

Abstract
IκBα is an inhibitor of NF-κB, a family of transcription factors that transactivate genes related to inflammation. Upon inflammatory stimuli, IκBα is rapidly degraded via the ubiquitin-proteasome pathway. While it is very clear that the SCFβ-TRCP ubiquitin ligase ubiquitinates IκBα upon stimulation, little is known about the postubiquitinational events of IκBα proteolysis. Here, we report that p97, a valosin-containing protein (also called VCP), plays an essential role in the postubiquitinational regulation of IκBα turnover after tumor necrosis factor alpha (TNF-α) or interleukin-1β (IL-1β) treatment. The ATPase activity of p97 is essential for its role in IκBα proteolysis. Moreover, we found that UFD1L and NPL4, two cofactors of p97, assist p97 to control the postubiquitinational regulation of IκBα. The p97-UFD1L-NPL4 protein complex specifically associates with ubiquitinated IκBα via the interactions between p97 and the SCFβ-TRCP ubiquitin ligase and between the polyubiquitin binding domain of UFD1L and polyubiquitinated IκBα. Furthermore, we observed that the postubiquitinational regulation of IκBα by the p97-UFD1L-NPL4 complex is important for NF-κB activation under stimuli.
INTRODUCTION
NF-κB is a family of transcription factors that regulate the inflammatory response. NF-κB is normally sequestered in the cytoplasm as an inactive form through association with its inhibitors, IκBs, including the most prominent one, IκBα (1). Inflammatory stimuli, such as cytokines, pathogens, and cellular stress, activate an IκB kinase (IKK) kinase complex that phosphorylates IκBα at serine-32 (Ser-32) and serine-36 (Ser-36) and promote its ubiquitination and subsequent degradation through the 26S proteasome (2–4). Consequently, NF-κB is released and translocated into the nucleus, where it activates a spectrum of proinflammatory genes.
In addition to phosphorylation, ubiquitination and deubiquitination play indispensable roles in NF-κB activation (2–4). The ubiquitination of IκBα is catalyzed by the SCFβ-TRCP ubiquitin ligase, a family member of the SCF ubiquitin ligase family, which consists of a small ring finger protein, Rbx1, a scaffold subunit, cullin 1 (Cul1), a linker protein, Skp1, and one of ∼70 human F-box proteins that determine the specificity of SCF ubiquitin ligases (5, 6). Among the SCFβ-TRCP ubiquitin ligases, β-TRCP protein binds directly to phosphorylated IκBα at Ser-32 and Ser-36 and ubiquitinates and triggers its degradation through the 26S proteasome (7–10). Two homologs of β-TRCP have been found in the human genome, and both of them are involved in IκBα ubiquitination (11).
While the role of ubiquitination in IκBα proteolysis is very clear, less is known about the postubiquitinational regulation of IκBα. Recent studies indicated that the postubiquitinational process is a tightly regulated process (12). Ubiquitinated proteins are often recognized by a family of polyubiquitin receptors which transport their targets to the proteasome and surrender them for degradation. Recognition is a critical step, because ubiquitin has seven lysine residues, each of which can be engaged in ubiquitin chain formation. Polyubiquitin chains with different linkages play distinctive roles in determining the fate of ubiquitinated proteins (12–14). Therefore, polyubiquitin chains can be likened to molecular ZIP codes, and polyubiquitin receptors function as mailmen who sort and deliver ubiquitinated proteins to different destinations according to their codes, i.e., the different linkages of polyubiquitin chains. Polyubiquitin chains formed through lysine-48 or lysine-11 of ubiquitin function as death signals (13, 15). Polyubiquitin receptors that shuttle substrates to the 26S proteasome often have distinctive ubiquitin binding domains (UBD) (12, 13).
The polyubiquitin receptor responsible for shuttling IκBα to the proteasome is unknown. It has been shown that overexpressed human FLIC-1 and FLIC-2 (also called ubiquilin-1 and ubiquilin-2), two polyubiquitin binding proteins, could hinder tumor necrosis factor alpha (TNF-α)-induced IκBα degradation (16). However, the actual role of these ubiquilin proteins in IκBα proteolysis is still unclear. Some evidence suggests that IκBα is transported by p97, a valosin-containing protein (VCP), which is involved in endoplasmic reticulum-associated protein degradation (ERAD) (17–20) and degradation of cytosolic and nuclear proteins (21–28). p97 was found in association with ubiquitinated IκBα (29), consistent with a role in transport, although no solid evidence yet exists to suggest this interaction is relevant to IκBα degradation. Recently, p97, together with its cofactor UBXD7, was found to control the degradation of HIF1α (22). p97, together with its cofactors, UFD1L and NPL4, was also found to regulate the turnover of Cdt1 and CD4 (21, 27). Moreover, p97 was shown to regulate protein turnover upon DNA damage response (21, 23, 26, 28). Using proteomic analysis, Alexandru et al. identified a family of p97 cofactors, many of which have a UBX domain that is important for their interaction with p97 (22). This raises a number of very important questions. Does p97 control postubiquitinational events in the degradation of IκBα? Does p97 recognize ubiquitinated IκBα through interaction with its cofactor, and if so, which one? Is the p97 protein complex important for NF-κB activation? Moreover, FAF1, a cofactor of p97, binds to β-TRCP2, also called FBW1B (22), so we want to ask whether postubiquitinational regulation of IκBα by p97 is coupled with ubiquitination of IκBα by its ubiquitin ligase, SCFβ-TRCP. Here, we provide genetic and biochemical evidence to show that IκBα is a bona fide substrate of p97. We found that p97 and its cofactors, UFD1L and NPL4, mediate the postubiquitinational regulation of IκBα by association with the SCFβ-TRCP ubiquitin ligase and ubiquitinated IκBα. Moreover, we found that the p97 protein complex is important for cytokine-induced NF-κB activation.
MATERIALS AND METHODS
Plasmids and plasmid transfection.
Open reading frames of human p97, IκBα, β-TRCP2, UFD1L, and NPL4 were amplified by PCR with Phusion high-fidelity DNA polymerase (New England Biolabs) from HeLa cDNAs that were prepared using a Superscript III cDNA synthesis kit (Invitrogen). Purified PCR products were cloned into the Gateway entry vector pENTR/D-Topo (Invitrogen). After verification of their sequences, the entry clones were shuttled to Gateway recipient vectors using LR Clonase (Invitrogen). Short interfering RNA (siRNA)-resistant rescue plasmids were generated using PCR-based mutagenesis.
Plasmids were transfected using Lipofectamine 2000 (Invitrogen).
Antibodies and other reagents.
The following antibodies were employed in this study: Cul1 (Invitrogen), Flag (Sigma), IκBα (C-21 rabbit antibody from Santa Cruz Biotechnology; monoclonal antibody from Cell Signaling), phosphoserine-32–IκBα (Cell Signaling), hemagglutinin (HA) and NPL4 (Santa Cruz), PSMB5 (Abcam), p97 (Cell Signaling and Bethyl Laboratories), Strep-tag (GeneScript), UFD1L (BD Biosciences), and ubiquitin (FK2; Millipore). The K48 and the K63 linkage-specific polyubiquitin antibodies and the β-TRCP antibody were reported previously (30, 31).
The anti-p97 antibody from Bethyl Laboratories and the monoclonal anti-IκBα antibody were employed for Western blotting, unless otherwise indicated.
Strep-Tactin resins, anti-Flag M2 affinity gel, and streptavidin beads are from Qiagen, Sigma-Aldrich, and Fisher Scientific, respectively.
Cell culture and retrovirus and lentivirus infection.
HeLa, U2OS, 293-TREX, and 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 5% CO2 at 37°C. The procedures of lentivirus and retrovirus production and infection were reported previously (32, 33). 293-TREX cells were purchased from Invitrogen. Doxycycline at 2 μg/ml was added to induce gene expression.
siRNA transfection.
siRNAs (200 pmol) from either Invitrogen or Sigma/Aldrich were transfected into HeLa, U2OS, or 293T cells (in 6-well dishes) using Lipofectamine RNAiMAX from Invitrogen. After 72 h, cells were left untreated or were treated with TNF-α or interleukin-1β (IL-1β) at 50 ng/ml for 10 min prior to lysis in SDS buffer. Extracts were quantified using a Bradford reagent (Bio-Rad) and subjected to SDS-PAGE and Western blotting.
Immunoprecipitation (IP) and protein purification.
Cells were harvested and lysed in extraction buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Nonidet-P40, 0.2 mM dithiothreitol, 10 mM β-glycerol phosphate, 10 mM p-nitrophenyl phosphate, 0.1 mM okadaic acid, 10 mM sodium fluoride, 5 μg/ml aprotinin, 1 μg/ml pepstatin, 1 mM leupeptin, and 0.1 mM phenylmethylsulfonyl fluoride), and the lysate was cleared by ultracentrifugation at 4°C. The extract was incubated with anti-Flag beads or antibody with protein A/G resin at 4°C for 1 h with slow rotation. Unbound proteins were washed away with extraction buffer four times, and bead-bound proteins were subjected to SDS-PAGE and Western blotting.
Biotinylated proteins were purified according to a previous report (34).
SCFβ-TRCP1 and SCFβ-TRCP2 were purified by a recombinant baculovirus method (35). Recombinant glutathione S-transferase (GST)–p97 proteins were expressed in DE3 Rosetta bacterial cells and purified with glutathione-Sepharose beads (GE Healthcare Life Sciences).
Reverse transcription-PCR (RT-PCR) and luciferase assay.
Total RNAs, isolated using TRIzol (Invitrogen), were employed to make cDNAs using Superscript III cDNA synthesis kit (Invitrogen). Cox2 messenger was amplified by PCR with limited cycles.
Luciferase assays were done using the dual-luciferase reporter assay system (Promega).
RESULTS
p97 depletion blocks the degradation but not ubiquitination of IκBα upon TNF-α stimulation.
It has been shown that p97 binds to ubiquitinated IκBα (29), implying that p97 is involved in the postubiquitinational regulation of IκBα. However, more loss-of-function and biochemical studies are needed to support this observation. To explore a potential role of p97 in cytokine-induced IκBα proteolysis, we employed an RNA interference (RNAi) approach to silence p97 in HeLa, a cervical carcinoma cell line that responds to TNF-α stimulation (Fig. 1A). IκBα was rapidly degraded in 10 min after addition of TNF-α (Fig. 1A, top, compare lanes 1 and 2). Knockdown of PSMB5 (Fig. 1A), a subunit of the 26S proteasome, did not affect the abundance of IκBα (Fig. 1A, top, compare lanes 1 and 3), suggesting that IκBα is a stable protein in HeLa cells under unstimulating conditions. The TNF-α-induced IκBα degradation, however, was blocked when PSMB5 was silenced (Fig. 1A, top, compare lanes 2 and 4), confirming that IκBα turnover induced by TNF-α treatment is mediated by the 26S proteasome. When p97 was silenced (Fig. 1A), IκBα expression was slightly decreased (Fig. 1A, top, compare lanes 1, 5, 7, 9, and 11), but the TNF-α-induced IκBα degradation was inhibited (Fig. 1A, top, compare lanes 2, 6, 8, 10, and 12). Four small interference RNA (siRNA) oligonucleotides targeting p97 produced similar phenotypes, suggesting that the observed RNAi phenotype was not an off-target event. Together, these data indicate that p97, like the 26S proteasome, is essential for IκBα proteolysis upon TNF-α treatment. Interestingly, we noticed a ladder of proteins of greater molecular mass than IκBα that also reacted with anti-IκBα antibody when either p97 or PSMB5 was silenced by RNAi (Fig. 1A, top, lanes 4, 6, 8, 10, and 12). These high-molecular-weight proteins also reacted with another anti-IκBα antibody from a different source (Fig. 1B) and with the anti-phospho-IκBα antibody targeting Ser-32 of IκBα (Fig. 1C), hinting that they are authentic IκBα proteins that are ubiquitinated but not degraded. Indeed, cosilencing of p97 and IκBα erased both low- and high-molecular-mass bands (Fig. 1D, compare lanes 4 and 6), further supporting the idea that high-molecular-mass IκBα proteins accumulated when the 26S proteasome or p97 was inactivated using the RNAi approach.
FIG 1.

p97 is required for TNF-α-induced IκBα degradation. HeLa cells were subjected to RNAi of PSMB5, p97, or β-TRCP as described in Materials and Methods. Western blotting was performed with anti-IκBα antibodies to show the dynamic change of IκBα under TNF-α treatments. The p97 antibody is from Cell Signaling. (A) Silencing of either PSMB5 or p97 blocked TNF-α-induced IκBα proteolysis. (B) The blot shown in panel A was stripped and then probed with a polyclonal anti-IκBα antibody. (C) Accumulation of phosphorylated IκBα upon either PSMB5 or p97 was depleted using siRNA. (D) Confirmation of both high- and low-molecular-mass IκBα proteins by cosilencing of p97 and IκBα. (E) β-TRCP depletion blocked TNF-α-induced IκBα ubiquitination. (F) The wild type and an ATPase-defective mutant, E578Q of p97, were delivered into HeLa cells using a lentiviral infection method. TNF-α was employed to trigger IκBα ubiquitination and degradation. A Western blotting method was employed to detect modified and unmodified IκBα.
In contrast to p97 depletion, silencing of β-TRCP, including both β-TRCP1 and β-TRCP2, did not accumulate high-molecular-mass IκBα proteins (Fig. 1E, compare lanes 4 and 6), indicating that p97 regulates the postubiquitinational events of IκBα upon TNF-α treatment.
p97 is an ATPase that belongs to the AAA1 ATPase family; therefore, it is interesting to determine whether its ATPase activity is important for its function in IκBα turnover. We constructed an E578Q mutant of p97 that is defective in its ATPase activity (36). We then expressed the wild type (WT) and the E578Q mutant in HeLa cells (Fig. 1F) and induced IκBα degradation with TNF-α treatment. We found that the wild-type p97 did not affect TNF-α-induced IκBα turnover (Fig. 1F, compare lanes 2 and 4); however, the E578Q mutant blocked the degradation but not ubiquitination of IκBα (Fig. 1F, compare lanes 2 and 6). p97 forms a homohexamer, and the E578Q mutant can interact with wild-type p97 (37). Therefore, our data demonstrate that the E578Q mutant functions as a dominant-negative one, and the ATPase activity of p97 is required for its role in TNF-α-induced IκBα turnover.
To determine whether those high-molecular-weight proteins are truly ubiquitinated IκBα, endogenous IκBα was immunoprecipitated using anti-IκBα antibody after TNF-α stimulation. As expected, IκBα proteins at high molecular weights were observed in IκBα immunoprecipitates, but only if p97 was previously silenced by RNAi (Fig. 2A). Those high-molecular-weight IκBα protein bands reacted with the antiubiquitin FK2 antibody that recognizes conjugated ubiquitin specifically (Fig. 2B). Moreover, those high-molecular-mass IκBα proteins were recognized by the K48 linkage-specific antibody (30) (Fig. 2C) but not by the K63 linkage-specific antibody (data not shown). These results support that IκBα is ubiquitinated but not degraded upon TNF-α stimulation if p97 is silenced.
FIG 2.

Accumulation of ubiquitinated IκBα under TNF-α treatments in p97-silenced cells. An IP-Western blotting approach was employed to confirm that the high-molecular-mass IκBα antibody-reactive bands are truly ubiquitinated IκBα. (A) Endogenous IκBα proteins were immunoprecipitated using a polyclonal anti-IκBα antibody in a native buffer and a monoclonal anti-IκBα antibody reacted with anti-IκBα immunoprecipitates. (B) Endogenous IκBα proteins were immunoprecipitated using the polyclonal anti-IκBα antibody in a native buffer and antiubiquitin FK2 antibody reacted with anti-IκBα immunoprecipitates. (C) The blot shown in panel B was stripped and probed with the K48 linkage-specific antibody. (D) Exogenous biotin-tagged IκBα (Bio-IκBα) was expressed in 293T cells and responded to TNF-α for destruction. (E) Ubiquitinated and unmodified Bio-IκBα proteins were precipitated using streptavidin beads under denaturing conditions, and the FK2 antibody reacted with precipitates. (F) The blot shown in panel E was stripped and then probed with the polyclonal anti-IκBα antibody. (G) The blot shown in panel F was stripped and probed with the K48 linkage-specific antibody. (H) Total ubiquitinated proteins were pulled down with streptavidin beads in a HeLa-biotin-ubiquitin cell line under denaturing conditions. Ubiquitinated IκBα was validated using the polyclonal anti-IκBα antibody.
We then considered the possibility that the ubiquitin conjugates detected in IκBα immunoprecipitates were due to ubiquitinated IκBα-binding proteins, because we did the anti-IκBα immunoprecipitation (IP) under native conditions. To further validate that the high-molecular-weight proteins were truly ubiquitinated IκBα, we expressed a biotin-tagged IκBα (Bio-IκBα) in 293T cells using retrovirus infection (Fig. 2D). We found that exogenous Bio-IκBα still responded to TNF-α treatment (Fig. 2D), although its expression was slightly higher than that of the endogenous one (Fig. 2D). This result indicates that the biotin tag did not alter the degradation pathway. We then purified Bio-IκBα with streptavidin beads under denaturing conditions as we previously reported (34). Mammalian cells contain only a few endogenous biotinylated proteins (38). Therefore, more specific results can be achieved. We detected ubiquitinated IκBα using the FK2 antibody (Fig. 2E). Small amounts of ubiquitinated Bio-IκBα were found upon TNF-α treatment (Fig. 2E, lane 2), whereas more ubiquitinated Bio-IκBα was accumulated in p97-depleted cells (Fig. 2E, compare lanes 2 and 4). We reconfirmed our observation by reprobing the same membrane with anti-IκBα antibody (Fig. 2F). Moreover, those high-molecular-mass proteins reacted with the K48 linkage-specific antibody (Fig. 2G) but not with the K63 linkage-specific antibody (data not shown). Interestingly, an FK2 and K48 antibody-reactive band appeared in p97-depleted but untreated cells (Fig. 2E and G, lane 3) but did not react with anti-IκBα antibody (Fig. 2F, lane 3), suggesting that some endogenous biotinylated proteins are conjugated with K48 polyubiquitin chains, and their degradation is controlled by p97 as well.
To examine whether endogenous IκBα was truly ubiquitinated, we treated a HeLa-biotin-ubiquitin cell line with TNF-α. In this cell line, the biotin-tagged ubiquitin was incorporated into polyubiquitin chains attached to ubiquitinated substrates. As we reported previously (34), total ubiquitinated proteins were purified using streptavidin beads under denaturing conditions. Western blotting with anti-IκBα antibody confirmed that ubiquitinated IκBα was detectable upon TNF-α treatment (Fig. 2H). More ubiquitinated IκBα was amassed in p97-silenced cells upon TNF-α treatment (Fig. 2H, compare lanes 2 and 4). Taken together, these data strongly support the conclusion that IκBα is ubiquitinated but not degraded when p97 is suppressed, demonstrating that p97 has no direct impact on ubiquitination but affects postubiquitinational processing of IκBα after TNF-α stimulation.
The postubiquitinational regulation of IκBα by p97 is most likely a common pathway in control of IκBα turnover.
IκBα proteolysis through the ubiquitin-proteasome pathway is induced by various stimulators (1, 4). However, signaling pathways upstream of the IKK kinase complex are diversified among different stimuli (39). To address whether p97-controlled postubiquitinational regulation of IκBα is a universal phenomenon, we depleted p97 in U2OS cells (Fig. 3A, middle), a human osteosarcoma line that responds to both TNF-α and IL-1β for IκBα turnover. IκBα was rapidly destroyed upon either TNF-α or IL-1β application in U2OS cells (Fig. 3A, top, compare lanes 1, 2, and 3). However, IκBα degradation was blocked when p97 was silenced regardless of which cytokine was applied (Fig. 3A, top, compare lanes 2, 3, 5, and 6). Once again, unlike siβ-TRCP, sip97 U2OS cells accumulated ubiquitinated IκBα (Fig. 3B), suggesting that p97 controls postubiquitinational regulation of IκBα in response to either TNF-α or IL-1β. A similar result was also found in TNF-α-treated 293T, a transformed human embryonic kidney cell line, when p97 was depleted by siRNA (Fig. 3C). Again, four independent siRNA oligonucleotides of p97 produced similar phenotypes. Taken together, we found that p97-mediated postubiquitinational regulation of IκBα is a common process that is required for IκBα proteolysis upon various cytokine treatments in different cell lines.
FIG 3.
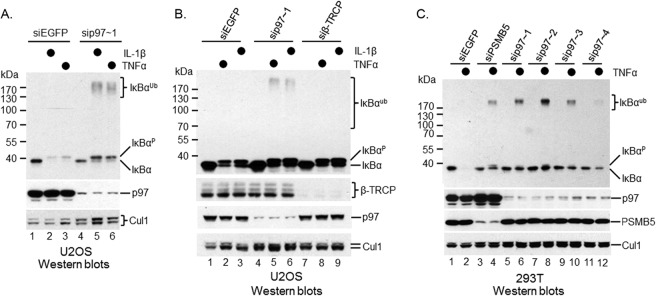
p97-mediated postubiquitinational regulation of IκBα is a common pathway of IκBα proteolysis. p97 was depleted using siRNA oligonucleotides in multiple cell lines. The function of p97 in postubiquitinational regulation of IκBα under either TNF-α or IL-1β was evaluated. The p97 antibody is from Cell Signaling. (A) sip97 blocked IκBα proteolysis in U2OS cells treated with either TNF-α or IL-1β. (B) β-TRCP depletion blocked TNF-α-induced IκBα ubiquitination, whereas silencing of p97 inhibited TNF-α-induced IκBα proteolysis in U2OS cells. (C) sip97 inhibited IκBα destruction in 293T cells treated with TNF-α.
p97 interacts with ubiquitinated IκBα.
p97 was found to associate with ubiquitinated IκBα (29). To confirm this observation, we expressed Flag/HA-tagged p97 (Flag/HA-p97) in a HeLa cell line using a lentivirus infection method. The exogenous Flag/HA-p97 was expressed at a level comparable to that of endogenous p97 (Fig. 4A). We then treated HeLa-Flag/HA-p97 cells with TNF-α to trigger IκBα proteolysis. To capture ubiquitinated IκBα, we added the proteasome inhibitor MG132 30 min before the TNF-α treatment. p97 and its associated proteins were immunoprecipitated using anti-HA antibody. Western blotting with anti-IκBα antibody showed that only ubiquitinated IκBα proteins were associated with p97 (Fig. 4B). This phenomenon was reconfirmed by IκBα depletion (Fig. 4B). Moreover, p97 did not interact with endogenous IκBα in unstimulated cells (Fig. 4C). Therefore, p97 can bind to IκBα, but only when it is ubiquitinated.
FIG 4.

p97 binds to ubiquitinated IκBα. An IP-Western blotting approach was employed to examine the association of p97 with ubiquitinated IκBα. (A) Flag/HA-p97 was expressed in HeLa cells. (B) Flag/HA-p97 was immunoprecipitated from HeLa-Flag/HA-p97 cells using anti-HA antibody. The association of p97 with ubiquitinated IκBα was verified using anti-IκBα antibody. (C) HeLa and HeLa-Flag/HA-p97 cells were treated with MG132 for 30 min and then with TNF-α for 10 min or no further treatment. Cells were lysed for IP with anti-HA antibody, followed by Western blotting using anti-IκBα antibody. (D) Endogenous IκBα was immunoprecipitated using a polyclonal anti-IκBα antibody and confirmed with anti-IκBα antibody. (E) The interaction between endogenous IκBα and p97 was confirmed by Western blotting using anti-p97 antibody.
To verify the interaction between p97 and IκBα at an endogenous level, we purified IκBα and its associated proteins using anti-IκBα antibody (Fig. 4D) and found that endogenous p97 associated with IκBα when TNF-α-induced degradation of IκBα was blocked by MG132 in HeLa cells (Fig. 4E). These data further support the hypothesis that p97 interacts with IκBα, especially ubiquitinated IκBα under stimuli.
p97 interacts with the SCFβ-TRCP ubiquitin ligase.
How p97 selects its substrates is still a mystery. p97 only binds to ubiquitinated IκBα, suggesting that IκBα alone is insufficient for substrate selection of p97. p97 binds to many ubiquitin ligases, including many members of the cullin/RING E3 family (22). To examine the interaction between p97 and SCFβ-TRCP ex vivo, we expressed a Flag/HA-tagged β-TRCP2 (Flag/HA-β-TRCP2) in the 293-TREX cell line (Invitrogen) using a retrovirus infection method. The exogenous Flag/HA-β-TRCP2 was induced with doxycycline (Fig. 5A) and isolated using anti-HA antibody (Fig. 5B). Western blotting with anti-p97 antibody showed that p97 associated with Flag/HA-β-TRCP2 (Fig. 5C). Of note, some Flag/HA-β-TRCP2 proteins were observed without doxycycline induction, suggesting that the expression of Flag/HA-β-TRCP2 was leaky in 293-TREX cells (Fig. 5A). Correspondingly, residual p97 was observed to associate with Flag/HA-β-TRCP2 without doxycycline treatment (Fig. 5C). To further confirm the interaction between p97 and β-TRCP, we expressed Strep-tagged p97 (Strep-p97) in 293T cells (293T-Strep-p97) using a lentiviral infection approach. Western blotting showed that the exogenous Strep-p97 was expressed at a level comparable to that of the endogenous p97 (Fig. 5D). We then expressed triple HA-tagged β-TRCP2 (HA3-β-TRCP2) in 293T-Strep-p97 cells using a transfection method (Fig. 5D). TNF-α was employed to trigger IκBα turnover (Fig. 5D). The exogenous Strep-p97 and its associated proteins were purified using Strep-Tactin resins (Fig. 5E). HA3-β-TRCP2 was found to associate with Strep-p97 (Fig. 5E). Together, our data demonstrate that p97 interacts with β-TRCP2, although we do not know if such an interaction is direct or indirect. Interestingly, such an interaction was enhanced upon TNF-α treatment (Fig. 5C and E, compare lanes 3 and 4).
FIG 5.

p97 binds to the SCFβ-TRCP ubiquitin ligase. An IP-Western blotting approach was employed to examine the association of p97 with the SCFβ-TRCP ubiquitin ligase. In vitro GST pulldown was applied to analyze the interaction between recombinant p97 and the SCFβ-TRCP ubiquitin ligase. (A) Flag/HA-β-TRCP2 was expressed in a 293-TREX cell line. Anti-HA antibody was used to show the induction of Flag/HA-β-TRCP2 by doxycycline. (B) Exogenous Flag/HA-β-TRCP2 was immunoprecipitated using anti-HA antibody. (C) The association of p97 with the SCFβ-TRCP2 ubiquitin ligase was verified using anti-p97 antibody. (D and E) 293T-Strep-p97 cells were transfected with either empty vector or vector expressing HA3-β-TRCP2. Seventy-two h later, cells were treated with TNF-α for 10 min or left untreated. Cells were lysed for Strep-Tactin pulldown, followed by Western blotting using anti-HA and anti-Strep antibodies. (F) Purified GST-p97 from bacteria was shown using the Coomassie blue staining method. (G) In vitro interaction between purified GST-p97 and the SCFβ-TRCP1 ubiquitin ligase using GST pulldown assay. (H) In vitro interaction between purified GST-p97 and the SCFβ-TRCP2 ubiquitin ligase using a GST pulldown assay.
To determine whether p97 binds to the SCFβ-TRCP ubiquitin ligase directly, we turned to an in vitro binding approach using recombinant proteins. We purified both SCFβ-TRCP1 and SCFβ-TRCP2 protein complexes via a procedure we developed for the purification of SCF ubiquitin ligases using a recombinant baculovirus method (35). We also purified GST-tagged p97 (GST-p97) using a bacterial expression system (Fig. 5F). We then employed an in vitro GST pulldown experiment to analyze the interaction between p97 and the SCFβ-TRCP ubiquitin ligase and found that ∼0.1% of either SCFβ-TRCP1 (Fig. 5G) or SCFβ-TRCP2 (Fig. 5H) binds to GST-p97, indicating that p97 binds to the SCFβ-TRCP ubiquitin ligase directly.
UFD1L and NPL4 are cofactors that regulate IκBα postubiquitination.
p97 binds to dozens of cofactors, some of which have been shown to assist p97's function in protein proteolysis (21–28, 40). To identify any p97 cofactors that could be involved in cytokine-induced IκBα turnover, we silenced either UFD1L or NPL4, two cofactors that have been reported to regulate CD4 proteolysis upon HIV infection (27). The ubiquitination of CD4 and IκBα is mediated by the same ubiquitin ligase, SCFβ-TRCP (4, 31). Given that p97 binds to SCFβ-TRCP (Fig. 5), it seemed conceivable that these two proteins were involved in IκBα postubiquitinational regulation. Indeed, when UFD1L was silenced by two independent siRNA oligonucleotides in HeLa cells (Fig. 6A), the degradation, but not ubiquitination, of IκBα upon TNF-α stimulation was inhibited (Fig. 6A). The ubiquitinated IκBα also reacted with the anti-phospho-IκBα antibody targeting Ser-32 of IκBα (Fig. 6B), indicating that UFD1L depletion mimicked the phenotype of knocking down p97. A similar phenotype was observed when UFD1L was silenced in 293T cells (Fig. 6C), indicating that the siUFD1L phenotype is not a cell line-specific event.
FIG 6.

UFD1L and NPL4 are required for IκBα proteolysis. An siRNA method was employed to deplete either UFD1L or NPL4 in multiple cell lines. Their effects in IκBα proteolysis under TNF-α treatment were evaluated using a Western blotting approach. (A) Ubiquitinated IκBα was accumulated in UFD1L-silenced HeLa cells treated with TNF-α. (B) Phosphorylated IκBα was accumulated in UFD1L-silenced HeLa cells treated with TNF-α. (C) Ubiquitinated IκBα was accumulated in UFD1L-silenced 293T cells treated with TNF-α. (D) Ubiquitinated IκBα was accumulated in NPL4-silenced HeLa cells treated with TNF-α. (E) Phosphorylated IκBα was accumulated in NPL4-silenced HeLa cells treated with TNF-α.
UFD1L forms a heterodimer with NPL4. Therefore, it is necessary to determine whether NPL4 is also involved in the postubiquitinational regulation of IκBα. We depleted NPL4 in HeLa cells using siRNA oligonucleotides (Fig. 6D) and observed that the degradation, but not ubiquitination, of IκBα was inhibited upon TNF-α treatment (Fig. 6D). Once again, ubiquitinated IκBα proteins reacted with the anti-phospho-IκBα antibody (Fig. 6E), suggesting that NPL4, like p97 and UFD1L, is important for the postubiquitinational regulation of IκBα under TNF-α treatment. Because UFD1L and NPL4 form a heterodimer, their expression could depend on each other. We then checked UFD1L expression in NPL4-silenced cells and noticed that UFD1L expression was reduced (Fig. 6D). Two independent siRNA oligonucleotides yielded similar phenotypes, suggesting it is unlikely to be an off-target event. Interestingly, NPL4 expression was not affected when UFD1L was silenced (Fig. 6A). Together, these data suggest that NPL4 is required for the proper expression of UFD1L, although the mechanism remains to be resolved.
Both UFD1L and NPL4 bind to ubiquitinated IκBα.
To better understand the role of UFD1L as the cofactor of p97 in the postubiquitinational regulation of IκBα, it was necessary to see whether UFD1L bound to ubiquitinated IκBα in a manner similar to that of p97. To do so, we expressed C-terminally Flag-tagged UFD1L (UFD1L-Flag) in HeLa cells (HeLa-UFD1L-Flag) using a retroviral infection approach. Consistent with p97 binding analysis, only ubiquitinated IκBα was found in anti-Flag immunoprecipitates (Fig. 7A), demonstrating that UFD1L, like p97, binds to ubiquitinated IκBα only.
FIG 7.
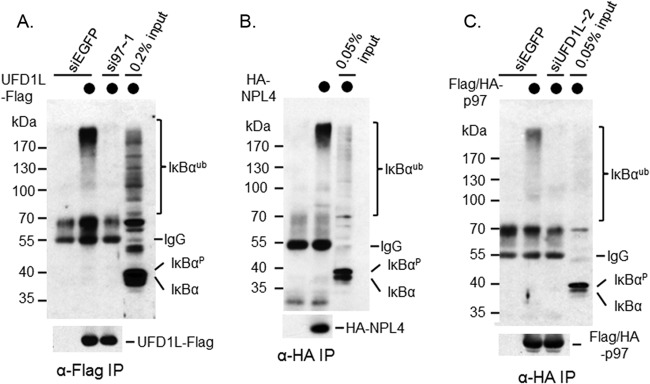
Each subunit of the p97 complex binds to ubiquitinated IκBα, and their interactions with ubiquitinated IκBα depend on each other. (A) Flag-UFD1L was expressed in HeLa cells and precipitated using anti-Flag M2 affinity gel. The association of UFD1L with ubiquitinated IκBα was confirmed by Western blotting. The interaction between ubiquitinated IκBα and Flag-UFD1L was attenuated when p97 was silenced. (B) HA-NPL4 was expressed in HeLa cells and precipitated using anti-HA antibody. The association of NPL4 with ubiquitinated IκBα was confirmed by Western blotting. (C) Flag/HA-p97 was expressed in HeLa cells and precipitated using anti-HA antibody. The association of p97 with ubiquitinated IκBα was confirmed by Western blotting. The interaction between ubiquitinated IκBα and Flag/HA-p97 was attenuated when UFD1L was silenced.
To determine whether NPL4 binds to ubiquitinated IκBα, we expressed HA-tagged NPL4 in HeLa cells (HeLa-HA-NPL4) using a retroviral infection approach. Consistent with both p97 and UFD1L binding analysis, only ubiquitinated IκBα appeared in anti-HA immunoprecipitates (Fig. 7B, top), indicating that NPL4, like p97 and UFD1L, interacts with ubiquitinated IκBα only.
Taken together, our data demonstrate that each subunit of the p97-UFD1L-NPL4 protein complex can associate with IκBα, but only when it is ubiquitinated.
Both p97 and UFD1L are important for the p97 complex to bind to ubiquitinated IκBα.
Having found that each subunit of the p97 protein complex binds to ubiquitinated IκBα, we next wanted to determine whether the binding of these subunits to ubiquitinated IκBα depends on each other. We observed that the interaction between ubiquitinated IκBα and p97 was almost abolished when UFD1L was silenced (Fig. 7C). Conversely, the interaction between ubiquitinated IκBα and UFD1L was almost reduced to the background level when p97 was depleted (Fig. 7A). Therefore, it is critical to maintain the p97 protein complex as an intact one so that the complex can recognize ubiquitinated IκBα.
The polyubiquitin binding domain of UFD1L is required for its function in IκBα turnover.
p97 contains no obvious polyubiquitin chain binding domain, whereas UFD1L has one at its N terminus, implying that the p97 protein complex uses the polyubiquitin chain binding domain (UBD) of UFD1L to recognize the polyubiquitin chains of IκBα. To prove it, we mutated five residues to alanines (A5 mutant) in the UBD of UFD1L (Fig. 8A). These residues were found to interact with polyubiquitin chain directly (41). We expressed the WT and the A5 mutant as siUFD1L-resistant cDNAs in HeLa cells using a lentiviral infection approach (Fig. 8B). Interestingly, when exogenous UFD1L proteins were expressed in HeLa cells, the expression of endogenous UFD1L was reduced (Fig. 8B, middle, compare lanes 1, 2, 5, 6, 9, and 10). To determine the function of the UBD of UFD1L, we silenced endogenous UFD1L (Fig. 8B) and then treated cells with TNF-α to trigger IκBα turnover. Depletion of UFD1L blocked IκBα degradation but not ubiquitination (Fig. 8B, top, compare lanes 2 and 4). The WT, but not the A5 mutant, of UFD1L rescued TNF-α-induced IκBα turnover (Fig. 8B), suggesting that the UBD of UFD1L plays an essential role in TNF-α-induced IκBα proteolysis.
FIG 8.

Polyubiquitin binding domain of UFD1L is important for its function in control of postubiquitination of IκBα in HeLa cells upon TNF-α treatment. (A) Sequence alignment between the wild type and the A5 mutant of human UFD1L. (B) The wild type and the A5 mutant of UFD1L were expressed in HeLa cells as Flag tag fusion proteins. A Western blotting approach was employed to show expressed proteins. Their effects in control of postubiquitination of IκBα were evaluated using an RNAi rescue approach. (C) Exogenous Flag-UFD1L was purified using anti-Flag antibody. Its interactions with ubiquitinated IκBα, p97, and NPL4 were shown by Western blotting.
To further characterize the function of the UBD of UFD1L, we treated these cells with MG132 for 30 min and then with TNF-α for 10 min for IP-Western blotting assay. As expected, although the wild-type UFD1L associated with significant amounts of ubiquitinated IκBα, such an interaction was largely abolished from HeLa-Flag-UFD1L-A5 cells (Fig. 8C). Of note, Flag-UFD1L-A5 proteins migrated a little bit more slowly than wild-type UFD1L ones (Fig. 8B and C), raising the question of whether the A5 mutant has lost its other functions. Using an IP-Western blotting approach, we found that the interactions of the A5 mutant of UFD1L with p97 and NPL4 were not affected (Fig. 8C). Therefore, although the A5 mutant has lost its polyubiquitin binding ability, its other functions are maintained.
Ubiquitinated IκBα still associates with RelA, a subunit of NF-κB.
IκBα is an inhibitor of NF-κB (2–4). However, it is still unclear whether the ubiquitination process of IκBα can trigger its separation from NF-κB under stimulation. To address this issue, we expressed a Flag-tagged IκBα in HeLa-biotin-ubiquitin cells (Fig. 9A). Similar to endogenous IκBα, exogenous Flag-IκBα responded to TNF-α treatment, as it migrated more slowly upon TNF-α stimulation (Fig. 9A). We then purified ubiquitinated Flag-IκBα using anti-Flag M2 affinity gel, followed by elution with Flag peptides and pulldown with streptavidin beads (Fig. 9B). We found that both RelA and p97 bound to ubiquitinated IκBα. These data indicate that ubiquitinated IκBα still inhibits NF-κB. Therefore, the postubiquitinational regulation of IκBα is important for NF-κB activation upon stimulation.
FIG 9.

p97 protein complex is important to TNF-α-induced NF-κB activation. IP and pulldown approaches were employed to determine the interactions of ubiquitinated IκBα with either p97 or RelA. Dual-luciferase and semiquantitative RT-PCR approaches were used to evaluate the function of the p97 protein complex in TNF-α-induced NF-κB activation. (A) Flag-IκBα was expressed in the HeLa-biotin-ubiquitin cell line. Cells were treated with MG132 for 30 min and then either with TNF-α for 10 min or without further treatment. The expression of IκBα, p97, RelA, and Cul1 were shown using Western blotting. (B) Cells from panel A were collected for IP using anti-Flag M2 affinity gel. The bound proteins were eluted using Flag peptides. Ubiquitinated IκBα was purified using streptavidin beads. A Western blotting approach was employed to detect p97, RelA, and ubiquitinated IκBα. (C) Dual-luciferase assay in sip97 HeLa cells. (D) Dual-luciferase assay in siUFD1L 293T cells. (E) Cox2 induction was reduced in sip97 HeLa cells using RT-PCR. (F) Cox2 induction was decreased in siUFD1L HeLa cells using RT-PCR.
p97 and UFD1L are important for the activation of NF-κB as a transcription factor.
Having found that the p97-UFD1L protein complex controls the postubiquitinational processing of IκBα proteolysis under cytokine stimuli, we next needed to examine the contribution of p97 and UFD1L in NF-κB activation. To do so, we cotransfected pGL4.32[luc2P/NF-κB-RE/Hygro] (a firefly luciferase reporter plasmid containing an NF-κB response element), pSV40-hRluc (a renilla luciferase control plasmid), and siRNA for either enhanced green fluorescent protein (EGFP) or p97 into HeLa cells. Seventy-two hours later, cells were treated with TNF-α for either 2 or 4 h before collection for an in vitro dual-luciferase assay. We found that the firefly/renilla luciferase ratio was rapidly enhanced over the course of TNF-α treatments in siEGFP cells, while it was significantly reduced in p97-silenced cells (Fig. 9C). These data show that the activation of NF-κB as a transcription factor is attenuated in p97-silenced cells. Similar results were observed in 293T cells when UFD1L was silenced (Fig. 9D).
To determine whether p97 is important for the activation of NF-κB-targeted genes, we analyzed the expression of Cox2, a bona fide transcriptional target of NF-κB, under cytokine stimulation (42). Using a semiquantitative RT-PCR approach, we observed that Cox2 mRNA rapidly increased in HeLa cells after TNF-α treatment (Fig. 9E, compare lanes 1 and 2). The induction of Cox2 was dependent on RelA, because the expression of Cox2 was significantly attenuated when RelA was inactivated (Fig. 9E, compare lanes 6 and 2), indicating that RelA, as one of the NF-κB transcription factors, is responsible for the transactivation of Cox2 in HeLa cells in response to TNF-α. p97 depletion did not affect the basal expression of Cox2 but repressed the induction of Cox2 by TNF-α (Fig. 9E, compare lanes 4 and 2). Similar results were achieved when UFD1L was silenced in HeLa cells (Fig. 9F). Given that NPL4 depletion decreases the UFD1L expression (Fig. 6D), we expect to observe a similar result if we deplete NPL4. Based on these data, we conclude that the p97-UFD1L-NPL4 protein complex is important for the activation of NF-κB as a transcription factor under stimulation.
DISCUSSION
IκBα is the true substrate of p97.
The majority of studies concerning the roles of ubiquitination in NF-κB activation after stimuli focus on the signaling events that lead to IκBα ubiquitination by the SCFβ-TRCP ubiquitin ligase (2–4). In general, all of the upstream signal pathways are converted to the activation of the IKK kinase complex that phosphorylates IκBα at serine-32 and serine-36 residues (2–4). These phosphorylation events of IκBα create a phospho-degron motif on IκBα that is recognized by the SCFβ-TRCP ubiquitin ligase. In coordination with E2 enzymes, such as UbcH5 and Cdc34, the SCFβ-TRCP ubiquitin ligase conjugates polyubiquitin chains on IκBα (43). Consequently, ubiquitinated IκBα is sent to and degraded by the 26S proteasome. Recent studies suggest that postubiquitinational regulation is an essential step during protein turnover through the ubiquitin-proteasome pathway (12). It also becomes increasingly clear that diverse pathways exist to regulate postubiquitinational events of different proteins.
One well-studied function of p97 is to mediate protein turnover via the ERAD pathway. It was not until recent years that the role of p97 in protein degradation pathways other than ERAD has been explored (21–28). It has been speculated that IκBα is a substrate of p97 (29); however, no comprehensive genetic and biochemical evidence was provided until our study. Using an RNAi approach, we found that cytokine-induced IκBα degradation, but not ubiquitination, was inhibited in p97-silenced cells (Fig. 1 and 3), indicating that p97 is involved in the postubiquitinational regulation of IκBα proteolysis. Using various biochemical approaches, we confirmed our observation that p97 does not affect the ubiquitination but does affect the turnover of IκBα under cytokine stimulations (Fig. 2). The pathway governed by p97 is most likely universal to all of the extracellular signals that lead to IκBα proteolysis via the ubiquitin-proteasome pathway, considering that divergent signaling events have been uncovered only upstream of the IKK kinase complex thus far (2–4). Indeed, IκBα degradation was blocked when p97 was inactivated in multiple cell lines treated with different cytokines (Fig. 1 and 3), further supporting our hypothesis.
p97 contains an ATPase domain. Our results demonstrate that the ATPase activity of p97 is essential to its role in cytokine-induced IκBα proteolysis (Fig. 1F), indicating that the postubiquitinational regulation of cytokine-induced IκBα turnover is an energy-dependent process. Our data also show that the p97 protein complex is important for cytokine-induced NF-κB activation (Fig. 9), implying that p97 is a good drug target to control NF-κB activation and inflammatory responses.
UFD1L and NPL4 are cofactors of p97 in postubiquitinational control of IκBα.
Current models suggest that ubiquitinated substrates need to be recognized by a polyubiquitin binding protein, i.e., polyubiquitin receptor, and delivered to the 26S proteasome for degradation (12). p97 alone is unlikely to fulfill the entire mission, although p97 is a big protein and forms a homohexamer. Considering that p97 binds to dozens of cofactors, many of which contain a ubiquitin-binding domain (22, 40, 41), it is unlikely that p97 is involved in direct interaction with polyubiquitin chains on substrates. Indeed, we found that depletion of either UFD1L or NPL4, two cofactors of p97, produced phenotypes similar to that of did p97 depletion (Fig. 6), suggesting that these two proteins are cofactors of p97 in regulation of postubiquitination of IκBα. Moreover, our preliminary data suggest that NPL4 does not contribute directly to the postubiquitinational control of IκBα; rather, it maintains the proper expression of UFD1L. Two pieces of evidence support our hypothesis. First, NPL4 depletion reduces UFD1L expression (Fig. 6D), whereas UFD1L depletion does not affect NPL4 expression (Fig. 6A). Second, the expression of exogenous UFD1L can decrease the expression of endogenous UFD1L (Fig. 8B). UFD1L and NPL4 form a heterodimer. Therefore, we speculate that exogenous UFD1L competes with endogenous UFD1L for NPL4 binding and triggers the degradation of free endogenous UFD1L. This could be a common phenomenon, because a similar observation was found in p97-mediated CD4 degradation (27) and Cdt1 turnover (21). Further studies are needed to confirm this observation. Nevertheless, our data support that UFD1L is the main cofactor of p97 to regulate postubiquitination of cytokine-induced IκBα proteolysis. Of note, UFD1, a yeast homolog of human UFD1L, contains a ubiquitin-binding domain that is capable of associating with polyubiquitin chains (40). We found that the polyubiquitin binding-defective mutant of UFD1L was defective in association with ubiquitinated IκBα (Fig. 8C) and was unable to promote the postubiquitination of IκBα under TNF-α treatment (Fig. 8B), although it still associates with p97 and NPL4 (Fig. 8C). Therefore, the polyubiquitin binding domain of human UFD1L plays an essential role in postubiquitinational regulation of IκBα.
Model for p97 and UFD1L in postubiquitinational regulation of IκBα.
Thus far, only a few protein substrates of p97 other than the ERAD substrates have been identified in mammalian systems. These proteins are so divergent that no notable common features can be detected among them. An important issue is how specificity is determined with regard to the function of the p97 protein complexes in postubiquitinational regulation. Interestingly, three p97 substrates, including IκBα, CD4, and MCL1, share the same ubiquitin ligase, SCFβ-TRCP, although multiple ubiquitin ligases have been identified to mediate the ubiquitination of MCL1 (44–46). Therefore, it is possible that ubiquitin ligases play important roles in the selection of specific postubiquitination pathways for their cognate substrates. p97 interacts with many ubiquitin ligases, most likely via its cofactors (22). p97 protein complexes bind to both β-TRCP2, an F-box protein in the SCFβ-TRCP2 E3 complex via FAF1 (22), and HUWE1, the ubiquitin ligase of MCL1 after UV irradiation (46), via either p47 or UBXD8 (22). We found that recombinant p97 binds to both purified SCFβ-TRCP1 and SCFβ-TRCP2 (Fig. 5G and H). Moreover, p97 interacts with β-TRCP2 in 293 and 293T cells (Fig. 5B and E). Overall, the interaction between p97 and the SCFβ-TRCP ubiquitin ligase is relatively weak. Whether its cofactors, such as UFD1L, can enhance its binding affinity to the SCFβ-TRCP ubiquitin ligase remains to be examined. UFD1L also associates with several UBA domain-containing cofactors, including FAF1, SAKS1, UBXD7, and UBXD8, but not p47, most likely via p97 (22, 47). Therefore, it is highly possible that UFD1L is a common cofactor of p97 for many protein substrates, while other cofactors function as specificity factors to lead the p97-dependent pathway for the postubiquitinational process of protein proteolysis. We noticed that significant amounts of ubiquitinated IκBα accumulated in either p97- or UFD1L-silenced cells, suggesting that polyubiquitin chains of IκBα have been protected from nonspecific deubiquitinating enzymes (DUBs). Considering that the human genome contains more than 90 DUB genes (48), it is conceivable that ubiquitinated substrates would be deubiquitinated if their polyubiquitin chains were not protected. Such a ubiquitin-binding protein could be involved in the first step of IκBα postubiquitination and should interact with p97 directly or indirectly. If this is the case, FAF1, UBXD7, UBXD8, and SAKS1 are among the leading candidates for p97 cofactors to initiate the postubiquitinational processing of IκBα under stimuli. Of them, FAF1 is an intriguing one, because it binds to β-TRCP2 (22). However, FAF1 is unlikely to be involved in IκBα degradation directly, because its overexpression inhibited IκBα degradation under TNF-α treatment (49, 50), potentially by disrupting the IKK complex assembly (51). In contrast, overexpression of FAF1 promotes the degradation of β-catenin by enhancing its polyubiquitination (52). Given that ubiquitination of both β-catenin and IκBα is regulated by the SCFβ-TRCP ubiquitin ligase (7, 10), further studies are needed to clarify the function of FAF1 in the postubiquitinational regulation of IκBα.
Our data demonstrate that the p97 complex interacts with IκBα only when it is ubiquitinated (Fig. 4 and 7). Apparently, such an interaction requires the intact p97 protein complex, because p97 depletion can disrupt the association of UFD1L with ubiquitinated IκBα and vice versa (Fig. 7). Our results also indicate that the UBD domain of UFD1L is important for UFD1L to interact with ubiquitinated IκBα and TNF-α-induced IκBα proteolysis (Fig. 8). Moreover, p97 binds to the SCFβ-TRCP ubiquitin ligase directly (Fig. 5). Together, these data suggest that the p97 protein complex utilizes its p97 subunit to interact with the SCFβ-TRCP ubiquitin ligase and its UFD1L subunit to recognize ubiquitinated IκBα, respectively. Therefore, we believe that the p97 complex plays a critical role in cytokine-induced IκBα proteolysis and NF-κB activation.
How the p97 complex delivers the ubiquitinated IκBα to the 26S proteasome is another important question in IκBα proteolysis. p97 and several p97 cofactors associate with the 26S proteasome (53, 54). However, it is still unclear if the p97 complex contacts the proteasome directly or via additional polyubiquitin receptors. It is worth mentioning that human ubiquilin-1 and ubiquilin-2, also called PLIC1 and PLIC2, are polyubiquitin receptors that can block IκBα proteolysis if overexpressed in human cells (16). Thus far, the actual role of ubiquilin proteins in IκBα postubiquitinational regulation remains elusive. We speculate that these ubiquitin binding proteins, such as FAF1 and ubiquilin proteins, function as nonspecific ubiquitin binding proteins to disrupt cohesive pathways of postubiquitinational regulation if they are overexpressed. The fact that the UBA domain fragment of FAF1 delayed the TNF-α-induced IκBα turnover better than the full-length FAF1 (50) further supports our hypothesis. If UFD1L is a common cofactor of p97-mediated postubiquitinational regulation in protein turnover, we propose that UFD1L interacts with the polyubiquitin receptors on the 26S proteasome directly or indirectly to surrender ubiquitinated IκBα to the 26S proteasome.
ACKNOWLEDGMENTS
We thank Linda Guarino for critical reading of the manuscript. We also thank Vishva Dixit and Klaus Strebel for providing antibodies.
This work is supported by a National Institutes of Health grant (R01GM102529) and the Welch Foundation (AU-1711) to J.J. W.Z. is supported by a National Institutes of Health grant (DK080236).
Footnotes
Published ahead of print 18 November 2013
REFERENCES
- 1.Hayden MS, Ghosh S. 2008. Shared principles in NF-κB signaling. Cell 132:344–362. 10.1016/j.cell.2008.01.020 [DOI] [PubMed] [Google Scholar]
- 2.Harhaj EW, Dixit VM. 2011. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 21:22–39. 10.1038/cr.2010.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu S, Chen ZJ. 2011. Expanding role of ubiquitination in NF-κB signaling. Cell Res. 21:6–21. 10.1038/cr.2010.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skaug B, Jiang X, Chen ZJ. 2009. The role of ubiquitin in NF-kappaB regulatory pathways. Annu. Rev. Biochem. 78:769–796. 10.1146/annurev.biochem.78.070907.102750 [DOI] [PubMed] [Google Scholar]
- 5.Cardozo T, Pagano M. 2004. The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5:739–751. 10.1038/nrm1471 [DOI] [PubMed] [Google Scholar]
- 6.Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. 2004. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 18:2573–2580. 10.1101/gad.1255304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z. 1999. HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene 18:2039–2046. 10.1038/sj.onc.1202760 [DOI] [PubMed] [Google Scholar]
- 8.Hatakeyama S, Kitagawa M, Nakayama K, Shirane M, Matsumoto M, Hattori K, Higashi H, Nakano H, Okumura K, Onoé K, Good RA, Nakayama K. 1999. Ubiquitin-dependent degradation of IkappaBalpha is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc. Natl. Acad. Sci. U. S. A. 96:3859–3863. 10.1073/pnas.96.7.3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kroll M, Margottin F, Kohl A, Renard P, Durand H, Concordet JP, Bachelerie F, Arenzana-Seisdedos F, Benarous R. 1999. Inducible degradation of IkappaBalpha by the proteasome requires interaction with the F-box protein h-betaTrCP. J. Biol. Chem. 274:7941–7945. 10.1074/jbc.274.12.7941 [DOI] [PubMed] [Google Scholar]
- 10.Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. 1999. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 13:270–283. 10.1101/gad.13.3.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. 2003. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev. Cell 4:799–812. 10.1016/S1534-5807(03)00154-0 [DOI] [PubMed] [Google Scholar]
- 12.Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78:477–513. 10.1146/annurev.biochem.78.081507.101607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grabbe C, Husnjak K, Dikic I. 2011. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 12:295–307. 10.1038/nrm3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda F, Crosetto N, Dikic I. 2010. What determines the specificity and outcomes of ubiquitin signaling? Cell 143:677–681. 10.1016/j.cell.2010.10.026 [DOI] [PubMed] [Google Scholar]
- 15.Rape M. 2010. Assembly of k11-linked ubiquitin chains by the anaphase-promoting complex. Subcell. Biochem. 54:107–115. 10.1007/978-1-4419-6676-6_9 [DOI] [PubMed] [Google Scholar]
- 16.Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM. 2000. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6:409–419. 10.1016/S1097-2765(00)00040-X [DOI] [PubMed] [Google Scholar]
- 17.Bar-Nun S. 2005. The role of p97/Cdc48p in endoplasmic reticulum-associated degradation: from the immune system to yeast. Curr. Top. Microbiol. Immunol. 300:95–125. 10.1007/3-540-28007-3_5 [DOI] [PubMed] [Google Scholar]
- 18.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. 2006. Central pore residues mediate the p97/P97 activity required for ERAD. Mol. Cell 22:451–462. 10.1016/j.molcel.2006.03.036 [DOI] [PubMed] [Google Scholar]
- 19.Raasi S, Wolf DH. 2007. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 18:780–791. 10.1016/j.semcdb.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 20.Vembar SS, Brodsky JL. 2008. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9:944–957. 10.1038/nrm2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raman M, Havens CG, Walter JC, Harper JW. 2011. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell 44:72–84. 10.1016/j.molcel.2011.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. 2008. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 134:804–816. 10.1016/j.cell.2008.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma R, Oania R, Fang R, Smith GT, Deshaies RJ. 2011. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell 41:82–92. 10.1016/j.molcel.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wójcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN. 2006. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 17:4606–4618. 10.1091/mbc.E06-05-0432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu S, Peng G, Wang Y, Fang S, Karbowski M. 2011. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 22:291–300. 10.1091/mbc.E10-09-0748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franz A, Orth M, Pirson PA, Sonneville R, Blow JJ, Gartner A, Stemmann O, Hoppe T. 2011. CDC-48/p97 coordinates CDT-1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol. Cell 44:85–96. 10.1016/j.molcel.2011.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magadán JG, Pérez-Victoria FJ, Sougrat R, Ye Y, Strebel K, Bonifacino JS. 2010. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 6:e1000869. 10.1371/journal.ppat.1000869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. 2011. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 18:1345–1350. 10.1038/nsmb.2188 [DOI] [PubMed] [Google Scholar]
- 29.Dai RM, Chen E, Longo DL, Gorbea CM, Li CC. 1998. Involvement of valosin-containing protein, an ATPase co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkappaBalpha. J. Biol. Chem. 273:3562–3573. 10.1074/jbc.273.6.3562 [DOI] [PubMed] [Google Scholar]
- 30.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. 2008. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134:668–678. 10.1016/j.cell.2008.07.039 [DOI] [PubMed] [Google Scholar]
- 31.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1:565–574. 10.1016/S1097-2765(00)80056-8 [DOI] [PubMed] [Google Scholar]
- 32.Jin J, Arias EE, Chen J, Harper JW, Walter JC. 2006. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23:709–721. 10.1016/j.molcel.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 33.Jin J, Li X, Gygi SP, Harper JW. 2007. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447:1135–1138. 10.1038/nature05902 [DOI] [PubMed] [Google Scholar]
- 34.Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. 2010. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40:22–33. 10.1016/j.molcel.2010.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin J, Ang XL, Shirogene T, Elledge S, Harper JW. 2005. Identification of substrates for F-box proteins. Methods Enzymol. 399(B):287–309 [DOI] [PubMed] [Google Scholar]
- 36.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. 2006. Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell 22:451–462. 10.1016/j.molcel.2006.03.036 [DOI] [PubMed] [Google Scholar]
- 37.Dalal S, Rosser MF, Cyr DM, Hanson PI. 2004. Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell 15:637–648. 10.1091/mbc.E03-02-0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chapman-Smith A, Cronan JE., Jr 1999. The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity. Trends Biochem. Sci. 24:359–363. 10.1016/S0968-0004(99)01438-3 [DOI] [PubMed] [Google Scholar]
- 39.Xu M, Skaug B, Zeng W, Chen ZJ. 2009. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol. Cell 36:302–314. 10.1016/j.molcel.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. 2005. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120:73–84. 10.1016/j.cell.2004.11.013 [DOI] [PubMed] [Google Scholar]
- 41.Park S, Isaacson R, Kim HT, Silver PA, Wagner G. 2005. Ufd1 exhibits the AAA-ATPase fold with two distinct ubiquitin interaction sites. Structure 13:995–1005. 10.1016/j.str.2005.04.013 [DOI] [PubMed] [Google Scholar]
- 42.Plummer SM, Holloway KA, Manson MM, Munks RJ, Kaptein A, Farrow S, Howells L. 1999. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene 18:6013–6020. 10.1038/sj.onc.1202980 [DOI] [PubMed] [Google Scholar]
- 43.Wu K, Kovacev J, Pan ZQ. 2010. Priming and extending: a UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol. Cell 37:784–796. 10.1016/j.molcel.2010.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. 2007. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 27:4006–4017. 10.1128/MCB.00620-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, Xiao Y, Christie AL, Aster J, Settleman J, Gygi SP, Kung AL, Look T, Nakayama KI, DePinho RA, Wei W. 2011. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471:104–109. 10.1038/nature09732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong Q, Gao W, Du F, Wang X. 2005. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121:1085–1095. 10.1016/j.cell.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 47.Hänzelmann P, Buchberger A, Schindelin H. 2011. Hierarchical binding of cofactors to the AAA ATPase p97. Structure 19:833–843. 10.1016/j.str.2011.03.018 [DOI] [PubMed] [Google Scholar]
- 48.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park MY, Jang HD, Lee SY, Lee KJ, Kim E. 2004. Fas-associated factor-1 inhibits nuclear factor-kappaB (NF-kappaB) activity by interfering with nuclear translocation of the RelA (p65) subunit of NF-kappaB. J. Biol. Chem. 279:2544–2549. 10.1074/jbc.M304565200 [DOI] [PubMed] [Google Scholar]
- 50.Song EJ, Yim SH, Kim E, Kim NS, Lee KJ. 2005. Human Fas-associated factor 1, interacting with ubiquitinated proteins and valosin-containing protein, is involved in the ubiquitin-proteasome pathway. Mol. Cell. Biol. 25:2511–2524. 10.1128/MCB.25.6.2511-2524.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park MY, Moon JH, Lee KS, Choi HI, Chung J, Hong HJ, Kim E. 2007. FAF1 suppresses IkappaB kinase (IKK) activation by disrupting the IKK complex assembly. J. Biol. Chem. 282:27572–27577. 10.1074/jbc.C700106200 [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Zhou F, van Laar T, Zhang J, van Dam H, Ten Dijke P. 2011. Fas-associated factor 1 antagonizes Wnt signaling by promoting β-catenin degradation. Mol. Biol. Cell 22:1617–1624. 10.1091/mbc.E10-12-0985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besche HC, Haas W, Gygi SP, Goldberg AL. 2009. Isolation of mammalian 26S proteasomes and p97/P97 complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48:2538–2549. 10.1021/bi802198q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bousquet-Dubouch MP, Baudelet E, Guérin F, Matondo M, Uttenweiler-Joseph S, Burlet-Schiltz O, Monsarrat B. 2009. Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome-interacting proteins. Mol. Cell. Proteomics 8:1150–1164. 10.1074/mcp.M800193-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]