS after G1 arrest. In all panels, error bars indicate standard deviations in at least three independent experiments.
Multiple signals affect Ndd1 recruitment during HU exposure.
Study of the behavior of Ndd1 in HU-treated mec1 and rad53 cells gave the impression that the regulatory mechanism in this case is more complex. We found that under these conditions a deletion of RAD53 could not suppress the transcriptional reduction as effectively as MMS exposure conditions could. Although a mec1 strain would give higher values of CLB2 RNA production than the rad53 strain, it still did not restore the levels to those obtained with no treatment (Fig. 4A). The corresponding measurements for Ndd1 chromatin recruitment indicated similar deficiencies in the suppression effects of the mutants (Fig. 4B; for the results of FACS analysis, see Fig. S4 in the supplemental material). To investigate any possible role of Tel1 and Chk1 in providing a bypass to the Mec1-Rad53 signals, we examined the effect of HU in mutants affecting the scaffolding elements Rad9 and Mrc1. For this purpose, we measured the CLB2 promoter occupancy rates of Ndd1 in an HU-treated rad9 mrc1 strain expressing either wild-type MRC1 or a checkpoint-defective MRC1 allele, mrc1AQ (24). Our results indicated that deletion of RAD9 with additional inactivation of Mrc1 could not suppress the HU-induced inhibition of Ndd1 recruitment (Fig. 4C). Overall, these results therefore support the notion that additional Mec1-independent signals contribute to the downregulation of G2/M-specific genes upon HU treatment.
FIG 4.

HU-induced inhibition of G2/M-specific transcription is only partially dependent on the Mec1-Rad53 pathway. (A) Deletion of MEC1 could partially suppress HU-induced inhibition of mitotic gene transcription, whereas RAD53 deletion had only a lesser effect. Northern blotting was used to analyze CLB2 mRNA isolated from sml1 (JV754), sml1 mec1 (JV753), and sml1 rad53 (SKY324) cells released into YEPD with or without HU after G1 arrest. (B) The kinetics of Ndd1 recruitment to G2/M promoters are partially reestablished in HU-treated mec1 cells, whereas in the rad53 mutant, Ndd1 remains absent from the promoter. Ndd1-HA ChIP followed by qPCR analysis targeted to the CLB2 promoter was performed with the lysates of sml1 (SKY276), sml1 mec1 (SKY277), and sml1 rad53 (SKY324) cells released into YEPD with or without HU after G1 arrest. (C) Deletion of either RAD9 or MRC1 could not suppress HU-mediated inhibition of Ndd1 recruitment to G2/M promoters. Ndd1-HA ChIP followed by conventional multiplex PCR analysis was performed with the lysates of mrc1 rad9 cells expressing either MRC1 (SKY301) or mrc1AQ (SKY303) released into YEPD with or without HU after G1 arrest. ALD3 and FUS1 promoter-specific primers were used as negative controls. In all panels, error bars indicate standard deviations in at least three independent experiments. WCE, whole-cell extract.
Identification of genotoxic stress-induced phosphorylations in Ndd1 by MS.
There are at least two possible mechanisms through which the negative regulation of Ndd1 could occur in a phosphorylation-dependent manner. Checkpoint kinases might prevent activating phosphorylation events on Ndd1 S/T-P motifs by downregulating Cdk activity. Alternatively, they could promote the phosphorylation of other sites which inhibit Ndd1 function. As mentioned previously, Cdk-dependent S/T-P phosphorylations are essential for the recruitment of Ndd1 to G2/M promoters (8, 9). To distinguish between these explanations, we performed an MS analysis of Ndd1 purified from sml1, sml1 mec1, and sm11 rad53 strains grown in YEPD with or without HU or MMS. To avoid extract-dependent modifications, purification of HTBeaq-tagged Ndd1 was performed by tandem affinity purification under denaturing conditions as described previously (25). Complete MS data for Ndd1 purified from various strains and conditions is provided in Table S1 in the supplemental material. As shown in the phosphorylation site map of Ndd1 (Fig. 5A), we identified 43 phosphorylation sites with a site probability of more than 75% within 89% sequence coverage. Sites which were also identified in previous studies (8, 11, 12, 16) were differentially represented. Of these 43 high-confidence sites, 14 were S/T-P sites and 29 were non-S/T-P sites. To identify the phosphorylation sites that were differentially regulated under certain conditions, we manually quantified them by evaluating and comparing the integrated peak areas of the corresponding peptide signals. Such a calculation for different S/T-P sites (serines 157, 168, 254, and 357 and threonines 179, 183, 265, 277, and 332) revealed that the level of phosphorylation of all sites, except S157, in MMS-treated sml1 cells was in the same range as that in untreated cells (Fig. 5B). In contrast, S157 was downregulated severalfold. In rad53 cells treated with MMS, S157 phosphorylation reverted back to normal levels. Furthermore, phosphorylation of S157, S254, and S357 was significantly downregulated in HU-treated cells. Interestingly, phosphorylation of S157 and S254 was still downregulated in HU-treated rad53 or mec1 cells, hinting at differences due to the stress regimen. More importantly, our data show that phosphorylation of S/T-P sites is not significantly influenced by the MMS treatment, suggesting that activating modifications are still in full operation. However, this cannot be said with respect to HU treatment, as we observed downregulation in some of them. Finally, quantification of several non-S/T-P sites (serines 25, 285, 286, 288, 384, 437, 440, 450, 454, 525, 527, and 530 and threonine 452) indicated that the phosphorylation of these sites was upregulated in HU- or MMS-treated sml1 cells (Fig. 5C). Not surprisingly, these phosphorylations were downregulated or missing in rad53 or mec1 cells treated in the same way. These measurements clearly show that genotoxic stress induces upregulation of multiple Mec1- and Rad53-dependent phosphorylations on Ndd1 independently of the status of activating modifications.
FIG 5.

Ndd1 is phosphorylated in a Mec1-Rad53-dependent manner upon genotoxic stress. (A) Ndd1 is a highly phosphorylated protein. A phosphorylation site map of Ndd1 derived by fusing the data from different MS runs obtained with Ndd1-HTBeaq purified from various strains and conditions is shown. Big and bold letters, phosphorylation sites identified in our studies; underlines, sites identified in previous studies (8, 11, 12, 16); green, serine and threonine phosphorylation sites; red, sites upregulated in HU- or MMS-treated sml1 cells; brown, sites whose phosphorylation was not altered significantly; blue, sites for which quantification data are not available. Sequence coverage is highlighted in a light blue background. (B and C) Relative quantification ratios of different phosphorylation sites (S/T-P and non-S/T-P) identified on Ndd1. Fold ratios of phosphorylated peptides over unphosphorylated peptides are provided (the value for am unphosphorylated peptide is set to 1).
To further substantiate a more central role of Rad53 in Ndd1 regulation, we tested whether one could obtain biochemical evidence for their interaction by coprecipitation experiments. A strain coexpressing Rad53-TAP and Ndd1-HA was synchronized and released into normal or MMS- or HU-containing medium. Extracts were prepared 60 min after release, and the presence of Ndd1-HA in the Rad53-TAP pulldowns was measured by Western blotting. A distinct positive interaction signal could be observed for Ndd1-HA under both stress-induced and uninduced conditions, not only providing supportive evidence for a close interaction but also suggesting a constitutive relationship between the two factors, as has been previously observed for other Rad53 interactors (Fig. 6) (29).
FIG 6.
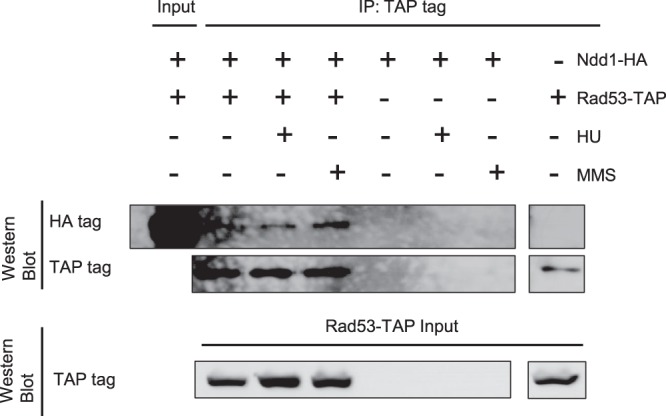
Rad53 interacts with Ndd1 under both physiological and genotoxic stress conditions. Coimmunoprecipitation of Ndd1 with Rad53. A RAD53-TAP strain (SKY361) expressing NDD1-HA on a centromeric plasmid (pAS40) was synchronized in G1 and released into YEPD with or without HU or MMS. At 60 min after release, samples were collected and the interaction of Rad53 with Ndd1 was examined by Rad53-TAP immunoprecipitation, followed by Ndd1-HA Western blotting. Two percent and 10% of the lysates were used as inputs for detecting Ndd1-HA and Rad53-TAP, respectively. Lysates from cells that did not express one of the tags were used as negative controls.
Multiple genotoxic stress-induced phosphorylations prevent Ndd1 recruitment and activation of G2/M-specific promoters.
To study the regulatory role of these induced sites, we generated sets of the respective alanine (S → A) or valine (T → V) substitutions in various combinations and named them Ndd1 checkpoint-defective (NDD1-CD) mutants (Fig. 7A). These mutant versions of Ndd1 were tested for viability, transcriptional activation, and recruitment to the CLB2 locus and recruitment to the GAL1 promoter via the Gal4-FHA domain fusion. None of the mutants showed significant defects for activation and recruitment under normal conditions (Fig. 7D and data not shown; see Fig. S5B in the supplemental material). Within this mutant Ndd1 collection, we identified Ndd1-CD-2A, which contained the S437/440A mutation and which exhibited higher binding to the CLB2 promoter upon MMS treatment (Fig. 7B). Notably, these two serine residues were conserved among the homologues of Ndd1 in various fungal species (Fig. 7C). We identified another mutant Ndd1-CD-8A which exhibited a slight increase in binding to the CLB2 promoter compared to that for the wild-type Ndd1 after MMS treatment (Fig. 7B). Finally, Ndd1-CD-10A, made by combining the mutations in Ndd1-CD-2A and -8A, was able to circumvent the MMS effect significantly in comparison to wild-type Ndd1 (Fig. 7D). Although the Ndd1-CD-10A mutant was more strongly recruited to the CLB2 promoter upon MMS treatment than wild-type Ndd1, the recruitment was still lower than that for untreated cells. Even CLB2 mRNA transcription was not completely restored by the Ndd1-CD-10A mutant after MMS treatment (see Fig. S5B in the supplemental material). The additional mutations made in the Ndd1-CD-11A mutant did not show any further additive effect compared to the effect seen in the Ndd1-CD-10A mutant (see Fig. S5C and D in the supplemental material). Strikingly, in MMS-treated cells, Ndd1-CD-10A was able to bind the GAL1-10 promoter with mediation by Gal4DBD-fkh21-306, with the kinetics almost being comparable to that for untreated cells (Fig. 7E). This difference in behavior between normal promoters and the artificial recruitment system gave a strong hint that the Ndd1-FHA domain interaction may not constitute an exclusive target of the checkpoint. Of course, we could have also missed some of the important phosphorylations on Ndd1, as identifying the complete phosphorylation status of a protein via MS studies is not always feasible. Moreover, Ndd1-CD-10A that had lost most of its responsiveness to MMS treatment was still not able to effectively bind to CLB2 upon HU treatment (Fig. 7D). It also could not suppress the loss of recruitment in the artificial Gal4-FHA domain system (Fig. 7E). This difference between HU and MMS treatment again indicates the involvement of additional signaling systems during replication stress.
FIG 7.

MMS-dependent phosphorylations of Ndd1 prevent its recruitment to the CLB2 promoter as well as its interaction with the FHA domain of Fkh2. (A) Model showing a series of Ndd1 checkpoint-defective mutants (NDD1-CD) used in the experiments whose results are presented in panels B, D, and E. (B) Conditional GAL promoter-driven NDD1 strains (based on strain JV323) expressing either NDD1-WT–HA, NDD1-CD-2A–HA, or NDD1-CD-8A–HA from endogenous promoters were synchronized in G1 and released into YEP-glucose with MMS. Ndd1 recruitment to the CLB2 promoter was examined by ChIP, followed by qPCR analysis. Error bars indicate standard deviations in three independent experiments. (C) Pairwise sequence alignment of various Ndd1 homologues indicates that S437 and S440 and the surrounding amino acids in the yeast S. cerevisiae Ndd1 are conserved in other yeast species. S. kluyveri, Saccharomyces kluyveri; E. gossypii, Eremothecium gossypii; C. albicans, Candida albicans; Z. rouxii, Zygosaccharomyces rouxii; K. lactis, Kluyveromyces lactis; K. thermotolerans, Kluyveromyces thermotolerans. (D and E) Recruitment of Ndd1-CD-10A mutant to the CLB2 promoter (D) as well as its FHA domain-mediated recruitment to the GAL1-10 promoter (E) is significantly reestablished upon MMS treatment but is still abolished in HU-treated cells. A conditional GAL promoter-driven NDD1 strain (JV323) expressing either NDD1-WT-HA and GAL4DBD-fkh21-306 or NDD1-CD-10A–HA and GAL4DBD-fkh21-306 was synchronized in G1 and released into YEP-glucose with or without MMS or HU. Ndd1 recruitment to the CLB2 and GAL1-10 promoters was examined by ChIP, followed by qPCR analysis. The data sets used for Ndd1-WT releases were the same as those used for the experiment whose results are shown in Fig. 2F. Error bars indicate standard deviations in at least two independent experiments.
Inhibition of G2/M-specific transcription during genotoxic stress is physiologically relevant.
Considering the role of Pds1 as the main checkpoint target for anaphase arrest, we wanted to address the question of how relevant the inhibition of G2/M transcription is for maintaining genomic integrity and cell viability during genotoxic stress. When expressed from the native promoter, the Ndd1-CD-10A mutation did not confer any increased sensitivity toward HU or MMS treatment (data not shown). However, when overexpressed under the control of the GAL1-10 promoter, Ndd1-CD-10A became toxic in HU- and MMS-treated cells, whereas it did not show any abnormal effects without such treatment (Fig. 8A). Ndd1-WT overexpression did not reveal any abnormal effects. We further tested whether Ndd1-CD-10A expression at native levels would possibly cause an increase in chromosome loss under HU or MMS treatment. As an indicator, we used a strain expressing a minichromosome (pSKY393) containing the ADE2 gene. Loss of this plasmid leads to the formation of pink sectors in otherwise white colonies. Indeed, a strain expressing Ndd1-CD-10A showed a significant increase in sector formation when grown on HU or MMS in comparison to that for a strain expressing Ndd1-WT (Fig. 8B). While both induction of spontaneous chromosome loss and increased cell death by Ndd1-CD-10A during MMS treatment are in accordance with our results described in the previous section, similar results with HU treatment seemed to contradict our earlier observations. However, one has to note that both physiological assays were done under conditions of chronic HU exposure, whereas G2/M expression and Ndd1 recruitment experiments were done with synchronous cultures over a limited time window. Thus, under chronic HU exposure conditions, the effects of Mec1-Rad53-induced phosphorylations seemed to be relevant for cell adaptation and recovery. The importance of Ndd1 downregulation during genotoxic stress was further emphasized in HU- or MMS-challenged mec1 mutants that exhibit premature spindle elongation and nuclear division (22). Under conditions of artificial Ndd1 depletion, these abnormal phenotypes were completely reversed (see Fig. S6 and S7 in the supplemental material).
FIG 8.
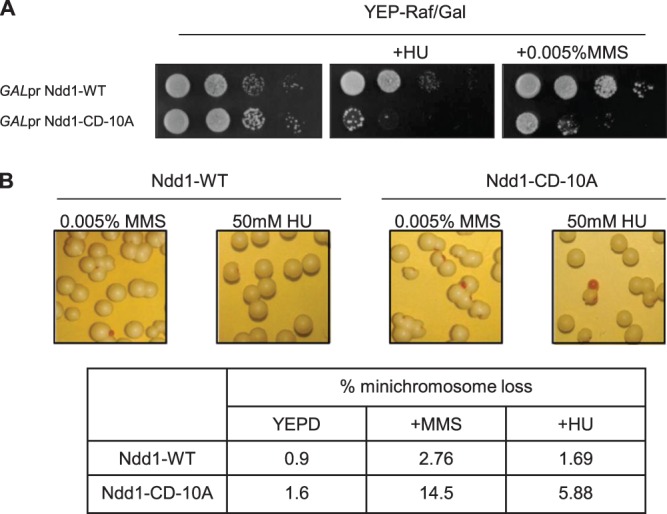
Ndd1-CD-10A increases genomic instability and sensitivity toward genotoxic stress. (A) Overexpression of the NDD1-CD-10A allele is detrimental for growth upon genotoxic stress. Tenfold serial dilutions of wild-type cells expressing either Ndd1-WT (pSKY379) or Ndd1-CD-10A (pSKY381) under the control of the GAL1-10 promoter were spotted on YEP-Raf-Gal plates with or without HU or MMS. Plates were photographed after 3 days of incubation at 30°C. (B) Ndd1-CD-10A expressed from an endogenous promoter increases spontaneous chromosome loss upon HU or MMS treatment. Cells expressing Ndd1-WT (SKY365) or Ndd1-CD-10A (SKY366) along with a circular minichromosome plasmid containing the ADE2 gene (pSKY393) were plated on YEP-glucose plates with or without MMS or HU. Percent minichromosome loss was determined by calculating the ratio of the number of colonies exhibiting pink sectors, indicating minichromosome loss, to the total number of colonies.
DISCUSSION
In this study, we have confirmed previous observations that connected DNA damage with low levels of expression of G2/M-specific genes. We extended these observations by showing that the lack of transcriptional activation must be mainly due to a block in the chromatin recruitment of the transcriptional activator Ndd1 (see the accompanying paper by Edenberg et al. [30]). Although a recent analysis suggested a link between Ndd1 modification and transcriptional changes during genotoxic stress, it did not provide any detailed mechanistic insights (16). Here we showed by mutational analysis of the modification sites that Rad53-dependent phosphorylation of Ndd1 indeed forms an important basis during MMS treatment. However, our analysis also indicated that an early block of DNA synthesis by HU treatment must induce additional regulatory branches (see the model in Fig. 9). These findings opened important questions not only about the structural basis of regulation but also about the physiological relevance of transcriptional regulation in coordinating and perhaps fine-tuning mitosis-specific events with respect to the S phase.
FIG 9.
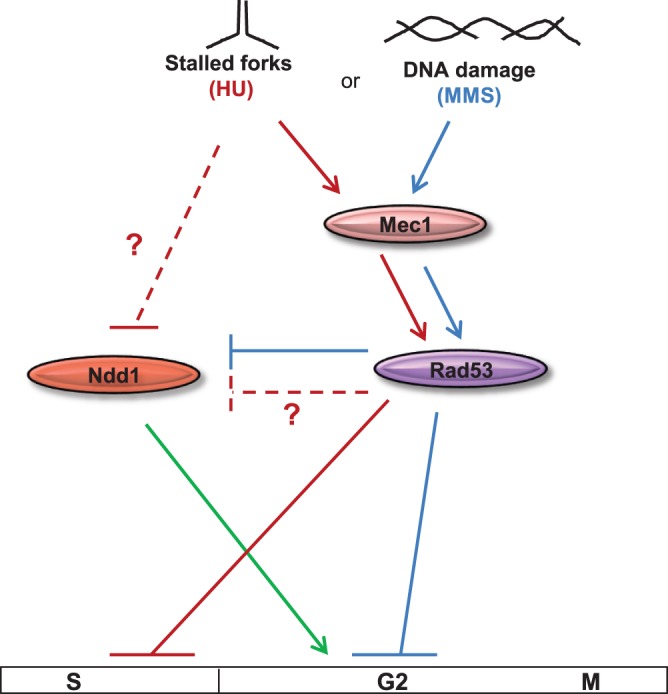
Model showing two distinct mechanisms by which Ndd1 activity is regulated upon genotoxic stress. During MMS-induced G2/M arrest, Ndd1 activity is inhibited solely in a Mec1-Rad53-dependent manner (blue lines). HU-induced deoxynucleoside triphosphate depletion blocks cell cycle progression in S phase (red lines) and inhibits Ndd1 activity by unknown mechanisms along with Mec1-Rad53.
Over the years, the highly regulated expression of G2/M-specific genes has revealed itself to be a complicated, multifaceted process. In its initial phase during G1/S transition, high levels of Cln-Cdc28 kinase activity remove the repressor Sin3/Rpd3 complex from G2/M promoters, thereby facilitating some albeit minor promoter activity that occurs independently of Ndd1 (5). Cln-Cdc28 activity also seems to be involved in the stabilization of the Ndd1 protein during late G1 (S. K. Yelamanchi, unpublished data). Neither of these two early steps, however, seems to be affected by genotoxic stress. However, our data with presynchronized cells are consistent with the proposal that the chromatin recruitment of Ndd1 by the forkhead factor Fkh2 is the main target of genotoxic stress. As the key experiment for this conclusion, we consider our test where an artificial Gal4-FHA domain fusion not only was able to bind Ndd1 in a cell cycle-regulated fashion but also responded to the genotoxic stress signals. The results clearly implicate the FHA-Ndd1 interaction and, as an extension, the phosphorylation status of Ndd1 as perhaps the predominant parameter of the regulation.
According to well-founded views, Ndd1 binding eventually leads to a rapid increase in Clb2-Cdc28 kinase activity after S phase via a positive-feedback mechanism in which Ndd1 phosphorylation by mitotic cyclin-dependent kinase (CDK) and increased affinity between the FHA domain and hyperphosphorylated Ndd1 are thought to play a central role (8, 10, 11). The question is therefore whether DNA damage or replication stress acts on the Ndd1 function indirectly via a decrease in its CDK-mediated phosphorylation or directly by causing an increase in modifications that interfere with binding. We have provided here an extensive and reproducible set of data on Ndd1 phosphorylation sites from wild-type and rad53 mutants treated or not treated with genotoxic stress. The data mainly support the second notion, although one has to be aware that it is still a major challenge to obtain reliable posttranslational modification information by mass spectrometry and also arrive at unambiguous quantitative interpretations. Nevertheless, in combination with our genetic analysis, we think that it is safe to conclude that the loss of transcription, at least by MMS-induced damage, must rely mainly on Rad53-dependent Ndd1 phosphorylation and not on the decrease in the activating modifications.
With regard to HU-attenuated G2/M transcription, the mass spectrometry results point to a combination of both mechanisms. This conclusion is supported by the genetic suppression data that suggest a more differentiated situation in the case of replication stress. Whereas mec1 rad53 cells or NDD1 alleles with alanine substitutions at genotoxically induced phosphorylation sites (e.g., NDD1-CD-10A) bypass the effects of MMS on G2/M transcription, this is not the case with the HU treatment. RAD53 deletions and NDD1-CD-10A have no significant effect on Ndd1 recruitment upon HU treatment, whereas mec1 cells can at least partially rescue G2/M transcription under these conditions, although one has to note that this suppression never reaches the levels observed without treatment. Thus, even though HU or MMS treatment effectively downregulates G2/M-specific transcription, one cannot escape the notion that differences exist in the mechanisms utilized to achieve this inhibition. There are several possibilities for the reasons why Rad53 deficiency or even Mec1 deficiency alone is not sufficient to suppress the HU effect. Among these possibilities are functional redundancies of the two genotoxic signaling cascades (Mec1 and Tel1) and their cooperation with additional signaling systems. For example, one might argue that active Tel1 in HU-treated mec1 cells activates Chk1 to an extent sufficient for repression of G2/M transcription. This argument is most likely invalid, because when cells of a rad9 mrc1AQ strain (a strain that is incapable of activating Rad53 and Chk1 [24]) were treated with HU, Ndd1 was still not recruited to G2/M promoters. Inhibition of Ndd1 recruitment to G2/M promoters in HU-treated cells could occur because cells are blocked very early in S phase in our experimental setup. Indeed, we have observed that the activating phosphorylations are slightly reduced in cells treated with HU. Nevertheless, we cannot yet exclude the possibility that HU treatment modulates additional cellular signal systems that contribute to a blockade in Ndd1 recruitment. Obvious candidates for this effect might be signals induced by morphogenetic abnormalities, as a substantial cross talk between their checkpoint signals and the Mec1-induced signals has been found to occur (29, 31, 32). For example, protein kinase C was shown to downregulate Ndd1 function (12).
What is the physiological significance of checkpoint regulation of G2/M transcription? During genotoxic stress, downregulation of G2/M-specific transcription substantially decelerates late cell cycle events from initiation of anaphase to mitotic exit, suggesting that the cell might obtain more time for necessary repair steps. Indeed, overriding the transcriptional block by Ndd1-CD-10A leads to increased genomic instability and low cell survival under conditions of chronic application of genotoxic stress. Finally, this identification of a connection between DNA damage and transcriptional attenuation in budding yeast aligns the checkpoint strategies of this organism with those documented for S. pombe and mammalian cells (13, 14).
Supplementary Material
ACKNOWLEDGMENTS
We are greatly indebted to David Toczyski for sharing unpublished data. We thank Maria Pia Longhese for mec1 and rad53 strains, Stephen Elledge for rad9 mrc1AQ strains, Franz Klein for minichromosome plasmid containing ADE2 gene, and the PRIDE team for their help in uploading MS data into ProteomeXchange. We also thank Dea Slade and Wolfgang Reiter for critical reading of the manuscript.
This study has been supported by an EC-FP7 grant (UNICELLSYS) and grants of the Austrian Science Foundation (FWF) F3411-B19 and the University of Vienna (IK 746001).
Footnotes
Published ahead of print 9 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01090-13.
REFERENCES
- 1.Boye E, Nordstrom K. 2003. Coupling the cell cycle to cell growth. EMBO Rep. 4:757–760. 10.1038/sj.embor.embor895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lew DJ, Reed SI. 1995. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J. Cell Biol. 129:739–749. 10.1083/jcb.129.3.739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Negishi T, Ohya Y. 2010. The cell wall integrity checkpoint: coordination between cell wall synthesis and the cell cycle. Yeast 27:513–519. 10.1002/yea.1795 [DOI] [PubMed] [Google Scholar]
- 4.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9:3273–3297. 10.1091/mbc.9.12.3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veis J, Klug H, Koranda M, Ammerer G. 2007. Activation of the G2/M-specific gene CLB2 requires multiple cell cycle signals. Mol. Cell. Biol. 27:8364–8373. 10.1128/MCB.01253-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koranda M, Schleiffer A, Endler L, Ammerer G. 2000. Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature 406:94–98. 10.1038/35017589 [DOI] [PubMed] [Google Scholar]
- 7.Loy CJ, Lydall D, Surana U. 1999. NDD1, a high-dosage suppressor of cdc28-1N, is essential for expression of a subset of late-S-phase-specific genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:3312–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds D, Shi BJ, McLean C, Katsis F, Kemp B, Dalton S. 2003. Recruitment of Thr 319-phosphorylated Ndd1p to the FHA domain of Fkh2p requires Clb kinase activity: a mechanism for CLB cluster gene activation. Genes Dev. 17:1789–1802. 10.1101/gad.1074103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pic-Taylor A, Darieva Z, Morgan BA, Sharrocks AD. 2004. Regulation of cell cycle-specific gene expression through cyclin-dependent kinase-mediated phosphorylation of the forkhead transcription factor Fkh2p. Mol. Cell. Biol. 24:10036–10046. 10.1128/MCB.24.22.10036-10046.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darieva Z, Pic-Taylor A, Boros J, Spanos A, Geymonat M, Reece RJ, Sedgwick SG, Sharrocks AD, Morgan BA. 2003. Cell cycle-regulated transcription through the FHA domain of Fkh2p and the coactivator Ndd1p. Curr. Biol. 13:1740–1745. 10.1016/j.cub.2003.08.053 [DOI] [PubMed] [Google Scholar]
- 11.Darieva Z, Bulmer R, Pic-Taylor A, Doris KS, Geymonat M, Sedgwick SG, Morgan BA, Sharrocks AD. 2006. Polo kinase controls cell-cycle-dependent transcription by targeting a coactivator protein. Nature 444:494–498. 10.1038/nature05339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darieva Z, Han N, Warwood S, Doris KS, Morgan BA, Sharrocks AD. 2012. Protein kinase C regulates late cell cycle-dependent gene expression. Mol. Cell. Biol. 32:4651–4661. 10.1128/MCB.06000-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu Z, Li J, Eshaghi M, Peng X, Karuturi RK, Liu J. 2007. Modulation of cell cycle-specific gene expressions at the onset of S phase arrest contributes to the robust DNA replication checkpoint response in fission yeast. Mol. Biol. Cell 18:1756–1767. 10.1091/mbc.E06-10-0928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor WR, Stark GR. 2001. Regulation of the G2/M transition by p53. Oncogene 20:1803–1815. 10.1038/sj.onc.1204252 [DOI] [PubMed] [Google Scholar]
- 15.Gasch AP, Huang M, Metzner S, Botstein D, Elledge SJ, Brown PO. 2001. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell 12:2987–3003. 10.1091/mbc.12.10.2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaehnig EJ, Kuo D, Hombauer H, Ideker TG, Kolodner RD. 2013. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 4:174–188. 10.1016/j.celrep.2013.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou L. 2013. Four pillars of the S-phase checkpoint. Genes Dev. 27:227–233. 10.1101/gad.213306.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. 2002. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36:617–656. 10.1146/annurev.genet.36.060402.113540 [DOI] [PubMed] [Google Scholar]
- 19.Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ. 2001. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3:958–965. 10.1038/ncb1101-958 [DOI] [PubMed] [Google Scholar]
- 20.Tinker-Kulberg RL, Morgan DO. 1999. Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev. 13:1936–1949. 10.1101/gad.13.15.1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. 1999. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286:1166–1171. 10.1126/science.286.5442.1166 [DOI] [PubMed] [Google Scholar]
- 22.Krishnan V, Nirantar S, Crasta K, Cheng AY, Surana U. 2004. DNA replication checkpoint prevents precocious chromosome segregation by regulating spindle behavior. Mol. Cell 16:687–700. 10.1016/j.molcel.2004.11.001 [DOI] [PubMed] [Google Scholar]
- 23.Baroni E, Viscardi V, Cartagena-Lirola H, Lucchini G, Longhese MP. 2004. The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol. Cell. Biol. 24:4151–4165. 10.1128/MCB.24.10.4151-4165.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osborn AJ, Elledge SJ. 2003. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 17:1755–1767. 10.1101/gad.1098303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiter W, Anrather D, Dohnal I, Pichler P, Veis J, Grotli M, Posas F, Ammerer G. 2012. Validation of regulated protein phosphorylation events in yeast by quantitative mass spectrometry analysis of purified proteins. Proteomics 12:3030–3043. 10.1002/pmic.201200185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nairz K, Klein F. 1997. mre11S—a yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes Dev. 11:2272–2290. 10.1101/gad.11.17.2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hieter P, Mann C, Snyder M, Davis RW. 1985. Mitotic stability of yeast chromosomes: a colony color assay that measures nondisjunction and chromosome loss. Cell 40:381–392. 10.1016/0092-8674(85)90152-7 [DOI] [PubMed] [Google Scholar]
- 28.Vizcaíno JA, Côté RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Pérez-Riverol Y, Reisinger F, Ríos D, Wang R, Hermjakob H. 2013. The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41:D1063–D1069. 10.1093/nar/gks1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smolka MB, Chen SH, Maddox PS, Enserink JM, Albuquerque CP, Wei XX, Desai A, Kolodner RD, Zhou H. 2006. An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J. Cell Biol. 175:743–753. 10.1083/jcb.200605081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edenberg ER, Vashisht A, Benanti JA, Wohlschlegel J, Toczyski DP. 2014. Rad53 downregulates mitotic gene transcription by inhibiting the transcriptional activator Ndd1. Mol. Cell. Biol. 34:725–738. 10.1128/MCB.01056-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enserink JM, Smolka MB, Zhou H, Kolodner RD. 2006. Checkpoint proteins control morphogenetic events during DNA replication stress in Saccharomyces cerevisiae. J. Cell Biol. 175:729–741. 10.1083/jcb.200605080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soriano-Carot M, Bano MC, Igual JC. 2012. The yeast mitogen-activated protein kinase Slt2 is involved in the cellular response to genotoxic stress. Cell Div. 7:1. 10.1186/1747-1028-7-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.