

Abstract
WNT signaling plays multiple roles in skeletal myogenesis during gestation and postnatal stages. The R-spondin (RSPO) family of secreted proteins and their cognate receptors, members of leucine-rich repeat-containing G protein-coupled receptor (LGR) family, have emerged as new regulatory components of the WNT signaling pathway. We previously showed that RSPO2 promoted myogenic differentiation via activation of WNT/β-catenin signaling in mouse myoblast C2C12 cells in vitro. However, the molecular mechanism by which RSPO2 regulates myogenic differentiation is unknown. Herein, we show that depletion of the LGR4 receptor severely disrupts myogenic differentiation and significantly diminishes the response to RSPO2 in C2C12 cells, showing a requirement of LGR4 in RSPO signaling during myogenic differentiation. We identify the transforming growth factor β (TGF-β) antagonist follistatin (Fst) as a key mediator of RSPO-LGR4 signaling in myogenic differentiation. We further demonstrate that Fst is a direct target of the WNT/β-catenin pathway. Activation and inactivation of β-catenin induced and inhibited Fst expression, respectively, in both C2C12 cells and mouse embryos. Specific TCF/LEF1 binding sites within the promoter and intron 1 region of the Fst gene were required for RSPO2 and WNT/β-catenin-induced Fst expression. This study uncovers a molecular cross talk between WNT/β-catenin and TGF-β signaling pivotal in myogenic differentiation.
INTRODUCTION
WNT signals, which are transmitted via multiple intracellular signaling cascades, including the canonical WNT/β-catenin pathway, play key roles in various aspects of skeletal myogenesis during gestation and postnatal stages (1, 2). During embryonic myogenesis, β-catenin function is essential for establishing the myogenic potential of the Pax3-positive progenitor cells within presomitic mesoderm cells and nascent somites (3). Different WNT proteins secreted from the surrounding tissue, such as dorsal neural tube and lateral ectoderm tissues, differentially regulate the myogenic program, especially the expression of myogenic determination factors, within somites (4–6). While activation of the WNT/β-catenin pathway by dorsal neural tube-derived WNT1 and WNT3A proteins preferentially induces Myf5 expression, demarcating epaxial muscle in somite explants, lateral mesoderm-derived WNT7A induces Pax3 and MyoD via the noncanonical WNT pathway to specify hypaxial muscle (5, 7). In addition, the WNT11 ligand expressed in the myotome regulates directional elongation of myofibers within the myotome in a β-catenin-independent manner without affecting myogenic differentiation (8).
In adult skeletal muscle, WNT/β-catenin signaling promotes myogenic differentiation of satellite cells (muscle-resident stem cells) by antagonizing mitogenic NOTCH signals through the inhibition of glycogen synthase kinase 3β (9). However, specific WNT ligands responsible for this regulation have not been identified. Activation of the WNT/β-catenin pathway is also associated with the CD45-positive stem cell population present in regenerating skeletal muscles (10). In addition, the WNT7A protein regulates self-renewal of satellite cells via the noncanonical WNT pathway in a fibronectin-dependent manner (11).
Members of the R-spondin (RSPO) family of secreted cysteine-rich proteins (RSPO1, -2, -3, and -4) activate the WNT/β-catenin signaling pathway at the receptor level in various cellular contexts (12, 13). The RSPO family proteins share two furin-like cysteine-rich (CR) domains followed by a single thrombospondin type I repeat (TSR) domain (12, 13). The CR domains are essential for activation of WNT/β-catenin signaling (14, 15). Interestingly, the RSPO proteins potentiate the activities of the WNT proteins in WNT/β-catenin signaling (14–16), indicating that RSPOs are important regulators of WNT/β-catenin signaling.
Several cognate receptors for the RSPO proteins have been identified. The leucine-rich repeat-containing G protein-coupled receptors 4, 5, and 6 (LGR4/5/6), markers for the intestinal and hair follicle stem cells (17, 18), were identified as RSPO receptors (19–22). Crystal structure analysis showed that the CR2 domain of RSPO1 directly interacts with the ectodomain of the LGR4 family receptors (23–25). Depletion of the LGR4 and LGR5 receptors in HEK 293T cells disrupts activation of WNT/β-catenin signaling by RSPOs and the synergy between RSPOs and WNTs, indicating that the LGR4 family receptors are active components of RSPO-induced WNT/β-catenin activation (19–21). In addition, the RSPO proteins were reported to bind to the extracellular domain of the LRP6 receptor, a coreceptor for canonical WNT signaling, in some studies (15, 26, 27). However, other studies failed to demonstrate RSPO-LRP6 binding (14, 19, 20, 28), leaving the role of LRP6 as an RSPO receptor inconclusive. The Frizzled receptors involved in both canonical and noncanonical WNT signaling do not directly bind the RSPO proteins (26, 29). Recently, RSPO1 was shown to inhibit the function of ZNRF3, a plasma membrane-bound E3 ubiquitin ligase which regulates the level of Frizzled and likely LRP6 receptors on the plasma membrane by ubiquitin-dependent degradation (30). RSPO1 binding to both LGR4 and ZNRF3 is suggested to induce a clearance of ZNFR3 on the plasma membrane (30), thereby resulting in an increase in the number of the available WNT receptors on the plasma membrane and a potentiation of WNT signaling activity. In addition to having a regulatory role in WNT/β-catenin signaling, RSPOs play a role in noncanonical WNT signaling. In Xenopus laevis embryos, RSPO3 and WNT5A cooperatively activate the noncanonical WNT signaling pathway through the Frizzled7 receptor (29). This activation depends on RSPO3 interaction with syndecan4 through the TSR domain of RSPO3. Taken together, the RSPO and LGR4 family proteins are a novel class of WNT signaling regulators that can activate the WNT pathway via mechanisms distinct from those of classical WΝΤ proteins.
We previously showed that the RSPO2 protein enhances myogenic differentiation and myocyte fusion in a WNT/β-catenin signaling-dependent manner in C2C12 myoblasts (31). In contrast, inhibition of both Rspo2 and Rspo3 gene expression by RNA interference (RNAi) significantly compromised myogenic differentiation (31). In this study, we investigate the molecular mechanism of RSPO function in promoting myogenic differentiation. We show that the LGR4 receptor plays an active role and mediates RSPO2 function during myogenic differentiation in C2C12 myoblast cells. Furthermore, we provide evidence that the TGF-β antagonist follistatin (FST) is a crucial mediator of RSPO-LGR4 function in myogenesis and that Fst gene transcription is directly regulated by a β-catenin/TCF4 transcription factor complex activated by the RSPO-LGR4 signaling cascade in vitro and in vivo. This study proposes signaling cross talk in which FST induced by the RSPO2–LGR4–β-catenin signaling cascade leads to inhibition of the TGF-β signaling pathway during skeletal myogenesis.
MATERIALS AND METHODS
Mice.
Myf5+/cre mice (32) were kindly provided by Thomas Gridley. Mice carrying conditional β-catenin loss-of-function (LOF) Ctnnb1flox (33) and gain-of-function (GOF) Ctnnb1flox(Ex3) (34) alleles were obtained from The Jackson Laboratory and Terry Yamaguchi, respectively. Mouse embryos were collected from pregnant, timed-mated female mice. The day of conception was designated embryonic day 0.5. Mice and embryos were genotyped using genomic DNA PCR as described elsewhere (32–34). Mice were housed in a pathogen-free air barrier facility, and animal handling and procedures were approved by the Maine Medical Center Institutional Animal Care and Use Committee.
Cell culture.
The mouse myoblast cell line C2C12 and human embryonic kidney 293T (HEK 293T) cells were obtained from the American Type Culture Collection (ATCC), Manassas, VA, and maintained in growth medium (Dulbecco's modified Eagle's medium [DMEM] containing 10% fetal bovine serum [FBS] and 1% penicillin-streptomycin) under 5% CO2 at 37°C. To induce myogenic differentiation, nearly confluent C2C12 cells were cultured in differentiation medium (DMEM containing 2% heat-inactivated horse serum and 1% penicillin-streptomycin) for up to 5 days. Fresh differentiation medium was replaced every 2 days. Recombinant FST, RSPO1, RSPO2, and WNT3A proteins were obtained from R&D Systems (Minneapolis, MN) and treated as indicated in the figure legends. 4-Hydroxytamoxifen (4-OHT; Sigma-Aldrich, St. Louis, MO) was added to the cells at a concentration of 1 μM.
Viral vectors and transduction.
Lentiviral vectors (pLKO.1 based) encoding short hairpin RNAs (shRNAs) specific to the mouse Lgr4, Fst, and control enhanced green fluorescent protein (eGFP) genes developed by the RNAi Consortium were obtained from Thermo Scientific (Lafayette, CO) and Sigma-Aldrich. cDNA encoding an active form of human β-catenin [β-cat(S33A)] fused to the ligand-binding domain of an estrogen receptor with a high affinity for tamoxifen (called ERT) was inserted into the pWPI lentiviral vector (obtained from Didier Trono). Lentiviruses were packaged in the recombinant viral vector facility at the MMCRI. C2C12 cells transduced with the shRNA viruses were selected in growth medium containing puromycin (2 μg/ml) to establish cell pools stably expressing shRNA. C2C12 cells were transduced with the β-cat(S33A)ERT lentivirus to nearly 100% transduction efficiency as determined by lentiviral-vector-derived GFP expression.
DNA and siRNA transfection.
Plasmid DNA transfection was performed using TransIt-LT1 reagent (Mirus Bio, Madison, WI) according to the manufacturer's instructions. Briefly, DNA TransIT mix was incubated with cells for 16 h, and the transfected cells were harvested at 2 days after transfection for luciferase assay. Small interfering RNAs (siRNAs) specific to the mouse Lgr4, Fst, and Ctnnb1 genes and nontargeting siRNA were obtained from Thermo Scientific/Dharmacon. siRNA was transfected into C2C12 cells using Lipofectamine RNAiMax reagent (Invitrogen, Carlsbad, CA) in a reverse-transfection format according to the manufacturer's protocol. Briefly, siRNA diluted in serum-free Opti-MEM I medium (Invitrogen) was mixed with the Lipofectamine reagent. The siRNA mixture was directly added to culture plates and incubated for 20 min at room temperature. C2C12 cells resuspended in growth medium without antibiotics were seeded into the culture plate containing the siRNA-Lipofectamine mixture (the final concentration of siRNA was 25 nM). Myogenic differentiation was induced at 2 days after transfection. The knockdown efficiency was determined by quantitative real-time PCR (qRT-PCR) at 2 days after transfection.
Microarray transcriptome analysis and qRT-PCR.
Total RNA was isolated from cultured cells using either an RNeasy plus kit (Qiagen, Carol Stream, IL) or TRIzol reagent (Invitrogen). For microarray analysis, RNA samples prepared in triplicate were analyzed using Affymetrix mouse gene ST1.0 arrays. Differential gene expression profiles were analyzed by the Bioconductor Program. Hybridization, scanning, and data processing were performed in the Genetics and Bioinformatics Core Facilities at the University of Vermont. For qRT-PCR, first-strand cDNA was synthesized using a ProtoScript first-strand cDNA synthesis kit (New England BioLabs, Ipswich, MA). Normally, 1 μg of total RNA was used for cDNA synthesis. cDNA in an amount equivalent to 20 ng of total RNA was used for qRT-PCR and in a customized RT2 Profiler PCR array (SA Biosciences, Valencia, CA). The PCR primer sequences and primer sets used in this study are listed in Tables S1 and S2 in the supplemental material, respectively.
Western blot analysis.
Cells were lysed with lysis buffer (10 mM Tris-Cl, pH 7.2, 2 mM EDTA, 150 mM NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, and 1% sodium deoxycholate) containing 50 mM sodium fluoride, 1 mM phenylmethanesulfonyl fluoride, 0.2 mM sodium vanadate, and protease inhibitor mixture (set V; EMD Chemicals, Gibbstown, NJ). The cell lysates (15 to 20 μg) were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. After the membranes were blocked with 5% skim milk in phosphate-buffered saline containing 0.1% Tween 20 for 1 h at room temperature, they were incubated with primary antibodies at 4°C overnight. Secondary antibodies conjugated with horseradish peroxidase (1:5,000 dilution; GE Healthcare/Life Sciences, Pittsburgh, PA) were applied to the membranes for 2 h at room temperature. The membranes were incubated with SuperSignal West Dura Luminol enhancer solution (Thermo Scientific/Pierce) and immediately exposed to X-ray film (Premium Autoradiography Film, Metuchen, NJ) for signal detection. Antibodies against β-catenin (1:500 dilution; BD Biosciences; San Jose, CA), FST (1:1,000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), and β-tubulin (1:1,000; Santa Cruz Biotechnology) were used.
Immunofluorescence staining.
Immunofluorescence staining was performed as previously described (31). Primary antibodies against myogenin (MYOG) (F5D, 1:100 dilution; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), myosin heavy chain (MyHC) (MF20, 1:200 dilution; Developmental Studies Hybridoma Bank), and β-catenin (1:100 dilution; BD Biosciences) and secondary anti-mouse IgG antibody conjugated with Cy3 (1:800 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA) were used. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Cell images were obtained using a Leica DFC340 FX digital camera attached to a Leica DM IRB inverted epifluorescence microscope.
Fst-luciferase reporters and luciferase assay.
Mouse Fst genomic DNAs spanning from bp −1846 to bp +20, containing the promoter and a portion of exon 1, and from bp +128 to bp +2168, containing the remaining exon 1 and intron 1, were PCR amplified. To create the Fst4-Luc reporter construct, the promoter/exon 1 and exon 1/intron 1 fragments were inserted into sequences upstream and downstream of the luciferase gene in the pGL2-basic vector (Promega, Madison, WI), respectively. Other Fst4-Luc derivatives were constructed similarly using shorter PCR-amplified Fst genomic DNA fragments. The promoter/exon 1 DNA containing mutations at the T1 and T2 sites was generated by ligating two PCR-amplified DNA fragments upstream and downstream of the T1/T2 sites, flanked with the EcoRI restriction enzyme site, thereby disrupting the T1/T2 sites with the EcoRI binding sequence.
Luciferase assays were performed using the dual-luciferase assay kit (Promega) according to the manufacturer's procedures. Renilla luciferase-thymidine kinase DNA (RL-TK) was used as a transfection control.
ChIP.
C2C12–β-cat(S33A)ERT cells (80% confluence) in two 60-mm dishes cultured in the absence or presence of 4-OHT overnight were fixed with 2% formaldehyde for 10 min at room temperature. Chromatin extraction and immunoprecipitation were performed according to the manufacturer's protocol using a ChIP-IT express chromatin immunoprecipitation (ChIP) kit (Active Motif, Carlsbad, CA). Anti-β-catenin antibody (Santa Cruz Biotechnology) and a negative-control anti-hemagglutinin (HA) mouse antibody (Sigma-Aldrich) were used for chromatin immunoprecipitation. The primer sequences used to amplify the Fst genomic DNA isolated from the immunoprecipitated chromatin are listed in Table S3 in the supplemental material.
In vitro protein translation and EMSA.
The TNT T7 transcription/translation system with rabbit reticulocyte lysate (Promega) was used to produce MYC-tagged human TCF4 protein. Synthesis of the TCF4 protein was confirmed by Western blotting using an anti-MYC antibody (1:1,000 dilution; Santa Cruz Biotechnology). Ten picomoles of synthetic double-stranded DNA oligonucleotide with a 5′ overhang was labeled with [γ-32P]dCTP (3,000 Ci/mmol; PerkinElmer, Waltham, MA) using the Klenow fragment of DNA polymerase I (New England BioLabs). For the electrophoretic mobility shift assays (EMSA), 2 μl of a reticulocyte lysate containing TCF4 was mixed with 0.4 pmol of radiolabeled probes in binding buffer [20-μl final volume, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.5), 7% glycerol, 0.05% Nonidet P-40, 0.1 mg/ml poly(dI-dC), 70 mM KCl, 50 mM dithiothreitol, 1 mM MgCl2]. For confirming the specificity of the DNA-TCF4 complex, a 50-fold excess of unlabeled double-stranded wild-type or mutant oligonucleotide or 2 μg of anti-MYC (Developmental Studies Hybridoma Bank) or anti-HA (Sigma-Aldrich) mouse antibody was added into the reaction mixture 15 min prior to the addition of the labeled probe. After incubation for 30 min at room temperature, the DNA-TCF4 complex was separated on a 5% nondenaturing polyacrylamide gel in a half-concentration of Tris-borate-EDTA buffer. Signal was detected by exposing the dried gel to X-ray film. The oligonucleotide sequences used in EMSA are listed in Table S4 in the supplemental material.
Whole-mount in situ hybridization.
Whole-mount in situ hybridization was performed on mouse embryos fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) as described previously (35). An antisense digoxigenin-incorporated RNA probe was synthesized from the plasmid containing an 850-bp cDNA containing the 5′ untranslated region and a portion of the protein coding sequence of the Fst RNA. Stained embryos were photographed with a Zeiss AxioCam digital camera under a Zeiss Stemi-SV6 stereomicroscope.
RESULTS
Downregulation of the LGR4 receptor suppresses myogenic differentiation in C2C12 myoblast cells.
To gain insight into the functional roles of the LGR4 family of RSPO receptors in skeletal myogenesis, we examined the RNA expression of the Lgr4 family genes in C2C12 mouse myoblast cells by qRT-PCR (Fig. 1A). Lgr4 RNA expression was mildly increased by day 1 of myogenic differentiation but decreased to a level similar to or slightly lower than that of undifferentiated cells by day 3 of differentiation. Lgr6 RNA expression also transiently increased by day 1 of differentiation. In contrast, Lgr5 expression continuously increased throughout differentiation. Compared to the ratio of housekeeping Gapdh RNA expression in exponentially growing, undifferentiated cells, Lgr4 transcripts seem most abundantly expressed among the family members (data not shown).
FIG 1.

Inhibition of Lgr4 expression by RNA interference disrupted myogenic differentiation in C2C12 cells. (A) Expression of the Lgr4 family of genes in C2C12 cells determined by qRT-PCR. U, undifferentiated, exponentially growing cells; D1 to D3, C2C12 cells cultured in differentiation medium for 1 to 3 days, respectively. Gene expression level was determined and normalized to Gapdh expression in triplicate samples. (B) Lgr4 expression in C2C12 cells transduced with lentiviruses bearing control eGFP- or Lgr4-specific shRNA determined by qRT-PCR. (C) Immunofluorescence staining for myogenic differentiation markers in control, Lgr4A-KD, and Lrg4B-KD C2C12 cells. Cells were cultured in differentiation medium for 1 day or 4 days. Cells were stained with anti-MYOG and MyHC primary antibodies followed by Cy3-conjugated secondary antibodies. Cell nuclei were counterstained with DAPI. (D and E) MYOG-positive nuclei or the nuclei in MyHC-positive cells were counted and are presented as percentages of the total number of nuclei as a differentiation index. (F) Distribution of MyHC-positive cells for a cell fusion index. MyHC-positive cells and fibers containing a single nucleus (Mono), two to five nuclei, and more than five nuclei were counted. The percentage for each group is presented. Error bars indicate standard errors of the means (SEM). P values were calculated by Student's t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not significantly different.
To determine the functions of the LGR4 receptor during myogenic differentiation in vitro, Lgr4 gene expression was inhibited by RNA interference in C2C12 cells. The knockdown (KD) efficiencies of the Lgr4 gene by two specific shRNAs (Lgr4A and Lgr4B) stably expressed via lentiviral transduction were determined to be approximately 60% and 85%, respectively, as measured by qRT-PCR (Fig. 1B). When both stable Lgr4-KD cell pools were induced for myogenic differentiation, they exhibited severe deficits in myogenic differentiation. The number of myogenin (MYOG)-positive cells was significantly reduced in Lgr4-KD cells on differentiation day 1 compared to that in control C2C12 cells transduced with eGFP-specific shRNA (Fig. 1C and D). Immunostaining for the myosin heavy-chain (MyHC) protein on differentiation day 4 also showed that both myogenic differentiation and cell fusion were significantly compromised in Lgr4-KD cells compared with those in control cells (Fig. 1C, E, and F). Together, these results showed that Lgr4 is essential for myogenic differentiation.
Depletion of the LGR4 receptor attenuates myogenesis promotion and WNT/β-catenin activation induced by RSPO2.
To determine whether the promyogenic activity of RSPO2 requires the LGR4 receptor, RSPO2 was added to control and Lgr4-KD cells under myogenic differentiation conditions. RSPO2 significantly promoted myogenic differentiation and cell fusion in control cells expressing eGFP shRNA, as we previously showed with naive C2C12 cells (Fig. 2A) (31). However, RSPO2 activity to enhance myogenic differentiation and myocyte fusion was significantly impaired in Lgr4-KD cells (Fig. 2B and C), suggesting that RSPO2 activity is mediated largely through the LGR4 receptor.
FIG 2.
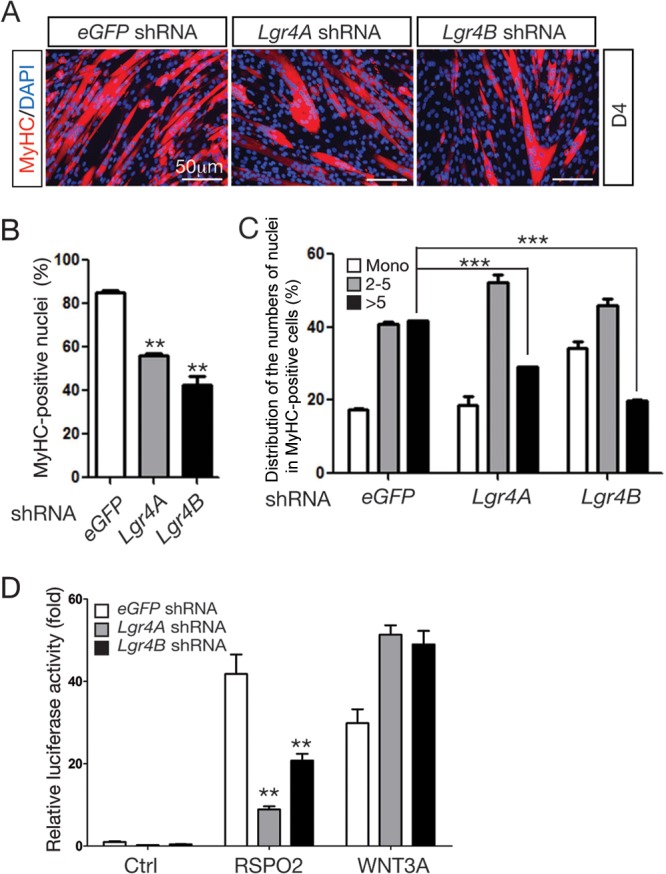
Depletion of Lgr4 expression attenuated RSPO2-enhanced myogenic differentiation and WNT/β-catenin signaling. (A) Immunofluorescence staining for the MyHC protein in differentiating control and Lgr4 KD C2C12 cells treated with RSPO2 (200 ng/ml) for 4 days. (B) The myogenic differentiation index was calculated by dividing the number of nuclei in MyHC-positive cells by the total number of nuclei and expressing the result as a percentage. (C) The cell fusion index was presented as a distribution of MyHC-positive cells based on their nucleus number. (D) Activation of WNT/β-catenin signaling was measured by the activity of the super TopFlash reporter. Control and Lgr4-KD C2C12 cells in 24-well plates were transfected with the TopFlash reporter (350 ng/well) and control Renilla luciferase reporter (150 ng/well) plasmid DNAs and stimulated with the RSPO2 (25 ng/ml) and WNT3A (50 ng/ml) proteins for 24 h. Luciferase activity was measured and normalized to the activity of control Renilla luciferase. Samples were prepared in triplicate. Error bars indicate SEM. P values were calculated by Student's t test. ***, P < 0.001; **, P < 0.01.
The promyogenic activity of RSPO2 depends on the activation of WNT/β-catenin signaling in C2C12 cells (31). The LGR4 family receptors are crucial for mediating RSPO-induced WNT/β-catenin signaling in various cell types (19–22). To determine whether activation of WNT/β-catenin by RSPO2 is defective in Lgr4-KD cells, we performed the TopFlash reporter assay to assess activation of the WNT/β-catenin pathway. While RSPO-mediated activation of the reporter gene was severely hampered in Lgr4-KD cells, WNT3A-mediated activation of the reporter was not affected (Fig. 2D). Thus, the LGR4 receptor is required specifically for RSPO2-mediated, but not WNT3A-mediated, activation of β-catenin to promote myogenesis.
RSPO2 action is restricted to the early stage of myogenic differentiation in C2C12 cells.
We previously showed that continuous treatment of the RSPO2 protein in differentiating C2C12 cells for up to 4 days significantly enhanced myogenic differentiation and myocyte fusion (31). However, it is not known whether RSPO2 activity mostly influences the early, late, or entire stage of myogenic differentiation. To determine which stage during myogenic differentiation is most sensitive to RSPO2 treatment, we treated C2C12 cells with the RSPO2 protein at different time points during myogenic differentiation (Fig. 3A). RSPO2 incubation during the first 24 h or 4 consecutive days resulted in equivalent myogenic effects (Fig. 3B to D). However, when RSPO2 was added at day 2 or later during differentiation, it did not produce any significant effect on myogenic differentiation and myocyte fusion (Fig. 3B to D). Therefore, RSPO2 exerts its effect during the first 24 h of the myogenic differentiation process in C2C12 cells.
FIG 3.

Enhanced myogenic differentiation by RSPO2 during the early stage of myogenic differentiation. (A) Differentiating C2C12 cells were treated with the RSPO2 proteins (200 ng/ml) at the different time points indicated. Treatment conditions a to e correspond to image labels and bars a to e in panels B to D. GM, growth medium; DM, differentiation medium; IF, immunofluorescence. (B) Images of MyHC immunofluorescence staining of RSPO2-treated differentiating C2C12 cells. (C) Percentages of MyHC-positive nuclei in total nuclei. (D) Distribution of MyHC-positive cells containing a single nucleus, 2 to 5 nuclei, and more than 5 nuclei in differentiating C2C12 cells treated with RSPO2. Experiments were performed in duplicate, and more than 1,000 total nuclei were counted. Error bars indicate SEM. P values were calculated by Student's t test. **, P < 0.01.
Identification of genes modulated by RSPO2.
To identify the molecular targets that may mediate RSPO2's effect on myogenic differentiation and myocyte fusion, we performed transcriptome analysis in differentiating C2C12 cells treated with RSPO2 or control bovine serum albumin (BSA) for 24 h. Additionally, we analyzed transcript expression in exponentially growing C2C12 cells cultured in the absence and presence of RSPO2 for 24 h.
In exponentially growing C2C12 cells, 15 upregulated and 18 downregulated genes were identified as having a >2-fold change with RSPO2 (see Table S5 in the supplemental material). In differentiating C2C12 cells, we identified 57 genes that were upregulated and 41 genes that were downregulated by 2-fold or more after RSPO2 treatment (see Table S6 in the supplemental material). Among the differentially regulated genes, 12 upregulated and 11 downregulated genes were common in both exponentially growing and differentiating C2C12 cells (Table 1).
TABLE 1.
Genes regulated by RSPO2 in C2C12 cells
| Gene symbol | Gene product | Fold inductiona |
|||
|---|---|---|---|---|---|
| Microarray |
qRT-PCR |
||||
| U1 | D1 | U1 | D1 | ||
| Axin2 | Axin2 | 2.502 | 3.912 | 37.372 | 21.363 |
| Car3 | Carbonic anhydrase 3 | 2.489 | 2.81 | ND | ND |
| Ch25h | Cholesterol 25-hydroxylase | 3.075 | 2.207 | ND | ND |
| Cxcr6 | Chemokine (C-X-C motif) receptor 6 | 2.572 | 2.33 | 5.616b,c | 6.755b,c |
| Enpp2 | Ectonucleotide pyrophosphatase/phosphodiesterase 2 | 2.919 | 7.945 | 7.931 | 3.258 |
| Fst | Follistatin | 2.986 | 4.786 | 10.549 | 10.342 |
| Il33 | Interleukin 33 | 2.869 | 2.388 | 5.484b | 4.303b |
| Lgr5 | Leucine-rich repeat containing G protein-coupled receptor 5 | 3.677 | 4.444 | 51.921b | 47.331b |
| Pfn2 | Profilin 2 | 2.387 | 4.677 | ND | ND |
| Qpct | Glutaminyl-peptide cyclotransferase (glutaminyl cyclase) | 2.103 | 2.441 | ND | ND |
| Tgfb2 | Transforming growth factor beta 2 | 2.404 | 2.715 | 4.524b | 3.173b |
| Tspan8 | Tetraspanin 8 | 3.35 | 3.782 | 5.683 | 4.398 |
| Aspn | Asporin | −2.447 | −2.894 | −2.583b | −1.877b |
| Cxcl12 | Chemokine (C-X-C motif) ligand 12 | −2.521 | −2.254 | −4.256b | −2.242b |
| Cxcl5 | Chemokine (C-X-C motif) ligand 5 | −2.414 | −3.609 | −14.507b | −21.22b |
| Enpp1 | Ectonucleotide pyrophosphatase/phosphodiesterase 1 | −2.531 | −2.509 | −2.699 | −1.927 |
| Enpp3 | Ectonucleotide pyrophosphatase/phosphodiesterase 3 | −2.241 | −2.646 | −2.061 | −1.766 |
| F3 | Coagulation factor III | −2.736 | −2.572 | ND | ND |
| Fgf7 | Fibroblast growth factor 7 | −3.772 | −3.745 | −4.801b | −6.012b |
| Hgf | Hepatocyte growth factor | −2.784 | −2.808 | −2.916b,c | −2.673b |
| Lphn2 | Latrophilin 2 | −2.682 | −2.367 | ND | ND |
| Ptn | Pleiotrophin | −2.761 | −3.871 | −6.058b | −1.561b,d |
| Rgs2 | Regulator of G protein signaling 2 | −2.49 | −4.442 | −2.223 | −1.7 |
P values of the fold induction of each gene are smaller than 0.01 except for the ones noted. U1, undifferentiated cells on day 1; D1, differentiated cells on day 1; ND, not determined.
qRT-CPR was performed using a customized RT2 profiler PCR array from SA Biosciences. The names of primer sets are listed in Table S4 in the supplemental material.
P < 0.05.
Not statistically significant.
Expression of 17 commonly regulated genes was further validated by qRT-PCR analyses (Table 1). A well-established WNT/β-catenin signaling target, Axin2, was induced approximately 37- and 21-fold by RSPO2 in undifferentiated and differentiating C2C12 cells, respectively, confirming that RSPO2 activates WNT/β-catenin signaling in these cells. Lgr5, a member of the RSPO receptor gene family, was upregulated by RSPO2. Lgr5 expression is positively regulated by WNT/β-catenin activation in intestinal stem cells (17), further confirming the activation of WNT/β-catenin signaling by RSPO2. Repression of the fibroblast growth factor 7 (Fgf7) and hepatocyte growth factor (Hgf) genes by RSPO2 suggests that RSPO2 inhibits the production of autocrine factors that promote proliferation of myogenic cells.
FST is a key mediator of RSPO2-LGR4 signaling.
Among the RSPO2 downstream target genes that we identified, we decided to further analyze the functional interaction between RSPO2 and the follistatin (Fst) gene because the FST protein plays significant positive regulatory roles during myogenic differentiation and myoblast fusion in vitro (36, 37) and in muscle hypertrophy in vivo (38) through inhibition of TGF-β family signaling.
We first analyzed Fst expression by qRT-PCR in C2C12 cells treated with RSPO2 during myogenic differentiation. Fst expression dramatically increased between day 1 and day 3 of myogenic differentiation in C2C12 cells (Fig. 4A). Fst transcript levels further increased approximately 8- to 10-fold in C2C12 cells treated with RSPO2 under both exponential-growth and differentiation conditions (Table 1 and Fig. 4A), consistent with the microarray data. RSPO2 treatment, however, did not cause any change in myostatin (Mstn) expression in the same samples (data not shown).
FIG 4.

Depletion of Fst compromised RSPO2-enhanced skeletal myogenesis in C2C12 cells. (A) Fst expression in undifferentiated (U) and differentiating (D1 and D3) C2C12 cells treated with control BSA (200 ng/ml) or RSPO2 (200 ng/ml) for 1 and 3 days was determined by qRT-PCR. Fst expression was normalized to Gapdh expression. Samples were prepared in triplicate. (B) Depletion of Fst expression by shRNA in C2C12 cells determined by qRT-PCR. Gapdh expression was used for normalization. Samples were tested in duplicate. (C to E) Immunofluorescence staining for MyHC expression in Fst-KD C2C12 cells and control eGFP-shRNA cells cultured in the presence of control BSA (200 ng/ml) and RSPO2 (200 ng/ml). Cells were harvested at differentiation day 5. (E) The percentages of nuclei in MyHC-positive cells relative to the total number of nuclei and percentages of MyHC-positive cells containing a single nucleus, 2 to 5 nuclei, 6 to 10 nuclei, and more than 10 nuclei were calculated. Experiments were performed in duplicate, and more than 1,000 total nuclei were counted. Error bars indicate SEM. P values were calculated by Student's t test. ***, P < 0.001; **, P < 0.01.
To investigate whether the Fst gene is a mediator of the myogenic activity of RSPO2 in C2C12 cells, we examined the effects of RSPO2 in C2C12 cells transduced with lentivirus encoding Fst-specific shRNA. We confirmed that Fst shRNA reduced endogenous Fst expression by approximately 80% compared to expression of control eGFP shRNA in C2C12 cells (Fig. 4B). Under myogenic differentiation conditions, Fst-KD cells displayed reduced myogenic differentiation and myocyte fusion (Fig. 4C to E). Furthermore, myogenic differentiation and myocyte fusion enhanced by RSPO2 were significantly compromised in Fst-KD cells (Fig. 4C to E).
To determine whether the LGR4 receptor is essential for RSPO2-induced Fst expression, Fst expression levels were analyzed in Lgr4-KD C2C12 cells cultured in the presence or absence of RSPO2. The Fst transcript level was reduced by approximately 70% in naive Lgr4-KD cells compared to control cells (Fig. 5A). When RSPO2 was added, Fst expression was significantly less effectively induced in Lgr4-KD cells than in control cells (Fig. 5A).
FIG 5.

Fst rescued defective skeletal myogenesis in Lgr4-KD C2C12 cells. (A) Fst expression in undifferentiated Lgr4-KD C2C12 cells treated with control BSA (200 ng/ml) or RSPO2 (200 ng/ml) for 1 day was determined by qRT-PCR. Fst expression was normalized to Gapdh expression. Samples were prepared in triplicate. (B to D) Exogenous FST protein rescued myogenic defects in Lgr4-KD C2C12 cells. Immunofluorescence staining for MyHC expression in Lgr4-KD C2C12 cells and control eGFP-shRNA cells cultured in the presence of control BSA (500 ng/ml) and FST (500 ng/ml). Cells were harvested at differentiation day 5. (E) The percentages of nuclei in MyHC-positive cells relative to the total number of nuclei and percentages of MyHC-positive cells containing a single nucleus, 2 to 5 nuclei, 6 to 10 nuclei, and more than 10 nuclei were calculated. Experiments were performed in duplicate, and more than 1,000 total nuclei were counted. Error bars indicate SEM. P values were calculated by Student's t test. **, P < 0.01; *, P < 0.05.
To determine whether exogenous FST can rescue myogenic-differentiation defects observed in Lgr4-KD C2C12 cells, Lgr4-KD cells were differentiated in the presence of the FST protein. FST treatment effectively rescued the differentiation defects almost to the level of control cells expressing eGFP shRNA (Fig. 5B to D). Taken together, these results strongly indicate that FST is a key downstream mediator of RSPO2-LGR4 signaling to promote myogenic differentiation and myocyte fusion.
Fst expression is regulated by the WNT/β-catenin pathway in C2C12 cells and mouse embryos.
RSPO2-enhanced myogenic differentiation is dependent mainly on activation of WNT/β-catenin signaling (31); therefore, we examined whether Fst expression is also regulated through activation of WNT/β-catenin signaling. The siRNA-mediated knockdown of the Ctnnb1 gene, encoding the β-catenin protein, significantly suppressed both the Fst transcript level and the protein expression induced by RSPOs in C2C12 cells (Fig. 6A and B). In addition, DKK1, a specific antagonist of WNT/β-catenin signaling, also effectively blocked RSPO-induced Fst expression (Fig. 6C).
FIG 6.

The WNT/β-catenin pathway regulates Fst expression in C2C12 cells and mouse embryos. (A) C2C12 cells were transiently transfected with control nontargeting (nt) siRNA or siRNA specific to the β-catenin gene. Transfected cells were incubated with RSPO1 (100 ng/ml) or RSPO2 (200 ng/ml) in differentiation medium for 1 day and harvested for total RNA isolation and qRT-PCR. Gapdh expression was used for normalization. Samples were prepared in duplicate. Error bars indicate SEM. P values were calculated by Student's t test. ***, P < 0.001. (B) Endogenous FST protein level was analyzed by Western blotting in C2C12 cells transiently transfected with β-catenin gene-specific siRNAs 1 day after myogenic differentiation was induced by RSPO1 (100 ng/ml) and RSPO2 (200 ng/ml). (C and D) Whole-mount in situ hybridization of Fst gene expression in mouse embryos at embryonic day 10.5. Mouse embryos with β-catenin GOF in Myf5-positive myogenic cells (Ctnnb1flox(Ex3)/+; Myf5Cre/+) and wild-type (wt) embryos (Ctnnb1+/+; Myf5Cre/+) were collected from matings between Myf5Cre/+ and Ctnnb1flox(Ex3)/+ mice. Mouse embryos with β-catenin LOF in Myf5-postive cells (Ctnnb1flox/flox; Myf5Cre/+) and wild-type embryos (Ctnnb1flox/+; Myf5Cre/+) were collected from matings between Myf5Cre/+; Ctnnb1flox/+ compound mice and Ctnnb1flox/flox mice. Dotted lines indicate the following embryonic structures: forelimb bud (FLB), hind limb bud (HLB), somite (Sm), and tail bud (TB). Sm1, Sm4, and Sm6 are indicated by black and blue arrows. Red arrows indicate somites at the forelimb level. Red and blue brackets indicate somites showing an Fst expression defect.
To investigate whether β-catenin regulates Fst expression in mouse embryos, we conditionally inactivated or activated the β-catenin function in Myf5-positive embryonic myogenic cells in vivo. Fst expression in somites closely overlaps Myf5 expression (39, 40), and Myf5 expression within somites is regulated by WNTs derived from the dorsal neural tube in cooperation with sonic hedgehog protein from the ventral neural tube and notochord (4–6, 41). Furthermore, the robust Rspo1 and Rspo3 expression in the dorsal neural tube overlaps Wnt1 and Wnt3a expression (42), raising a strong possibility that the Fst gene in Myf5-positive cells is under the regulation of the WNT/β-catenin pathway, activated by both WNTs and RSPOs.
Activation of β-catenin in Myf5-positive myogenic cells increased Fst expression within somites (red arrows in two upper panels of Fig. 6D) and produced ectopic expression within the most rostral somites (red brackets in two upper panels of Fig. 6D). In contrast, inactivation of β-catenin in the same Myf5-positive cells significantly abolished Fst expression (Fig. 6D, lower panels). Interestingly, inhibition of Fst expression is not observed in nascent somites (somites 1 to 5) and become evident in more-mature somites (e.g., somite 6 and somites at the interlimb level, depicted by blue arrows and brackets, respectively, in the two lower panels in Fig. 6D). Thus, we conclude that activation of WNT/β-catenin signaling is necessary and sufficient to activate Fst expression in myogenic cells in vitro and in vivo.
Fst is a direct transcription target of the β-catenin/TCF transcription factor complex.
To determine whether Fst is a direct target of the β-catenin/TCF transcription activator complex, we analyzed Fst expression in C2C12 cells expressing a tamoxifen-inducible form of the constitutively active β-catenin protein [C2C12–β-cat(S33A)ERT]. Treatment with 4-OHT, an active tamoxifen analog, induced immediate nuclear translocation of the overexpressed β-catenin protein within 1 h in cultured cells (Fig. 7A). Fst and Axin2 transcripts were effectively induced after 4 h of treatment with 4-OHT, strongly suggesting that Fst may be a direct transcriptional target of the β-catenin/TCF complex (Fig. 7B).
FIG 7.

The Fst gene is a direct transcriptional target of WNT/β-catenin signaling. (A) Tamoxifen-dependent nuclear translocation of the β-cat(S33A)ERT protein. 293T cells transduced with a lentivirus encoding the β-cat(S33A)ERT construct were treated with 4-OHT (1 μM) for 1 h and immunostained for the β-catenin protein. Arrows indicate nuclear β-cat(S33A)ΕRΤ protein. (B) Gene expression in C2C12 cells stably transduced with a β-cat(S33A)ERT lentivirus, determined by qRT-PCR. Cells were treated with 4-OHT for 4 h and harvested for RNA isolation. RNA expression was normalized to Gapdh RNA expression. Samples were collected in duplicate. Error bars indicate SEM. P values were calculated by Student's t test. ***, P < 0.001. (C) Schematic structures of the mouse Fst gene and Fst-luciferase (Luc) reporter gene constructs. A 4.0-kb DNA sequence of the mouse Fst gene containing the promoter, exon 1, and intron 1 was analyzed for transcription factor binding sites using MatInspector 4.0, and a number of conserved TCF/LEF1-binding elements (TBEs) were identified. Exon 1 and potential TBEs (labeled from T1 to T11) are indicated. Confirmed TBEs (black lines) in regions labeled A and B are indicated. The luciferase gene was inserted immediately downstream of the start codon of the Fst gene. (D) Detection of β-catenin binding to the Fst gene by ChIP. C2C12-β-cat(S33A)ERT cells were treated with 4-OHT (1 μM) for 24 h to induce activation of β-catenin. Untreated cells were used as a negative control. Chromatin was immunoprecipitated with anti-β-catenin and control anti-HA antibodies (Ab), respectively. PCR was performed using the primer sets spanning the potential TBEs on genomic DNA extracted from the immunoprecipitated chromatin. Genomic DNA isolated from chromatin prior to immunoprecipitation (input) was used as a control for PCR efficiency and accuracy. (E) Binding of TCF4 to the TBEs of the Fst gene in EMSA. 32P-labeled double-stranded oligonucleotides containing individual TBEs (T1 to T11 and TAxin2) were incubated with in vitro-translated human TCF4 protein with an N-terminal MYC tag. For competition, a 50-fold excess amount of unlabeled double-stranded oligonucleotides was added into the binding reaction mixture. DNA-TCF4 complexes and unbound free probe are indicated with brackets, and a nonspecific band is marked with an asterisk. mut, mutant. (F) Luciferase reporter assay. C2C12 cells in 24-well plates were transfected with the Fst-Luc reporter constructs (200 ng/well) and treated with RSPO2 (50 ng/ml) and WNT3A conditional medium (10%, vol/vol) for 48 h. (G) C2C12 cells in 24-well plates were cotransfected with the Fst-Luc reporters (100 ng/well) and β-cat(S33A) and TCF4 expression plasmids (100 ng each/well). Luciferase activities in triplicate samples were normalized to the activity of the cotransfected Renilla luciferase reporter (50 ng/well). Error bars indicate SEM.
Search for transcription factor binding sites resulted in 11 conserved TCF/LEF1 binding elements (TBEs) within the 4.0 kbp of the DNA sequence of the mouse Fst gene spanning from the 5′ upstream region to the first-intron region (Fig. 7C). We performed ChIP assays using anti-β-catenin antibody in chromatin preparations from untreated and 4-OHT-treated C2C12–β-cat(S33A)ERT cells. Among 8 primer sets spanning one or two TBEs, we found that 5 primer sets produced robust PCR products from DNA isolated from the chromatin complex immunoprecipitated with the β-catenin antibody in a 4-OHT-dependent manner (Fig. 7D). The two tandem TBEs in the promoter region (T1 and T2) and five TBEs localized within intron 1 (T7 to T11) were identified as potential β-catenin/TCF binding sites.
To further examine whether the TCF4 protein binds these identified sites, we performed EMSA using double-stranded oligonucleotides containing each TBE. In vitro-translated TCF4-Myc proteins specifically bound to the T1/T2 and T8 sites with an affinity comparable to that of a known TBE derived from the Axin2 gene promoter (Fig. 7E) (43). The specificity of binding was verified with competition with an excess amount of unlabeled double-stranded oligonucleotides specific to the wild-type or mutant TBE (Fig. 7E) and by disruption of the TCF4-DNA complex by preincubation with anti-MYC antibodies (data not shown). While we did not observe significant TCF4 binding to the T7 site, we observed weak but specific binding of TCF4 to the T9, T10, and T11 sites.
To determine the function of the identified TBEs in RSPO2-induced Fst expression, the 4.0 kbp of DNA containing all identified TBEs was linked to the luciferase reporter gene (named Fst4-Luc in Fig. 7C) and tested for its response to WNT/β-catenin signaling activation. This reporter was effectively induced by RSPO2 or WNT3A treatment (Fig. 7F) and by coexpression of the TCF4 and β-catenin(S33A) proteins in C2C12 cells (Fig. 7G). When T1 and T2 elements in the Fst promoter region were either deleted (Fst4ΔΑ-Luc) or mutated [Fst4(mutT1/T2)-Luc] in the Fst4-Luc reporter construct, approximately 70 to 90% of the reporter activity induced by RSPO2, WNT3A, or coexpression of β-cat(S33A)/TCF4 disappeared, whereas deletion of the T7 to T11 elements in the first intron region of the Fst gene (Fst4ΔB-Luc) generated an approximately 50-to-60% reduction of the reporter activity (Fig. 7F and G). The reporter lacking all five TBEs (Fst4ΔAB-Luc) failed to be induced by RSPO2, WNT3A, and β-cat(S33A)/TCF4 (Fig. 7F and G). Taking these data together, we conclude that the identified TBEs within the Fst gene are the functional binding sites for WNT/β-catenin signaling and that the Fst gene is a direct downstream target of WNT/β-catenin signaling.
DISCUSSION
The Lgr4 receptor mediates RSPO signals during myogenic differentiation.
The RSPO proteins enhance myogenic differentiation and myocyte fusion in a WNT/β-catenin-dependent manner in mouse C2C12 myoblast cells (31). The recent discovery that proteins of the LGR4 family are RSPO receptors (19–22) raises questions of whether the LGR4 family members mediate the myogenic activities of the RSPO proteins in C2C12 cells. Our gene expression analysis of the Lgr4 family members identified that Lgr4 is the most abundantly expressed gene in both undifferentiated and differentiating C2C12 cells. Depletion of Lgr4 expression by RNA interference severely compromised the onset and progression of myogenic differentiation, suggesting that the LGR4 receptor plays a crucial role in the myogenic differentiation of C2C12 cells.
We previously reported that inhibition of the Rspo2 and Rspo3 genes by siRNA significantly compromised myogenic differentiation in C2C12 cells (31). Therefore, the differentiation defects observed in Lgr4-KD cells may be due to the defect in the mediation of endogenous RSPO signaling. In support of this notion, RSPO2 treatment of Lgr4-KD cells generated a significantly reduced myogenic response compared to that of control cells. Consistently, the WNT/β-catenin signaling reporter assay confirmed that RSPO-induced activation of the WΝΤ/β-catenin pathway was largely defective in Lgr4-KD cells but that WNT/β-catenin activation by WNT3A was basically intact. These results indicate that myogenic-differentiation defects associated with LGR4 depletion is linked to a failure of mediating endogenous RSPO signals.
Is the LGR4 receptor engaged only in RSPO-dependent signaling during myogenic differentiation? It is reported that loss of the Lgr4 gene attenuates embryonic and postnatal bone formation by regulating the cyclic AMP (cAMP)-protein kinase A (PKA)-ATF4 pathway in osteoblasts (44). So far, no direct evidence indicates that the RSPO proteins regulate the cAMP-PKA-ATF4 pathway through the LGR4 receptor. Recently, it was also demonstrated that overexpression of the LGR5 receptor induced the G12/13-Rho GTPase pathway independently of RSPOs (45). Therefore, future investigation of whether the deficits in myogenic differentiation observed in Lgr4-KD cells is also linked to the disruption of the RSPO-independent GPCR pathway is warranted.
Fst is a direct target for WNT/β-catenin signaling in myogenic cells.
The FST protein, an antagonist of the TGF-β family proteins (46, 47), plays positive roles in skeletal myogenesis. The FST protein enhances myogenic differentiation and myocyte fusion in vitro (36, 37). Mice lacking the Fst gene show reduced intercostal and diaphragm muscles at birth (48), and Fst gene haploinsufficiency is associated with reduced muscle mass in adult mice (38). Furthermore, overexpression of the Fst gene in skeletal muscle in transgenic mice or administration of Fst-bearing recombinant adenoviruses results in a significant muscle mass increase (49–51). These FST functions are associated mainly with antagonizing myostatin (MSTN), a member of the TGF-β family and a strong inhibitor of skeletal muscle growth (52, 53), and activin (38). In addition, FST regulates the mTOR pathway in muscle hypertrophy independently of MSTN inhibition (54).
Despite Fst's critical roles in skeletal muscle, the regulatory mechanism of its expression in skeletal myogenesis is not well known. Inhibition of histone deacetylase by trichostatin A induces Fst expression mediated by MYOD and CREB transcription factors in myogenic cells in vitro (36). While there are a few reports suggesting that Fst expression depends on WNT signaling in other biological contexts (55, 56), no concrete molecular connection between WNT/β-catenin signaling and Fst expression has been established in skeletal myogenesis.
Our study provides compelling evidence that FST is a key mediator of myogenic activity of RSPO-LGR4 signaling via activation of WNT/β-catenin signaling and that the mouse Fst gene is a direct target of β-catenin signaling in myoblasts in vitro and in vivo. Activation and blockade of β-catenin signaling resulted in induction and inhibition, respectively, of the Fst gene or FST protein expression in C2C12 cells and mouse embryos.
We identified functional TBEs in the promoter and the intron 1 region of the mouse Fst gene using a combination of ChIP, EMSA, and a Fst gene reporter assay. The tandemly positioned T1/T2 sites located in the 1.6-kbp region (region A) upstream from the transcription start site of the Fst gene are the most critical TBEs mediating RSPO2- or WNT-activated β-catenin signaling, which represents approximately 70% of the reporter activity. In EMSA, T1/T2 DNA formed a strong complex with TCF4. DNA sequences of both sites are identically conserved in human, rat, and mouse genomes, suggesting that T1/T2 TBEs are likely to play the same role in mammals. Interestingly, the T1/T2 DNA-TCF4 complex displayed a mobility similar to that of the complex of TCF4 and Axin2 DNA, which contains only one TBE, suggesting that TCF4 likely binds either T1 or T2, not both.
Among the TBEs within the intron 1 region (region B) identified by ChIP assay, only the T8 site, which is highly conserved in human, rat, and mouse genomes, efficiently bound the TCF4 protein in EMSA. The other TBEs showed either poor or no binding by TCF4 in vitro. This discrepancy between in vivo ChIP and in vitro EMSA results may be explained by a few possibilities. First, multiple TCF4 proteins may bind the T8 site and other TBEs simultaneously in a cooperative manner in cells; however, this cooperative binding may not occur in vitro. Second, these weak or nonbinding sites may have a high affinity for other members of the TCF/LEF1 family, all of which can form a functional complex with the activated β-catenin protein in cells. Finally, because the average DNA size of the fragmented chromatin is about 500 bp and the distances between PCR amplification primer pairs spanning TBEs are less than 200 bp, it is possible that chromatin immunoprecipitated via β-catenin binding to the T8 site contains neighboring TBE DNA sequences. It is noteworthy that the T8 site is located approximately midway between the T7 and T11 sites. The first two possibilities are unlikely, because the Fst luciferase reporter lacking region B (Fst4ΔB-Luc) still yielded approximately 50% of the Fst4-Luc reporter, suggesting that the function of TBEs within region B is relatively minor and arguing against the possibility that the multiple TBEs within region B are all functional. Considering these findings together, we conclude that the T1/T2 sites within region A and the T8 site within region B are functional targets for WNT/β-catenin signaling in muscle differentiation. Interestingly, a TBE within the rat Fst promoter identical to the nonfunctional T4 site of the mouse Fst promoter is determined to be a target for WNT/β-catenin in human embryonic carcinoma cells (55), suggesting the possibility of a context-dependent TBE use.
Our study suggests the strong possibility of FST-mediated cross talk between the WNT/β-catenin and TGF-β signaling pathways during myogenic differentiation. Considering the robust positive role of FST in adult muscle hypertrophy by antagonizing TGF-β signaling, it would be noteworthy to investigate whether RSPO-LGR4 signaling and activation of the WNT/β-catenin pathway can also induce Fst expression in adult skeletal muscle and whether it regulates muscle hypertrophy; therefore, RSPO and LGR4 may provide new therapeutic targets for muscle-wasting diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lucy Liaw and Kyuson Yun for comments and suggestions and Thomas Gridley, Terry Yamaguchi, and Didier Trono for providing mouse lines and a lentiviral vector.
This research was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (5R01 AR055278 to J.K.Y.) and the National Institute of General Medical Sciences (NIGMS) (8P20 GM103465 to J.K.Y.; program director, D. Wojchowski). The core facilities that support this work were funded by the NIGMS (bioinformatics core grant 8P20 GM103465 to D. Wojchowski; recombinant viral vector and DNA analysis core grant 8P30 GM103392 to R. Friesel).
Footnotes
Published ahead of print 16 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01285-13.
REFERENCES
- 1.von Maltzahn J, Chang NC, Bentzinger CF, Rudnicki MA. 2012. Wnt signaling in myogenesis. Trends Cell Biol. 22:602–609. 10.1016/j.tcb.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cossu G, Borello U. 1999. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 18:6867–6872. 10.1093/emboj/18.24.6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutcheson DA, Zhao J, Merrell A, Haldar M, Kardon G. 2009. Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for beta-catenin. Genes Dev. 23:997–1013. 10.1101/gad.1769009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munsterberg AE, Kitajewski J, Bumcrot DA, McMahon AP, Lassar AB. 1995. Combinatorial signaling by Sonic hedgehog and Wnt family members induces myogenic bHLH gene expression in the somite. Genes Dev. 9:2911–2922. 10.1101/gad.9.23.2911 [DOI] [PubMed] [Google Scholar]
- 5.Tajbakhsh S, Borello U, Vivarelli E, Kelly R, Papkoff J, Duprez D, Buckingham M, Cossu G. 1998. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development 125:4155–4162 [DOI] [PubMed] [Google Scholar]
- 6.Munsterberg AE, Lassar AB. 1995. Combinatorial signals from the neural tube, floor plate and notochord induce myogenic bHLH gene expression in the somite. Development 121:651–660 [DOI] [PubMed] [Google Scholar]
- 7.Brunelli S, Relaix F, Baesso S, Buckingham M, Cossu G. 2007. Beta catenin-independent activation of MyoD in presomitic mesoderm requires PKC and depends on Pax3 transcriptional activity. Dev. Biol. 304:604–614. 10.1016/j.ydbio.2007.01.006 [DOI] [PubMed] [Google Scholar]
- 8.Gros J, Serralbo O, Marcelle C. 2009. WNT11 acts as a directional cue to organize the elongation of early muscle fibres. Nature 457:589–593. 10.1038/nature07564 [DOI] [PubMed] [Google Scholar]
- 9.Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. 2008. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2:50–59. 10.1016/j.stem.2007.10.006 [DOI] [PubMed] [Google Scholar]
- 10.Polesskaya A, Seale P, Rudnicki MA. 2003. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell 113:841–852. 10.1016/S0092-8674(03)00437-9 [DOI] [PubMed] [Google Scholar]
- 11.Bentzinger CF, Wang YX, von Maltzahn J, Soleimani VD, Yin H, Rudnicki MA. 2013. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 12:75–87. 10.1016/j.stem.2012.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lau WB, Snel B, Clevers HC. 2012. The R-spondin protein family. Genome Biol. 13:242. 10.1186/gb-2012-13-3-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin YR, Yoon JK. 2012. The R-spondin family of proteins: emerging regulators of WNT signaling. Int. J. Biochem. Cell Biol. 44:2278–2287. 10.1016/j.biocel.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W. 2004. R-Spondin2 is a secreted activator of Wnt/beta-catenin signaling and is required for Xenopus myogenesis. Dev. Cell 7:525–534. 10.1016/j.devcel.2004.07.019 [DOI] [PubMed] [Google Scholar]
- 15.Nam JS, Turcotte TJ, Smith PF, Choi S, Yoon JK. 2006. Mouse cristin/R-spondin family proteins are novel ligands for the Frizzled 8 and LRP6 receptors and activate beta-catenin-dependent gene expression. J. Biol. Chem. 281:13247–13257. 10.1074/jbc.M508324200 [DOI] [PubMed] [Google Scholar]
- 16.Kim KA, Kakitani M, Zhao J, Oshima T, Tang T, Binnerts M, Liu Y, Boyle B, Park E, Emtage P, Funk WD, Tomizuka K. 2005. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 309:1256–1259. 10.1126/science.1112521 [DOI] [PubMed] [Google Scholar]
- 17.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. 2007. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449:1003–1007. 10.1038/nature06196 [DOI] [PubMed] [Google Scholar]
- 18.Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, Toftgard R. 2008. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nature genetics 40:1291–1299. 10.1038/ng.239 [DOI] [PubMed] [Google Scholar]
- 19.Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. 2011. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. U. S. A. 108:11452–11457. 10.1073/pnas.1106083108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, Stange DE, van Es JE, Guardavaccaro D, Schasfoort RB, Mohri Y, Nishimori K, Mohammed S, Heck AJ, Clevers H. 2011. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476:293–297. 10.1038/nature10337 [DOI] [PubMed] [Google Scholar]
- 21.Glinka A, Dolde C, Kirsch N, Huang YL, Kazanskaya O, Ingelfinger D, Boutros M, Cruciat CM, Niehrs C. 2011. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 12:1055–1061. 10.1038/embor.2011.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruffner H, Sprunger J, Charlat O, Leighton-Davies J, Grosshans B, Salathe A, Zietzling S, Beck V, Therier M, Isken A, Xie Y, Zhang Y, Hao H, Shi X, Liu D, Song Q, Clay I, Hintzen G, Tchorz J, Bouchez LC, Michaud G, Finan P, Myer VE, Bouwmeester T, Porter J, Hild M, Bassilana F, Parker CN, Cong F. 2012. R-Spondin potentiates Wnt/beta-catenin signaling through orphan receptors LGR4 and LGR5. PLoS One 7:e40976. 10.1371/journal.pone.0040976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng WC, de Lau W, Forneris F, Granneman JC, Huch M, Clevers H, Gros P. 2013. Structure of stem cell growth factor R-spondin 1 in complex with the ectodomain of its receptor LGR5. Cell Rep. 3:1885–1892. 10.1016/j.celrep.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Huang B, Zhang S, Yu X, Wu W, Wang X. 2013. Structural basis for R-spondin recognition by LGR4/5/6 receptors. Genes Dev. 27:1339–1344. 10.1101/gad.219360.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen PH, Chen X, Lin Z, Fang D, He X. 2013. The structural basis of R-spondin recognition by LGR5 and RNF43. Genes Dev. 27:1345–1350. 10.1101/gad.219915.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei Q, Yokota C, Semenov MV, Doble B, Woodgett J, He X. 2007. R-spondin1 is a high affinity ligand for LRP6 and induces LRP6 phosphorylation and beta-catenin signaling. J. Biol. Chem. 282:15903–15911. 10.1074/jbc.M701927200 [DOI] [PubMed] [Google Scholar]
- 27.Li SJ, Yen TY, Endo Y, Klauzinska M, Baljinnyam B, Macher B, Callahan R, Rubin JS. 2009. Loss-of-function point mutations and two-furin domain derivatives provide insights about R-spondin2 structure and function. Cell Signal. 21:916–925. 10.1016/j.cellsig.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binnerts ME, Kim KA, Bright JM, Patel SM, Tran K, Zhou M, Leung JM, Liu Y, Lomas WE, III, Dixon M, Hazell SA, Wagle M, Nie WS, Tomasevic N, Williams J, Zhan X, Levy MD, Funk WD, Abo A. 2007. R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP6. Proc. Natl. Acad. Sci. U. S. A. 104:14700–14705. 10.1073/pnas.0702305104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohkawara B, Glinka A, Niehrs C. 2011. Rspo3 binds syndecan 4 and induces Wnt/PCP signaling via clathrin-mediated endocytosis to promote morphogenesis. Dev. Cell 20:303–314. 10.1016/j.devcel.2011.01.006 [DOI] [PubMed] [Google Scholar]
- 30.Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, Mao X, Ma Q, Zamponi R, Bouwmeester T, Finan PM, Kirschner MW, Porter JA, Serluca FC, Cong F. 2012. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485:195–200. 10.1038/nature11019 [DOI] [PubMed] [Google Scholar]
- 31.Han XH, Jin YR, Seto M, Yoon JK. 2011. A WNT/beta-catenin signaling activator, R-spondin, plays positive regulatory roles during skeletal myogenesis. J. Biol. Chem. 286:10649–10659. 10.1074/jbc.M110.169391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tallquist MD, Weismann KE, Hellstrom M, Soriano P. 2000. Early myotome specification regulates PDGFA expression and axial skeleton development. Development 127:5059–5070 [DOI] [PubMed] [Google Scholar]
- 33.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. 2001. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128:1253–1264 [DOI] [PubMed] [Google Scholar]
- 34.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. 1999. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18:5931–5942. 10.1093/emboj/18.21.5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin YR, Turcotte TJ, Crocker AL, Han XH, Yoon JK. 2011. The canonical Wnt signaling activator, R-spondin2, regulates craniofacial patterning and morphogenesis within the branchial arch through ectodermal-mesenchymal interaction. Dev. Biol. 352:1–13. 10.1016/j.ydbio.2011.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E, Minetti G, Zhao P, Hoffman EP, Puri PL, Sartorelli V. 2004. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev. Cell 6:673–684. 10.1016/S1534-5807(04)00107-8 [DOI] [PubMed] [Google Scholar]
- 37.Link BA, Nishi R. 1997. Opposing effects of activin A and follistatin on developing skeletal muscle cells. Exp. Cell Res. 233:350–362. 10.1006/excr.1997.3575 [DOI] [PubMed] [Google Scholar]
- 38.Lee SJ, Lee YS, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K, Cohn RD, Barton ER. 2010. Regulation of muscle mass by follistatin and activins. Mol. Endocrinol. 24:1998–2008. 10.1210/me.2010-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feijen A, Goumans MJ, van den Eijnden-van Raaij AJ. 1994. Expression of activin subunits, activin receptors and follistatin in postimplantation mouse embryos suggests specific developmental functions for different activins. Development 120:3621–3637 [DOI] [PubMed] [Google Scholar]
- 40.Amthor H, Connolly D, Patel K, Brand-Saberi B, Wilkinson DG, Cooke J, Christ B. 1996. The expression and regulation of follistatin and a follistatin-like gene during avian somite compartmentalization and myogenesis. Dev. Biol. 178:343–362. 10.1006/dbio.1996.0223 [DOI] [PubMed] [Google Scholar]
- 41.Borello U, Berarducci B, Murphy P, Bajard L, Buffa V, Piccolo S, Buckingham M, Cossu G. 2006. The Wnt/beta-catenin pathway regulates Gli-mediated Myf5 expression during somitogenesis. Development 133:3723–3732. 10.1242/dev.02517 [DOI] [PubMed] [Google Scholar]
- 42.Nam JS, Turcotte TJ, Yoon JK. 2007. Dynamic expression of R-spondin family genes in mouse development. Gene Expr. Patterns 7:306–312. 10.1016/j.modgep.2006.08.006 [DOI] [PubMed] [Google Scholar]
- 43.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. 2002. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22:1172–1183. 10.1128/MCB.22.4.1172-1183.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo J, Zhou W, Zhou X, Li D, Weng J, Yi Z, Cho SG, Li C, Yi T, Wu X, Li XY, de Crombrugghe B, Hook M, Liu M. 2009. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 136:2747–2756. 10.1242/dev.033571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon MS, Park BO, Kim HM, Kim S. 2013. Leucine-rich repeat-containing G-protein coupled receptor 5/GPR49 activates G-Rho GTPase pathway. Mol. Cells 36:267–272. 10.1007/s10059-013-0173-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel K. 1998. Follistatin. Int. J. Biochem. Cell Biol. 30:1087–1093. 10.1016/S1357-2725(98)00064-8 [DOI] [PubMed] [Google Scholar]
- 47.Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R, Patel K. 2004. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev. Biol. 270:19–30. 10.1016/j.ydbio.2004.01.046 [DOI] [PubMed] [Google Scholar]
- 48.Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. 1995. Multiple defects and perinatal death in mice deficient in follistatin. Nature 374:360–363. 10.1038/374360a0 [DOI] [PubMed] [Google Scholar]
- 49.Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, Murakami T, Uezumi A, Takeda S, Noji S, Sunada Y, Tsuchida K. 2008. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 22:477–487. 10.1096/fj.07-8673com [DOI] [PubMed] [Google Scholar]
- 50.Kota J, Handy CR, Haidet AM, Montgomery CL, Eagle A, Rodino-Klapac LR, Tucker D, Shilling CJ, Therlfall WR, Walker CM, Weisbrode SE, Janssen PM, Clark KR, Sahenk Z, Mendell JR, Kaspar BK. 2009. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci. Transl. Med. 1:6ra15. 10.1126/scitranslmed.3000112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SJ, McPherron AC. 2001. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. U. S. A. 98:9306–9311. 10.1073/pnas.151270098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McPherron AC, Lawler AM, Lee SJ. 1997. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387:83–90. 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- 53.McPherron AC, Lee SJ. 1997. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. U. S. A. 94:12457–12461. 10.1073/pnas.94.23.12457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. 2012. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J. Cell Biol. 197:997–1008. 10.1083/jcb.201109091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willert J, Epping M, Pollack JR, Brown PO, Nusse R. 2002. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2:8. 10.1186/1471-213X-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao HH, Matzuk MM, Jorgez CJ, Menke DB, Page DC, Swain A, Capel B. 2004. Follistatin operates downstream of Wnt4 in mammalian ovary organogenesis. Dev. Dyn. 230:210–215. 10.1002/dvdy.20042 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.