

Abstract
Activated inflammatory macrophages can express indoleamine 2,3-dioxygenase (IDO) and thus actively deplete their own tryptophan supply; however, it is not clear how amino acid depletion influences macrophage behavior in inflammatory environments. In this report, we demonstrate that the stress response kinase GCN2 promotes macrophage inflammation and mortality in a mouse model of septicemia. In vitro, enzymatic amino acid consumption enhanced sensitivity of macrophages to the Toll-like receptor 4 (TLR4) ligand lipopolysaccharide (LPS) with significantly increased interleukin 6 (IL-6) production. Tryptophan withdrawal induced the stress response proteins ATF4 and CHOP/GADD153; however, LPS stimulation rapidly enhanced expression of both proteins. Moreover, LPS-driven cytokine production under amino acid-deficient conditions was dependent on GCN2, as GCN2 knockout (GCN2KO) macrophages had a significant reduction of cytokine gene expression after LPS stimulation. To test the in vivo relevance of these findings, monocytic-lineage-specific GCN2KO mice were challenged with a lethal dose of LPS intraperitoneally (i.p.). The GCN2KO mice showed reduced inflammatory responses, with decreased IL-6 and IL-12 expression correlating with significant reduction in animal mortality. Thus, the data show that amino acid depletion stress signals (via GCN2) synergize with proinflammatory signals to potently increase innate immune responsiveness.
INTRODUCTION
Macrophages (Mϕ) are involved in many aspects of immune function, including innate clearance and effector responses, immune regulation, and adaptive tolerance (1–3). Mϕ activation and responsiveness are governed by a complex input of signals. Active receptor-driven inputs such as cytokine and pattern recognition receptors have been extensively examined; however, there are reasons to believe that external and internal environmental inputs affect important aspects of Mϕ biology. Mϕ recruited to an inflammatory lesion are exposed to many environmental stresses that engage cellular signaling pathways. One of the best-described environmental circuits results from low oxygen tension and induction of hypoxia-inducible factor 1α driving a hypoxic responsive program that increases expression of genes responsible for glycolysis and vasculogenesis (4). Similarly, induction of enzymatically driven amino acid deprivation would be expected to invoke a significant stress response within the cell. Indoleamine 2,3-dioxygenase (IDO) is a heme-containing intracellular enzyme that degrades tryptophan (Trp) to a series of catabolic products known collectively as kynurenines (5). IDO is induced in response to inflammatory stimuli in a variety of cells, including Mϕ, dendritic cells (DCs), and stromal epithelia (6), and is generally described as a suppressor of immune function dampening inflammatory cytokine production, inhibiting adaptive T cell responses, and promoting FoxP3+ T-regulatory cell (Treg) function (7). However, current data suggest that IDO may promote inflammatory cytokine production in certain contexts (8, 9). In particular, it was recently described that IDO induction promoted interleukin 6 (IL-6) production and mortality in a mouse model of endotoxemia (10). IDO is believed to modulate cell behavior via two primary mechanisms. (i) As a tryptophan-catabolizing enzyme, IDO lowers intracellular and microenvironmental tryptophan concentrations, engaging an amino acid-sensing pathway that activates the so-called integrated stress response (ISR) (11). This is associated with effector T cell proliferative arrest and apoptosis as well as FoxP3+ Treg activation (12). (ii) The kynurenines, produced by IDO, act on the aryl hydrocarbon receptor to suppress Th17 cell development, promote regulatory T cell differentiation, and likely modulate cytokine responses to a variety of stimuli (13–15).
Mechanistically, it is not known how amino acid depletion impacts Mϕ function in inflamed tissue microenvironments. Internally, amino acid deficiencies are detected by the serine/threonine kinase general control nonderepressible 2 (GCN2). GCN2 phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2), and the resulting changes in mRNA translation profoundly alter the transcriptional profile, eliciting the ISR and promoting expression of genes that counter the amino acid deficit (16). However, recent observations suggest that genes downstream of GCN2 impact inflammatory potential. In particular, C/EBP homologous protein 10 (CHOP-10; herein referred to as CHOP) dimerizes with C/EBP-β (also known as nuclear factor of IL-6 transcription [NF-IL-6]), potentially regulating expression of several cytokines (17–19).
In this study, we examined the impact of IDO expression or amino acid (i.e., tryptophan) deficiency on the Mϕ response to inflammatory stimuli. We found that IDO significantly enhanced lipopolysaccharide (LPS)-mediated IL-6 production in a manner dependent on amino acid deprivation-mediated stress. Moreover, we show evidence that tryptophan withdrawal stress, rather than functioning as a metabolic block for macrophage activity, serves as an important second signal activating the GCN2 pathway to augment proinflammatory cytokine production, thereby notably enhancing the pathological response to systemic challenge with a lethal dose of LPS.
MATERIALS AND METHODS
Mice.
C57BL6/J (B6), B6.Eif2ak4tm1.2Dron (GCN2−/−), and B6.LysM-Cre mice were obtained from The Jackson Laboratory. B6.Ddit3tm1Dron (CHOP−/−) and B6.Eif2ak4tm1.1Dron (GCN2flox/flox) mice were obtained from a colony maintained under specific-pathogen-free conditions in the Georgia Regents University animal facilities in accordance with Institutional Animal Care and Use Committee guidelines. To induce endotoxemia, mice (8 to 12 weeks) were injected intraperitoneally (i.p.) with 100 μl of phosphate-buffered saline (PBS) or LPS (15 mg/kg of body weight; the 50% lethal dose [LD50] is 2.5 mg/kg intravenously [i.v.]) (20) from Escherichia coli (Sigma; serotype O55:B5) in 100 μl of PBS.
Cell culture.
RAW 264.7 cells (ATCC) were grown in tryptophan-free RPMI 1640 medium (HyClone) supplemented with 20 mg/ml of albumin (Fisher Scientific), insulin-transferrin-selenium G supplement (Invitrogen), chemically defined lipid concentrate (Invitrogen), and 0.3 mg/liter of l-glutamine (Cellgro) and maintained in a 95% air–5% CO2 humidified atmosphere. Peritoneal exudate macrophages (PEMs) were isolated from peritoneal cavity lavage of mice 3 days after i.p. injection of 1 ml of 3% thioglycolate (Invitrogen). l-Tryptophan or a mixture of l-kynurenine, 3-hydroxyanthranilic acid, and quinolinic acid (Sigma) was prepared as a 25 mM stock and used to supplement the above-described medium at a final concentration of 25 μM. RAW 264.7 cells and PEMs were stimulated with 1 μg/ml of LPS (Sigma; serotype O55:B5) for the periods indicated below. This concentration was chosen because it likely reflects the approximate concentration of LPS in serum when mice are injected with the LD50 immediately after i.v. administration (21).
Lentiviral transduction.
IDO1 cDNA was cloned into pEGFPN1 (Clontech). The resulting IDO1-green fluorescent protein (IDO1-GFP) fusion protein was subsequently cloned into the lentiviral vector pLenti (a gift from Yukai He). Lentivirus was produced by transient transfection of the IDO1-enhanced GFP (IDO1-eGFP) pLenti construct into HEK293T cells along with packaging plasmids (pMDLg/pRRE, pRSV-Rev, and pMD2.VSV-G) using Expressfect transfection reagent (Denville Scientific). Virus-containing medium was collected 72 h posttransfection and filtered, and virus titers were determined. For lentiviral infection, RAW 264.7 cells (5 × 105 cells/ml) in RPMI 1640 containing 10% fetal bovine serum (FBS) were seeded in 24-well plates. The cells were infected with lentiviral vectors at a multiplicity of infection (MOI) of 10 in the presence of 8 μg/ml of Polybrene.
Cytokine measurements.
RNA from cells or tissue was purified using RNeasy RNA purification kits (Qiagen), and 250 ng of RNA was reverse transcribed using a random-hexamer cDNA reverse transcription kit (Clontech). For the PCR, 1 μl of cDNA was amplified and PCR was done using IQ Sybr green super mix (Bio-Rad) on an iQ5 real-time PCR detection system (Bio-Rad). Results were analyzed with the accompanying software according to the manufacturer's instructions. PCR for mouse β-actin, IDO1, CHOP, IL-6, IL-12p40, and tumor necrosis factor alpha (TNF-α) was done using previously described primers (3). Cytokine protein concentrations in plasma and cell culture medium were analyzed by enzyme-linked immunosorbent assay (ELISA) (eBiosciences).
In one set of experiments, PEMs were pretreated with tunicamycin (Sigma) at 2 μg/ml for 3 h and stimulated with 1 μg/ml of LPS for 8 h. Transcription levels of CHOP and IL-6 were measured by semiquantitative PCR (sqPCR) as described above.
Immunoblotting.
Western blotting and immunoprecipitation were done according to a previously published methodology (22). Specific antibodies against phospho-p38 mitogen-activated protein kinase (MAPK) and native p38 MAPK (Thr180/Tyr182), phospho- and native p44/42 MAPK (Thr202/Tyr204), phospho- and native IRF-3 (Ser396), NF-κB p65, IκBα, and β-actin were purchased from Cell Signaling Technology. Specific antibodies against activating transcription factor 4 (ATF4) and CHOP were purchased from Santa Cruz Biotechnology. Anti-C/EBPβ antibody recognizing both the liver-enriched transcriptional activating protein (LAP) and liver-enriched transcription inhibitory protein (LIP) isoforms was purchased from Abcam (clone 1H7).
In one set of experiments, mouse splenocytes were stained with anti-F4/80–phycoerythrin (PE) antibody (eBiosciences). PE-labeled cells were then purified using anti-PE microbeads (MACS; Miltenyi Biotec), and ATF4 protein level was analyzed by Western blotting as described above.
NF-κB p65 nuclear translocation.
Cells were cultured on chamber slides (Thermo Scientific) overnight in defined RPMI medium and fixed with PBS containing 4% paraformaldehyde for 10 min at room temperature. Cells were permeabilized with PBS containing 0.1% Triton X-100 and 2% FBS. After incubation with anti-NF-κB p65 (Santa Cruz Biotechnology), the cells were washed with PBS and incubated with secondary fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch). Slides were mounted with Prolong Gold antifade with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Fluorescent images were captured using a Zeiss LSM 510 Meta confocal microscope equipped with 1 argon and 2 HeNe lasers, using an HC PL APO lens at 20×/0.70 IMM CORR and 40×/1.3 oil and 90% glycerol (MP Biomedicals).
Flow cytometry.
PEMs were stained with a phycoerythrin-conjugated anti-TLR4 antibody (eBioscience) and subjected to flow cytometry on a FACScanto flow cytometer (BD Bioscience). Data were analyzed with FlowJo software (Tree Star). For sorting of splenic cell subsets, mice were injected with 15 mg/kg of LPS, and 3 h postinjection, spleens were collected and injected with 100 U of collagenase (Sigma) in 2 ml of PBS and incubated for 30 min at 37°C in 5 ml of PBS containing 400 U/ml of collagenase. From the digest, single-cell suspensions were generated and incubated with anti-F4/80 (eBioscience), anti-CD19, anti-Ly6G, and anti-CD11c (BD Pharmingen). The cells were sorted on a Dako Cytomation MoFlo cell sorter as previously described (3).
Kynurenine and Trp measurements by reverse-phase HPLC.
High-performance liquid chromatography (HPLC) analysis was performed as previously described (10). Briefly, for serum measurements of kynurenine and Trp, 100-μl serum samples were diluted with equal volume of 30 mM sodium acetate (NaAc), pH 4.0, and incubated at room temperature for 2 min. Fifty microliters (1/4 volume) of 30% trichloroacetic acid (TCA) was then added, and sample mixtures were incubated on ice for 5 min before being spun in a precooled (4°C) centrifuge at 10,000 × g. Supernatants were then collected for chromatography. For cell culture supernatants, 100 μl of culture medium was diluted with 10 μl (1/10 volume) of 150 mM NaAc, pH 4.0, and incubated at room temperature for 2 min. TCA was added as described above, and sample mixtures were incubated on ice for 5 min. For HPLC, 50 μl of sample was loaded by autosampler (Beckman Coulter model 508) and separated on a C18 (Shimadzu) column using the following conditions: mobile phase A, 2.5% acetonitrile in 15 mM NaAc, pH 4.0; mobile phase B, 100% acetonitrile; and flow rate, 1.2 ml/min. The mobile phase was delivered with a Beckman Coulter model 126 solvent module at a flow rate of 1.2 ml per minute. Kyurenine was detected with a Beckman Coulter model 166 detector at 360 nm, and tryptophan was detected using a Jasco FP-1520 fluorescent detector at an excitation wavelength of 285 nm and an emission wavelength of 365 nm.
Polysome analysis.
Polysome analysis was done as previously described (23, 24). Briefly, 107 cells were homogenized in 2 ml of buffer A (25 mM Tris, 25 mM NaCl, 5 mM MgCl2, 50 μg/ml of cycloheximide, 0.2 mg/ml of heparin, 1% Triton X-100; pH 7.5) with 10 strokes through a 26-gauge needle. The homogenate was centrifuged and decanted into another tube, and an equal volume of buffer B (4 volumes of buffer A diluted with 1 volume of 1 M MgCl2) was added. After incubation on ice for 1 h, 4 ml of total mixture was layered over 2-ml sucrose pads (0.2 M sucrose, 25 mM Tris, 25 mM NaCl, 100 mM MgCl2; pH 7.5) and centrifuged for 10 min at 27,000 × g, and the pellet containing polysome-associated mRNA was resuspended in RLT buffer (Qiagen). The supernatant was decanted into another tube, and an equal volume of 3 M LiCl was added, incubated at −20°C for 2 h, and then centrifuged at 27,000 × g for 10 min. Polysome-associated and free mRNAs were isolated using commercially available kits according to the manufacturer's directions (Qiagen), and sqPCR analysis was done as described above.
Image and statistical analysis.
Image analysis for relative Western blot band intensity was done using NIH IMAGEJ software. Means, standard deviations, and unpaired Student t test results were used to analyze the data. When comparing two groups, a P value of ≤0.05 was considered to be significant. Survival data were analyzed with Kaplan-Meier survival plots followed by the log rank test.
RESULTS
IDO promotes IL-6 production in response to LPS.
IDO-mediated regulation of T cells is well documented; however, how IDO influences Mϕ or DC behavior in response to septic inflammatory mediators remains an understudied aspect of the biology. To examine this issue, we transduced an Mϕ line (RAW 264.7) with an IDO-GFP fusion lentiviral construct, generating Mϕ populations with constitutive IDO expression and activity. In IDO+ macrophages, production of kynurenine, the rate-limiting step in IDO-mediated tryptophan (Trp) consumption, was increased 28-fold compared to that in control transfected macrophages (Fig. 1B). Moreover, tryptophan levels in IDO+ cultures were undetectable by HPLC, indicating that the IDO expressed was active, resulting in rapid consumption of free tryptophan in the culture medium (Fig. 1B). To test the impact of IDO on the response to inflammatory stimuli, IDO1-GFP cultures were stimulated with LPS and cytokine production was measured. The RAW Mϕ did not produce detectable IL-12p40 or IL-1β after LPS stimulation; however, IL-6 was significantly increased in IDO1-GFP+ cultures compared to that in GFP lentivirus-transduced controls (Fig. 1C). In contrast, LPS-driven TNF-α induction was unchanged by IDO expression.
FIG 1.
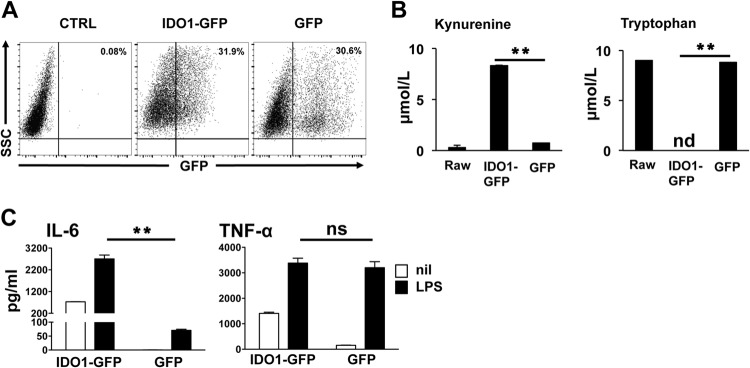
IDO enhances IL-6 production in macrophages. (A) RAW 264.7 macrophages were transduced with a lentivirus encoding an IDO1-GFP fusion protein, and 72 h later, IDO expression was assessed by flow cytometry as a function of GFP+ cells. Panels are representative of at least 5 samples per group. (B) Spontaneous kynurenine production and Trp consumption in IDO+ and control macrophage culture supernatants were assessed by HPLC as described in the text. (C) IDO+ and GFP+ control cultures were stimulated with LPS (1 μg/ml) for 18 h, and culture supernatants were tested by ELISA to determine concentrations of IL-6 and TNF-α. Bars represent the mean values for triplicate samples ± the standard deviations. **, P < 0.01 as determined by Student's t test. ns, not significant. Experiments were repeated at least three times, with similar results.
IDO can regulate cellular responses by Trp depletion and the production of immunologically active Trp catabolites (i.e., kynurenines). Thus, we next asked if IDO affects IL-6 production by one or both of these mechanisms. When Trp concentrations were titrated in peritoneal exudate macrophage (PEM) cultures, we found that Trp-free medium replicated the effects found in IDO-expressing RAW cells, with a significant increase in IL-6 protein production (Fig. 2A). However, even at low concentrations (0.156 μM), the presence of Trp did not affect LPS-driven IL-6 production, suggesting that very low concentrations of Trp are required for modulation of IL-6. Similar to the results with IDO-expressing Mϕ, titration of Trp in the culture medium had no impact on TNF-α production (Fig. 2A). In contrast to RAW cells, PEMs produced significant IL-12p40 when stimulated with LPS; however, unlike results with IL-6, titration of Trp in the culture to 0.313 μM or lower significantly reduced IL-12p40 production (Fig. 2A). Thus, taken together, the data suggest that restriction of IDO-driven reduction of Trp availability modifies the LPS-stimulated inflammatory response in a differential pattern that is cytokine species specific.
FIG 2.

Tryptophan depletion enhances IL-6 synthesis at the mRNA level. (A) PEMs were cultured overnight in defined media with titrated concentrations of Trp and stimulated with 1 μg/ml of LPS. Eighteen hours later, culture supernatants were collected and analyzed for production of cytokines by ELISA at the indicated time points. Trp concentrations tested were 2.5, 1.25, 0.625, 0.313, 0.156, and 0 μM. (B) PEMs were cultured with increasing concentrations of a mixture of kynurenines with or without Trp and stimulated with LPS as described above. Eighteen hours later cell culture supernatants were measured by ELISA for IL-6. (C) PEMs were cultured in media with or without Trp and stimulated with LPS as for panel A. RNA was extracted at the indicated time points, and the cytokine message was quantified by sqPCR as described in Materials and Methods. (D) PEMs treated as for panel C were stimulated with 1 μg/ml of LPS, and free versus polysome-bound IL-6 and TNF-α messages were determined by sqPCR. All bars or plot points (A to C) are the mean values for triplicate samples ± the standard deviations. *, P ≤ 0.05, and **, P < 0.01, as determined by Student's t test. Experiments were repeated at least three times, with similar results.
The addition of a mixture of three immunologically active tryptophan catabolites (kynurenine, 3-hydroxyanthranilic acid, and quinolinic acid) had no impact on IL-6 production in the presence or absence of Trp, suggesting that IDO-driven Trp depletion alone is responsible for increased IL-6 production (Fig. 2B). When cytokine mRNA was assessed by sqPCR, we found that the IL-6 message was increased greater than 10-fold over controls by the removal of Trp from the culture media, an effect that was sustained for at least 24 h post-LPS stimulation (Fig. 2C). In contrast, while TNF-α mRNA was increased 4 h after LPS treatment in the absence of Trp, the increase was only 25% compared to that in controls and was negligible by 8 h post-LPS addition (Fig. 2C). Similarly, IL-12p40 mRNA was increased in the absence of Trp 8 h after LPS stimulation, but the difference was 2-fold or less (Fig. 2C). Thus, the data suggest that IDO-driven Trp depletion increased IL-6 production by enhancing gene transcription.
The cytokine data presented in Fig. 2 suggested a dissociation between message accumulation and protein production after LPS stimulation, as the IL-6 message increased 10-fold in the absence of Trp but IL-6 protein production increased 2- to 3-fold. Metabolic stress can affect translation via modulation of eIF2 activity; thus, we reasoned that Trp withdrawal in Mϕ may limit IL-6 translation, manifesting as a decrease in the ratio of ribosome-bound versus unbound IL-6 mRNA. Supporting this, we observed a rapid decrease in polysome-bound IL-6 mRNA in Mϕ after LPS stimulation under Trp-free conditions, indicative of free message accumulation in the cell (Fig. 2D); however, when Mϕ were cultured in medium containing Trp, LPS stimulation rapidly increased the ratio of polysome-associated IL-6 mRNA. In contrast, TNF-α, which was not sensitive to Trp removal from culture medium (Fig. 2A), showed a time-dependent increase in polysome-associated message after LPS stimulation regardless of the availability of Trp (Fig. 2D). Thus, the data suggest that IL-6 translation is differentially impacted by the lack of Trp availability in the microenvironment.
LPS enhances the Trp depletion-mediated stress response.
Reduction in intracellular amino acid stores is sensed by the ISR kinase GCN2, which is activated by uncharged tRNAs and phosphorylates its only known target protein, α subunit of eukaryotic initiation factor 2 (eIF2α). When Trp was removed from Mϕ cultures, there was a time-dependent increase in phospho-eIF2α, with an 8-fold increase in phosphorylation after 6 h of culture (Fig. 3A). CHOP (GADD153) is an approximately 20-kDa protein which is induced by a variety of stress agents, including endoplasmic reticulum (ER) and amino acid depletion stress by PERK- and GCN2-dependent mechanisms, respectively (12, 25). Accordingly, we found a time-dependent increase in CHOP corresponding to the pattern of eIF2α phosphorylation, suggesting that Trp removal drives a stress response in otherwise quiescent Mϕ (Fig. 3A). This response was completely dependent on GCN2, as GCN2 knockout (GCN2KO) Mϕ did not phosphorylate eIF2α or induce CHOP expression in the absence of Trp (Fig. 3A). LPS stimulation has been reported to induce eIF2α phosphorylation after stimulation in cell lines (26); however, under Trp-free culture conditions, LPS did not increase detectable phospho-eIF2α, suggesting that GCN2 kinase activity functions independently of inflammatory stimuli in PEMs (Fig. 3B). Similarly, Mϕ cultured in media containing Trp did not show significant phospho-eIF2α, and LPS stimulation did not result in significant changes in the phosphorylation state (Fig. 3B).
FIG 3.

LPS stimulation enhances GCN2-pathway activation in the absence of Trp. (A) PEMs with the GCN2 genotype indicated were cultured in Trp-free media, and lysates were collected and probed for eIF2α phosphorylation and CHOP and ATF4 expression by Western blotting as described in Materials and Methods. (B) PEMs were cultured in media containing the Trp concentrations indicated for 6 h and stimulated with 1 μg/ml of LPS. At the indicated time points, cell lysates were tested for changes in the phosphorylation of eIF2α. (C) ATF4 protein levels were assessed by Western blotting in PEMs described for panel B. (D and E) PEMs were cultured under Trp-free conditions for 12 h, followed by addition of LPS (1 μg/ml). Twelve hours after addition of LPS, PEMs were examined for CHOP expression at the message level or ATF4 or CHOP or protein level by Western blotting. Bars in panel E represent the mean values for triplicate samples ± the standard deviations. *, P = 0.001 as determined by Student's t test. Western blots are representative of at least three experiments showing similar results. All experiments were repeated three or more times, with similar results.
GCN2-mediated eIF2α phosphorylation increases translation of a nodal transcriptional mediator of the GCN2 stress response, activating transcription factor 4 (ATF4) (16). In PEMs, ATF4 protein production under Trp-free conditions was enhanced by LPS stimulation, which rapidly induced ATF4 protein accumulation (2-fold increase within 30 min and greater than 4-fold at 24 h [Fig. 3C]). In contrast, LPS had a minimal effect on ATF4 concentrations under Trp+ culture conditions up to 24 h after stimulation, showing that ATF4 induction requires both Trp withdrawal stress and LPS stimulation (Fig. 3C). Furthermore, expression of ATF4 was dependent on GCN2 signaling, as GCN2KO macrophages failed to express ATF4 in the absence of Trp (Fig. 3A) and showed a large reduction in ATF4 after LPS stimulation under Trp-free conditions (Fig. 3D).
Since CHOP expression is induced by GCN2 in an ATF4-dependent manner (27, 28), we reasoned that CHOP production would be enhanced by LPS-mediated activation comparable to the increase in ATF4 translation. Accordingly, LPS stimulation of Mϕ under Trp-free conditions induced CHOP mRNA 10-fold, reflected by increased protein in the cultures (Fig. 3E). Moreover, paralleling the dependence of ATF4 on GCN2 for expression, GCN2KO Mϕ failed to induce significant CHOP after LPS stimulation (Fig. 3E). These data demonstrate that GCN2 signaling is requisite for activation of the ISR when intracellular amino acid availability is limited and indicate that LPS-driven activation of primary Mϕ enhances the ISR by potentiating ATF4 translation.
Trp depletion stress and LPS-induced signal transduction.
Since activation of GCN2 alters protein synthesis, we examined if Trp withdrawal altered components of signal transduction machinery required for IL-6 induction. Basal surface expression of TLR4 was not altered in Trp-free cultures (Fig. 4A), and LPS stimulation resulted in similar patterns of downregulation of TLR4, suggesting that low Trp stress did not affect TLR4 expression. Furthermore, Trp withdrawal stress did not affect IRF3 or MAPK (p38 or p42/44 ERK) phosphorylation (Fig. 4B). Thus, Trp withdrawal likely influences IL-6 production by a mechanism independent of initial signal transduction elicited by LPS stimulation.
FIG 4.
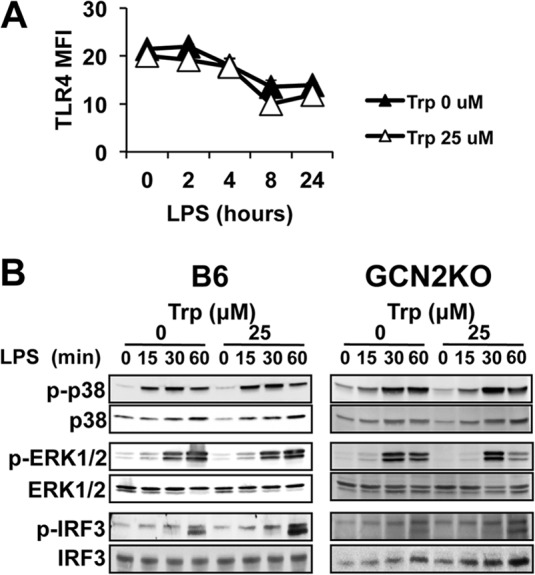
TLR4-mediated signal transduction is not affected by Trp withdrawal stress. PEMs were cultured for 18 h in defined media with or without Trp and stimulated with 1 μg/ml of LPS. (A) At the indicated time points, surface expression of TLR4 was determined by flow cytometric analysis. MFI, mean fluorescence. Plot points are the mean values for triplicate samples ± the standard deviations. The experiment was repeated three times, with similar results. (B) PEMs treated as for panel A were stimulated with 1 μg/ml of LPS. At the indicated time points, whole-cell lysates were probed via Western blotting for the presence of the phosphoproteins or total proteins indicated. Western blots are representative of four experiments showing similar results.
NF-κB signaling plays an indispensable role in the cellular response to LPS and is required for IL-6 production in Mϕ (29). NF-κB can signal via a canonical RelA/p65 pathway and a noncanonical RelB/p52 pathway. LPS does not induce significant noncanonical signaling (30–32), and we did not observe LPS-induced p100 cleavage in the presence or absence of Trp, suggesting that Trp withdrawal does not affect this aspect of NF-κB signal transduction. Likewise, Trp withdrawal did not affect overall p65 levels (Fig. 5A), suggesting that canonical NF-κB signaling was intact. Activation of p65 requires modification and degradation of the inhibitory IκBα subunit, and we found that Trp withdrawal decreased IκBα protein in Mϕ by 12 h, an effect that was dependent on GCN2 (Fig. 5B). IκBα mRNA was increased in the absence of Trp, suggesting that transcriptional inhibition was not the cause for the decrease in IκBα protein in PEMs (Fig. 5C). Conversely, polysome analysis showed that IκBα mRNA associated with ribosomes was reduced by 50% between 3 and 6 h after the removal of tryptophan (Fig. 5D). This reduction in ribosome-associated IκBα mRNA resulted in a reduced ability to synthesize new IκBα after LPS-mediated degradation of the protein (Fig. 5E). Based on these results, we predicted that increased nuclear translocation of p65 would occur under Trp-depleted conditions. Consistent with this, immunofluorescent staining demonstrated constitutive nuclear localization of p65 in Trp-starved PEMs, while controls exhibited a primarily cytoplasmic localization pattern (Fig. 5F). This was in contrast to GCN2KO Mϕ, which showed a primarily cytoplasmic localization pattern of p65 in the absence of Trp (Fig. 5F). Taken together, the data suggest that amino acid withdrawal enhances NF-κB activity, by reduction of IκB translation reducing negative regulation of p65.
FIG 5.

Trp withdrawal stress inhibits IκBα translation by a GCN2-dependent mechanism. (A) PEMs were cultured in Trp-free media for 16 h, and NF-κB p65 was measured in whole-cell lysates by Western blotting. (B) IκBα protein levels were determined by Western blotting at the indicated times after removal of Trp from the culture media. (C) PEMs were cultured under Trp withdrawal conditions for 16 h, and the IκBα relative message was determined by sqPCR. Bars represent the mean values for triplicate samples ± standard deviations. (D) At the indicated time points after removal of Trp from culture medium, the relative ratio of polysome (i.e., ribosome) bound versus unbound transcript for IκBα was determined by sqPCR. Plot points represent the relative value for pooled samples. (E) PEMs treated as for panel A were stimulated with 1 μg/ml of LPS, and IκBα was measured in whole-cell lysates collected at the indicated time points. (F) PEMs with the GCN2 genotype indicated were cultured for 12 h under Trp-free conditions, and cellular localization of NF-κB p65 was determined by indirect immunofluorescence. In one group the PEMs were stimulated with 1 μg/ml of LPS for 30 min prior to fixation and staining (right panels). Western blots and immunofluorescence are representative images of multiple experiments showing similar results. All experiments were repeated three or more times, with similar results.
GCN2 is required for cytokine gene expression in the absence of Trp.
The data above suggested that stress signals resulting from the lack of amino acid availability as a consequence of enzymatic consumption might play a role in Mϕ as a second signal augmenting inflammatory potential in response to pyrogenic stimulation. Since GCN2 signaling is a major cellular response to amino acid depletion, we hypothesized that increased IL-6 production in Mϕ in the absence of Trp may require GCN2 activity. Analysis of IL-6 mRNA kinetics revealed that the IL-6 message was consistently at least 40-fold lower in GCN2KO versus wild-type PEMs at any given time point after LPS-driven activation in the absence of Trl, suggesting that GCN2 signaling is critical for IL-6 gene expression under limited-amino-acid conditions (Fig. 6A). Similarly, LPS-driven TNF-α and IL-12p40 message induction was drastically reduced in the absence of GCN2, indicating a critical role in LPS-induced transcriptional responsiveness when available Trp is reduced. In agreement with the mRNA data, GCN2KO MΦs showed a 40-fold reduction in IL-6 protein production after LPS stimulation (Fig. 6B). Likewise, LPS-driven IL-12p40 was reduced nearly 6-fold and TNF-α protein was reduced by approximately 40% in the absence of GCN2 function (Fig. 6B). CHOP can dimerize with several members of the CCAAT/enhancer binding protein (C/EBP) family and may regulate cytokine production by a direct, transcriptionally mediated effect on cytokine promoters (17, 18, 33–35). Thus, we tested the involvement of CHOP in low Trp-driven enhancement of IL-6. We found that similar to GCN2KO Mϕ, CHOPKO Mϕ had a pronounced defect on LPS-driven IL-6 message induction under Trp-free conditions, suggesting that GCN2 mediates its effect on IL-6 transcription by induction of CHOP (Fig. 6C). This was reflected at the protein level, as LPS-stimulated CHOPKO Mϕ failed to produce significant IL-6 (Fig. 6D). In contrast, while CHOP deficiency did not significantly affect TNF-α mRNA induction kinetics, there was a reduction in TNF-α protein accumulation to a degree similar to that with GCN2KO Mϕ (Fig. 6D). However, while CHOP deficiency resulted in a minor decrease in IL-12p40 mRNA induced by LPS, the absence of CHOP did not impact IL-12p40 protein production (Fig. 6D).
FIG 6.

LPS-induced induction of cytokine message is dependent on GCN2 in the absence of Trp. (A and C) PEMs with the indicated genotype were cultured in Trp-free media for 6 h and stimulated with 1 μg/ml of LPS. At the indicated time points, RNA was extracted and the cytokine message was examined by sqPCR. (B and D) Twenty-four-hour culture supernatants from PEMs treated with LPS as described for panels A and C were analyzed by ELISA for the cytokines indicated. For panels A to D, plot points and bars represent the mean values for triplicate samples ± the standard deviations. (E) Western blot analysis for expression of C/EBPβ in PEMs cultured as for panel A and stimulated with 1 μg/ml of LPS for the indicated time points. (F) PEMs cultured for 6 h with or without Trp and activated with 1 μg/ml of LPS for 24 h. CHOP from native whole-cell lysates was immunoprecipitated and probed for the presence of C/EBPβ by Western blotting as described in Materials and Methods. Arrows, LAP and LIP isoforms of C/EBPβ. *, P ≤ 0.05, and **, P < 0.01, as determined by Student's t test. ns, not significant. Experiments were repeated four times, with similar results.
Members of the C/EBP family play an essential role in IL-6 transcription. In particular, C/EBPβ (nuclear factor of IL-6 transcription [NF-IL-6]) is required for IL-6 production in Mϕ (36). The C/EBPβ gene is translated into three different protein isoforms: the 38-kDa and 34-kDa liver-enriched transcriptional activating proteins (LAPs), which function as activators of transcription, and the liver-enriched transcription inhibitory protein (LIP; 20 kDa), which lacks most of the transactivation domain and acts as a dominant negative transcriptional repressor. Sharma et al. reported that IDO activity increased expression of LIP in TREX cells (37). However, Trp deprivation did not alter the ratio of long to short isoforms or overall C/EBPβ levels in PEMs (Fig. 6E), suggesting that modulation of C/EBPβ expression is not involved in enhanced Mϕ IL-6 transcription in the absence of Trp. CHOP can form heterodimers with C/EBPβ, and it has been proposed this interaction may modulate IL-6 transcriptional activity, although the exact function of CHOP in this context is not well understood (34, 35). We found that immunoprecipitation of CHOP from Trp withdrawal stressed Mϕ whole-cell lysates enriched for both long- and short-isoform C/EBPβ (Fig. 6F). However, we did not see the preferential association with either inhibitory or activating isoforms that had been described in previous studies using various cell lines (34, 35, 38). Nevertheless, the data support the hypothesis that CHOP may enhance IL-6 production by interacting with C/EBPβ.
To test if increased IL-6 mRNA induction was a general feature of the ISR, we treated PEMs with tunicamycin (TN) to induce ER stress and examined CHOP and IL-6 message induction by LPS. Treatment with TN alone was sufficient to induce a 100-fold induction of CHOP message over controls, and LPS stimulation in the presence of TN induced a further, significant increase in CHOP mRNA, suggesting that LPS enhanced the ER stress response (Fig. 7A). Moreover, in agreement with the data from IDO-expressing and Trp-starved Mϕ, TN-treated PEMs showed a significantly increased basal IL-6 message (Fig. 7B). The addition of LPS produced a synergistic effect resulting in a 50-fold increase in IL-6 mRNA over TN treatment alone and a 150-fold increase over the addition of LPS alone (Fig. 7B). Thus, the data suggest that ISR activation from both ER and amino acid depletion stress can enhance LPS-driven cytokine transcription.
FIG 7.
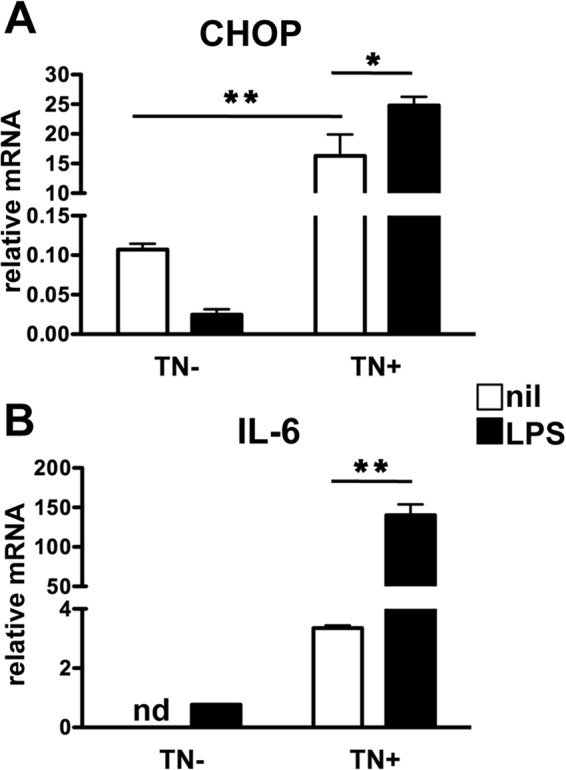
ER stress synergizes with LPS-driven IL-6 message induction. PEMs were cultured in complete media with or without 2 μg/ml of TN for 3 h, followed by stimulation with 1 μg/ml of LPS for 8 h. Samples were then collected, and the relative messages for CHOP (A) and IL-6 (B) were determined by sqPCR. Bars represent mean values for triplicate samples ± standard deviations. *, P ≤ 0.05, and **, P < 0.01, as determined by Student's t test. nd, not detected. The experiment was repeated twice, with similar results.
GCN2-mediated signaling potentiates cytokine production and lethal endotoxemia.
IDO is considered immunosuppressive, inhibiting antigen-presenting cell cytokine production and lymphocyte effector function. However, our data suggest that IDO expression enhances IL-6 production in response to microbial signals by a GCN2-dependent signaling mechanism in vitro. Thus, we next tested whether GCN2 function controlled Mϕ IL-6 production in vivo using a mouse model of LPS-mediated sepsis. However, since GCN2 is active in most cell types, to isolate the effect of Mϕ GCN2 activity, we generated monocytic lineage-specific GCN2KO mice (LysM-CRE GCN2flox/flox). LPS is a canonical IDO induction agent, and LPS injection i.p. induced a rapid increase (i.e., 14.5-fold after 3 h) in splenic IDO mRNA (Fig. 8A) and a significant, time-dependent increase in the serum kynurenine/Trp ratio (Fig. 8B), demonstrating rapid and significant IDO induction after LPS administration. LPS administration was associated with activation of the stress response and resulted in significant upregulation of splenic CHOP mRNA (3-fold at 3 h [Fig. 8C]). Moreover, in vivo CHOP induction was dependent on monocytic GCN2 activity, as it was blocked by disruption of GCN2 function (Fig. 8C). Consistent with the in vitro data, LysM-CRE GCN2flox/flox mice showed a 40% reduction in serum IL-6 compared to control animals, with no significant difference in serum TNF-α observed (Fig. 8D). Unexpectedly, LPS-driven systemic IL-12p40 in the serum was reduced greater than 2-fold, suggesting that in vivo GCN2 function is required for IL-12 as well as IL-6 induction. IDO message was induced greater than 10-fold 3 h after LPS injection in sorted splenic F4/80+ Mϕ and CD11c+ dendritic cells (DCs), an effect that was not significantly affected by GCN2 abrogation (Fig. 8E). However, paralleling the serum cytokine data, disruption of GCN2 reduced IL-6 message induction in response to LPS more than 3-fold in F4/80+ and CD11c+ cells, although IL-6 mRNA was consistently higher in sorted F4/80+ Mϕ (Fig. 8E). Similarly, in agreement with the serum cytokine data, IL-12p40 message induction was abrogated to a highly significant degree in the absence of GCN2 function in both sorted Mϕ and DCs (Fig. 8E). TNF-α was primarily expressed by F4/80+ Mϕ in the spleen, and correlating with TNF-α production in circulation, GCN2 ablation did not significantly affect expression (Fig. 8E). We then examined ATF4 protein in splenic, MACS-sorted, F4/80+ Mϕ. In agreement with the in vitro data, LPS induced a rapid increase in ATF4 detectable by Western blotting (Fig. 8F); however, in Mϕ lacking GCN2, basal ATF4 protein was reduced by 50% compared to that in littermate controls and LPS exposure failed to increase ATF4 levels. Thus, the data suggest that GCN2 is required for LPS-driven ATF4 expression and cytokine production.
FIG 8.

Deletion of GCN2 in LysM-CRE GCN2flox/flox mice reduces systemic IL-6 and IL-12p40 production and protects mice from LPS-induced endotoxemic mortality. (A) Whole splenic IDO1 message was determined in B6 mice as described in the text 3 h after i.p. LPS administration. (B) Serum kynurenine/Trp ratios were determined by HPLC at the indicated time points after i.p. injection of LPS as for panel A. (C) Total splenic CHOP message was determined by sqPCR 3 h after i.p. LPS injection. (D) Serum cytokine levels were determined by ELISA at the indicated time points after i.p. injection of 15 mg/kg of LPS. (E) F4/80+ Mϕ and CD11c+ DCs were sorted by FACS 3 h after injection with LPS as for panel A, and the relative message for the indicated mRNA species compared was determined by sqPCR. (F) Splenocytes from mice treated as for panel A were pooled (n = 3), and F4/80+ macrophages were purified via MACS columns. Purified macrophages were analyzed by Western blotting for the presence of the proteins indicated. (G) Mice of the indicated genotype were injected with 15 mg/kg of LPS i.p., and mortality over a 100-h period was determined. For panels A to D, n = 5 mice/group; for panel E, n = 3 mice/group. For panel F, n = 10 mice/group. *, P ≤ 0.05, and **, P < 0.01, as determined by Student's t test. For panel F, P = 0.006, determined as described in Materials and Methods. Experiments were repeated at least three times, with similar results.
IDO blockade has been reported to protect mice from septic mortality (8). Our data strongly suggested that Trp deprivation-mediated stress, rather than inhibiting function, enhanced LPS-driven cytokine production in Mϕ by activation of a GCN2 signaling cascade. Thus, we reasoned that LysM-CRE GCN2flox/flox mice would be protected from septic lethality in a similar manner. In agreement with this, LysM-CRE GCN2flox/flox mice showed markedly improved survival after systemic challenge with a lethal dose of LPS, which resulted in 100% mortality by 90 h postadministration in littermate controls (Fig. 8G). Thus, taken as a whole, the data reveal an important and novel role for GCN2 stress as an activating signal that is required for potent, sustained cytokine production in inflammatory environments.
DISCUSSION
IDO is an IFN-responsive, Trp-consuming enzyme that is rapidly induced in inflammatory settings; thus, Trp consumption-mediated cellular stress is likely a common condition of inflammation. However, the key mechanisms transmitting Trp withdrawal stress signals or how these pathways impact DCs or Mϕ responsiveness are only partially understood. Jung et al. reported that IL-6 induced by systemic LPS administration was partially dependent on IDO and pharmacological or genetic ablation of IDO reduced serum IL-6 and IL-12 levels, improving endotoxemic mortality rates, with follow-up studies showing similar results in the cecal ligation puncture model (8, 39). Similarly, stroma-derived IDO was shown to enhance cancer metastasis by potentiating IL-6 production and myeloid-derived suppressor cell expansion, and monocytes treated with gamma interferon (IFN-γ) and LPS produced IL-6 by an IDO-dependent mechanism (9). Cumulatively, these reports indicate that IDO can promote inflammatory pathology under certain conditions, although the exact conditions required are not known at present.
The data presented here suggest that IDO influences cytokine production primarily by consuming intracellular Trp, as Trp withdrawal replicated the effects observed in IDO+ Mϕ. Trp is one of eight essential amino acids which cannot be synthesized de novo and is the rarest of all amino acids in terms of storage and overall abundance in the body (40, 41). Thus, increases in Trp consumption either via protein biosynthesis or via consumption in IDO-dependent enzymatic processes will likely result in rapid depletion of Trp stores. Our data presented in Fig. 1 demonstrated that IDO expression in a minority of cells in an Mϕ population (i.e., 30%) results in rapid Trp consumption from culture medium, driving increased IL-6 production. While it is unlikely that IDO-dependent consumption of Trp will deplete extracellular Trp in most circumstances, it is reasonable to assume that cytoplasmic stores of the amino acid will be rapidly depleted in IDO+ cells within, creating an intracellular environment with low available Trp and subsequent activation of the ISR.
The response to Trp withdrawal is primarily mediated by GCN2, a latent kinase that is activated by increased uncharged tRNAs as a result of deficiency in cellular amino acid pools (42). Activated GCN2 phosphorylates eIF2α, lowering GDP-GTP exchange on eIF2, thereby attenuating ribosomal assembly on mRNA and reducing global translation. However, rare mRNAs encoding transcriptional activators of genes involved in metabolism and amino acid synthesis and transport are translationally activated (28, 43). Our data would argue that in Mϕ (and likely DCs), the GCN2 signaling pathway serves an additional purpose to enhance inflammatory responses by increasing expression of certain cytokines. It is not clear how GCN2 modulates cytokine production in antigen-presenting cells; however, our data indicate that the downstream transcription factor CHOP is a key factor in this process. We find a strong dependence of IL-6 production on GCN2-mediated CHOP expression, as disruption of either gene abrogates IL-6 mRNA induction coupled with Trp depletion. Since CHOP is associated with C/EBPβ, a transcription factor critical for IL-6 production, it is likely that C/EBPβ-CHOP complexes are required for IL-6 promoter activity under stress, a notion further supported by our observations that CHOPKO and GCN2KO Mϕ failed to sustain IL-6 message levels compared to wild-type Mϕ under similar conditions.
Phosphorylation of eIF2a contributes to NF-κB activation (44), and GCN2 may serve as an upstream kinase in this process (44, 45). Our data show that Trp withdrawal inhibits translation of IκBα while not affecting RelA/p65 levels. While basal IκB is relatively stable, it is very quickly degraded after receptor-driven activation, and its rapid resynthesis is an important negative-feedback mechanism. GCN2 activation appears to restrict translational ability by reducing message association with the ribosome, thereby augmenting basal NF-κB nuclear trafficking. Though GCN2-dependent activation by Trp depletion appears to be critical in this process, the relative contributions of this NF-κB-mediated process and the aforementioned CHOP-mediated process to cytokine induction remain to be determined.
The in vivo results were consistent with the in vitro data suggesting an important role for GCN2-mediated stress signals in the response to systemic LPS. Moreover, the observation that splenic CHOP induction is lost in LysM-CRE GCN2flox/flox mice suggests that stress elicited by LPS is mechanistically dependent on GCN2. In vivo, GCN2 appeared critical for IL-12p40 production, as LysM-CRE GCN2flox/flox mice showed reduced message and protein induction. This is divergent from our in vitro results; however, it is in agreement with the study by Jung et al., who showed that IDO blockade reduced IL-12 levels in toxemic mice (8). Moreover, IL-12 production was dependent on GCN2, in agreement with the hypothesis that amino acid withdrawal stress augments the LPS-induced cytokine storm.
Inflammatory Mϕ are activated, terminally differentiated cells with limited proliferative responses. Our data suggest that in this context, GCN2 synergizes with other inflammatory signals to fine-tune the Mϕ response. This is in contrast to other cell types, such as naive lymphocytes, wherein GCN2 likely acts as a potent metabolic inhibitor. In contrast to this, resting FoxP3+ Tregs can acquire regulatory phenotypes rapidly and without division in a process dependent on the T cell receptor, negative costimulation, and GCN2 activation (7). Thus, like Mϕ, Tregs in this context do not proliferate and GCN2 may serve an alternate function as an “accessory signal,” promoting essential cellular responses to activation. Consequently, the requirement for proliferation is likely an important determinant factor in the metabolic effects of GCN2 activation on a cell population.
In conclusion, our data demonstrate a novel stress-dependent pathway regulating Mϕ cytokine production. While the complete mechanistic details are not known yet, given the ubiquitous nature of inflammation-driven stress and the potential for differential regulation in septic and sterile inflammatory situations, these findings may have broad implications for our understanding of environmental stress in inflammation and immunity.
ACKNOWLEDGMENTS
This work was supported by an intramural grant from Georgia Regents University (T.L.M.), grant AI092213 from NIH/NIAID (T.L.M.), and Wellcome Trust Principal Research Fellowship and EU FP7 BetaBat grant (no. 277713) (D.R.).
Footnotes
Published ahead of print 18 November 2013
REFERENCES
- 1.McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. 2011. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 117:5403–5412. 10.1182/blood-2010-11-320028 [DOI] [PubMed] [Google Scholar]
- 2.Murray PJ, Wynn TA. 2011. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11:723–737. 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, Munn DH, Mellor AL, Karlsson MC, McGaha TL. 2012. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A. 109:3909–3914. 10.1073/pnas.1117736109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aragonés J, Elorza A, Acosta-Iborra B, Landazuri MO. 2011. Myeloid hypoxia-inducible factors in inflammatory diseases. Crit. Rev. Immunol. 31:1–13. 10.1615/CritRevImmunol.v31.i1.10 [DOI] [PubMed] [Google Scholar]
- 5.McGaha TL, Huang L, Lemos H, Metz R, Mautino M, Prendergast GC, Mellor AL. 2012. Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol. Rev. 249:135–157. 10.1111/j.1600-065X.2012.01149.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munn DH, Mellor AL. 2013. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 34:137–143. 10.1016/j.it.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. 2007. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 117:2570–2582. 10.1172/JCI31911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jung ID, Lee MG, Chang JH, Lee JS, Jeong YI, Lee CM, Park WS, Han J, Seo SK, Lee SY, Park YM. 2009. Blockade of indoleamine 2,3-dioxygenase protects mice against lipopolysaccharide-induced endotoxin shock. J. Immunol. 182:3146–3154. 10.4049/jimmunol.0803104 [DOI] [PubMed] [Google Scholar]
- 9.Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, Boulden J, Sutanto-Ward E, Soler AP, Laury-Kleintop LD, Mandik-Nayak L, Metz R, Ostrand-Rosenberg S, Prendergast GC, Muller AJ. 2012. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2:722–735. 10.1158/2159-8290.CD-12-0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang L, Lemos HP, Li L, Li M, Chandler PR, Baban B, McGaha TL, Ravishankar B, Lee JR, Munn DH, Mellor AL. 2012. Engineering DNA nanoparticles as immunomodulatory reagents that activate regulatory T cells. J. Immunol. 188:4913–4920. 10.4049/jimmunol.1103668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munn DH, Mellor AL. 2007. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 117:1147–1154. 10.1172/JCI31178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. 2005. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22:633–642. 10.1016/j.immuni.2005.03.013 [DOI] [PubMed] [Google Scholar]
- 13.Desvignes L, Ernst JD. 2009. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31:974–985. 10.1016/j.immuni.2009.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fallarino F, Grohmann U, Vacca C, Orabona C, Spreca A, Fioretti MC, Puccetti P. 2003. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 527:183–190. 10.1007/978-1-4615-0135-0_21 [DOI] [PubMed] [Google Scholar]
- 15.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC, Puccetti P. 2006. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 176:6752–6761 [DOI] [PubMed] [Google Scholar]
- 16.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099–1108. 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 17.Caristi S, Piraino G, Cucinotta M, Valenti A, Loddo S, Teti D. 2005. Prostaglandin E2 induces interleukin-8 gene transcription by activating C/EBP homologous protein in human T lymphocytes. J. Biol. Chem. 280:14433–14442. 10.1074/jbc.M410725200 [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. 2005. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 280:21763–21772. 10.1074/jbc.M501759200 [DOI] [PubMed] [Google Scholar]
- 19.Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. 2011. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. U. S. A. 108:6561–6566. 10.1073/pnas.1008942108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawes AS, Rock CS, Keogh CV, Lowry SF, Calvano SE. 1992. In vivo effects of the antiglucocorticoid RU 486 on glucocorticoid and cytokine responses to Escherichia coli endotoxin. Infect. Immun. 60:2641–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faggioni R, Fantuzzi G, Villa P, Buurman W, van Tits LJ, Ghezzi P. 1995. Independent down-regulation of central and peripheral tumor necrosis factor production as a result of lipopolysaccharide tolerance in mice. Infect. Immun. 63:1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGaha TL, Phelps RG, Spiera H, Bona C. 2002. Halofuginone, an inhibitor of type-I collagen synthesis and skin sclerosis, blocks transforming-growth-factor-beta-mediated Smad3 activation in fibroblasts. J. Investig. Dermatol. 118:461–470. 10.1046/j.0022-202x.2001.01690.x [DOI] [PubMed] [Google Scholar]
- 23.Feldman S, Richardson J, Freeman C, Pollock S, Berger J, Kaplan F, Rhoa G. 1974. In vitro assessment of in vivo absorption of drug complexes. J. Pharm. Sci. 63:454–456. 10.1002/jps.2600630334 [DOI] [PubMed] [Google Scholar]
- 24.Palmiter RD. 1974. Magnesium precipitation of ribonucleoprotein complexes. Expedient techniques for the isolation of undergraded polysomes and messenger ribonucleic acid. Biochemistry 13:3606–3615 [DOI] [PubMed] [Google Scholar]
- 25.Oyadomari S, Mori M. 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11:381–389. 10.1038/sj.cdd.4401373 [DOI] [PubMed] [Google Scholar]
- 26.Clavarino G, Claudio N, Dalet A, Terawaki S, Couderc T, Chasson L, Ceppi M, Schmidt EK, Wenger T, Lecuit M, Gatti E, Pierre P. 2012. Protein phosphatase 1 subunit Ppp1r15a/GADD34 regulates cytokine production in polyinosinic:polycytidylic acid-stimulated dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 109:3006–3011. 10.1073/pnas.1104491109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11:619–633. 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 28.Kilberg MS, Shan J, Su N. 2009. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 20:436–443. 10.1016/j.tem.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Broser M, Rom WN. 1994. Activation of the interleukin 6 gene by Mycobacterium tuberculosis or lipopolysaccharide is mediated by nuclear factors NF-IL6 and NF-kappa B. Proc. Natl. Acad. Sci. U. S. A. 91:2225–2229. 10.1073/pnas.91.6.2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann M, Wohlleben G, Chuvpilo S, Kistler B, Wirth T, Serfling E, Schimpl A. 1996. CD40, but not lipopolysaccharide and anti-IgM stimulation of primary B lymphocytes, leads to a persistent nuclear accumulation of RelB. J. Immunol. 157:4862–4869 [PubMed] [Google Scholar]
- 31.Ohmori Y, Tebo J, Nedospasov S, Hamilton TA. 1994. Kappa B binding activity in a murine macrophage-like cell line. Sequence-specific differences in kappa B binding and transcriptional activation functions. J. Biol. Chem. 269:17684–17690 [PubMed] [Google Scholar]
- 32.Xie QW, Kashiwabara Y, Nathan C. 1994. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 269:4705–4708 [PubMed] [Google Scholar]
- 33.Endo M, Mori M, Akira S, Gotoh T. 2006. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 176:6245–6253 [DOI] [PubMed] [Google Scholar]
- 34.Gao H, Schwartz RC. 2009. C/EBPzeta (CHOP/Gadd153) is a negative regulator of LPS-induced IL-6 expression in B cells. Mol. Immunol. 47:390–397. 10.1016/j.molimm.2009.09.002 [DOI] [PubMed] [Google Scholar]
- 35.Hattori T, Ohoka N, Hayashi H, Onozaki K. 2003. C/EBP homologous protein (CHOP) up-regulates IL-6 transcription by trapping negative regulating NF-IL6 isoform. FEBS Lett. 541:33–39. 10.1016/S0014-5793(03)00283-7 [DOI] [PubMed] [Google Scholar]
- 36.Natsuka S, Akira S, Nishio Y, Hashimoto S, Sugita T, Isshiki H, Kishimoto T. 1992. Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood 79:460–466 [PubMed] [Google Scholar]
- 37.Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, Mellor AL, He Y, Munn DH. 2009. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 113:6102–6111. 10.1182/blood-2008-12-195354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiribau CB, Gaccioli F, Huang CC, Yuan CL, Hatzoglou M. 2010. Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol. Cell. Biol. 30:3722–3731. 10.1128/MCB.01507-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yim HS, Choi KM, Kim B, Jung ID, Park YM, Kang YK, Lee MG. 2013. Effect of 1-methyl-d-tryptophan and the adoptive transfer of dendritic cells on polymicrobial sepsis induced by cecal content injection. Microbiol. Immunol. 57:633–639. 10.1111/1348-0421.12081 [DOI] [PubMed] [Google Scholar]
- 40.Reilly JG, McTavish SF, Young AH. 1997. Rapid depletion of plasma tryptophan: a review of studies and experimental methodology. J. Psychopharmacol. 11:381–392. 10.1177/026988119701100416 [DOI] [PubMed] [Google Scholar]
- 41.Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, Dougherty DM. 2009. L-tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int. J. Tryptophan Res. 2:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wek SA, Zhu S, Wek RC. 1995. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol. 15:4497–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wek RC, Jiang HY, Anthony TG. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34:7–11. 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- 44.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. 2003. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 23:5651–5663. 10.1128/MCB.23.16.5651-5663.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang HY, Wek RC. 2005. GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem. J. 385:371–380. 10.1042/BJ20041164 [DOI] [PMC free article] [PubMed] [Google Scholar]