

Abstract
Sulfotransferase (SULT)-mediated sulfation represents a critical mechanism in regulating the chemical and functional homeostasis of endogenous and exogenous molecules. The cholesterol sulfotransferase SULT2B1b catalyzes the sulfoconjugation of cholesterol to synthesize cholesterol sulfate (CS). In this study, we showed that the expression of SULT2B1b in the liver was induced in obese mice and during the transition from the fasted to the fed state, suggesting that the regulation of SULT2B1b is physiologically relevant. CS and SULT2B1b inhibited gluconeogenesis by targeting the gluconeogenic factor hepatocyte nuclear factor 4α (HNF4α) in both cell cultures and transgenic mice. Treatment of mice with CS or transgenic overexpression of the CS-generating enzyme SULT2B1b in the liver inhibited hepatic gluconeogenesis and alleviated metabolic abnormalities both in mice with diet-induced obesity (DIO) and in leptin-deficient (ob/ob) mice. Mechanistically, CS and SULT2B1b inhibited gluconeogenesis by suppressing the expression of acetyl coenzyme A (acetyl-CoA) synthetase (Acss), leading to decreased acetylation and nuclear exclusion of HNF4α. Our results also suggested that leptin is a potential effector of SULT2B1b in improving metabolic function. We conclude that SULT2B1b and its enzymatic by-product CS are important metabolic regulators that control glucose metabolism, suggesting CS as a potential therapeutic agent and SULT2B1b as a potential therapeutic target for metabolic disorders.
INTRODUCTION
Sulfation is the metabolic introduction of a sulfonate group (SO3−1) from the universal sulfonate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) into an acceptor molecule. The sulfation substrates range from biomacromolecules to small biological molecules. Regulation by sulfation represents an important mechanism for controlling the chemical and functional homeostasis of endogenous and exogenous molecules (1). Examples of the biological consequences of sulfation include estrogen and androgen deprivation (2–4), thyroid hormone deactivation (5), bile acid detoxification (6), and deactivation of the liver X receptor (LXR)-activating oxysterols (7).
Sulfation is catalyzed by sulfotransferases (SULTs), a family of conjugating enzymes. Among SULTs, the cholesterol sulfotransferase SULT2B1b preferentially catalyzes the sulfoconjugation of cholesterol to synthesize cholesterol sulfate (CS) (8, 9), although this enzyme has additional substrates (10). Cholesterol sulfation is an important part of hepatic cholesterol metabolism, whose dysregulation is known to affect the severity of nonalcoholic fatty liver disease (11). SULT2B1b is expressed in multiple tissues, including the liver (9, 12, 13). Although the mouse liver does not have high basal expression of SULT2B1b (9), hepatic expression of SULT2B1b is highly inducible, as in response to the constitutive androstane receptor (CAR) ligand 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) (13).
Cholesterol sulfate, the enzymatic by-product of SULT2B1b, is a predominant steroid sulfate in the circulation. The concentrations of cholesterol sulfate in human plasma range from 134 to 254 μg/ml (12, 14, 15). Cholesterol sulfate is also present in various body fluids and tissues, including urine, bile, seminal plasma, skin, adrenal glands, kidney, and liver (12, 16). Despite its prevalence and abundance, the physiological role of CS remains to be defined. In vitro studies have shown that CS is a natural agonist for the retinoic acid-related orphan receptor α (RORα) (17). It has been suggested that CS may play a role in the immune response and that a shortage of CS in the fetus may contribute to the development of autism (18). Cholesterol sulfate is also a major cell surface substance that is essential for cell membrane function (19).
Metabolic syndrome, often manifested as obesity and insulin-resistant type 2 diabetes, is a major health concern (20). The dysregulation of glucose and lipid metabolism plays an important pathogenic role in the development of metabolic syndrome. High cholesterol is a common consequence of obesity. Although the activity of SULT2B1b in catalyzing cholesterol sulfation has been documented, the roles of SULT2B1b and its enzymatic by-product CS in energy metabolism and metabolic syndrome remain largely unknown. Recent studies have suggested that SULT2B1b has antilipogenic activity. We reported that adenoviral overexpression of SULT2B1b decreased serum and hepatic lipid levels by suppressing the LXR-sterol regulatory element binding protein 1c (SREBP-1c)-mediated lipogenic pathway (7). The expression and induction of SULT2B1b have been suggested to play an important role in the CAR-mediated suppression of lipogenic gene expression (13). Increased gluconeogenesis is a major pathogenic event in type 2 diabetes (21). However, it has not been reported whether and how CS and SULT2B1b inhibit gluconeogenesis, and how this inhibition may contribute to the relief of metabolic syndrome.
In this study, we showed that CS and SULT2B1b inhibited gluconeogenesis by targeting hepatocyte nuclear factor 4α (HNF4α). Treatment of mice with CS or transgenic overexpression of SULT2B1b inhibited hepatic gluconeogenesis and alleviated metabolic abnormalities in dietary and genetic mouse models of obesity and type 2 diabetes. Mechanistically, CS and SULT2B1b inhibited gluconeogenesis by suppressing the acetylation of the gluconeogenic factor HNF4α and perturbing the nuclear translocation of HNF4α. Our study established CS as an important metabolic regulator in controlling glucose metabolism and energy homeostasis, pointing to CS as a potential therapeutic agent and to SULT2B1b as a potential therapeutic target for metabolic disorders.
MATERIALS AND METHODS
Cell culture, transient transfection, and lentiviral infection.
Hepa1-6 cells were purchased from the American Type Culture Collection. Normal human hepatocytes were obtained from the Liver Tissue Procurement and Distribution System, University of Pittsburgh, Pittsburgh, PA. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS). Transfection was performed using Lipofectamine 2000 reagent from Invitrogen (Carlsbad, CA). The glucose-6-phosphatase (G6pase) luciferase reporter plasmid was a gift from Richard M. O'Brien (Vanderbilt University Medical School). The lentiviral vector expressing HNF4α RNA interference (RNAi) (shHNF4α) (22) was a gift from Stephen A. Duncan (Medical College of Wisconsin). The expression vectors for the enzyme-dead SULT2B1b mutant (Mut-SULT2B1b) (23) and the acetylation-resistant HNF4α mutant (HNF4α-M4) (24) were constructed as described previously.
Glucose production assay.
Cells were washed three times with phosphate-buffered saline (PBS) to remove glucose and were incubated in glucose production buffer (glucose-free DMEM [pH 7.4] containing 20 mM sodium lactate and 2 mM sodium pyruvate without phenol red). After 4 h, 500 μl of medium was removed and was centrifuged at 15,000 × g for 10 min. The glucose content of the supernatant was measured with a glucose assay kit from Sigma. Glucose concentrations were normalized to cellular protein concentrations.
Immunofluorescence.
Cells were seeded on sterile glass slides, fixed with 4% paraformaldehyde in PBS, and incubated overnight with an anti-FLAG antibody at 4°C. Cells were then incubated with fluorescein-labeled goat anti-rabbit IgG and were visualized by using a fluorescence microscope.
Animals and animal diet.
To construct the TetRE-SULT2B1b transgene, the cDNA of human SULT2B1b was subcloned into the tetracycline-responsive element (TetRE) transgene cassette. Microinjection to produce TetRE-SULT2B1b mice was performed at the University of Pittsburgh Transgenic Core Facility. TetRE-SULT2B1b mice were bred with fatty acid binding protein (FABP)-tetracycline transcriptional activator (tTA) mice (25) to generate double-positive transgenic (TG) mice. ob-TG mice were generated by breeding the TetRE-SULT2B1b and FABP-tTA transgenes into the ob/ob background. Age-matched littermates were used for all experiments. ob/ob and ob-TG mice were maintained on a chow diet. When necessary, wild-type (WT) and TG mice were fed a high-fat diet (HFD) (catalog no. S3282; Bio-Serv, Frenchtown, NJ). Hydrodynamic transfection of mouse liver was performed as described previously (26). Adenoviruses (Ad) expressing short hairpin RNA (shRNA) against HNF4α (Ad-shHNF4α) or β-galactosidase (Ad-shLacZ) (27) were gifts from Yanqiao Zhang (Northeastern Ohio Universities College of Medicine). Adenoviruses were injected through tail veins at a dose of 1010 PFU per mouse. Mice were analyzed 1 week after viral injection. The use of mice in this study complied with relevant federal guidelines and institutional policies.
Serum and liver tissue chemistry.
Levels of triglycerides (catalog no. 2100-430; Stanbio), cholesterol (catalog no. 1010-430; Stanbio), and insulin (catalog no. 90080; Crystal Chem) in serum were measured using commercial assay kits. The concentration of acetyl coenzyme A (acetyl-CoA) in the liver was measured using the PicoProbe acetyl-CoA assay kit (ab87546; Abcam, Cambridge, MA).
GTT, ITT, PTT, and hyperinsulinemic-euglycemic clamp.
For the glucose tolerance test (GTT), overnight-fasted mice were given an intraperitoneal (i.p.) injection of glucose (2 mg/g of body weight). For the insulin tolerance test (ITT), 6-h-fasted mice were given an i.p. injection of insulin (1 U/kg of body weight). For the pyruvate tolerance test (PTT), overnight-fasted mice received an i.p. injection of sodium pyruvate (2 g/kg of body weight). Blood glucose concentrations were determined with a glucometer at baseline and at 30, 60, 90, and 120 min after injection. Hyperinsulinemic-euglycemic clamps were performed on conscious mice as we have described previously (28).
Immunoprecipitation, Western blot analysis, and the ChIP assay.
Immunoprecipitation and Western blot analysis were performed as described previously (29). The primary antibodies used included antibodies against HNF4α (catalog no. 3113), forkhead box protein O1 (FoxO1) (catalog no. 2880S), AKT (catalog no. 9272S), phosphorylated AKT (p-AKT) (catalog no. 9611S), acetylated lysine (catalog no. 9441S), FLAG (catalog no. 2368), and insulin receptor substrate 1 (IRS1) (catalog no. 2382), all purchased from Cell Signaling (Danvers, MA). The antibodies against acetyl-CoA synthetase (Acss) (sc-85258) and SULT2B1b (sc-67103) were purchased from Santa Cruz (Santa Cruz, CA). Western blotting images were quantified with ImageJ software (http://rsb.info.nih.gov/ij/). The chromatin immunoprecipitation (ChIP) assay was performed as we have described previously (30).
Gene expression analysis.
Total RNA was extracted using the TRIzol reagent from Invitrogen. SYBR green-based real-time PCR was performed with the ABI 7500 real-time PCR system. Gene expression was normalized to the expression of the cyclophilin gene.
Statistical analysis.
All the data are expressed as means ± standard deviations (SD). Differences were evaluated by the unpaired two-tailed Student t test (GraphPad Prism, version 4.0). The criterion for statistical significance was a P value of <0.05.
RESULTS
CS and SULT2B1b inhibited gluconeogenesis in hepatic cells, and SULT2B1b was induced in obese mice and during the transition from the fasted to the fed state.
To evaluate the effect of cholesterol sulfate (CS) on glucose homeostasis, we first measured glucose production in Hepa1-6 mouse hepatoma cells that were either left untreated or treated with CS. A 24-h CS treatment (5 μM) had little effect on the basal glucose level, but it substantially inhibited forskolin (FSK)-stimulated glucose production (Fig. 1A). Gene expression analysis showed that treatment of Hepa1-6 cells with CS inhibited the expression of glucose-6-phosphatase (G6pase) and phosphoenolpyruvate carboxykinase (Pepck), two key gluconeogenic enzymes, in a dose-dependent manner (Fig. 1B). This inhibition of FSK-responsive glucose production (Fig. 1C) and of G6pase and Pepck gene expression (Fig. 1D) by CS was also observed in human primary hepatocytes.
FIG 1.

Cholesterol sulfate (CS) and SULT2B1b inhibited gluconeogenesis in hepatic cells, and SULT2B1b was induced in obese mice and during the transition from the fasted to the fed state. (A) Glucose production in Hepa1-6 cells treated with a vehicle (VEH) or 5 μM CS for 24 h. (B) Relative expression of G6pase and Pepck mRNAs in Hepa1-6 cells treated with a vehicle or with CS at 2.5 or 5 μM for 24 h, as determined by real-time PCR analysis. (C) Glucose production in human primary hepatocytes (HPH) treated with a vehicle or 5 μM CS for 24 h. (D) Relative expression of G6pase and Pepck mRNAs in HPH treated with a vehicle or with CS at 2.5 or 5 μM for 24 h. (E) Glucose production in Hepa1-6 cells transfected with the indicated plasmids and combinations. Cells were maintained either in normal FBS or in FBS treated with dextran-coated charcoal (DCC FBS). (F) Glucose production in Hepa1-6 cells transfected with a vector, SULT1E1, or SULT2A1. (G) The hepatic expression of SULT2B1b mRNA (top) and protein (bottom) in HFD-fed WT mice and chow-fed ob/ob mice was measured by real-time PCR. (H) Hepatic expression of SULT2B1b mRNA (top) and protein (bottom) in fasted and refed mice. The relative expression of SULT2B1b protein in panels G and H is given below the bands. Results are means ± SD for three independent experiments or for 5 mice per group. *, P < 0.05; **, P < 0.01.
CS is known to be synthesized from cholesterol through the enzymatic activity of SULT2B1b. We went on to determine whether SULT2B1b could also inhibit gluconeogenesis. In this experiment, Hepa1-6 cells were transiently transfected with an empty vector or SULT2B1b before the measurement of FSK-responsive glucose production. As shown in Fig. 1E, overexpression of SULT2B1b inhibited glucose production when cells were maintained in normal fetal bovine serum (FBS), but this inhibition was abolished when cells were maintained in FBS treated with dextran-coated charcoal (DCC FBS) that was free of lipids and cholesterol. We also showed that the inhibitory effect of SULT2B1b depended on its enzymatic activity, since an enzyme-dead mutant of SULT2B1b (Mut-SULT2B1b) failed to inhibit glucose production (Fig. 1E). Mut-SULT2B1b was created by mutating the PAPS-binding domain of SULT2B1b (23). The inhibitory effect of SULT2B1b was also abolished when cells were cotransfected with steroid sulfatase (STS), an enzyme capable of desulfonating CS (Fig. 1E). The inhibition appeared to be SULT2B1b specific, because cotransfection with either of two other SULT isoforms, the estrogen sulfotransferase SULT1E1 and the hydroxysteroid sulfotransferase SULT2A1, failed to inhibit glucose production (Fig. 1F).
In investigating the pathophysiological role of SULT2B1b in energy metabolism in vivo, we found that hepatic expression of endogenous SULT2B1b mRNA and protein was induced both in mice fed a high-fat diet (HFD) and in chow-fed ob/ob mice (Fig. 1G). Moreover, hepatic expression of SULT2B1b mRNA and protein was induced during the transition from the fasted to the fed state (Fig. 1H), suggesting that this pathway may be involved in the suppression of gluconeogenesis in the fasting-to-feeding transition.
CS inhibited the gluconeogenic activity of HNF4α.
Peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), forkhead box protein O1 (FoxO1), and hepatocyte nuclear factor 4α (HNF4α) are three important positive regulators of gluconeogenesis (31). To determine whether they are the targets of the inhibitory effect of CS on gluconeogenesis, we transfected them individually into Hepa1-6 cells together with a G6pase promoter luciferase reporter gene that is responsive to PGC1α, FoxO1, and HNF4α (32). As shown in Fig. 2A, cotransfection of PGC1α, FoxO1, or HNF4α resulted in the activation of the reporter gene, as expected. Treatment of transfected cells with CS impaired HNF4α-responsive reporter activity in a dose-dependent manner but had little effect on the activities of PGC1α and FoxO1. The same pattern of CS-induced, HNF4α-specific effect was observed when the expression of the endogenous G6pase gene in transfected cells was measured (Fig. 2B). These results suggested that CS might have inhibited gluconeogenesis by its specific inhibition of HNF4α. To verify these results in vivo, we performed hydrodynamics-based transfection of mouse livers with HNF4α (33). One hour after transfection, the HNF4α-transfected mice were divided into two groups, one of which received an i.p. injection of CS (25 mg/kg) while the other received the vehicle. Mice were sacrificed 10 h after CS injection. As shown in Fig. 2C, transfection of HNF4α induced hepatic expression of G6pase and Pepck, as expected, but this induction was abolished in CS-treated mice. In agreement with the pattern of gluconeogenic gene expression, fasting blood glucose levels were elevated in vehicle-treated HNF4α-transfected mice but not in CS-treated HNF4α-transfected mice (Fig. 2D). A chromatin immunoprecipitation (ChIP) assay showed that CS inhibited the recruitment of HNF4α onto the G6pase gene promoter in the mouse liver (Fig. 2E).
FIG 2.

CS inhibited the gluconeogenic activity of HNF4α. (A and B) Relative G6pase promoter luciferase reporter activity (A) and G6pase mRNA expression (B) in Hepa1-6 cells transfected with the indicated vectors. The cells were treated with a vehicle or with CS at 2.5 or 5 μM for 24 h. Results are means ± SD for three independent experiments. (C to E) Expression of hepatic G6pase and Pepck mRNAs (C), fasting blood glucose levels (D), and results of a ChIP assay of the livers of mice (E) after hydrodynamic transfection of the liver with the indicated vectors. The graph in panel E shows the quantification of ChIP assay results after normalization to input. n, 6 mice per group. (F) HNF4α mRNA expression and glucose production in Hepa1-6 cells infected with a lentivirus expressing HNF4α RNAi (shHNF4α) or a control RNAi (shNT). (G) (Left) The expression of HNF4α protein in shNT and shHNF4α cells was determined by Western blotting. (Right) Quantification of Western blotting results. Results are means ± SD for three independent experiments. *, P < 0.05; **, P < 0.01.
To further confirm the role of HNF4α, we infected Hepa1-6 cells with a lentivirus expressing HNF4α RNAi (shHNF4α) (22) before treatment with CS. The efficiency of HNF4α knockdown was confirmed both by real-time PCR (Fig. 2F, left) and by Western blotting (Fig. 2G). In control RNAi (shNT)-transfected cells, FSK-responsive glucose production was inhibited by CS. HNF4α knockdown was sufficient to inhibit FSK-responsive glucose production, whereas treatment with CS had little further effect (Fig. 2F, right). These results indicated that CS treatment and HNF4α knockdown were equally efficient at suppressing glucose production, suggesting that HNF4α inhibition might have accounted for the inhibitory effect of CS.
CS improved insulin sensitivity in mice with diet-induced or genetic obesity.
Increased gluconeogenesis is a major pathogenic event in the development of obesity and type 2 diabetes (21). The inhibition of gluconeogenesis by CS prompted us to determine whether CS can improve insulin sensitivity. Two type 2 diabetes models were used: mice with diet-induced obesity (DIO) (induced by 12 weeks of HFD feeding) and ob/ob mice. In both models, mice received i.p. injections of a vehicle or CS (25 mg/kg) 3 times per week for 2 weeks. We found that CS treatment decreased blood glucose levels in both DIO and ob/ob mice (Fig. 3A). CS treatment also improved insulin sensitivity, as evidenced by improved glucose tolerance test (GTT) (Fig. 3B) and insulin tolerance test (ITT) (Fig. 3C) performance, in both models. CS-treated DIO and ob/ob mice also showed inhibition of the expression of gluconeogenic and lipogenic genes and of HNF4α target genes (Fig. 3D). The CS treatment also caused significant upregulation of the lipolytic enzyme hormone-sensitive lipase (Hsl) and the glycolytic enzyme glycokinase (Gck) in both models (Fig. 3D).
FIG 3.

CS improved insulin sensitivity in mice with diet-induced or genetic obesity. (A to D) Either C57BL/6J mice fed a high-fat diet for 12 weeks to induce diet-induced obesity (DIO) or 12-week-old ob/ob mice maintained on a chow diet were used. The mice were then treated with a vehicle (VEH) or CS for 2 weeks before analysis. Shown are blood glucose levels (A), results of glucose tolerance tests (GTT) (B) and insulin tolerance tests (ITT) (C), and hepatic gene expression (D) in DIO (left) and ob/ob (right) mice. The percentages in panel C are relative variations from the time 0 min, which was arbitrarily set as 100%. Results are means ± SD; n, 4 mice per group. *, P < 0.05.
Transgenic overexpression of SULT2B1b in the liver improved metabolic function and alleviated insulin resistance in DIO and ob/ob mice.
To examine the chronic effect of increased CS levels in vivo, we generated transgenic mice expressing the CS-producing enzyme SULT2B1b in the liver by using the Tet-Off transgenic system as outlined in Fig. 4A. We first generated TetRE-SULT2B1b transgenic mice expressing human SULT2B1b under the control of the tetracycline response element (TetRE). TetRE-SULT2B1b mice were then bred with FABP-tTA transgenic mice, expressing the tetracycline transcriptional activator (tTA) under the control of the rat fatty acid binding protein (FABP) gene promoter, which directs the expression of the transgene to the liver (25). In this system, double transgenic (TG) mice will express transgenic SULT2B1b, whereas doxycycline (Dox) administration will silence the transgene expression. Hepatic expression of transgenic SULT2B1b in TG mice was confirmed by Northern blotting and a cholesterol sulfation assay (Fig. 4B). The level of CS circulating was markedly increased in TG mice, as expected (Fig. 4C).
FIG 4.

Transgenic overexpression of SULT2B1b in the liver improved metabolic function. (A) Strategy for the creation of transgenic mice expressing SULT2B1b in the liver. PCMV, human cytomegalovirus (CMV) immediate-early promoter. (B) Northern blot analysis (left) and a SULT2B1b enzymatic assay using cholesterol as the substrate (right) were performed for wild-type (WT) and transgenic (TG) mice. (C) Levels of circulating CS. (D and E) Body weights (D) and fat-to-body weight ratios (E) of mice fed an HFD for the indicated periods. (F) H&E staining of the liver, white adipose tissue (WAT), and brown adipose tissue (BAT) in HFD-fed mice. (G) Relative expression of leptin mRNA in the WAT of HFD-fed mice. (H) Hepatic mRNA expression of gluconeogenic and lipogenic genes and HNF4α target genes in mice fed a chow diet or an HFD. (I) (Left) Expression of Srebp-1c protein as shown by Western blotting. (Right) Quantification of Western blotting data. (J) Recruitment of HNF4α (left) and FoxO1 (right) onto the G6pase gene promoter in the livers of HFD-fed mice as determined by ChIP assays. Bar graphs show the quantification of ChIP results. Results are means ± SD; n, 6 to 8 mice per group. *, P < 0.05; **, P < 0.01.
When analyzing the metabolic phenotype, we found no significant differences in body weight, fat mass, GTT results, ITT results, or fasting insulin or glucose levels between chow-fed WT mice and TG mice (data not shown). However, when challenged with an HFD, the TG mice gained less body weight (Fig. 4D) and had lower fat-to-body weight ratios (Fig. 4E). Table 1 summarizes the major changes in metabolic parameters in TG mice, including decreases in food intake, liver weight, liver and serum bile acid levels, concentrations of triglycerides in the liver and cholesterol in the serum, fasting glucose and insulin levels, and homeostatic model assessment of insulin resistance (HOMA-IR), as well as an increased serum leptin level and increased O2 consumption during the dark phase. The inhibition of hepatic steatosis and the decreased sizes of adipocytes in white adipose tissue (WAT) and brown adipose tissue (BAT) in TG mice were shown by staining with hematoxylin and eosin (H&E) (Fig. 4F). In agreement with the increased level of leptin in serum, the level of leptin mRNA expression in WAT, the major leptin-producing tissue, was markedly increased in TG mice (Fig. 4G). When hepatic gene expression was analyzed, we found that the mRNA expression of gluconeogenic and lipogenic genes and HNF4α target genes was suppressed in HFD-fed TG mice relative to that in their WT counterparts (Fig. 4H). The expression of Hsl and Gck was increased in HFD-fed TG mice (Fig. 4H). The decreased protein expression of SREBP-1c, a master lipogenic transcriptional factor, was confirmed by Western blotting (Fig. 4I), a finding consistent with our previous report (7). A ChIP assay on mouse livers showed that the recruitment of HNF4α, but not that of FoxO1, onto the G6pase gene promoter was decreased in HFD-fed TG mice (Fig. 4J).
TABLE 1.
Metabolic profiles of HFD-fed WT and TG mice
| Characteristic | Value for mice |
|
|---|---|---|
| WT | TGa | |
| Food intake (g/g of body wt/wk) | 0.62 ± 0.044 | 0.59 ± 0.044* |
| Liver wt (g) | 1.29 ± 0.061 | 1.05 ± 0.035* |
| Liver bile acid level (nmol/g of liver) | 473.9 ± 30.74 | 163.7 ± 83.90* |
| Serum bile acid level (μmol/liter) | 8.02 ± 0.82 | 3.717 ± 1.96* |
| Liver triglyceride concn (mg/g liver) | 74.58 ± 11.16 | 37.30 ± 8.75* |
| Serum cholesterol concn (mg/dl) | 109.1 ± 4.46 | 85.20 ± 2.93* |
| Fasting glucose level (mg/dl) | 130.3 ± 31.03 | 81.40 ± 7.82** |
| Serum insulin level (ng/ml) | 9.98 ± 2.59 | 6.96 ± 1.51* |
| HOMA-IR | 3.97 ± 0.48 | 1.516 ± 0.43* |
| Serum leptin level (ng/ml) | 17.43 ± 10.11 | 30.09 ± 1.75* |
| Oxygen consumption (dark phase) (ml/kg/h) | 2,813 ± 349.4 | 3,221 ± 570.6* |
Asterisks indicate significant differences from the WT (*, P < 0.05; **, P < 0.01).
When insulin sensitivity was examined, we found that HFD-fed TG mice showed improved performance on the pyruvate tolerance test (PTT), GTT, and ITT (Fig. 5A). The hyperinsulinemic-euglycemic clamp revealed that the HFD-fed TG mice had an increased glucose infusion rate (Fig. 5B) and marked suppression of hepatic glucose production (Fig. 5C), also suggestive of improved insulin sensitivity. The improved insulin sensitivity in the livers of TG mice was further supported by their increased insulin-responsive induction of tyrosine phosphorylation of insulin receptor substrate 1 (IRS1) and AKT (Fig. 5D). Chronic inflammation in the adipose tissue and liver is known to play a pathogenic role in the development of insulin resistance (30). In agreement with their improved insulin sensitivity, HFD-fed TG mice showed a favorable pattern of cytokine expression in the WAT (Fig. 5E) and liver (Fig. 5F), including decreased expression of the M1 macrophage-derived proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and ApoE, increased expression of the M2 macrophage-derived anti-inflammatory cytokines Arg1 and Mrc2, and decreased expression of overall macrophage markers Cd68 and F4/80. The metabolic benefit was transgene dependent, because the effects of the transgene on body weight, body composition, GTT and ITT performance, and hepatic gene expression were all abolished in TG mice treated with Dox, which efficiently silenced transgene expression (data not shown).
FIG 5.

Overexpression of SULT2B1b protected mice from diet-induced insulin resistance in the DIO and ob/ob models. (A to F) Mice were fed an HFD. (A) Pyruvate tolerance tests (PTT), glucose tolerance tests (GTT), and insulin tolerance tests (ITT). (B and C) Glucose infusion rate (B) and hepatic glucose production (C) during the hyperinsulinemic-euglycemic clamp procedure. (D) (Left) Insulin-stimulated phospho-IRS1 and phospho-AKT as shown by Western blotting. (Right) Bar graph showing quantification of the Western blot results. (E and F) mRNA expression of proinflammatory genes (M1), anti-inflammatory genes (M2), and macrophage marker genes in the WAT (E) and liver (F). (G to I) ob-TG mice were created by breeding the TG mice into the ob/ob background. Both ob/ob and ob-TG mice were maintained on a chow diet. (G) PTT, GTT, and ITT. (H) (Left) Insulin-stimulated phospho-IRS1and phospho-AKT as shown by Western blotting. (Right) Bar graph showing quantification of the Western blot results. (I) Hepatic gene expression as measured by real-time PCR analysis. Results are means ± SD; n, 4 to 8 mice per group. *, P < 0.05; **, P < 0.01. The percentages in panels A and G are relative variations from the time 0 min, which was arbitrarily set as 100%.
The metabolic benefit of the SULT2B1b transgene was also observed in ob/ob mice. In this experiment, the SULT2B1b transgene was bred into the ob/ob background, and the resulting mice were termed ob-TG mice. Both the ob/ob and ob-TG mice were maintained on a chow diet. Compared to their ob/ob counterparts, ob-TG mice showed decreased fasting glucose and insulin levels and decreased HOMA-IR (data not shown), findings consistent with those observed for HFD-fed TG mice (Table 1). The ob-TG mice also exhibited improved PTT, GTT, and ITT performance (Fig. 5G), increased insulin-responsive tyrosine phosphorylation of IRS1 and AKT in the liver (Fig. 5H), and inhibition of the expression of gluconeogenic and lipogenic genes and HNF4α target genes (Fig. 5I). The expression of Hsl and Gck was increased in ob-TG mice (Fig. 5I). These phenotypes were consistent with those observed in HFD-fed TG mice. Interestingly, we found that total body weight, body composition, food intake, oxygen consumption, and hepatic steatosis did not differ for ob/ob and ob-TG mice (data not shown).
The SULT2B1b-induced alleviation of insulin resistance may have been achieved by inhibiting HNF4α activity.
Having shown that HNF4α is the likely target of CS in inhibiting gluconeogenesis, we wanted to determine whether the metabolic benefit of the SULT2B1b transgene was also achieved through targeting HNF4α. In this experiment, we knocked down HNF4α expression in the mouse liver by using a recombinant adenovirus that expresses an shRNA against HNF4α (shHNF4α) (27). Ad-shHNF4α or the control, Ad-shLacZ, was introduced by tail vein injection into WT and TG mice that had been fed an HFD for 12 weeks. Mice were analyzed 1 week after infection. The efficiency of HNF4α knockdown was confirmed both by Western blotting (Fig. 6A) and by real-time PCR (data not shown). Ad-shLacZ-infected TG mice showed lower fasting glucose levels (Fig. 6B) and lower G6pase gene expression levels (Fig. 6C) than their WT counterparts. WT-shHNF4α mice showed inhibition of fasting glucose (Fig. 6B) and G6pase gene expression (Fig. 6C) similar to those observed in TG-shLacZ mice. HNF4α knockdown had little effect on the inhibitory activity of TG. A similar pattern of shHNF4α effect was observed when the GTT (Fig. 6D) and ITT (Fig. 6E) were evaluated. These results indicated that the SULT2B1b transgene and HNF4α knockdown were equally efficient at suppressing gluconeogenesis and improving insulin sensitivity, suggesting that HNF4α inhibition might have accounted for the inhibitory effect of SULT2B1b.
FIG 6.
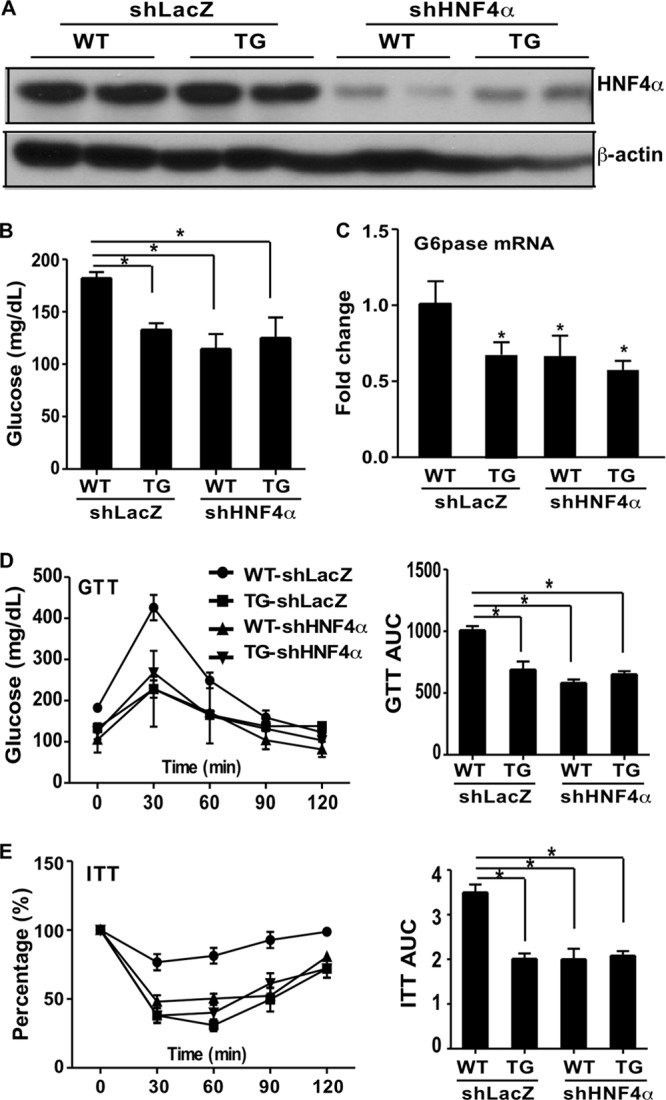
The SULT2B1b-induced alleviation of insulin resistance may have been achieved by inhibiting the activity of HNF4α. An adenovirus encoding shHNF4α or the control, shLacZ, was introduced by tail vein injection into WT and TG mice that had been fed an HFD for 12 weeks. Mice were analyzed 1 week after infection. (A) HNF4α protein expression as shown by Western blotting. (B) Fasting blood glucose levels. (C) Relative hepatic expression of G6pase mRNA. (D and E) GTT (D) and ITT (E) results. The percentage in panel E is relative variation from the time 0 min, which was arbitrarily set as 100%. Quantifications of the areas under the curve (AUC) are shown on the right. Results are means ± SD; n, 4 mice per group. *, P < 0.05.
The molecular mechanism by which CS and SULT2B1b inhibit HNF4α.
As a transcriptional factor, HNF4α must enter the nucleus in order to realize its transcriptional activity. In investigating the molecular mechanism by which CS and SULT2B1b inhibit HNF4α, we found that CS and SULT2B1b inhibited the nuclear translocation of HNF4α. When transfected into COS7 cells, the HNF4α protein was localized mostly in the nucleus, as revealed by immunofluorescence (Fig. 7A). However, upon cotransfection of SULT2B1b or treatment with CS, HNF4α translocated from the nucleus to the cytoplasm. The effect of SULT2B1b on the subcellular distribution of HNF4α in COS7 cells was further confirmed by cytosolic-nuclear fractionation and Western blot analysis (Fig. 7B). In the livers of WT mice, endogenous HNF4α was detected in both the cytoplasm and nuclei of hepatocytes, but HNF4α became exclusively cytoplasmic in TG mice, as shown by immunofluorescence (Fig. 7C) and cytosolic-nuclear fractionation (Fig. 7D).
FIG 7.

Molecular mechanism by which CS and SULT2B1b inhibit HNF4α. (A) COS7 cells were transfected with FLAG-HNF4α. The cells were either cotransfected with SULT2B1b or treated with CS. The subcellular distribution of HNF4α was visualized by immunofluorescence staining with an anti-FLAG antibody (red). 4′,6-Diamidino-2-phenylindole (DAPI) (blue) was used for nuclear counterstaining. (B) Western blot analysis of HNF4α protein levels in nuclear and cytoplasmic fractions of COS7 cells transfected with an empty vector (Vec) or SULT2B1b. The purity of the nuclear and cytoplasmic fractions was confirmed by immunoblotting with an antibody against histone H3 (nuclear fraction marker) and an antibody against tubulin (cytoplasmic fraction marker). (C and D) The subcellular distribution of endogenous HNF4α in the livers of HFD-fed WT and TG mice was assessed by immunofluorescence (C) and Western blot analysis (D). (E) COS7 cells were transfected with FLAG-tagged WT HNF4α or the acetylation-resistant mutant HNF4α-M4. The cells were treated with a vehicle (Veh) or with CS. The subcellular distribution of HNF4α or HNF4α-M4 was visualized by immunofluorescence staining with an anti-FLAG antibody. (F) COS7 cells were transfected with FLAG-HNF4α (top) or FoxO1 (bottom). The cells were either cotransfected with SULT2B1b or treated with CS. Cell lysates were immunoprecipitated with an anti-FLAG (top) or anti-FoxO1 (bottom) antibody before being immunoblotted with antibodies against acetylated lysine and FLAG (top) or against acetylated lysine and FoxO1 (bottom). (G) Liver lysates were immunoprecipitated with an anti-HNF4α antibody before being immunoblotted with antibodies against acetylated lysine and HNF4α. (H) Relative expression of Acss mRNA in COS7 cells treated with CS or transfected with SULT2B1b (left) or in the livers of HFD-fed WT and TG mice (right). (I) Expression of Acss protein as measured by Western blotting. (J) Concentrations of acetyl-CoA in liver tissue. (K) Relative hepatic expression of Acss in fasted and refed mice. (L and M) Glucose production (L) and G6pase mRNA expression (M) in Hepa1-6 cells transfected with the indicated vectors and either left untreated or treated with 5 μM CS for 24 h. (N) Subcellular distribution of FLAG-HNF4α in COS7 cells transfected with the indicated vectors and either left untreated or treated with 5 μM CS for 24 h. HNF4α was visualized by immunofluorescence with an anti-FLAG antibody. Results are means ± SD for three independent experiments. *, P < 0.05; **, P < 0.01.
The subcellular localization of HNF4α can be regulated by acetylation-dependent nuclear retention (24). Indeed, mutation of four acetylation target lysine residues to alanines (HNF4α-M4) abolished the gluconeogenic activity of HNF4α (33; also data not shown), and this acetylation-resistant HNF4α mutant became constitutively and exclusively cytoplasmic regardless of CS treatment (Fig. 7E). We then asked whether CS treatment caused the regulation of the acetylation of HNF4α. Immunoprecipitation of HNF4α-transfected cells showed that cotransfection with SULT2B1b or treatment with CS dramatically reduced the level of acetylation of HNF4α (Fig. 7F). Of note, the expression of HNF4α protein (Fig. 7F) and mRNA (data not shown) was not different in SULT2B1b-transfected and CS-treated cells. The decreased acetylation appeared to be HNF4α specific, because the acetylation of transfected FoxO1 was not affected (Fig. 7F). Decreased acetylation of HNF4α was also observed in the livers of TG mice, in which the total HNF4α protein levels were not affected (Fig. 7G).
Acetyl-CoA is essential for the acetylation of HNF4α, and Acss catalyzes the formation of acetyl-CoA (33). We found that the expression of acetyl-CoA synthetase (Acss) mRNA was significantly decreased in SULT2B1b-transfected or CS-treated Hepa1-6 cells, as well as in TG mice (Fig. 7H). The suppression of Acss protein expression in TG mice was confirmed by Western blotting (Fig. 7I) and was further supported by the decreased level of acetyl-CoA, the enzymatic by-product of Acss, in the liver (Fig. 7J). We also found that the expression of Acss was significantly decreased during the transition from the fasted to the fed state (Fig. 7K), suggesting that this regulation may be involved in the suppression of gluconeogenesis in the fasting-to-feeding transition. To determine the functional relevance of Acss downregulation, we investigated whether forced expression of Acss could abolish the inhibitory effect of CS on gluconeogenesis. Indeed, cotransfection of Hepa1-6 cells with Acss was sufficient to abolish the inhibitory effect of CS on HNF4α-induced glucose production (Fig. 7L) and G6pase gene expression (Fig. 7M). Immunofluorescence showed that cotransfection of Acss was also efficient in reversing the HNF4α nuclear exclusion effect of CS (Fig. 7N).
DISCUSSION
In this study, we showed that CS decreased gluconeogenic gene expression and alleviated insulin resistance. Given that the liver plays a critical role in the regulation of glucose metabolism and that approximately 20 to 25% of total cholesterol is formed in the liver (35), we investigated the chronic effect of CS on hepatic glucose metabolism by using transgenic mice overexpressing SULT2B1b in the liver. We showed that transgenic overexpression of SULT2B1b had gluconeogenesis-inhibiting and insulin resistance-relieving effects similar to those of CS. Hepatic expression of SULT2B1b was induced during the transition from the fasted to the fed state, suggesting that the effect of SULT2B1b is physiologically relevant. The induction of SULT2B1b in obese mice may represent a protective response to metabolic abnormalities.
One of our most interesting findings is that CS and SULT2B1b inhibited gluconeogenesis by targeting HNF4α. HNF4α is a key regulator of gluconeogenesis through its positive regulation of G6pase and Pepck (31), and liver-specific deletion of HNF4α resulted in decreased blood glucose levels (36). We showed that CS and SULT2B1b specifically inhibited HNF4α-mediated gluconeogenesis. The idea that HNF4α is the target of CS and SULT2B1b in their inhibition of gluconeogenesis was also supported by the knockdown of HNF4α in cell cultures and in TG mice. We showed that CS treatment or SULT2B1b overexpression and HNF4α knockdown were equally efficient in suppressing gluconeogenesis. Knockdown of HNF4α also prevented further suppression of gluconeogenesis by CS or SULT2B1b. These results together suggested that HNF4α inhibition might have accounted for the inhibitory effects of CS and SULT2B1b.
An even more intriguing finding is that CS and SULT2B1b inhibited HNF4α activity by regulating the acetylation and subcellular distribution of HNF4α. Acetylation is required for the nuclear retention of HNF4α, preventing its active export to the cytoplasm and increasing the level of HNF4α DNA binding (24). We showed that HNF4α translocated from the nucleus to the cytoplasm and that HNF4α acetylation decreased in CS-treated or SULT2B1b-transfected cells and in SULT2B1b TG mice. In our investigation of the mechanism for the decreased HNF4α acetylation, we found that the expression of Acss, an enzyme that catalyzes the formation of acetyl-CoA from coenzyme A and acetate (33), was downregulated in CS-treated or SULT2B1b-transfected cells and in TG mice. Acetyl-CoA is an essential for the acetylation of HNF4α (24). The downregulation of Acss may have been accounted for by the decreased expression of Srebp-1c, a positive regulator of Acss (32, 37) and a target of inhibition by SULT2B1b (see below). We also found Acss downregulation during the transition from the fasted to the fed state, suggesting that the regulation of Acss is also physiologically relevant.
In addition to the inhibition of gluconeogenesis, the antilipogenic activity of SULT2B1b may also have contributed to the overall metabolic benefit of the transgene. We have reported previously that adenoviral overexpression of SULT2B1b suppressed the LXR- and Srebp-1c-mediated lipogenic pathways (7), which we reasoned was due to the sulfation and deactivation of oxysterols, the endogenous LXR agonists (7, 38). Adenoviral overexpression of SULT2B1b in the mouse liver decreased the Srebp-1c protein level (7, 39). Indeed, our SULT2B1b TG mice showed inhibition of hepatic steatosis and downregulation of Srebp-1c. We reason that the downregulation of Srebp-1c may have accounted for the suppression of the Acss gene, an SREBP-1c target gene (33, 37). Although the antilipogenic activity of SULT2B1b has been reported (7), our results represent the first report that CS also inhibits lipogenesis. The antilipogenic activity of SULT2B1b was also associated with an increased level of circulating CS. Cholesterol sulfate has been reported as a natural agonist of RORα (17). Indeed, we showed that hepatic expression of RORα target genes, such as the Cyp7b1, Bmal1, and Ikk genes, was induced in TG mice (data not shown). We have reported previously that RORα can trans suppress the lipogenic activity of LXR (40). Based on these observations, it is tempting to speculate that increased production of CS and activation of RORα may represent a novel mechanism by which SULT2B1b inhibits lipogenesis. RORα is also known for its anti-inflammatory activity (41). The activation of RORα was consistent with our observation that inflammation was suppressed in the WAT and livers of TG mice. Future studies using RORα-null mice will help to further define the role of RORα in the metabolic benefits of CS and SULT2B1b.
Although most of the metabolic benefits of the transgene observed in the DIO model and in ob/ob mice were consistent with each other, one obvious discrepancy is that the transgene inhibited obesity in the DIO model but had little effect on the body weight and body composition of ob/ob mice. In addition, although lipogenic gene expression was suppressed in ob-TG mice, we did not observe significant relief of hepatic steatosis in this genotype. An interesting observation was that the HFD-fed TG mice showed increased expression of leptin in WAT and increased serum leptin levels despite having less fat mass. Leptin is a key regulator of energy homeostasis. Chronic leptin administration causes suppression of food intake, downregulation of hepatic lipogenic gene expression, regulation of peripheral lipid metabolism, and promotion of fatty acid oxidation (42). Indeed, food intake was suppressed and energy expenditure was increased in HFD-fed TG mice. In contrast, ob/ob mice are leptin deficient, and we did not observe differences in food intake, body weight, oxygen consumption, and hepatic steatosis between ob/ob and ob-TG mice (data not shown). Although the mechanism by which SULT2B1b induces the expression of leptin remains to be defined, our results suggested that leptin is a potential effector of SULT2B1b in inhibiting obesity and improving metabolic function.
In HFD-fed TG mice, we also observed significant decreases in bile acid levels in the liver and serum (Table 1). We cannot exclude the possibility that the metabolic benefit of the transgene was also been contributed by bile acid-sensing nuclear receptors, such as the farnesoid X receptor (FXR), pregnane X receptor (PXR), and vitamin D receptor (VDR) (43).
In summary, our study established CS as an important metabolic regulator in controlling glucose metabolism and energy homeostasis, pointing to CS as a potential therapeutic agent and to SULT2B1b as a potential therapeutic target for metabolic disorders.
ACKNOWLEDGMENTS
This study was supported in part by NIH grants DK083953 and HD073070 (to W.X.). W.X. is the Joseph Koslow Endowed Chair in Pharmaceutical Sciences at University of Pittsburgh School of Pharmacy. Normal human hepatocytes were obtained through the Liver Tissue Procurement and Distribution System, Pittsburgh, PA, which was funded by NIH contract N01-DK-7-0004/HHSN267200700004C.
We thank Yanqiao Zhang for the adenovirus expressing shHNF4α, Stephen A. Duncan for the lentivirus expressing HNF4α RNAi, Richard M. O'Brien for the G6pase luciferase reporter gene, and Henry Dong (University of Pittsburgh) for the FoxO1-expressing vector.
Footnotes
Published ahead of print 25 November 2013
This paper is dedicated to the memory of Victor A. Fung.
REFERENCES
- 1.Strott CA. 2002. Sulfonation and molecular action. Endocr. Rev. 23:703–732. 10.1210/er.2001-0040 [DOI] [PubMed] [Google Scholar]
- 2.Gong HB, Guo P, Zhai Y, Zhou J, Uppal H, Jarzynka MJ, Song WC, Cheng SY, Xie W. 2007. Estrogen deprivation and inhibition of breast cancer growth in vivo through activation of the orphan nuclear receptor liver X receptor. Mol. Endocrinol. 21:1781–1790. 10.1210/me.2007-0187 [DOI] [PubMed] [Google Scholar]
- 3.Lee JH, Gong H, Khadem S, Lu Y, Gao X, Li S, Zhang J, Xie W. 2008. Androgen deprivation by activating the liver X receptor. Endocrinology 149:3778–3788. 10.1210/en.2007-1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang B, Cheng QQ, Ou ZM, Lee JH, Xu MS, Kochhar U, Ren SR, Huang M, Pflug BR, Xie W. 2010. Pregnane X receptor as a therapeutic target to inhibit androgen activity. Endocrinology 151:5721–5729. 10.1210/en.2010-0708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visser TJ. 1994. Role of sulfation in thyroid hormone metabolism. Chem. Biol. Interact. 92:293–303. 10.1016/0009-2797(94)90071-X [DOI] [PubMed] [Google Scholar]
- 6.Saini SPS, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren SR, Moore DD, Evans RM, Xie W. 2004. A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol. Pharmacol. 65:292–300. 10.1124/mol.65.2.292 [DOI] [PubMed] [Google Scholar]
- 7.Bai Q, Zhang X, Xu L, Kakiyama G, Heuman D, Sanyal A, Pandak WM, Yin L, Xie W, Ren S. 2012. Oxysterol sulfation by cytosolic sulfotransferase suppresses liver X receptor/sterol regulatory element binding protein-1c signaling pathway and reduces serum and hepatic lipids in mouse models of nonalcoholic fatty liver disease. Metabolism 61:836–845. 10.1016/j.metabol.2011.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Javitt NB, Lee YC, Shimizu C, Fuda H, Strott CA. 2001. Cholesterol and hydroxycholesterol sulfotransferases: identification, distinction from dehydroepiandrosterone sulfotransferase, and differential tissue expression. Endocrinology 142:2978–2984. 10.1210/en.142.7.2978 [DOI] [PubMed] [Google Scholar]
- 9.Shimizu C, Fuda H, Yanai H, Strott CA. 2003. Conservation of the hydroxysteroid sulfotransferase SULT2B1 gene structure in the mouse: pre- and postnatal expression, kinetic analysis of isoforms, and comparison with prototypical SULT2A1. Endocrinology 144:1186–1193. 10.1210/en.2002-221011 [DOI] [PubMed] [Google Scholar]
- 10.Fuda H, Javitt NB, Mitamura K, Ikegawa S, Strott CA. 2007. Oxysterols are substrates for cholesterol sulfotransferase. J. Lipid Res. 48:1343–1352. 10.1194/jlr.M700018-JLR200 [DOI] [PubMed] [Google Scholar]
- 11.Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou HP, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ. 2012. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 15:665–674. 10.1016/j.cmet.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strott CA, Higashi Y. 2003. Cholesterol sulfate in human physiology: what's it all about? J. Lipid Res. 44:1268–1278. 10.1194/jlr.R300005-JLR200 [DOI] [PubMed] [Google Scholar]
- 13.Dong B, Saha PK, Huang WD, Chen WL, Abu-Elheiga LA, Wakil SJ, Stevens RD, Ilkayeva O, Newgard CB, Chan L, Moore DD. 2009. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. U. S. A. 106:18831–18836. 10.1073/pnas.0909731106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamasawa N, Tamasawa A, Takebe K. 1993. Higher levels of plasma cholesterol sulfate in patients with liver cirrhosis and hypercholesterolemia. Lipids 28:833–836. 10.1007/BF02536238 [DOI] [PubMed] [Google Scholar]
- 15.Veares MP, Evershed RP, Prescott MC, Goad LJ. 1990. Quantitative determination of cholesterol sulphate in plasma by stable isotope dilution fast atom bombardment mass spectrometry. Biomed. Environ. Mass Spectrom. 19:583–588. 10.1002/bms.1200191002 [DOI] [PubMed] [Google Scholar]
- 16.Drayer NM, Lieberman S. 1967. Isolation of cholesterol sulfate from human aortas and adrenal tumors. J. Clin. Endocrinol. Metab. 27:136–139. 10.1210/jcem-27-1-136 [DOI] [PubMed] [Google Scholar]
- 17.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. 2004. Crystal structure of the human RORα ligand binding domain in complex with cholesterol sulfate at 2.2 Å. J. Biol. Chem. 279:14033–14038. 10.1074/jbc.M400302200 [DOI] [PubMed] [Google Scholar]
- 18.Seneff S, Davidson R, Mascitelli L. 2012. Might cholesterol sulfate deficiency contribute to the development of autistic spectrum disorder? Med. Hypotheses 78:213–217. 10.1016/j.mehy.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto K, Miyazaki K, Higashi S. 2010. Cholesterol sulfate alters substrate preference of matrix metalloproteinase-7 and promotes degradations of pericellular laminin-332 and fibronectin. J. Biol. Chem. 285:28862–28873. 10.1074/jbc.M110.136994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson AM, Olefsky JM. 2013. The origins and drivers of insulin resistance. Cell 152:673–684. 10.1016/j.cell.2013.01.041 [DOI] [PubMed] [Google Scholar]
- 21.DeFronzo RA. 2004. Pathogenesis of type 2 diabetes mellitus. Med. Clin. North Am. 88:787–835. 10.1016/j.mcna.2004.04.013 [DOI] [PubMed] [Google Scholar]
- 22.DeLaForest A, Nagaoka M, Si-Tayeb K, Noto FK, Konopka G, Battle MA, Duncan SA. 2011. HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development 138:4143–4153. 10.1242/dev.062547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu K, Driscoll WJ, Koh YC, Strott CA. 1994. A P-loop related motif (GxxGxxK) highly conserved in sulfotransferases is required for binding the activated sulfate donor. Biochem. Biophys. Res. Commun. 204:1178–1185. 10.1006/bbrc.1994.2587 [DOI] [PubMed] [Google Scholar]
- 24.Soutoglou E, Katrakili N, Talianidis I. 2000. Acetylation regulates transcription factor activity at multiple levels. Mol. Cell 5:745–751. 10.1016/S1097-2765(00)80253-1 [DOI] [PubMed] [Google Scholar]
- 25.Zhou J, Zhai YG, Mu Y, Gong HB, Uppal H, Toma D, Ren SR, Evans RM, Xie W. 2006. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 281:15013–15020. 10.1074/jbc.M511116200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song YK, Liu F, Zhang G, Liu D. 2002. Hydrodynamics-based transfection: simple and efficient method for introducing and expressing transgenes in animals by intravenous injection of DNA. Methods Enzymol. 346:92–105. 10.1016/S0076-6879(02)46050-8 [DOI] [PubMed] [Google Scholar]
- 27.Yin L, Ma H, Ge X, Edwards PA, Zhang Y. 2011. Hepatic hepatocyte nuclear factor 4α is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler. Thromb. Vasc. Biol. 31:328–336. 10.1161/ATVBAHA.110.217828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J, He J, Shi X, Stefanovic-Racic M, Xu M, O'Doherty RM, Garcia-Ocana A, Xie W. 2012. Sex-specific effect of estrogen sulfotransferase on mouse models of type 2 diabetes. Diabetes 61:1543–1551. 10.2337/db11-1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He J, Hu B, Shi X, Weidert ER, Lu P, Xu M, Huang M, Kelley EE, Xie W. 2013. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 33:2047–2055. 10.1128/MCB.01658-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wada T, Ihunnah CA, Gao J, Chai X, Zeng S, Philips BJ, Rubin JP, Marra KG, Xie W. 2011. Estrogen sulfotransferase inhibits adipocyte differentiation. Mol. Endocrinol. 25:1612–1623. 10.1210/me.2011-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wada T, Gao J, Xie W. 2009. PXR and CAR in energy metabolism. Trends Endocrinol. Metab. 20:273–279. 10.1016/j.tem.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 32.Kim DH, Perdomo G, Zhang T, Slusher S, Lee S, Phillips BE, Fan Y, Giannoukakis N, Gramignoli R, Strom S, Ringquist S, Dong HH. 2011. FoxO6 integrates insulin signaling with gluconeogenesis in the liver. Diabetes 60:2763–2774. 10.2337/db11-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sone H, Shimano H, Sakakura Y, Inoue N, Amemiya-Kudo M, Yahagi N, Osawa M, Suzuki H, Yokoo T, Takahashi A, Iida K, Toyoshima H, Iwama A, Yamada N. 2002. Acetyl-coenzyme A synthetase is a lipogenic enzyme controlled by SREBP-1 and energy status. Am. J. Physiol. Endocrinol. Metab. 282:E222–E230. 10.1152/ajpendo.00189.2001 [DOI] [PubMed] [Google Scholar]
- 34. Reference deleted.
- 35.Dietschy JM. 1984. Regulation of cholesterol metabolism in man and in other species. Klin. Wochenschr. 62:338–345. 10.1007/BF01716251 [DOI] [PubMed] [Google Scholar]
- 36.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. 2001. Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21:1393–1403. 10.1128/MCB.21.4.1393-1403.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luong A, Hannah VC, Brown MS, Goldstein JL. 2000. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 275:26458–26466. 10.1074/jbc.M004160200 [DOI] [PubMed] [Google Scholar]
- 38.Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. 2007. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 5:73–79. 10.1016/j.cmet.2006.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu L, Bai Q, Rodriguez-Agudo D, Hylemon PB, Heuman DM, Pandak WM, Ren S. 2010. Regulation of hepatocyte lipid metabolism and inflammatory response by 25-hydroxycholesterol and 25-hydroxycholesterol-3-sulfate. Lipids 45:821–832. 10.1007/s11745-010-3451-y [DOI] [PubMed] [Google Scholar]
- 40.Wada T, Kang HS, Jetten AM, Xie W. 2008. The emerging role of nuclear receptor RORα and its crosstalk with LXR in xeno- and endobiotic gene regulation. Exp. Biol. Med. (Maywood) 233:1191–1201. 10.3181/0802-MR-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delerive P, Monte D, Dubois G, Trottein F, Fruchart-Najib J, Mariani J, Fruchart J-C, Staels B. 2001. The orphan nuclear receptor RORα is a negative regulator of the inflammatory response. EMBO Rep. 2:42–48. 10.1093/embo-reports/kve007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klok MD, Jakobsdottir S, Drent ML. 2007. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: a review. Obes. Rev. 8:21–34. 10.1111/j.1467-789X.2006.00270.x [DOI] [PubMed] [Google Scholar]
- 43.Li T, Chiang JY. 2012. Bile acid signaling in liver metabolism and diseases. J. Lipids 2012:754067. 10.1155/2012/754067 [DOI] [PMC free article] [PubMed] [Google Scholar]